

Abstract
Purpose:
Approved therapies for EGFR exon 20, HER2 mutations, and NRG1 fusions are currently lacking for non-small cell lung cancer and other cancers. Tarloxotinib is a prodrug that harnesses tumor hypoxia to generate high levels of a potent, covalent pan-HER tyrosine kinase inhibitor, tarloxotinib-E, within the tumor microenvironment. This tumor-selective delivery mechanism was designed to minimize the dose limiting toxicities that are characteristic of systemic inhibition of wild type EGFR.
Experimental Design:
Novel and existing patient-derived cell lines and xenografts harboring EGFR exon 20 insertion mutations, ERBB2 (HER2) mutations and amplification, and NRG1 fusions were tested in vitro and in vivo with tarloxotinib to determine its impact on cancer cell proliferation, apoptosis and cell signaling.
Results:
Tarloxotinib-E inhibited cell signaling and proliferation in patient-derived cancer models in vitro by directly inhibiting phosphorylation and activation of EGFR, HER2, and HER2/HER3 heterodimers. In vivo, tarloxotinib induced tumor regression or growth inhibition in multiple murine xenograft models. Pharmacokinetic analysis confirmed markedly higher levels of tarloxotinib-E in tumor tissue than plasma or skin. Finally, a patient with lung adenocarcinoma harboring a HER2 exon 20 p.A775_G776insYVMA mutation demonstrated a dramatic clinical response to tarloxotinib.
Conclusion:
Experimental data with tarloxotinib validate the novel mechanism of action of a hypoxia-activated prodrug in cancer models by concentrating active drug in the tumor vs. normal tissue and this activity can translate into clinical activity in patients.
Introduction
Members of the ErbB family of receptor tyrosine kinases have long been implicated as oncogenes in numerous cancer types. Several studies have delineated the proliferation signals generated by ErbB receptors within cancer cells (1). The ErbB family consists of four members: the epidermal growth factor receptor (EGFR or HER1), HER2 (ERBB2), HER3 (ERBB3), and HER4 (ERBB4). Under physiologic conditions, ligand binding induces receptor hetero- or homodimerization which initiates a signaling network that includes the Ras/Raf/MAPK and the phosphatidylinositol 3-kinase (PI3K)/Akt pathways, which are key intracellular pathways that govern fundamental cellular processes including proliferation, cell migration, metabolism, and survival (2).
EGFR activating mutations occur in a range of 10–40% of non-small cell lung cancer (NSCLC) (3), of these ~85–90% are in-frame deletions in exon 19 or L858R. These mutations are responsive to first (erlotinib and gefitinib), second (afatinib and dacomitinib) and third (osimertinib) generation EGFR tyrosine kinase inhibitors (TKIs), which are US FDA approved for these mutations (4–8). EGFR exon 20 insertions are also activating and account for 5–9% of EGFR mutations driving NSCLC (9,10). EGFR exon 20 insertion mutations represent a combination of in-frame insertions and/or duplications of 3 to 21 base pairs, predominantly clustered between codon 763 and 774 (10).
Approved EGFR TKIs have a wide therapeutic window for EGFR mutations such as L858R, endowed by a higher affinity of the drug combined with a lower affinity for ATP for the mutant receptor (11). In contrast, EGFR exon 20 insertion mutations, with the exception of EGFR p.A763_Y764insFQEA, show similar affinity to both EGFR TKIs and ATP as does wild-type (WT) EGFR (9,12). As a result of this disparate biochemistry, approved EGFR tyrosine kinase inhibitors, including potent, covalent, pan-HER inhibitors such as afatinib, have little activity against EGFR exon 20 mutations (13,14). The doses of standard EGFR TKIs that could effectively inhibit EGFR exon 20 are likely to cause significant toxicity related to inhibition of WT EGFR, narrowing the therapeutic window and limiting effective drug exposure in patients. There are currently no approved therapies for EGFR exon 20 insertions.
EGFR is not the only ErbB-related gene that has oncogenic alterations with a limited therapeutic window, ERBB2 and NRG1 have proven difficult to target in NSCLC. ERBB2 mutations, most commonly insertions in exon 20 similar to EGFR and gene amplification occur in NSCLC but respond poorly to pan-HER TKIs such as dacomitinib and others, likely as a consequence of dose-limiting inhibition of WT EGFR (15,16). Gene fusions involving neuregulin 1 (NRG1) represent a recently identified class of oncogenes first identified in NSCLC (17), but now recognized to occur across numerous tumor types including breast, ovarian, and pancreatic cancers (18). Neuregulin 1 is one of the heregulin family growth factors that binds HER3 and induces dimerization, frequently with HER2, followed by transphosphorylation of the receptors and the activation of downstream signaling pathways (17,19). Currently there are no approved therapies for ERBB2 oncogenic alterations in NSCLC or for tumors bearing NRG1 fusions.
Based on a lack of available clinical strategies for EGFR exon 20 insertions and HER2 alterations in NSCLC as well as NRG1 fusions, we sought to exploit a novel mechanism of action to generate a favorable therapeutic window in patients where EGFR toxicity may be limiting for standard therapeutic approaches. Tarloxotinib (named for targeting low oxygen) was designed as a hypoxia-activated prodrug (Supplementary Figure S1) that bears a permanent positive charge rendering it less able to transit into cells to interact with the kinase domain of EGFR, HER2 and HER4. Tarloxotinib, once reduced by a single electron to the nitro radical anion intermediate, can act as a direct oxygen sensor. In the presence of physiological concentrations of oxygen, back-oxidation of the nitro radical anion occurs to regenerate the intact prodrug and superoxide in a futile redox cycle. However, when oxygen concentrations are low (hypoxia), the nitro radical anion is sufficiently long-lived to fragment, releasing a potent, irreversible pan-ErbB (EGFR, HER2, and HER4) TKI referred to as tarloxotinib-effector (tarloxotinib-E; Supplementary Figure S1) (20,21). Hypoxia is common to most tumors and has been correlated with tumor progression, resistance to therapy and poor clinical outcome (22). In NSCLC, most target lesions were demonstrated to be hypoxic using the hypoxia PET imaging radiotracer 18F-HX4 (23). Prior work demonstrates that the prodrug tarloxotinib has very weak inhibitory activity against WT EGFR in cells (24) but can be converted to the active metabolite tarloxotinib-E in a hypoxic tumor environment. This mechanism would generate a therapeutic window by achieving inhibitory doses of the pan-HER inhibitor with activity against both WT and mutant forms of EGFR, HER2 and/or HER4 in the tumor microenvironment while sparing oxygenated normal tissues from WT EGFR inhibition (20). Prior phase 1 clinical trials established the maximum tolerated dose as 150 mg/m2 once weekly via intravenous administration (25).
In this work, we demonstrate the preclinical activity of tarloxotinib in vitro and in vivo using novel patient-derived tumor models harboring oncogenic EGFR exon 20 insertion mutations as well as cancer models with ERBB2 alterations and NRG1 fusions. We also describe the dramatic tumor response in a patient with NSCLC enrolled on the phase II trial of tarloxotinib (NCT03805841) whose tumor harbors an ERBB2 p.Y772_A775dup (YVMA) insertion mutation, validating that the novel mechanism of action of the hypoxia-activated prodrug translates into clinical activity in patients.
Material and Methods
Cell lines and Reagents
Supplementary Table 1 summarizes the cell lines and their respective oncogenes. H1781, Calu-3, H2170, A431 and H661 cell lines were acquired from the UC Denver Tissue Culture Core. MDA-MB-175vIII (ATCC HTB-25) were purchased from ATCC. Cell lines were validated by fingerprinting and mycoplasma tested. All cell lines were cultured in RPMI-1640 supplemented with 10% FBS. Gefitinib and afatinib were purchased from Selleck Chemicals (Houston, TX). Tarloxotinib and tarloxotinib-E were designed and synthesized by Jeff Smaill and Adam Patterson (Auckland Cancer Society Research Center; Auckland, New Zealand) and provided by Rain Therapeutics. Antibodies used were as follows: AKT pS437 (4058), total AKT (2920), ERK pT202/Y204 (9101), total ERK (9107), EGFR pY1068 (2234), total EGFR (2232), HER2 Y1221 (2243), HER3 Y1222 (4784) all from Cell Signaling Technologies. Total EGFR (610017) and total HER2 (610161) from BD Transduction Laboratories. Total HER3 and GAPDH (MAB374) from EMD Millipore.
