

Chlamydia trachomatis is an obligate intracellular pathogen that causes sexually transmitted disease. In women, chlamydial infections may cause pelvic inflammatory disease (PID), ectopic pregnancy, and infertility. The role of antibodies in protection against a primary Chlamydia infection is unclear and was a focus of this work. Using the C. muridarum mouse infection model, we show that intestinal mucosa is infected via intranasal (i.n.) or per-oral (p.
KEYWORDS: Chlamydia, vaccine, mucosa, vaccination, IgA, antibodies, female reproductive tract, mucosal vaccines, neutralizing antibodies
ABSTRACT
Chlamydia trachomatis is an obligate intracellular pathogen that causes sexually transmitted disease. In women, chlamydial infections may cause pelvic inflammatory disease (PID), ectopic pregnancy, and infertility. The role of antibodies in protection against a primary Chlamydia infection is unclear and was a focus of this work. Using the C. muridarum mouse infection model, we show that intestinal mucosa is infected via intranasal (i.n.) or per-oral (p.o.) Chlamydia inoculation and that unlike the female reproductive tract (FRT) mucosa, it halts systemic Chlamydia dissemination. Moreover, p.o. immunization or infection with Chlamydia confers protection against per-vaginal (p.v.) challenge, resulting in significantly decreased bacterial burden in the FRT, accelerated Chlamydia clearance, and reduced hydrosalpinx pathology. In contrast, subcutaneous (s.c.) immunization conferred no protection against the p.v. challenge. Both p.o. and s.c. immunizations induced Chlamydia-specific serum IgA. However, IgA was found only in the vaginal washes and fecal extracts of p.o.-immunized animals. Following a p.v. challenge, unimmunized control and s.c.-s.c.-immunized animals developed Chlamydia-specific intestinal IgA yet failed to develop IgA in the FRT, indicating that IgA response in the FRT relies on the FRT to gastrointestinal tract (GIT) antigen transport. Vaginal secretions of p.o.-immunized animals neutralize Chlamydia in vivo, resulting in significantly lower Chlamydia burden in the FRT and Chlamydia transport to the GIT. We also show that infection of the GIT is not necessary for induction of protective immunity in the FRT, a finding that is important for the development of p.o. subunit vaccines to target Chlamydia and possibly other sexually transmitted pathogens.
INTRODUCTION
The occurrence of new sexually transmitted infections (STIs) worldwide has been on the rise, with over 1 million new cases reported daily (1). In the United States, the situation is just as concerning, with significant increases in new cases of chlamydia, gonorrhea, and syphilis (2). Young adults (aged 15 to 24) account for around half of the new STIs. Between 2014 and 2018, chlamydial infections in the United States increased by 19%, with infection rates two times higher in women than in men (2). In women, about 80% of chlamydial infections are asymptomatic and may lead to the development of pelvic inflammatory disease (PID), chronic pelvic pain, hydrosalpinx, and tubal infertility (3–5). In addition, women infected with Chlamydia are at a higher risk of acquiring HIV infections and developing human papillomavirus (HPV)-associated cervical cancer (6, 7). Despite extensive efforts, the development of effective mucosal vaccines against STIs, including Chlamydia, has been for the most part unsuccessful, with the exception of the vaccine against HPV, which is administered parenterally (8). Parenteral immunization, however, induces mainly systemic immunity and protection against some mucosally acquired pathogens, such as influenza virus, HPV, and poliovirus. In contrast, mucosal immunization induces systemic and local mucosal immunity, which is essential for protection against most mucosal pathogens, including Chlamydia.
Animal studies have been instrumental in identifying protective immune responses to chlamydial infections and have guided vaccine development. Overall, for resolution of an infection and the development of immunity to reinfection, CD4+ T cells and cytokines, such as gamma interferon (IFN-γ) and interleukin 12 (IL-12), have been shown to be essential (9–12), while CD8+ T cells do not appear to be important (10–13). Although antibodies induced by most vaccines are key to their effectiveness, whether and how Chlamydia-specific antibodies confer protection against a primary infection is unclear (14, 15). Chlamydial infections induce Chlamydia-specific antibodies (16, 17), which could aid in protective immunity by neutralizing Chlamydia elementary bodies (EBs) in the female reproductive tract (FRT) mucosa, by activating the complement, as well as other cellular effector functions. Chlamydia exists in two different forms, namely, as infectious EBs and as noninfectious, intracellular reticulate bodies (RBs). Therefore, antibodies specific for antigens of the extracellular EB stage could serve to neutralize EBs and activate a number of other antibody-mediated effector functions. In guinea pigs, resistance to a secondary infection is decreased when humoral immunity is suppressed (18) and the transfer of convalescent-phase serum to naive animals significantly reduces bacterial shedding from the FRT (19). This indicates that antibodies play a role in controlling Chlamydia infections. Studies done with mice that lack B cells or IgA specifically have revealed no clear role for B cells or IgA in the resolution of a primary genital Chlamydia infection, as mice lacking B cells or IgA resolve infections at a rate similar to that of controls (15, 20, 21). Adoptive transfer of convalescent-phase sera does not protect mice against a primary infection; however, it does protect against a secondary infection (14). In women, secretory IgA (sIgA) in the FRT was shown to be inversely correlated with Chlamydia burden (22), indicating that IgA plays an important role in protection against this pathogen.