Cell Line Derivation
Written informed consent was obtained from the patients prior to collection of the patients’ tumor samples. The consent form and protocol were reviewed and approved by the Colorado Multiple Institutional Review Board. The patient-derived EGFR exon20 insertion cell lines presented here originated from malignant pleural effusions or ascites fluid. Total cells were isolated from the patient’s fluid by centrifugation followed by lyses of the red blood cell via a hypotonic solution from Lonza ACK Lysing Buffer (Walkerville,MD). The remaining nucleated cells were then cultured overnight in RPMI-1640 supplemented with 10% FBS in tissue culture dishes to initially select out any adherent stromal cells. The non-adherent cell fraction was further enriched for tumor cells by subtracting out any immune cells using a CD45 magnetic bead depletion kit (Stem Cell Technology, Cambridge, MA). The refined tumor cell mixture was then cultured in RPMI-1640 with 10%FBS until outgrowth of tumor cells. Tumor cells containing the patient tumor sample matched EGFR Exon 20 insertion were confirmed via custom-capture, targeted NGS (38). Cell lines were designated CUTO14 (CU Thoracic Oncology), CUTO17 and CUTO18. The patient from which CUTO14 was derived was previously treated with carboplatin and pemetrexed chemotherapy only (no prior EGFR-directed therapies). The patient from which CUTO17 was derived was previously treated with cisplatin/pemetrexed, pembrolizumab, then a low dose of an investigational EGFR TKI on a phase 1 clinical trial for less than one month. The patient from which CUTO18 was derived was previously treated with erlotinib and an investigational agent on a clinical trial (best response of disease progression), carboplatin and pemetrexed, then a low dose of an investigational EGFR TKI on a phase 1 clinical trial for less than one month.
Lentivirus shRNA production and cell transduction
Production of lentivirus was performed by co-transfecting pCMV-VSV-G and pCMVΔR8.2 into 293T cells along with Non-Targeted shRNA Control (SHC002), or two EGFR shRNA (Functional Genomics Facility, University of Colorado; Aurora, CO) using Mirus TransIT-293 reagent according to the manufacturer’s protocol. Viral supernatants were collected 48hrs after transfection and added to different cell lines.
Immunoblotting
Cells were lysed in T-PER Tissue Protein Extraction Reagent (ThermoScientific, MA) supplemented with Halt Protease and Phosphatase Inhibitor Cocktail. Proteins were resolved by SDS-PAGE and analyzed by Western blot using the indicated primary antibodies. Protein detection was achieved by imagining with an Odyssey Imager and Image Studio software (LI-COR Biotechnology).
Kinase profiling
Kinase profiling of tarloxotinib-E was performed by Reaction Biology (Malvern, PA). Tarloxotinib-E was tested against a panel of kinases in 10-dose IC50 duplicate mode with a 4-fold serial dilution starting at 100 uM. Reactions were carried out with 33P-ATP for 2 hrs. The remaining radioactive phosphorylated substrate was measured. IC50 values were obtained using Prism Software (GraphPad).
Proliferation Assays
Proliferation for the EGFR shRNA experiments was evaluated using the IncuCyte Live-cell Analysis System. Briefly, CUTO14, CUTO17, and CUTO18, cells were plated in a 96 well, infected with the above lentiviral vectors and the plate was transferred to the IncuCyte. The IncuCyte system enables automated quantification of cell proliferation by automatically gathering and analyzing images through time. Growth curves using the IncuCyte were generated by confluence imaging every four hours by triplicate for a total of 72 hours. Data was analyzed using IncuCyte Zoom software to evaluate proliferation base on the percentage of confluency per well.
Proliferation assays to measure drug effect were performed in media supplemented with 10% FBS. Cell were seeded at a density of 2 × 103 cells per well in a 96-well plate and treated with the indicated doses of inhibitors for 72hrs. MTS assays were performed according to the manufacturer’s instructions (CellTiter 96 Aqueous One Solution Cell Proliferation Assay, Promega). Each experiment was performed in triplicate and repeated at least three times. Data was analyzed in Prism software (GraphPad).
Apoptosis assays
The IncuCyte caspase 3/7 green apoptosis assay reagent was employed to detect apoptosis in real time. Cell were seeded at a density of 2 × 103 cells per well in a 96-well plate, the following day cells were treated with the indicated doses of inhibitors and the apoptosis reagent. Plates were immediately placed in the IncuCyte System, after 30 min to allow the plate to warm scanning was started. Pictures were taken every 4 hours for a total of 4 days. The analysis of the image was done using the IncuCyte software.
Xenograft studies
All studies involving animals were approved by the Institutional Animal Care and Use Committee Office of the University of Colorado Anschutz Medical Campus. Cells were re-suspended in 1:1 (v/v) media and matrigel for subcutaneous flank implantation (3 × 106 cells/0.1 ml). Tumor growth was monitored once a week by bilateral caliper measurements; once tumors reached 0.15–0.25 cm3, mice were randomized into vehicle or treatment groups (7–10 mice per group, with two flank tumors each mouse). Dose regimens were as follows: afatinib, dosed 6mg/kg once daily by oral gavage for 4 weeks; tarloxotinib, dosed 48 mg/kg or 26 mg/kg once a week by intraperitoneal injection for 4 weeks and vehicle (20% v/v 2-hydroxypropyl-Beta-cyclodextrin). Tumor growth inhibition from start of treatment was assessed by comparison of the mean change in tumor volume for the control and treated groups. Statistical significance was evaluated using an ANOVA test (Graph Pad Prism). Mice were weighed at least once a week during treatment and percent of weight change was calculated.
Immunofluorescence and quantification
Slides were de-paraffinized in xylene and rehydrated in graded concentrations of ethanol before antigen retrieval (EnVision FLEX Target retrieval solution high pH) in a pressure cooker at 121°F for 10 minutes. Next, the slides were cooled for 20 minutes before washing in TBS-0.1%Tween. They were then treated with Duolink blocking solution (Sigma) for 30 minutes at 37oC. The samples were incubated with primary antibodies: hypoxyprobe kit Mab1 antibody and cytokeratin (abcam ab217916) overnight at 4oC followed by fluorescent tag secondary antibodies (Biotium, 488 donkey anti-mouse and Invitrogen alexa 647) plus DAPI (Thermofisher) for 1 hour at room temperature. Slides were then washed and mounted (Fluoroshield mountain media, Abcam).
Slides were analyzed using an IX83 Olympus microscope, pictures were taken at 20X and quantification was done using cellSens (Olympus) and ImageJ software. Each tumor image was separated in channels (red:cytokeratin, green:hypoxia and blue:DAPI). Area of each channel was measured, and percent was calculated based on the total area of the tumor. Data is presented as average ± SEM.
Pharmacokinetics studies
Tarloxotinib (48 mg/kg) was administered by intraperitoneal injection once. At the indicated times after the dose mice were euthanized by CO2 inhalation and cervical dislocation. Blood samples were obtained directly from the heart, heparinized blood was immediately centrifuged at 800g for 10 minutes. The supernatant layer of the blood was collected as plasma.
For tumor tissue was also collected at the indicated times. Tumor tissue was excised, divided in half by scalpel and one half was snap frozen in liquid nitrogen while the second was fixed in formalin for other studies. Tissue in liquid nitrogen was used for analysis. Here, plasma (10 μL) or frozen tissue (~100 mg) was mixed with 4 volumes of ice-cold acetonitrile containing deuterated internal standards (ISDs). Tissue samples were subject to additional steps of homogenisation (Tissuelyser II, Qiagen) and extraction (Heidolph Multi Reax, Germany). All samples were centrifuged and clear supernatants mixed (1:2) with 45mM ammonium formate buffer pH 4.5 before loading (Agilent 1100 autosampler) onto LC system (5μm Zorbax SB-C18 column, Agilent) with mobile phase 0.01% formic acid in 80% acetonitrile-20% water, v/v (solvent A), and 0.01% formic acid in water (solvent B); linear gradient with 9 min run time. Mass spectrometric detection (Agilent 6410 triple quadrupole) utilized positive electrospray ionization parameters optimized for positively charged ions representing the ([M+H]+) for tarloxotinib, tarloxotinib-E, and their respective ISDs. Agilent MassHunter software (v.4.04.00) was used for data acquisition and chromatographic peak integration with standard curves (0.005 μmol/L to 30 μmol/L) prepared in mouse plasma to estimate the concentration of tarloxotinib, tarloxotinib-E in the samples.