The mouse-specific pathogen Chlamydia muridarum is widely used for studying pathogenesis and immune responses because it induces a long-term FRT pathology similar to the one caused by Chlamydia trachomatis in women (23, 24). In both animals and humans, Chlamydia establishes long-term infections in the gastrointestinal tract (GIT), which present with no obvious pathology (25–27). In mice, infection of the GIT by C. muridarum induces immunity to a C. muridarum per-vaginal (p.v.) challenge (27). However, a deliberate infection with a nonattenuated pathogen poses inherent risks and would be perceived unsafe by the public and therefore would be an unacceptable form of immunization. This opens up the possibility that per-oral (p.o.) immunization with noninfectious chlamydial antigens may be an effective approach for the development of vaccines to target Chlamydia and possibly other STIs. We hypothesized that immune responses in the FRT may be induced in the gut-associated lymphoid tissue (GALT). Based on previous reports that mucosal priming followed by subcutaneous (s.c.) boosting induces long-lasting mucosal and systemic immunity (28–30), we hypothesized that p.o. priming with inactivated (killed) Chlamydia EBs followed by s.c. boosting would induce immunity to Chlamydia challenge in the FRT. Here, we address these hypotheses and show that inactivated Chlamydia EBs administered to the GIT are just as effective as live Chlamydia in inducing protection against Chlamydia challenge in the FRT. Moreover, we show that IgA secretion in the FRT is induced following immunization or infection of the GIT and not the FRT. FRT IgA effectively neutralizes Chlamydia and impedes its invasion in the FRT lamina propria and systemic dissemination.
RESULTS
C. muridarum infects the GIT following p.o. (intragastric) or i.n. administration.
Although known for its ability to cause STIs and long-term pathology in humans, C. trachomatis also infects and persists in the GIT (31, 32). In mice, the nonpathogenic infection of the GIT by C. muridarum can occur following a p.o. exposure (e.g., via coprophagy or grooming). However, host cells can also spread Chlamydia from the FRT to the GIT internally (33), and this systemic spread of Chlamydia and infection of the spleen specifically were proposed to be critical for the long-term hydrosalpinx pathology in the FRT (33). On the other hand, p.o. and intranasal (i.n.) infections with Chlamydia induce protective immunity to a primary Chlamydia challenge in the FRT (27, 34). In light of these observations, we hypothesized that like p.o. inoculation, i.n. inoculation with live Chlamydia EBs leads to the colonization of the GIT and that GALT represents a firewall that prevents Chlamydia systemic dissemination. To address these hypotheses, mice were infected i.n. or p.o. (intragastrically) with live C. muridarum EBs and Chlamydia titers were examined in ceca, mesenteric lymph nodes (MLNs), and the spleen for up to 21 days postinfection (dpi). Following p.o. or i.n. inoculation, Chlamydia infected the GIT, with ceca of all inoculated animals becoming positive by 7 or 21 dpi, respectively (Fig. 1A and B). The delayed Chlamydia spread to the GIT after the i.n. inoculation was likely due to the lower Chlamydia dose used for i.n. than for p.o. inoculation (104 versus 107 inclusion-forming units [IFU], respectively). While at 7, 14, and 21 dpi p.o. or i.n., a small proportion of MLNs were positive for live Chlamydia (Fig. 1C and D), no Chlamydia was detected in spleens of p.o.- or i.n.-infected mice at any time point examined (Fig. 1E and F). Within ceca, chlamydial inclusions were observed in the epithelium, as well as in the subepithelial compartment (Fig. 2A to C). These findings indicate that unlike the FRT (33, 35), GIT mucosa effectively contains Chlamydia and halts its spread to the deeper tissues, such as the spleen. To examine whether p.o. immunization with killed Chlamydia EBs induces protection in the FRT, two studies were conducted (Fig. 2D). In the first study, groups of mice were p.o. fed live or killed EBs and then s.c. boosted with killed EBs [PO(L)-SC(K) and PO(K)-SC(K) mice]. Another group was s.c. primed and boosted with killed EBs [SC(K)-SC(K) mice], as this form of immunizations induces systemic but not mucosal immunity and allows for examination of the role of serum and mucosal antibodies in protection against a p.v. Chlamydia challenge. In the second study, a similar prime-boost immunization approach was used, with the difference that mice were intragastrically inoculated to ensure that antigen was administered to the GALT alone. Both p.o. and intragastric immunizations are referred to as p.o. immunizations. In the second study, we also added an immunization group in which mice were p.o. infected with live Chlamydia, but were s.c. boosted with phosphate-buffered saline (PBS) [PO(L)-PBS mice]. Control mice were administered sterile PBS p.o. and s.c. (PBS-PBS mice). In both studies, immunized and unimmunized controls were p.v. challenged with 2 × 105 live Chlamydia EBs at day 42 after priming (2 weeks after boosting immunization). Six weeks after the p.v. challenge (day 84), FRT pathology and Chlamydia clearance and burden in the FRT were evaluated (Fig. 2D). We confirmed that the stock of paraformaldehyde (PFA)-treated and sonicated EBs used for p.o. immunizations in both studies had no viable organisms by culturing the stock in HeLa cells and passaging the infection 6 times. In addition, we determined the titers of rectal swabs of PO(L)-SC(K) and PO(K)-SC(K) mice to ensure that PO(K)-SC(K) mice were not infected prior to p.v. challenge. Mice p.o. immunized with live EBs had positive rectal swabs at 14, 28, and 42 after p.o. priming, as well as 14, 28, and 42 dpi p.v. (Fig. 2E). In contrast, none of the mice p.o. immunized with killed EBs had positive rectal titers prior to p.v. challenge (Fig. 2E).
FIG 1.