Mice were given 6 mg/kg of afatinib by oral gavage and blood was collected at the indicated times following the protocol previously described. Analysis of the plasma was done by the CU Cancer Center Pharmacology Shared Resource (PharmSR).
Clinical Trial
NCT03805841 is an ongoing multi-center phase 2 study evaluating the efficacy of tarloxotinib in selected patients with oncogenic alterations in EGFR, HER2, HER4 or NRG1 and metastatic or advanced NSCLC or other solid tumors. The study is approved by Institutional Review Boards at all institutions that enroll patients, and eligible patients provided written informed consent to participate. The study is sponsored by Rain Therapeutics and is conducted in accordance with the in compliance with Good Clinical Practice (GCP) and with the Declaration of Helsinki.
Results
Novel in vitro models of EGFR exon20 insertions
A goal of this study was to determine whether tarloxotinib can overcome clinical resistance of several oncogene classes including EGFR exon 20 insertion mutations, but the lack of preclinical models, such as patient-derived cell lines or mouse models has hindered this effort. To address this need, we successfully derived and maintained three unique cancer cell lines bearing EGFR exon 20 insertions. The three cell lines were confirmed to have 3 different exon 20 insertion mutations: EGFR p.A767_V769dupASV (CUTO14), p.N771_773dupNPH (CUTO17), and p.S768_D770dupSVD (CUTO18) (Supplementary Table S1). The mutations in each cell line matched the known mutation from the patients’ tumors obtained by clinical sequencing (not shown). There is significant diversity in EGFR exon 20 insertion mutations (Supplementary Figure S2A), but these three EGFR exons 20 insertion mutations represent the most common in NSCLC, accounting for up to 50% of all EGFR exon 20 insertion mutations (10).
These patient-derived cell line models allowed us to directly test whether cellular proliferation and survival is dependent on these EGFR oncogenes. To do so, we introduced shRNAs to decrease EGFR expression and evaluate cell proliferation. We confirmed the reduction of protein expression by performing immunoblots of EGFR (Supplemental Figure S2B). EGFR knockdown in CUTO14, CUTO17, and CUTO18 resulted in a reduction in proliferation compared to non-targeted controls (NTC) for each of the EGFR exon 20 cell lines, but not an EML4-ALK+ cell line (Supplemental Figure S2C).
Tarloxotinib-E but not tarloxotinib is a potent inhibitor of EGFR and HER2
First, we wanted to test the potency of the active form of the prodrug, tarloxotinib-E, in cancer models bearing oncogenic alterations in the ERBB gene family under normal oxygen conditions. To verify the on-target effect of tarloxotinib-E, we evaluated EGFR phosphorylation (pEGFR) in cancer cells bearing EGFR exon 20 insertions after treatment with tarloxotinib-E. Our results show that in the cell lines CUTO14, CUTO17, and CUTO18, tarloxotinib-E inhibits phosphorylation of the receptor at relatively low concentrations (Fig. 1A and Supplementary Fig. S3A and B). Gefitinib (a 1st generation inhibitor), and osimertinib (a 3rd generation inhibitor) showed inhibition at higher concentrations. Afatinib (a 2nd generation inhibitor) had a similar effect to tarloxotinib-E, which was anticipated given the structural similarities. When we analyzed signaling pathways commonly activated by EGFR, we observed that tarloxotinib-E inhibited the phosphorylation of ERK1/2 (pERK1/2) and AKT (pAKT) at similar concentrations to pEGFR inhibition, consistent with the role of EGFR exon 20 insertions as a dominant oncogene driver in these cells that directly activates canonical cancer signaling MAPK and AKT pathways (Fig. 1A and Supplementary Fig. S3A and B). Similarly, gefitinib, afatinib, and osimertinib inhibited pERK1/2 and pAKT at concentrations that inhibited pEGFR, but generally required higher concentrations than tarloxotinib-E.
Figure 1. Tarloxotinib-E inhibits ErbB family member phosphorylation and downstream signaling in EGFR-, HER2- or NRG1-driven cell lines.

Cells were treated with the indicated doses (nmo/L) of afatinib, gefitinib, osimertinib or tarloxotinib-E (active drug) for 2 hours, lysed and analyzed by immunoblot for the indicated proteins. A) CUTO14 cell line (EGFR exon 20 insertion mutation). B) H1781 (HER2 exon 20 insertion mutation). C) MDA-MB-175vII (NRG1 fusion cell line). Representative images of blots are shown. Experiments were performed in triplicate.
We also treated NSCLC cell lines with HER2 alterations including H1781 (ERBB2 p. G776Ins V_G/C), CALU-3 and H2170 (both with ERBB2 gene amplification) with tarloxotinib-E (26–28). These models also demonstrated inhibition of pHER2 with tarloxotinib-E at lower doses than gefitinib, afatinib, and osimertinib (Fig. 1B and Supplementary Fig. 3C and D). Notably, there was a clear reduction of pAKT at similar concentrations required for pHER2 inhibition, but not pERK1/2, with all of the inhibitors tested.
NRG1 gene fusions have recently been identified as oncogenic drivers in NSCLC (17) and other cancer types (18). We therefore tested whether tarloxotinib-E could effectively inhibit signaling in the breast cancer cell line MDA-MB-175vIII, which harbors a DOC4-NRG1 gene fusion (29). Tarloxotinib-E effectively inhibited phosphorylation of both HER2 and HER3 at a concentration similar to afatinib but at lower doses than gefitinib and osimertinib (Fig. 1C). Inhibition of pERK1/2 and pAKT was achieved at similar concentrations required to inhibit pHER2/pHER3 suggesting that these signaling pathways are directly linked.
The prodrug, tarloxotinib, was engineered to be far less effective against EGFR, HER2 and HER4 in cells to spare any on-target activity in non-tumor tissues. In A431 cells, which express WT EGFR, the IC50 for inhibition of pEGFR was 2 nmol/L for tarloxotinib-E compared to 201 nmol/L for tarloxotinib demonstrating an approximate 100-fold difference in potency (Supplementary Fig. S4A). In the presence of ligand (EGF), the IC50 of EGFR phosphorylation was 8 nmol/L. Similarly, in Calu-3 cells which express WT HER2, the IC50 for inhibition of pHER2 was 18 nmol/L for tarloxotinib-E compared to nearly 3 μmol/L for tarloxotinib showing a near 155-fold difference in potency. Accordingly, significantly higher levels of tarloxotinib were also required to inhibit pEGFR in CUTO14 cells and pHER2 or pHER3 in H1781, Calu-3 and MDA-MB-175vIII cells (Supplementary Fig. S4B). In vitro kinase data (Supplementary Table 2) demonstrates that tarloxotinib-E is also a potent inhibitor of HER4, therefore we tested tarloxotinib-E and tarloxotinib in H661 cells which express high levels of HER4. Tarloxotinib-E inhibited phosphorylation of HER4 at approximately 10 nmol/L, which correlated with a reduction of ERK1/2 phosphorylation. Gefitinib, afatinib and osimertinib reduced phosphorylation at higher concentrations compared to tarloxotinib-E (Supplementary Fig. S4C).
Tarloxotinib-E but not tarloxotinib is a potent inhibitor of cellular proliferation and inducer of apoptosis under normoxic conditions
Following demonstration that tarloxotinib-E inhibits EGFR and HER2 (and HER3) phosphorylation in a variety of oncogene-driven models, we then evaluated whether tarloxotinib-E inhibits cell proliferation in these cell lines. The three EGFR exon 20 insertion mutation cell lines CUTO14/17/18 demonstrated increased sensitivity to tarloxotinib-E compared to gefitinib, afatinib and osimertinib (Fig. 2A, and Table 1). The prodrug tarloxotinib, however, has significantly diminished activity on these cell lines under normal oxygen conditions and only inhibited proliferation at very high concentrations, in the range of 3–10μM (Fig. 2A, and Table 1). On average, the prodrug was ~65 times less potent than the active drug consistent with the intentional design of tarloxotinib (Fig. 2A and Table 1).
Figure 2. Tarloxotinib-E inhibits proliferation of cell lines harboring EGFR, HER2, or NRG1 oncogenes.