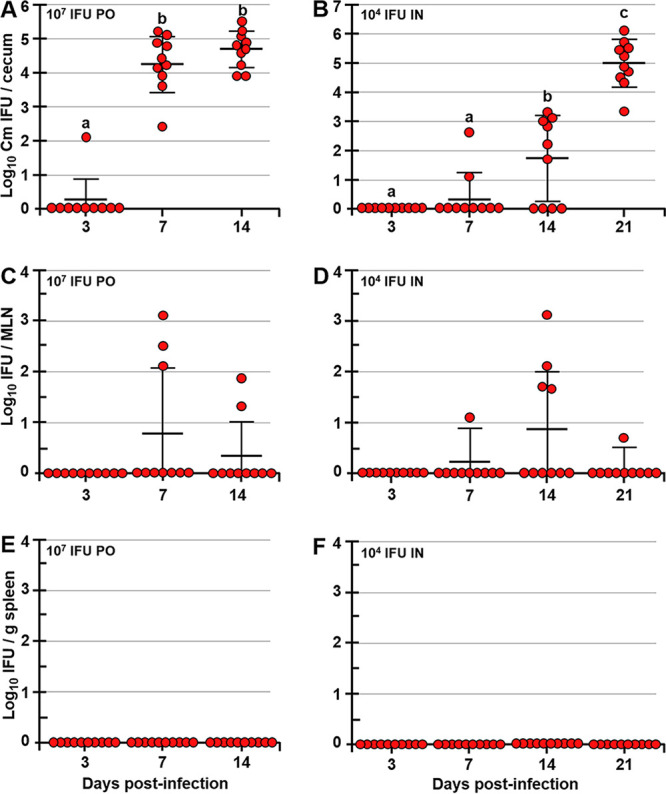
Following p.o. or i.n. administration, Chlamydia infects the GIT of mice but does not disseminate to the spleen. Shown are C. muridarum titers in ceca (A and B), MLNs (C and D), and spleens (E and F) of p.o.- or i.n.-infected mice. Mice were infected p.o. via gastric gavage with 107 or i.n. with 104 IFU of Chlamydia and at 3, 7, 14, and 21 dpi, and Chlamydia titers in tissues were determined. Chlamydia titers are expressed as log10 number of IFU per cecum, per MLN, or per gram of spleen. Data are representative of those from two separate experiments and are expressed as means ± SDs. Group means were separated using Tukey’s multiple-comparison test and were declared significantly different at a P value of <0.05 (n = 10 mice per time point). Group means that do not share a lowercase letter are significantly different.
FIG 2.

Chlamydia inclusions in ceca of p.o.-infected mice and immunization and challenge timeline. Shown are chlamydial inclusions in ceca of mice p.o. infected with 107 IFU of C. muridarum at 7 dpi. Tissues were stained with antibodies specific for Chlamydia antigens IncA (A), LOS (B), or MOMP (C) (green) and actin-binding phalloidin-Alexa Fluor 350 (blue). (D) Timeline of prime-boost immunizations and p.v. challenge with C. muridarum. Mice were primed p.o. or s.c. with live or killed Chlamydia EBs at day 0 and then s.c. boosted at day 28 with killed EBs. Two weeks later (day 42), mice were p.v. challenged with 2 × 105 IFU of C. muridarum. Six weeks after p.v. challenge (day 84), the studies were terminated and FRT pathology and Chlamydia burdens were determined. (E) Rectal swab titers of PO(K)-SC(K) and PO(L)-SC(K) mice collected at days 14, 28, and 42 prior to and 14, 28, and 42 after p.v. challenge with 2 × 105 IFU of Chlamydia.
Serum and intestinal antibody responses following immunization and p.v. Chlamydia challenge.
Prime-boost immunization influences the type, location, and the longevity of the immune response. Thus, the use of various prime-boost strategies allowed us to separately examine the role of systemic and mucosal antibodies in immunity to a Chlamydia challenge in the FRT. We hypothesized that the GALT, rather than the FRT, is the inductive site of IgA secretion not only in the GIT but also in the FRT. To address this hypothesis, serum IgG1 and IgG2c, as well as mucosal IgA responses in the FRT and the GIT, were examined. At day 28 (4 weeks after priming) none of the immunized mice from either study developed serum IgG1 titers (Fig. 3A and D). After boosting immunization (day 42), only SC(K)-SC(K) mice developed serum IgG1, while after p.v. challenge both unimmunized controls (PBS-PBS) and SC(K)-SC(K) mice developed serum IgG1 titers (Fig. 3A and D). In contrast, p.o.-immunized mice [PO(L)-SC(K), PO(K)-SC(K), and PO(L)-PBS mice] did not develop Chlamydia-specific serum IgG1 at any time point (Fig. 3A and D). Animals that were immunized p.o. with live Chlamydia or s.c. with inactivated Chlamydia developed serum IgG2c titers after priming immunization (days 28), while mice primed p.o. with inactivated Chlamydia, much like unimmunized controls, did not develop detectable serum IgG1 or IgG2c after priming or s.c. boosting (Fig. 3A, B, D, and E). After the p.v. challenge, all immunized and unimmunized animals developed serum IgG2c (Fig. 3B and E). As expected, only mice that were primed mucosally (p.o.) with live or inactivated Chlamydia developed intestinal IgA titers after priming and boosting (day 28 and 42), while all groups developed intestinal IgA following the p.v. challenge (Fig. 3C and F). Animals p.o. primed with inactivated Chlamydia developed measurable serum IgG2c antibodies only after the p.v. challenge (Fig. 3B and E), although they did develop Chlamydia-specific intestinal IgA after priming (Fig. 3C and F). IgA titers in fecal extracts of these mice were lower than in mice primed with live Chlamydia (Fig. 3C and F), likely indicating an inefficient uptake of larger noninfectious Chlamydia antigens, as opposed to the findings using antigen-carrying nanoparticles (28, 36). The presence of intestinal IgA in mice that were not inoculated p.o. with live Chlamydia [PBS-PBS, SC(K)-SC(K), and PO(K)-SC-(K) mice] at day 84 (6 weeks postchallenge) was likely due to Chlamydia dissemination to the GIT after p.v. challenge (33), or acquisition of infection via grooming or coprophagy (Fig. 3C and F).
FIG 3.

Serum and mucosal antibody titers following p.o. immunization or challenge with C. muridarum. Antibody titers in sera and intestinal extracts of mice 4 weeks after priming (day 28), 2 weeks after boosting (day 42), and 6 weeks after p.v. challenge (day 84). Two studies were conducted. In the first study, animals were fed live or killed Chlamydia EBs via a pipette, while in the second study (D to F), Chlamydia live or killed EBs were administered by gastric gavage, in order to preclude sublingual immunization. Samples from five animals from treatment group of each study were pooled and Chlamydia-specific IgG1, IgG2c, and intestinal IgA were analyzed by ELISA. Titers are expressed as log10 values of IgG1, IgG2c, or IgA antibodies detected in serum (A, B, D, and E) or fecal extracts (C and F) of mice following priming, boosting, and p.v. challenge with C. muridarum. Titers represent the highest dilutions of samples showing an absorbance value at 405 nm that is twice that of the negative control.
p.o. immunization does not affect GIT colonization by Chlamydia, but it enhances Chlamydia clearance in the FRT.
Although all p.o.-immunized mice developed Chlamydia-specific intestinal sIgA prior to p.v. challenge, the presence of these antibodies did not affect Chlamydia clearance from the GIT, as cecal Chlamydia titers were comparable among all groups in both studies (Fig. 4A and B). In contrast, mice p.o. immunized with live or inactivated EBs exhibited significantly lower Chlamydia titers in vaginal swabs starting at day 3 postchallenge and cleared the FRT Chlamydia weeks faster than SC(K)-SC(K) mice and unimmunized controls (Fig. 4C and D). In this regard, mice p.o. primed with live EBs and s.c. boosted with killed EBs [PO(L)-SC(K) mice] cleared the FRT Chlamydia faster than mice that were p.o. infected only and had significantly lower vaginal swab titers for up to day 28 postchallenge (Fig. 4C and D). Chlamydia titers in vaginal swabs of SC(K)-SC(K) mice were about 2 logs higher than for PO-SC-immunized mice at 3 dpi and were not different from the titers of unimmunized controls, indicating that serum antibodies provide no protection against primary infection. Across two studies, all SC(K)-SC(K) mice and unimmunized controls (PBS-PBS mice) (20/20) failed to completely clear FRT Chlamydia, even at day 42 post-p.v. challenge, and had comparable Chlamydia titers in vaginal swabs (Fig. 4C and D). Overall, mice primed p.o. with live or killed EBs and then boosted s.c. with killed EBs cleared Chlamydia from the FRT faster than mice from other immunization groups, including mice that were infected p.o. with live EBs alone (Fig. 4C and D).
FIG 4.

Immunization does not affect Chlamydia loads in ceca, but it enhances Chlamydia clearance from the FRT following a p.v. challenge. (A and B) Chlamydia titers in ceca of unimmunized and immunized mice at 6 weeks after p.v. challenge with 2 × 105 IFU of C. muridarum. (C and D) Chlamydia titers in vaginal swabs of mice at days 3, 7, 14, 21, 35, and 42 after p.v. challenge with 2 × 105 IFU of C. muridarum. Chlamydia titers are expressed as log10 number of IFU per cecum or per swab. Data are expressed as means ± SDs. Group means were separated using Tukey’s multiple-comparison procedures and were declared significantly different at a P value of <0.05. Means that do not share a lowercase letter are significantly different from each other.
p.o. infection or immunization with killed Chlamydia confers protection against p.v. Chlamydia challenge.
To examine whether p.o. immunization confers protection against a primary p.v. Chlamydia challenge, we examined hydrosalpinx pathology and Chlamydia burdens in the uteri and combined ovary and oviduct tissues. In the first study, we found that p.o. priming with live or killed Chlamydia, followed by s.c. boosting [PO(L)-SC(K) and PO(K)-SC(K) mice], resulted in significantly reduced hydrosalpinx scores and Chlamydia burdens in the ovaries/oviducts and uteri compared to those of unimmunized controls and SC(K)-SC(K) animals (Fig. 5A to C). In the second study, p.o.-primed mice had significantly lower hydrosalpinx pathology than did unimmunized controls and SC(K)-SC(K)-immunized animals (Fig. 5). Although not statistically significant, p.o.-primed s.c.-boosted mice had numerically lower hydrosalpinx scores and Chlamydia burdens in the ovaries/oviducts than did mice p.o. primed only [PO(L)-PBS mice]. In addition, p.o.-s.c.-immunized mice had significantly lower bacterial burdens in the uteri than did mice that were p.o. primed with live Chlamydia only [PO(L)-PBS mice], as well SC(K)-SC(K) mice and unimmunized controls (PBS-PBS mice). Although p.o. infection alone did confer protection against Chlamydia challenge, p.o.-s.c. prime-boost immunization resulted in the lowest hydrosalpinx pathology and bacterial burdens in the upper FRT (Fig. 5D to F).
FIG 5.

p.o. priming with live or killed C. muridarum significantly reduces FRT pathology and Chlamydia loads in ovaries/oviducts and uteri of mice following a p.v. challenge. (A and D) Hydrosalpinx scores and Chlamydia loads in ovaries/oviducts (B and E) and uteri (C and F) of C57BL/6J mice at 6 weeks after p.v. challenge with 2 × 105 IFU of C. muridarum. Chlamydia titers are expressed as log10 number of IFU per ovary/oviduct or uterus. Data are expressed as means ± SDs. Group means were separated using Tukey’s multiple-comparison procedures and were declared significantly different at a P value of <0.05. Means that do not share a lowercase letter are significantly different from each other.
Serum antibodies neutralize Chlamydia in vitro.