Dose response curves of cell proliferation of A) CUTO14, CUTO17, and CUTO18 (EGFR ex20ins); B) H1781 (HER2 ex20ins), H2170 and Calu-3 (HER2 amp) and C) MDA-MB-175VIII (breast cancer, DOC4-NRG1 fusion) and H661 (HER4 overexpression). Cells were treated with tarloxotinib (prodrug) or tarloxotinib-E (active drug) for 72 hours and analyzed by MTS assay. Experiments were done in triplicate; mean ± SEM is plotted.
Table 1.
Summary of IC50 (nmol/L) values
| Cell Lines | Gefitinib | Afatinib | Osimertinib | Tarloxotinib | Tarloxotinib-E | Tarloxotinib/ Tartoxot nib-E fold difference |
|---|---|---|---|---|---|---|
| EGFR Ex20ins | ||||||
| CUT014 | 3741±5 | 110.9+30.2 | 303+15.2 | 4645±37.8 | 72.2+49.9 | 64.3 |
| CUT017 | 4197±6.6 | 219.7±20.6 | 426±21.5 | 3090±7.9 | 48.1 ±4.8 | 64.2 |
| CUT01S | >10000 | 841.3±5.8 | 647±6.7 | >10000 | 158,4±3.2 | 63.1 |
| HER2 | ||||||
| H17S1 (ex20ins) | 4168+5 | 66+2.5 | 406+47.2 | 816+13.9 | 15±1.6 | 54.4 |
| Calu-3 (amp) | 1324±7.8 | 31 ±11.4 | 188±35.2 | 3252.7±2.7 | 2 ±4 | 162.5 |
| H2170 (amp) | 1156±7.3 | 11±2.9 | 113±4.4 | 588±6.7 | 4±10.1 | 147 |
| NRG1 | ||||||
| MDA-MB-175VII | 404±6.7 | 1.2±3.1 | 37±6 9 | 307±4.2 | 0.3+3 | 1023.3 |
| HER4 | ||||||
| H661 | >10000 | 2S11±5.5 | 4518±2 | >10000 | 667±2 5 | 15 |
Similarly, when we evaluated the effects of tarloxotinib-E on the proliferation of HER2 altered cells, we also observed higher sensitivity compared to gefitinib, afatinib and osimertinib (Fig. 2B, and Table 1). Indeed, the effect of tarloxotinib-E was significantly more potent in the HER2 altered cell lines compared to the EGFR exon 20 cell lines. Tarloxotinib was up to 160 times less potent than tarloxotinib-E in the HER2 models (Fig. 2B, and Table 1). Similar analysis was performed using the NRG1 fusion model MDA-MB-175vIII and proliferation inhibition was achieved at less than 1 nmol/L with potency superior to the other EGFR/HER2 inhibitors (Fig. 2C and Table 1). Finally, analysis of the proliferation of HER4 cell line H661, show inhibition at 670 nmol/L. As in other models, tarloxotinib was far less potent, and all the other TKI evaluated required higher concentrations to inhibit proliferation (Fig. 2C, and Table 1).
We also assessed whether tarloxotinib-E induced apoptosis in cell lines harboring oncogenes that activate ErbB signaling pathways in addition to inhibiting cell proliferation. Cells were treated with 1 μmol/L of the indicated TKIs followed by measurement of caspase 3/7 activation. The EGFR and HER2 exon 20 mutant cells show an increase in caspase 3/7 activation after 12 hours of treatment with tarloxotinib-E and this persisted after 24 hours of treatment (Supplementary Fig. S5). Cells harboring the NRG1 fusion showed a modest effect at 12 hours, but a marked increase in caspase 3/7 after 24 hours or 48 hours of treatment with tarloxotinib-E.
Collectively, these data suggest that the active metabolite tarloxotinib-E is a potent inhibitor of cell proliferation and can induce apoptosis in cancer models harboring alterations that utilize the EGFR/HER2 (and HER3 signaling pathways) in vitro, whereas the prodrug has markedly diminished activity in these models under normoxic conditons.
Tarloxotinib inhibits tumor growth of ErbB-dependent xenograft models.
Prior work using plasma pharmacokinetic analyses of area under the curve (AUC0–24h) for free-drug had estimated the human equivalent dose (HED) of the recommended phase 2 dose of tarloxotinib (150 mg/m2 IV once weekly) used in two prior human clinical trials (NCT02449681 and NCT02454842) as well as the current, ongoing phase 2 clinical trial (NCT03805841) as 48 mg/kg intraperitoneal dosing once weekly in mice (20,24). To compare with a well-known pan-HER, we used afatinib. The FDA approved dose of afatinib is 40 mg (PO, once daily) providing a plasma AUCtotal for afatinib of 324 ng*hr/mL (30). The measured human plasma protein bound fraction for afatinib is 91.79% (Supplementary Fig. 6A, B) corresponding to an AUCfree of 27 ng*hr/mL. Mouse plasma protein bound fraction for afatinib was 94.23%. In order to replicate an AUCfree of 27 ng*hr/mL in mice a plasma AUCtotal of 468 ng*hr/mL is therefore required. Extrapolation using a linear regression model (Supplementary Fig. 6C) generated from pharmacokinetic parameters estimated using non-compartmental analysis on Phoenix Winnollin estimates a dose of 6 mg/kg is required to achieve a total AUC of 468 ng*hr/mL in nude mice (Supplementary Fig. 6E, D). Therefore, a dose of 6 mg/kg was considered to emulate human equivalent dose (HED) for afatinib in nude mice.
To evaluate the efficacy of tarloxotinib in vivo, CUTO14 and CUTO17 cells were implanted into mice (CUTO18 cells did not establish a xenograft). Mice were randomized to receive vehicle, afatinib (6 mg/kg daily by oral gavage), or tarloxotinib (48 mg/kg via intraperitoneal injection once weekly) for 4 weeks. In CUTO14 xenografts, afatinib did not reduce tumor burden and tumor volumes were comparable to the vehicle group. However, tarloxotinib demonstrated rapid and significant tumor shrinkage (p <0.005) (Fig. 3A, Supplemental Fig. S7A). To determine whether tarloxotinib would induce tumor reduction in much larger tumors (~500–600 mm3), mice in the vehicle group were treated with tarloxotinib using the same 48 mg/kg IP once weekly dose. Again, rapid and significant tumor shrinkage was observed (Supplementary Fig. S7B). In CUTO17 cells, we observed tumor growth inhibition in mice treated with tarloxotinib for four weeks (p <0.02) (Fig. 3A).
Figure 3. Tarloxotinib inhibits tumor growth of EGFR exon 20 mutant, HER2 gene altered, or NRG1 fusion models in vivo.

The percent change from baseline tumor volume was graphed for nude mice inoculated subcutaneously with the indicated cell lines; A) CUTO14 or CUTO17 (EGFR ex20ins); B) H1781 (HER2 ex20ins) or Calu-3 cells (HER2 amp). C) PDX model (OV-10–0050) with a CLU-NRG1 fusion. Mice were treated with vehicle, tarloxotinib (26 mg/kg or 48 mg/kg, once weekly, IP) and afatinib (6 mg/kg, daily, PO) for 4 weeks. Mean ± SEM is plotted. Statistical analysis was made using 2-way-ANOVA, *p<0.005, **p<0.02.
In order to determine the effects of tarloxotinib on tumors with HER2 dependence, cells harboring a HER2 exon 20 mutation (H1781) or HER2 amplification (Calu-3) were similarly utilized in xenograft studies and showed that treatment with tarloxotinib also inhibited tumor growth (p <0.005) (Fig. 3B, Supplemental Figure S7A).
Finally, we tested tarloxotinib in a patient-derived xenograft of ovarian cancer (OV-10–0050) which harbors the CLU-NRG1 gene fusion. Tarloxotinib showed a rapid and near complete response, whereas afatinib did not alter tumor growth compared to vehicle (Fig. 3C). There was no significant loss of body weight during the treatment with tarloxotinib or other therapies for any of the models evaluated (Supplementary Figure S8).
Pharmacological evidence for a therapeutic window in tarloxotinib treated tumor bearing mice
Given the proposed novel mechanism of tumor-specific, hypoxia-induced activation, we sought to quantify drug levels in various compartments for both the prodrug and the active metabolite. Following a single dose of tarloxotinib (48 mg/kg) the total mean exposure (AUC0–168) of OV-10–0050 tumor tissue was 1276 μmol-hr/kg. Prodrug distribution was greater (140%) in murine skin (1792 μmol-hr/kg), reflecting the vascular nature of cutaneous tissues (Fig. 4). In contrast, total exposure (concentration-time) to the metabolite tarloxotinib-E was 313% greater for tumor than skin (595 vs 190 μmol-hr/kg), a pharmacological advantage that is not observed in mice with afatinib (31). Overall, in OV-10–0050 tumor tissue 46.7% of prodrug was converted to active metabolite over 7 days, whereas in skin only 10.6% underwent apparent conversion during the same period. In tumor tissue, exposure to tarloxotinib-E metabolite decreased steadily with time, remaining above 1 μM for 7 days. Relative sparing of total cutaneous exposure to metabolite was associated with a rapid drop in exposure concentration below 1 μM by 24 hours. Apparent cutaneous exposure kinetics were paralleled by circulating plasma levels of tarloxotinib-E suggesting that a proportion of the tarloxotinib-E concentrations detected may be associated with direct exposure via the circulation at early time-points rather than in situ generation. This would be consistent with evidence of modest hypoxia in cutaneous structures (32). These results demonstrate tumor-enriched drug conversion, which could generate a therapeutic window in patients by relative sparing normal tissues from exposure to tarloxotinib-E and thus WT EGFR inhibition.
Figure 4. Pharmacological profile of tarloxotinib and tarloxotinib-E in mice bearing the PDX model (OV-10–0050).

After single dose of tarloxotinib, tumor, skin and blood were collected at 2, 24 and 168 hours. Tarloxotinib and tarloxotinib-E concentration was measured for each time point. Mean ± SEM is plotted.
Tumor Hypoxia in Xenograft Models
Tarloxotinib was designed to fragment to tarloxotinib-E under hypoxic conditions to take advantage of this pathophysiological condition observed in the microenvironment of many tumors. We therefore measured hypoxia in CUTO14 xenografted tumors treated with tarloxotinib or vehicle. As shown above, tarloxotinib resulted in decreased tumor size for both CUTO14 and H1781 (Fig. 3A and B). Pimonidazole is a 2-nitroimidazole that is reduced in hypoxic environments. In hypoxic cells, reduced pimonidazole binds to -SH-containing proteins, peptides and amino acids. We used an antibody that binds to these covalent adducts allowing their detection by fluorescence microscopy (33). Cytokeratin staining of CUTO14 tumors at the termination of the experiment demonstrated that tumor size underestimated the effect of tarloxotinib (Fig. 5A, B). In vehicle treated tumors, more than 50% of the area stained positive for cytokeratin indicative of tumor cells, whereas less than 20% of the total area of tarloxotinib treated tumors was occupied by cytokeratin-positive tumor cells. Pimonidazole staining of tumors demonstrated a similar hypoxic fraction for CUTO14 and H1781 in vehicle and tarloxotinib treated groups (less than 10%) (Fig. 5C).
Figure 5. Tumor hypoxia levels in xenograft models.
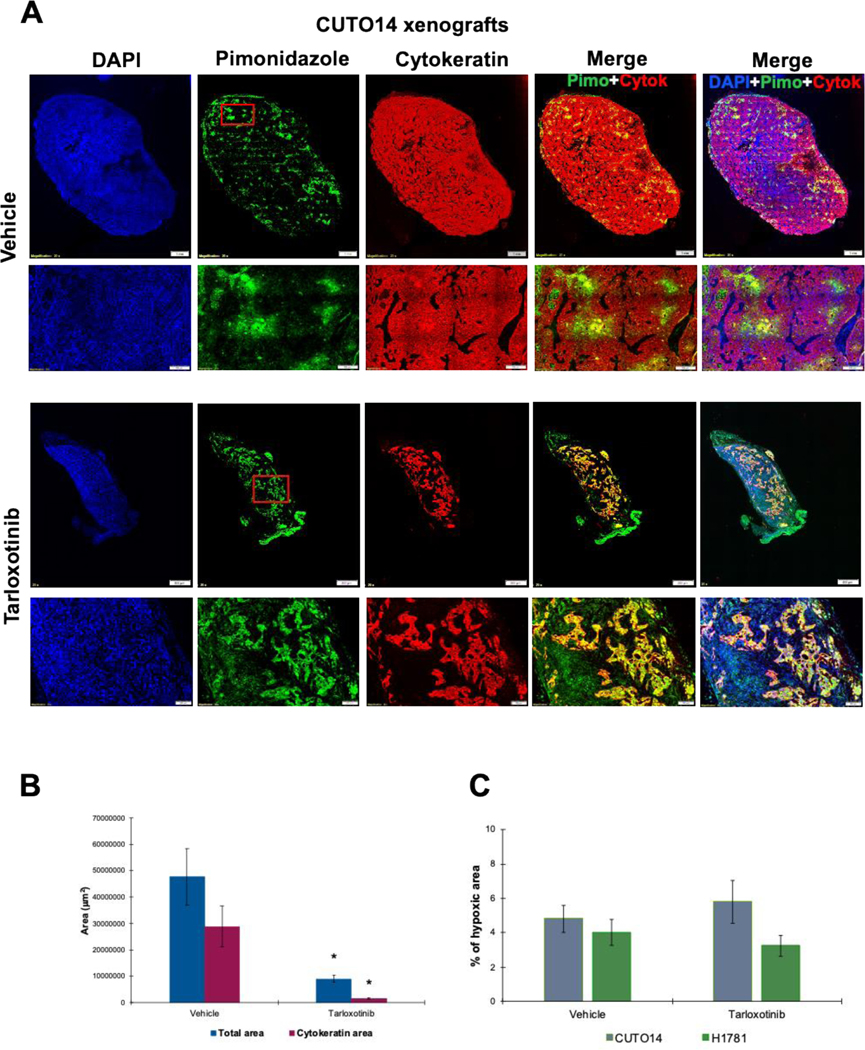
Mice were dose with pimonidazole HCl (60 mg/kg, IP) one hour before tumor collection. Tissue was fixed, paraffin embedded and process for immunofluorescence. The primary antibody Mab1 binds to protein adducts of pimonidazole in hypoxic cells (green). Cytokeratin staining was used to evaluate tumor content (red). All images were capture at 20× using an Olympus IX83 microscope. Scale bars are 1mm or 500μm as indicated in the figures. A) Images representing vehicle and treated CUTO14 xenografts. Red square in green channel indicates the higher magnification area. B) Total area and cytokeratin positive area in vehicle and treated CUTO14 xenografts. * p< 0.005. C) Percent of total area positive hypoxia staining in CUTO14 and H1781 xenografts. Mean ± SEM is plotted.
Response of a NSCLC patient with a HER2 mutation to tarloxotinib
A 25-year-old male never-smoker developed a cough that persisted despite treatment with antibiotics and bronchodilators. Initial diagnostic scans showed a large right hilar mass, lymphangitic disease throughout the right lung, lymphadenopathy involving the neck, mediastinum, and para-aortic area, a lesion in the right adrenal gland, and widespread osseous metastases. A brain MRI showed multiple metastases. Bronchoscopic biopsy revealed lung adenocarcinoma. He received the first cycle of carboplatin (AUC 5) and pemetrexed (500 mg/m2) followed by whole brain radiotherapy. He received cycle 2 carboplatin (AUC 5) and pemetrexed (500 mg/m2) with pembrolizumab (200 mg). Shortly after the treatment, he was hospitalized with bilateral segmental pulmonary emboli and deep vein thromboses, requiring anticoagulation. Restaging scans showed an enlarging right upper lobe mass, bilateral adrenal metastases, and new bone lesions, consistent with rapid disease progression. Analysis of circulating tumor DNA (ctDNA) showed a HER2 exon 20 mutation (p.A775_G776insYVMA), which was confirmed by next-generation sequencing (NGS) on the tumor tissue.
Given the presence of a HER2 mutation and a lack of US FDA approved therapies for this subset of oncogene mutations, the patient was referred for consideration of enrollment into the phase II trial of tarloxotinib (NCT03805841). The patient was found eligible for the trial and provided written informed consent. The baseline CT showed a large right hilar mass, right pleural effusion, and bilateral adrenal lesions as well as multiple mediastinal lymph nodes (Fig. 6A, data not shown). After initiation of tarloxotinib at 150 mg/m2 IV weekly, the patient experienced rapid improvement in his cough, dyspnea, and bony pain with decreased opioid requirement. A CT scan performed prior to cycle 2 showed a partial response with a marked decrease in size of the right hilar mass, right pleural effusion, and bilateral adrenal gland lesions (Fig. 6B). A confirmatory, follow-up CT scan done prior to cycle 4 demonstrated continued response to treatment (Fig. 6C). Tarloxotinib was well tolerated; treatment related adverse events that the patient experienced included rash, alanine aminotransferase (ALT) elevation, and asymptomatic QTc prolongation (all grade 1). The patient subsequently experienced disease progression, yielding a duration of response of 5 months.