Serum antibodies, dominated by IgG isotypes, were shown to be protective against secondary chlamydial infections (14). To examine the ability of serum and intestinal antibodies to neutralize Chlamydia, HeLa cells were infected with EBs that had been preincubated with heat-inactivated serum or fecal extracts of immunized or unimmunized controls. Four weeks after immunization (day 28), sera of mice primed p.o. with live EBs or s.c. with killed EBs had neutralizing activity; however, sera of p.o.-primed mice had significantly higher neutralizing ability (Fig. 6A and D). After s.c. boosting (day 42), there were no differences in neutralization ability among the sera of p.o.- and s.c.-primed mice (Fig. 6B and E). Following the p.v. challenge, sera of all mice exhibited neutralizing ability; however, sera of mice p.o. primed with live or killed EBs had significantly higher neutralizing ability than the sera of SC(K)-SC(K) mice or unimmunized controls (PBS-PBS mice) (Fig. 6C and F). In contrast, fecal extracts of mice from all groups failed to neutralize EBs to any significant extent (neutralization, <40%). The lack of significant in vitro neutralizing activity for intestinal IgA might have been due to the extensive dilution of these antibodies in buffers during preparation of fecal extracts. Since Chlamydia does infect the GIT for prolonged periods in spite of the presence of IgA, IgA might not play an important role in Chlamydia clearance after the GIT is already infected. Moreover, it shows that secreted intestinal IgA does not prevent GIT infection following p.v. challenge, seen in PO(K)-SC(K) animals. This is likely because in these mice FRT-GIT Chlamydia spread occurs internally via migration of infected host cells (33).
FIG 6.

Serum from immunized and p.v. challenged animals neutralizes Chlamydia EBs in vitro. Neutralizing activity of sera from immunized and unimmunized control mice collected after priming (day 28) (A and D), boosting (day 42) (B and E) and p.v. challenge (C and F) was determined. Pooled serum samples were collected from the first (A to C) and second (D to F) studies. Group means were separated by Tukey’s multiple-comparison procedures. Data are shown as means ± SDs and were declared statistically significant at a P value of <0.05. Means that do not share a lowercase letter are significantly different from each other.
p.o. immunization or infection induces IgA secretion in the FRT.
Using enzyme-linked immunosorbent assay (ELISA), we failed to detect Chlamydia-specific IgA or IgG titers in vaginal washes collected throughout two studies. This could be due to collection of vaginal washes over 4 consecutive days (in 40 μl of PBS each day), which might have led to excessive dilution of mucosal secretions. Instead, Western blot analysis of serum and vaginal washes was used to detect Chlamydia-specific or major outer membrane protein (MOMP)-specific antibodies. As shown by ELISA, PO(L)-SC(K) mice developed serum IgA, IgG, and intestinal IgA after priming (day 28), boosting (day 42), and challenge (day 84) (Fig. 7A). Most importantly, these mice also developed Chlamydia-specific IgA in vaginal washes at all time points examined (Fig. 7A). PO(K)-SC(K) mice developed significant serum IgA and IgG only after p.v. challenge (day 84); however, they did develop intestinal and FRT IgA, much like PO(L)-SC(K) animals (Fig. 7A and B). SC(K)-SC(K) mice developed serum IgA and IgG at all time points examined (days 28, 42, and 84); however, they developed intestinal IgA only after p.v. challenge (day 84) and failed to develop detectable IgA in vaginal washes (Fig. 7C). Unimmunized control mice (PBS-PBS mice) had Chlamydia-specific serum IgA and IgG, as well as intestinal IgA, antibodies after the p.v. challenge (day 84). Similar to SC(K)-SC(K) mice, unimmunized controls had no detectable IgA in vaginal washes at any time point (Fig. 7D). Mice that were p.o. infected with Chlamydia without an s.c. boost [PO(L)-PBS mice] had serum and mucosal antibody profiles identical to those of PO(L)-SC(K) mice and were not included in the figure. A substantial amount of serum and mucosal antibodies appear to be specific for chlamydial MOMP antigen (molecular weight [MW], ∼40 kDa). To confirm this, serum and vaginal washes collected after boosting (day 42) or challenge (day 84) from both studies were analyzed by Western blotting using C. muridarum MOMP antigen (37). As shown using whole EBs, PO(L)-SC(K), PO(L)-PBS, and SC(K)-SC(K) mice developed MOMP-specific serum IgG at day 42 and all mice developed serum IgG following a p.v. challenge (Fig. 8A and B). In contrast, all p.o.-primed animals [PO(L)-PBS, PO(L)-SC(K), and PO(K)-SC(K) mice] were positive for MOMP-specific FRT IgA at days 42 and 84, while SC(K)-SC(K) mice and unimmunized controls had no detectable MOMP-specific IgA in the FRT after boosting (day 42) or p.v. challenge (day 84) (Fig. 8C and D).
FIG 7.

Chlamydia-specific serum, intestinal, and FRT antibodies at 4 weeks after priming (day 28), 2 weeks after boosting (day 42), and 6 weeks after p.v. challenge (day 84). For Western blot analysis, nitrocellulose membranes containing whole Chlamydia antigen were incubated with pooled serum or vaginal wash samples overnight. Serum or vaginal wash IgG or IgA antibodies bound to Chlamydia antigens on the membrane were detected with AP-conjugated rabbit anti-mouse IgG or IgA antibodies.
FIG 8.

MOMP-specific antibodies in sera and vaginal washes of immunized and control mice 2 weeks after boosting (day 42) and 6 weeks after p.v. challenge (day 84). MOMP-specific serum IgG (A and B) and vaginal wash IgA (C and D) antibodies after boosting immunization (day 42) (A and C) and after p.v. challenge (day 84) (B and D) are shown. Membranes harboring MOMP antigen were incubated overnight with pooled sera or vaginal washes (collected from two studies), and then MOMP-specific IgG and IgA antibodies were detected with AP-conjugated rabbit anti-mouse IgG or IgA antibodies.
FRT IgA neutralizes Chlamydia in vivo and reduces its FRT to GIT dissemination.