Figure 6. Radiologic response to tarloxotinib.

Baseline imaging obtained prior to dosing with tarloxotinib (150 mg/m2 IV weekly) showed bulky right hilar mass, right pleural effusion, and bilateral adrenal gland lesions (red arrows) (A). A marked tumor response with decreased size of right hilar mass, right pleural effusion, and lesions in the adrenal glands was observed at week 4 (B) and week 12 (C).
Discussion
Oncogene-targeted therapies have markedly improved clinical outcomes for patients with NSCLC whose tumors bear oncogenic alterations in ALK, ROS1, BRAF, NTRK, MET and most EGFR mutations. However, a lack of selectivity between multiple EGFR/HER-family oncogenes and WT EGFR has made this strategy challenging for certain subsets of patients bearing oncogenes that activate the ErbB family of RTKs. Standard EGFR inhibitors exhibit on-target toxicities associated with inhibition of WT EGFR in critical normal tissues such as the skin and gastrointestinal tract inducing rash and diarrhea, respectively, among other side effects. The challenge of successful inhibition of EGFR exon 20 insertion mutations, unlike EGFR del 19 or L858R, is likely explained by the retention of high affinity for ATP and low affinity for most EGFR TKIs (9). Tarloxotinib is a prodrug designed to have weak cellular activity against EGFR, HER2, HER4 and their mutant forms, while the released active metabolite is a potent cellular pan-HER inhibitor. In this work, we harnessed the novel mechanism of action of the prodrug tarloxotinib to circumvent the inherent relative lack of selectivity that hinders the clinical utility of established and emerging EGFR and HER2 small molecule inhibitors. Tarloxotinib-E is a potent pan-HER inhibitor without inherent mutant selectivity, instead relying on selective activation within the tumor environment, leveraging the presence of hypoxia in malignant, but not normal tissues. Thus, while tarloxotinib-E is more potent than all of the EGFR/pan-HER TKIs tested in this study (Table 1), a direct comparison of potency may underestimate the potential for significantly higher tumor exposure given the unique mechanism of action which can lead to higher intratumor concentration (Figure 4) than is likely achieved by standard TKIs. To this point, tarloxotinib-E induces similar rates of apoptosis compared to afatinib in patient-derived cell lines at a concentration of 1 μM (Supplemental Figure S5). However the Cmax for afatinib in patients is only 38 ng/mL (78 nM) at steady state using the US FDA approved dose of 40mg PO daily (39), whereas tumor concentrations of tarloxotinib-E are sustained above 1 μM for one week after dosing of tarloxotinib at the HED (Figure 4).
Human derived cell lines and other human-derived cancer models have facilitated the dissection of oncogene signaling biology and drug development; thus, accelerating the prediction of drug resistance mechanisms in lung cancer and other malignancies. A large number of human-derived lung cancer cell lines are available and generally represent many of the oncogenes observed in lung adenocarcinoma, however at the outset of this study, no models that harbored EGFR exon 20 insertions existed. To address this limitation, we established three unique cell lines harboring EGFR exon 20 insertions: CUTO14, CUTO17 and CUTO18. Knockdown of EGFR demonstrated dependence on this oncogene in these three cell lines, establishing the rationale for inhibition of EGFR in patients whose tumors harbor EGFR exon 20 insertions.
The EGFR TKIs used in this study, including first (gefitinib), second (afatinib) and third (osimertinib) generation inhibitors demonstrated some degree of dose dependent inhibition of the primary oncogene or signaling pathway in the human cell line models used here. The canonical downstream signaling pathways, MAPK and PI3K/AKT, showed concordant inhibition with the upstream RTK inhibition. Tarloxotinib-E was generally the most potent inhibitor of pEGFR, pHER2, pHER3 and pHER4 when compared to other TKIs. The use of patient derived cell lines (CUTO-14, CUTO-17 and CUTO-18) demonstrated differential IC50s consistent with differential binding of EGFR TKIs.
Similar to target inhibition, dose dependent reduction of cellular proliferation by tarloxotinib-E was observed in all three EGFR exon 20 cell lines, all three HER2 models, the NRG1 fusion cell line, and a cell line expressing high levels of HER4. Notably, the prodrug tarloxotinib was consistently and significantly less potent than the active metabolite tarloxotinib-E in these cellular models reflecting the weak capacity of this molecule to inhibit mutant or wild-type forms of the HER family of RTKs in cells. Most importantly, tarloxotinib demonstrated considerably less activity on WT EGFR (and WT HER2) allowing for the potential to spare patient toxicity.
Dosing of multiple EGFR, HER2 or NRG1 mutant xenograft or PDX models with the prodrug tarloxotinib demonstrated marked tumor growth inhibition or tumor regression. The pharmacology of tumor and blood exposure indicated that the active metabolite tarloxotinib-E was enriched in the tumor with much lower levels found in the blood and skin, consistent with the proposed mechanism of action for this prodrug. In contrast, afatinib, a potent EGFR TKI across various in vitro assays in our models, demonstrated no evidence of tumor growth inhibition when dosed at the HED in vivo. This finding was consistent with the poor clinical activity of afatinib observed in NSCLC patients with EGFR exon 20 insertion in the LUX-Lung studies (13). Our analysis of hypoxic regions in tumor xenografts demonstrated that hypoxic fractions <10% can generate significant intratumoral concentration of the active pan-HER tumor inhibitor tarloxotinib-E and tumor inhibition, either by TKI diffusion, regional hypoxia, and/or hypoxia changes over time. The prodrug tarloxotinib has a prolonged half-life in tumor tissue (Figure 4) and thus allows for activation to tarloxotinib-E with variance in hypoxia with time and likely leads to the maintenance of tarloxotinib-E levels in the tumor over the 7-day dosing interval.
Finally, we demonstrated a confirmed, objective tumor response in a patient with metastatic NSCLC harboring a HER2 exon 20 insertion mutation dosed with tarloxotinib on the RAIN-701 phase II clinical trial. This tumor response occurred with minimal on-target EGFR-related toxicity, exemplified by the absence of diarrhea and only a mild rash. This trial is currently accruing NSCLC patients with HER2 activating mutations and patients with any tumor type (tumor agnostic) harboring NRG1 gene fusions (as well as other rare EGFR, HER2 and HER4 fusions). Recently presented clinical trial data demonstrated a relative lack of efficacy in the EGFR exon 20 cohort compared to the HER2 cohort (40). This is likely explained by the data presented here, given that the EGFR exon 20 models required approximately 10-fold more tarloxotinib-E to achieve IC50 compared to the HER2 models (Table 1). Although challenging to incorporate in clinical trials, imaging of patients using novel hypoxia radiotracers via PET scan in future studies using such methods may further our understanding of the relationship between tumor hypoxic fraction and anti-tumor response with tarloxotinib. Other emerging methods to detect hypoxia include oxygen-enhanced MRI and oncoradiomics.
The work here demonstrates that a strategy employing a hypoxia-activated prodrug can achieve anti-tumor response while circumventing the on-target toxicity associated with systemically administered TKIs. Poziotinib has demonstrated anti-tumor activity in NSCLC patients with EGFR and HER2 exon 20 insertion mutations, but with high levels of EGFR-related toxicity, including rash and diarrhea requiring dose reductions in a significant number of patients (34,35). This clinical finding is likely explained by the very high potency of poziotinib against WT EGFR (36). Consistent with this phenomenon, TAK-788 (mobocertinib) shows only a narrow therapeutic window between EGFR or HER2 exon 20 mutants, which also likely explains the similarly high levels of EGFR-related side effects observed in phase 1 clinical trial of this agent (37). Other ongoing clinical trials are using antibody approaches which include a HER2-ADC (fam-trastuzumab deruxtecan-nxki) and an EGFR-MET bispecific antibody (amivantamab). Unlike many other clinical development candidates for EGFR exon 20 or HER2, tarloxotinib establishes a therapeutic window, but using tumor hypoxia rather than binding specificity.
In summary, we have demonstrated that tarloxotinib is a novel hypoxia-activated prodrug that demonstrates enhanced conversion in tumors to tarloxotinib-E, a potent pan-HER inhibitor. Tarloxotinib was demonstrably active in a number of EGFR/HER-driven tumor models that currently do not have approved therapies. Finally, this drug induced a marked tumor response in a patient with a HER2 exon 20 insertion demonstrating proof of concept for this novel prodrug strategy.
Supplementary Material
Statement of translational relevance.
Targeting lung cancers with EGFR exon 20 and ERBB2 mutations has been unsuccessful due to the lack of a therapeutic window for inhibitors that induce side effects in normal tissues. Tarloxotinib circumvents this problem by using tumor hypoxia to generate high levels of a potent pan-HER inhibitor within the tumor.
Acknowledgements
We thank the patients that donated tissue for cell line isolation and to the patients and their families that are participating in these clinical trials.
Funding for this study was provided by Developmental Therapeutics Program, University of Colorado Cancer Center Support Grant (NIH/NCI P30CA046934), the University of Colorado Lung SPORE (NIH/NCI P50CA058187) and the Health Research Council of New Zealand project grant (14/290). Partial salary support (JBS, AVP) was provided by the Cancer Society Auckland Northland.
ATL holds ownership interest (including patents) in Molecular Abbott. VGT, Rain Therapeutics employee (stock options). JBS and AVP, tarloxotinib patent, license and stock options Rain Therapeutics. CK, (advisor) Novartis, (funding) AstraZeneca, BMS, Novartis, Regeneron, Tesaro, Karyopharm, Debiopharm, Altor Bioscience. SVL, (Advisory board / consultant) AstraZeneca, Boehringer-Ingelheim, Bristol-Myers Squibb, Celgene, G1 Therapeutics, Genentech, Guardant Health, Inivata, Janssen, Lilly, Loxo, Merck, PharmaMar, Pfizer, Regeneron, Takeda. (Research grant (institution)) from Alkermes, AstraZeneca, Bayer, Blueprint, Bristol-Myers Squibb, Corvus, Genentech, Lilly, Lycera, Merck, Molecular Partners, Pfizer, Rain Therapeutics, RAPT, Spectrum, Turning Point Therapeutics. RCD, Rain Therapeutics, Blueprint Medicines, Anchiano, Green Peptide, Genentech/Roche, Bayer, AstraZeneca (Advisory Board); Rain Therapeutics, Foundation Medicine, Abbott Molecular, Black Diamond, Pearl River, Voronoi (Intellectual Property Licensing); Rain Therapeutics (Stock Ownership).
Footnotes
COI: AEB, AED, SS, and MRB, these authors declare no conflicts of interest.
References
- 1.Burden S, Yarden Y. Neuregulins and their receptors: a versatile signaling module in organogenesis and oncogenesis. Neuron 1997;18(6):847–55 doi 10.1016/s0896-6273(00)80324-4. [DOI] [PubMed] [Google Scholar]
- 2.Hynes NE, MacDonald G. ErbB receptors and signaling pathways in cancer. Curr Opin Cell Biol 2009;21(2):177–84 doi 10.1016/j.ceb.2008.12.010. [DOI] [PubMed] [Google Scholar]
- 3.Herbst RS, Heymach JV, Lippman SM. Lung cancer. N Engl J Med 2008;359(13):1367–80 doi 10.1056/NEJMra0802714. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Mok TS, Wu YL, Thongprasert S, Yang CH, Chu DT, Saijo N, et al. Gefitinib or carboplatin-paclitaxel in pulmonary adenocarcinoma. N Engl J Med 2009;361(10):947–57 doi 10.1056/NEJMoa0810699. [DOI] [PubMed] [Google Scholar]
- 5.Wu YL, Zhou C, Hu CP, Feng J, Lu S, Huang Y, et al. Afatinib versus cisplatin plus gemcitabine for first-line treatment of Asian patients with advanced non-small-cell lung cancer harbouring EGFR mutations (LUX-Lung 6): an open-label, randomised phase 3 trial. Lancet Oncol 2014;15(2):213–22 doi 10.1016/S1470-2045(13)70604-1. [DOI] [PubMed] [Google Scholar]
- 6.Wu YL, Cheng Y, Zhou X, Lee KH, Nakagawa K, Niho S, et al. Dacomitinib versus gefitinib as first-line treatment for patients with EGFR-mutation-positive non-small-cell lung cancer (ARCHER 1050): a randomised, open-label, phase 3 trial. Lancet Oncol 2017;18(11):1454–66 doi 10.1016/S1470-2045(17)30608-3. [DOI] [PubMed] [Google Scholar]
- 7.Soria JC, Ohe Y, Vansteenkiste J, Reungwetwattana T, Chewaskulyong B, Lee KH, et al. Osimertinib in Untreated EGFR-Mutated Advanced Non-Small-Cell Lung Cancer. N Engl J Med 2018;378(2):113–25 doi 10.1056/NEJMoa1713137. [DOI] [PubMed] [Google Scholar]
- 8.Rosell R, Carcereny E, Gervais R, Vergnenegre A, Massuti B, Felip E, et al. Erlotinib versus standard chemotherapy as first-line treatment for European patients with advanced EGFR mutation-positive non-small-cell lung cancer (EURTAC): a multicentre, open-label, randomised phase 3 trial. Lancet Oncol 2012;13(3):239–46 doi 10.1016/S1470-2045(11)70393-X. [DOI] [PubMed] [Google Scholar]
- 9.Yasuda H, Park E, Yun CH, Sng NJ, Lucena-Araujo AR, Yeo WL, et al. Structural, biochemical, and clinical characterization of epidermal growth factor receptor (EGFR) exon 20 insertion mutations in lung cancer. Sci Transl Med 2013;5(216):216ra177 doi 10.1126/scitranslmed.3007205. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Arcila ME, Nafa K, Chaft JE, Rekhtman N, Lau C, Reva BA, et al. EGFR exon 20 insertion mutations in lung adenocarcinomas: prevalence, molecular heterogeneity, and clinicopathologic characteristics. Mol Cancer Ther 2013;12(2):220–9 doi 10.1158/1535-7163.MCT-12-0620. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Yun CH, Boggon TJ, Li Y, Woo MS, Greulich H, Meyerson M, et al. Structures of lung cancer-derived EGFR mutants and inhibitor complexes: mechanism of activation and insights into differential inhibitor sensitivity. Cancer Cell 2007;11(3):217–27 doi 10.1016/j.ccr.2006.12.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Yasuda H, Kobayashi S, Costa DB. EGFR exon 20 insertion mutations in non-small-cell lung cancer: preclinical data and clinical implications. Lancet Oncol 2012;13(1):e23–31 doi 10.1016/S1470-2045(11)70129-2. [DOI] [PubMed] [Google Scholar]
- 13.Yang JC, Sequist LV, Geater SL, Tsai CM, Mok TS, Schuler M, et al. Clinical activity of afatinib in patients with advanced non-small-cell lung cancer harbouring uncommon EGFR mutations: a combined post-hoc analysis of LUX-Lung 2, LUX-Lung 3, and LUX-Lung 6. Lancet Oncol 2015;16(7):830–8 doi 10.1016/S1470-2045(15)00026-1. [DOI] [PubMed] [Google Scholar]
- 14.Leduc C, Merlio JP, Besse B, Blons H, Debieuvre D, Bringuier PP, et al. Clinical and molecular characteristics of non-small-cell lung cancer (NSCLC) harboring EGFR mutation: results of the nationwide French Cooperative Thoracic Intergroup (IFCT) program. Ann Oncol 2017;28(11):2715–24 doi 10.1093/annonc/mdx404. [DOI] [PubMed] [Google Scholar]