To examine the in vivo neutralizing ability of secreted IgA, Chlamydia EBs were incubated with vaginal washes collected from PO(L)-SC(K), PBS-PBS, and naive (unimmunized and unchallenged) mice and then were used to p.v. infect naive mice. Starting as early as 3 dpi and up to 7 dpi, naive mice infected with EBs incubated with vaginal wash of PO(L)-SC(K) mice exhibited significantly lower vaginal swab titers than did naive mice infected with EBs incubated in vaginal wash of naive or PBS-PBS (challenged) mice (Fig. 9A). Moreover, mice infected with EBs incubated with vaginal wash of PO(L)-SC(K) mice exhibited no enlargement of iliac lymph nodes (ILNs) (Fig. 9B) compared to those of mice infected with EBs incubated with vaginal wash of PBS-PBS and naive mice (Fig. 9C and D), indicating lower infectivity of neutralized EBs in the FRT resulting in reduced Chlamydia transport to and burden in the ILNs. Examination of Chlamydia titers in the ILNs, spleen, and GIT at 7 dpi revealed that mice infected with EBs in vaginal wash of PO(L)-SC(K) mice had lower Chlamydia burdens in the ILNs and spleen (although not statistically significant) and significantly lower Chlamydia titers in ceca (Fig. 9E to G). A second study of a similar design revealed that neutralization of EBs with vaginal wash of PO(L)-SC(K) mice reduced Chlamydia burden in the FRT of naive mice by ∼1 log within 24 h (P < 0.2) and by ∼1.5 to 2 logs by days 2 (P < 0.0002), 3 (P < 0.0007), and 7 (P < 0.0001) after p.v. challenge compared to EBs neutralized with vaginal washes of PBS-PBS (unimmunized and challenged) or naive (unimmunized and unchallenged) mice (Fig. 9H). As in the first study, there were no differences in Chlamydia titers of ILNs and spleen at 7 dpi among groups (Fig. 9I and J). Chlamydia titers in ceca of mice infected with EBs neutralized in vaginal wash of PO(L)-SC(K) mice were significantly lower than titers of other groups (Fig. 9K).
FIG 9.

IgA antibodies in vaginal washes of PO-immunized mice neutralize C. muridarum EBs and reduce systemic Chlamydia spread. (A and H) Chlamydia titers in vaginal swabs of naive mice prior to p.v. infection (day 0) and at 1, 2, 3, and 7 dpi p.v. with 106 IFU of EBs that were neutralized with either vaginal wash of naive (unimmunized and unchallenged) mice or PBS-PBS (unimmunized and p.v. challenged) and PO(L)-SC(K) mice collected 6 weeks postchallenge (day 84). (B and D) Example images of ILNs of mice infected with EBs neutralized with vaginal washes of PO(L)-SC(K) (B), PBS-PBS (C), or naive (unimmunized and unchallenged) (D) mice collected 6 weeks post-p.v. challenge (day 84). (E to G and I to K) Chlamydia titers in ILNs, spleens, and ceca of naive mice at 7 dpi p.v. with neutralized EBs as described for panel A. Data are expressed as means ± SDs. Means were separated using Tukey’s multiple-comparison procedures and are declared significantly different at a P value of <0.05 (n = 6 and n = 5). Means that do not share a lowercase letter are significantly different from each other.
DISCUSSION
The continuously increasing rates of STIs, including chlamydial infections worldwide, have brought a renewed attention and urgency for the development of effective vaccines to target these pathogens (38). Chlamydial infections are acquired mucosally; thus, an effective vaccine that induces mucosal (local) and systemic immunity would be ideal. However, the variability in immune responses in the FRT due to hormonal fluctuations and the lack of organized lymphoid tissues and antigen-sampling M cells render the FRT mucosa a poor site for immunization. Immunization via other mucosal sites, such as i.n. immunization with live EBs, does protect against Chlamydia challenge in the FRT and is routinely used as a control in animal vaccine studies (34, 39, 40). i.n. immunizations, however, may have detrimental effects on the central nervous system and may require adjuvants and multiple antigen administrations; thus, they are impractical and likely unsafe (41, 42). Therefore, p.o. immunization may be the most effective and practical route of immunization against this pathogen. In support of this notion are reports that in mice GIT infections with Chlamydia confer protection against p.v. and i.n. challenges (27, 43). Here, we show that within a week of i.n. inoculation, Chlamydia infects the GIT mucosa, which likely contributes to the protective immunity observed in i.n.-immunized mice (34, 39, 40). As shown by others (26, 27), infection of the GIT by Chlamydia does not present with signs of inflammation or pathology, while chlamydial FRT infections result in local inflammation (44), pathogen dissemination to the spleen and the GIT (33, 35, 45), and long-term upper FRT pathology, which is in large part attributed to the induction of “pathogenic” CD8+ T cells (46, 47). The very distinct immune responses that occur at these mucosal sites (GIT versus FRT) led us to hypothesize that the initial encounter of the host with this pathogen determines the long-term outcome of infection. Thus, if following p.v. infection Chlamydia disseminates systemically, pathogenic CD8+ T cells are generated, which then contribute to the long-term disease sequelae. If, on the other hand, a host’s first experience with Chlamydia is via the GIT mucosa, then infection of the spleen does not occur and immune responses induced in the GALT are protective in the FRT. Indeed, although GIT infections persist for prolonged periods, Chlamydia does not reach the spleen, indicating that the GALT serves as a firewall that limits Chlamydia systemic dissemination.
The importance of CD4+ T cells in protection against Chlamydia challenge is well established. Mucosa-associated lymphoid tissues are the inducible sites of Chlamydia-specific CD4 T cells, which then provide protection to Chlamydia challenge in the FRT (48). The role of B cells and antibodies, on the other hand, is poorly understood. The synergistic action of antibodies and CD4+ T cells was demonstrated in early studies in which depletion of CD4+ T cells prolonged the resolution of a secondary infection, while depletion of CD4+ T cells in B cell-deficient mice resulted in failure resolve the secondary infection (15). B cells have been implicated in the expansion of Chlamydia-specific CD4 T cells in the FRT, as well as in Chlamydia systemic dissemination (49), which is inhibited by passive transfer of immune serum (50). Based on our previous work (28), we hypothesized that IgA secretion in the FRT is induced in the GALT and protects against primary Chlamydia infection and that Chlamydia need not be viable to induce immunity in the FRT (29, 30, 51). Prime-boost immunization strategies used in this study were based on reports that mucosal priming followed by systemic boosting confers superior protection against Chlamydia challenge (40) and enhances the magnitude and duration of antibody responses (28–30). We show that the type of Chlamydia antigen (live or inactivated/killed), the route of infection (p.o. or p.v.), and prime-boost immunization approaches affect the magnitude, diversity, and location of antibody secretion. In agreement with reports that serum antibodies do not protect against primary infection (14), we show that sera of SC(K)-SC(K) mice with an antigen cognition profile and neutralizing ability similar to those of sera of PO(L)-SC(K) animals (Fig. 7) conferred no protection against p.v. challenge. In addition, PO(K)-SC(K) mice had no substantial serum antibodies prior to challenge yet exhibited significantly lower Chlamydia burden in the FRT, starting at 3 dpi, which is an insufficient time for induction of serum antibodies.
As expected, all p.o.-primed mice developed Chlamydia-specific intestinal IgA (26, 27), which did not clear Chlamydia from the GIT, as there were no differences in cecal Chlamydia titers among groups (Fig. 4A and B). In contrast, unimmunized controls and SC(K)-SC(K) mice developed intestinal IgA only after p.v. challenge (Fig. 3, 7, and 8). Most importantly, all p.o.-immunized mice also had Chlamydia-specific IgA in vaginal washes after priming, and in contrast, unimmunized and SC(K)-SC(K) mice had no detectable Chlamydia-specific IgA at any time point (Fig. 7 and 8). Others reported the presence of Chlamydia-specific IgA in vaginal washes within 1 to 4 weeks of infection (52–54), which is in apparent disagreement with our results. This could be because in these studies, ELISAs were used for measuring IgA (53, 54), which in our hands lacked the sensitivity to detect low IgA concentrations in diluted vaginal secretions and was unreliable. While in one study (54), the presence of Chlamydia-specific IgA in vaginal washes was shown by Western blotting at 50 dpi p.v., others did not examine the specificity of IgA in p.v.-infected mice (52); thus, it is difficult to compare our results with the results of these particular studies. If Chlamydia-specific IgA is present in the FRT a few weeks after an infection, its concentrations are likely very low and biologically insignificant for protection against primary infection. Western blot analysis, used in this study, allows detection of small amounts of antibodies, as well as the specificity of antibodies to Chlamydia antigens. Importantly, Chlamydia-specific IgA profiles in the FRT prior to p.v. challenge mirror the intestinal IgA profiles, indicating that GALT is the inductive site for IgA secretion in the FRT. Our report that p.v. antigen administration induces intestinal, but not FRT, IgA antibodies (28), as observed in SC(K)-SC(K) and unimmunized mice after p.v. challenge, supports this notion. In unimmunized controls, Chlamydia disseminates internally to the GIT (33), thus inducing serum IgA and IgG, as well as IgA secretion in the GIT. Serum IgA is not the source of intestinal IgA, as SC(K)-SC(K) mice developed serum IgA after priming yet exhibited intestinal IgA only after p.v. Chlamydia challenge (Fig. 7C). The lack of detectable IgA in FRT secretions of SC(K)-SC(K) mice, in spite of IgA presence in serum, and the presence of IgA in FRT secretions of PO(K)-SC(K) mice, in spite of the lack of serum IgA, indicates that IgA is produced by local B cells in the FRT. Others have shown that expression of polymeric immunoglobulin receptor (pIgR), IgA, and IgG secretion in the FRT is upregulated during Chlamydia infection (52). However, since the specificity of IgA and IgG antibodies was not examined, their significance for Chlamydia neutralization or clearance is unclear. The lack of Chlamydia-specific IgA in vaginal washes of unimmunized mice after p.v. infection additionally explains why B cell- and IgA-deficient mice clear primary Chlamydia infection at rates similar to those of wild-type mice (15, 20).
We argue that secreted Chlamydia-specific IgA in the FRT is critical for protection against Chlamydia infection. Neutralization of Chlamydia EBs in the FRT by IgA is the likely reason for the drastic reduction of the infectious burden in naive mice as early as 1 to 3 dpi. In addition, neutralization of EBs hinders systemic dissemination of Chlamydia, reflected in significantly reduced Chlamydia titers in ceca at 7 dpi. Neutralization of Chlamydia in the FRT by secreted IgA can additionally inhibit Chlamydia ascension to the upper FRT, thus diminishing or preventing the inflammatory response and tissue damage. In women, C. trachomatis-specific IgA found in FRT secretions is critically important for pathogen clearance (22), which is in agreement with our findings. This is not to suggest that CD4+ T cells are not critical for protective immunity, including humoral immunity provided by B cells in the form of antibodies.