- 15.Kris MG, Camidge DR, Giaccone G, Hida T, Li BT, O’Connell J, et al. Targeting HER2 aberrations as actionable drivers in lung cancers: phase II trial of the pan-HER tyrosine kinase inhibitor dacomitinib in patients with HER2-mutant or amplified tumors. Ann Oncol 2015;26(7):1421–7 doi 10.1093/annonc/mdv186. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Mazieres J, Barlesi F, Filleron T, Besse B, Monnet I, Beau-Faller M, et al. Lung cancer patients with HER2 mutations treated with chemotherapy and HER2-targeted drugs: results from the European EUHER2 cohort. Ann Oncol 2016;27(2):281–6 doi 10.1093/annonc/mdv573. [DOI] [PubMed] [Google Scholar]
- 17.Fernandez-Cuesta L, Plenker D, Osada H, Sun R, Menon R, Leenders F, et al. CD74-NRG1 fusions in lung adenocarcinoma. Cancer Discov 2014;4(4):415–22 doi 10.1158/2159-8290.CD-13-0633. [DOI] [PubMed] [Google Scholar]
- 18.Jonna S, Feldman RA, Swensen J, Gatalica Z, Korn WM, Borghaei H, et al. Detection of NRG1 Gene Fusions in Solid Tumors. Clin Cancer Res 2019;25(16):4966–72 doi 10.1158/1078-0432.CCR-19-0160. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Muscarella LAaR A NRG1: a cinderella fusion in lung cancer. Lung Cancer Management 2018;6(4):121–3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Silva S VJ, G C, A M, B M, G A, et al. Preclinical efficacy of tarloxotinib bromide (TH-4000), a hypoxia-activated EGFR/HER2 inhibitor: rationale for clinical evaluation in EGFR mutant, T790M-negative NSCLC following progression on EGFR-TKI therapy. [abstract}. 2015. [Google Scholar]
- 21.Lu GL, Jaiswal J, L’ee H, Ashoorzadeh A, Maroz A, Squire CJ, et al. Optimization of substituted pyrido[3,4-d]pyrimidine derivatives as hypoxia-activated prodrugs of irreversible inhibitors of the epidermal growth factor receptor family: The discovery of tarloxotinib bromide. In preparation. [Google Scholar]
- 22.Bhandari V, Hoey C, Liu LY, Lalonde E, Ray J, Livingstone J, et al. Molecular landmarks of tumor hypoxia across cancer types. Nat Genet 2019;51(2):308–18 doi 10.1038/s41588-018-0318-2. [DOI] [PubMed] [Google Scholar]
- 23.Zegers CM, van Elmpt W, Wierts R, Reymen B, Sharifi H, Ollers MC, et al. Hypoxia imaging with [(1)(8)F]HX4 PET in NSCLC patients: defining optimal imaging parameters. Radiother Oncol 2013;109(1):58–64 doi 10.1016/j.radonc.2013.08.031. [DOI] [PubMed] [Google Scholar]
- 24.Jackson V, Silva S, Abbattista M, Guise C, Bull M, Ashoorzadeh A, et al. Preclinical rationale for the ongoing Phase 2 study of the hypoxia-activated EGFR-TKI tarloxotinib bromide (TH-4000) in patients with advanced squamous cell carcinoma of the head and neck (SCCHN) or skin (SCCS). . In: Proceedings of the AACR-NCI-EORTC International Conference: Molecular Targets and Cancer Therapeutics; 2015 Nov 5–9; Boston, MA Philadelphia (PA): AACR; Mol Cancer Ther 2015;14(12 Suppl 2):Abstract nr A662015. [Google Scholar]
- 25.Liu SV CA, B C, DR C, GD E, G B, et al. Phase 2 study or tarloxotinib bromide (TRLX) in patients (pts) with EGFR-mutatnt, T790M-negative NSCLC progressing on an EGFR TKI. ASCO Annual meeting June 3–7, Chicago, Illinois2016. [Google Scholar]
- 26.Blanco R, Iwakawa R, Tang M, Kohno T, Angulo B, Pio R, et al. A gene-alteration profile of human lung cancer cell lines. Hum Mutat 2009;30(8):1199–206 doi 10.1002/humu.21028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Shigematsu H, Takahashi T, Nomura M, Majmudar K, Suzuki M, Lee H, et al. Somatic mutations of the HER2 kinase domain in lung adenocarcinomas. Cancer Res 2005;65(5):1642–6 doi 10.1158/0008-5472.CAN-04-4235. [DOI] [PubMed] [Google Scholar]
- 28.Ise N, Omi K, Nambara D, Higashiyama S, Goishi K. Overexpressed HER2 in NSCLC is a possible therapeutic target of EGFR inhibitors. Anticancer Res 2011;31(12):4155–61. [PubMed] [Google Scholar]
- 29.Schaefer G, Fitzpatrick VD, Sliwkowski MX. Gamma-heregulin: a novel heregulin isoform that is an autocrine growth factor for the human breast cancer cell line, MDA-MB-175. Oncogene 1997;15(12):1385–94 doi 10.1038/sj.onc.1201317. [DOI] [PubMed] [Google Scholar]
- 30.Wind S, Schnell D, Ebner T, Freiwald M, Stopfer P. Clinical Pharmacokinetics and Pharmacodynamics of Afatinib. Clin Pharmacokinet 2017;56(3):235–50 doi 10.1007/s40262-016-0440-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Slobbe P, Windhorst AD, Stigter-van Walsum M, Schuit RC, Smit EF, Niessen HG, et al. Development of [18F]afatinib as new TKI-PET tracer for EGFR positive tumors. Nucl Med Biol 2014;41(9):749–57 doi 10.1016/j.nucmedbio.2014.06.005. [DOI] [PubMed] [Google Scholar]
- 32.Evans SM, Schrlau AE, Chalian AA, Zhang P, Koch CJ. Oxygen levels in normal and previously irradiated human skin as assessed by EF5 binding. J Invest Dermatol 2006;126(12):2596–606 doi 10.1038/sj.jid.5700451. [DOI] [PubMed] [Google Scholar]
- 33.Raleigh JA, Chou SC, Arteel GE, Horsman MR. Comparisons among pimonidazole binding, oxygen electrode measurements, and radiation response in C3H mouse tumors. Radiat Res 1999;151(5):580–9. [PubMed] [Google Scholar]
- 34.Heymach J, Negrao M, Robichaux J, Carter B, Patel A, Altan M, et al. A Phase II Trial of Poziotinib in EGFR and HER2 exon 20 Mutant Non-Small Cell Lung Cancer (NSCLC). Journal of Thoracic Oncology 2018;13(10):S323–S4 doi DOI 10.1016/j.jtho.2018.08.243. [DOI] [Google Scholar]
- 35.Heymach JV, Negrao MV, Robichaux JP, Carter BW, Patel A, Altan M, et al. Phase II trial of poziotinib for EGFR and HER2 exon 20 mutant NSCLC. IASLC 19th World Conference on Lung Cancer September 23–26, Toronto, Canada2018. [Google Scholar]
- 36.Lee Y, Kim TM, Kim DW, Kim S, Kim M, Keam B, et al. Preclinical Modeling of Osimertinib for NSCLC With EGFR Exon 20 Insertion Mutations. J Thorac Oncol 2019;14(9):1556–66 doi 10.1016/j.jtho.2019.05.006. [DOI] [PubMed] [Google Scholar]
- 37.Doebele RC, Riely GJ, Spira AI, Horn L, Piotrowska Z, Costa DB, et al. First report of safety, PK, and preliminary antitumor activity of the oral EGFR/HER2 exon 20 inhibitor TAK-788 (AP32788) in non–small cell lung cancer (NSCLC). Journal of Clinical Oncology 2018;36(15_suppl):9015- doi 10.1200/JCO.2018.36.15_suppl.9015. [DOI] [Google Scholar]
- 38.McCoach CE, Le AT, Gowan K, Jones K, Schubert L, Doak A, et al. Resistance Mechanisms to Targeted Therapies in ROS1(+) and ALK(+) Non-small Cell Lung Cancer. Clin Cancer Res 2018;24(14):3334–47 doi 10.1158/1078-0432.CCR-17-2452. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Wind S, Schmid M, Erhardt J, Goeldner RG, Stopfer P. Pharmacokinetics of afatinib, a selective irreversible ErbB family blocker, in patients with advanced solid tumours. Clin Pharmacokinet 2013;52(12):1101–9 doi 10.1007/s40262-013-0091-4. [DOI] [PubMed] [Google Scholar]
- 40.Liu SV, Villaruz LC, Lee VHF, Zhu VW, Baik CS, Sacher A, et al. First analysis of RAIN-701: Study oftarloxotinib in patients with non-small cell lung cancer (NSCLC) EGFR Exon 20 insertion, HER2-activating mutations & other solid tumours with NRG1/ERBB gene fusions. ESMO Virtual Congress September 19–21, 2020. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.