Our findings can be summed up in four main conclusions: (i) infection via the GIT or the FRT mucosa induces Chlamydia-specific serum IgG, with p.o. infection inducing primarily IgG2c and p.v. infection inducing IgG1 and IgG2c; (ii) p.o. immunization or infection induces IgA secretion in the GIT and the FRT; (iii) p.v. infection induces serum and intestinal IgA but fails to induce IgA secretion in the FRT; and (iv) secreted FRT IgA antibodies neutralize Chlamydia and are critically important for protection against Chlamydia challenge. Mucosal sites differ in their physiological functions, and consequently, the immune responses reflect these functions. Unlike the FRT (33), GALT halts the systemic dissemination of Chlamydia and induces IgA secretion locally (in the GIT) and at a distant site (the FRT). Thus, GALT appears to be the bona fide mucosal site for induction of IgA secretion in the FRT. Secreted IgA neutralizes Chlamydia in the FRT, thus lowering its infectivity, accelerating its clearance, and reducing its systemic spread. Importantly, infection with and replication of Chlamydia in the GIT are not necessary for induction of protective immunity in the FRT. This work will open the doors for the development of p.o. subunit vaccines to target sexually transmitted pathogens, as well as for beginning to understand how the GALT regulates the immune responses in the FRT.
MATERIALS AND METHODS
Ethics statement.
All animal experiments were carried out in strict accordance with the recommendations of the Guide for the Care and Use of Laboratory Animals (55). All procedures involving animals were approved by the Southern Illinois University’s Institutional Animal Care and Use Committee (protocols 15-035 and 15-041).
Animals and housing.
Six- to 8-week-old C57BL/6 female mice were purchased from the Jackson Laboratory (Bar Harbor, ME). Animals were housed in ventilated cages under specific-pathogen-free conditions with food and water provided ad libitum. To prevent infection of unimmunized controls and mice p.o. immunized with killed EBs, cages were changed one at a time under a biosafety flow hood.
Cell and bacterial culture conditions.
HeLa 229 cells (human cervical carcinoma epithelial cells) were grown in Dulbecco’s modified Eagle medium (DMEM) supplemented with 10% fetal bovine serum (DMEM-10) and 50 μg/ml of gentamicin in a humidified incubator at 37°C and 5% CO2. Chlamydia muridarum strain Nigg was propagated in and purified from HeLa 229 cells as described previously (56). Confluent HeLa cell monolayers were infected with C. muridarum by centrifugation for 1 h at 545 × g and room temperature (RT) (or by rocking for 2 h at 37°C) and incubated in infection medium (DMEM-10 supplemented with nonessential amino acids and 1 μg/ml of cycloheximide). At 40 to 42 h postinfection, HeLa cells were ruptured by brief sonication and cell debris was pelleted by low-speed centrifugation (500 × g for 10 min at 4°C). EBs were pelleted from the supernatant by high-speed centrifugation (10,000 × g for 30 min at 4°C) and purified on a Percoll gradient by centrifugation at 30,000 × g, as described previously (57). All C. muridarum EB stocks were stored at −80°C in sucrose-phosphate-buffered glutamic acid (SPG) until used.
Inactivation of C. muridarum.
Gradient-purified C. muridarum EBs were thawed on ice and then fixed in 4% paraformaldehyde (PFA) for 15 min. Fixed EBs were pelleted by high-speed centrifugation (13,000 × g) and washed three times in PBS. After the final wash, EBs were resuspended in PBS and sonicated on ice in order to generate smaller nanoparticle-sized antigen. To confirm that EBs were not infectious, aliquots of 200 μl (containing 1 × 108 IFU) taken from the prepared stock (which had 5 × 108 IFU/ml) were used to infect HeLa cell monolayers in triplicates, and the infection was expanded six times. No IFU were detected in HeLa cell monolayers infected with PFA-inactivated C. muridarum EBs.
Immunization of mice with C. muridarum.
Two studies were conducted in which mice were p.o. or s.c. primed with either viable (live) or inactivated (killed) C. muridarum EBs. In the first study, mice were fasted for 4 h and p.o. fed 1 × 106 IFU of live or 1 × 107 IFUs of killed C. muridarum in 20 μl of SPG (via a pipette tip). In the second study the same doses of live or killed Chlamydia EBs were administered via a gastric gavage using a round-tip needle to ensure immunization via GALT and not sublingually. For s.c. immunizations, mice were injected with 1 × 107 IFU of killed Chlamydia on their backs. To examine the dynamics of GIT infection, separate groups of mice were infected via gastric gavage with 1 × 107 or i.n. with 1 × 104 IFU of Chlamydia. For i.n. infections, mice were lightly anesthetized and infected with a total volume of 5 μl by applying 2.5 μl of the inoculum to each nare with a pipette tip.
Per-vaginal C. muridarum challenge.
In order to synchronize their estrus cycles, mice were s.c. injected with 2.5 mg of medroxyprogesterone acetate (MPA; Henry Schein) 5 days prior to p.v. challenge with 2 × 105 C. muridarum IFU delivered in 10 μl of SPG (at day 42 post-priming immunization). To monitor Chlamydia shedding, vaginal swabs were collected prior to p.v. infection, 3 days after the infection, and weekly thereafter until the study was terminated. To ensure consistency in immunization and p.v. challenge doses, all animals were primed, boosted, and p.v. challenged from a single, pretitered C. muridarum stock.
Collection of sera, fecal extracts, and vaginal washes.
Blood, fecal extracts, and vaginal washes were collected prior to immunization and weekly thereafter for the duration of the studies. Blood was collected via the tail vein, and plasma was separated by centrifugation. Fecal extracts were prepared as described previously (28, 29) and stored at −20°C until analysis. Vaginal washes were collected in 40 μl of PBS daily over 4 days. Samples were first collected from unimmunized controls (PBS-PBS mice), PO(K)-SC(K) mice, and then from groups that were p.o. infected with live Chlamydia [PO(L)-PBS and PO(L)-SC(K) mice]. This was done to preclude accidental infection of unimmunized controls (PBS-PBS mice) and PO(K)-SC(K) mice prior to p.v. challenge.
Determination of Chlamydia-specific serum and mucosal antibody titers and Western blot analysis of antibody specificity.
Antibody titers in sera, fecal extracts, and vaginal washes of mice were measured using ELISAs. EBs were rendered nonviable as described above, and flat-bottomed 96-well plates were coated with EB antigen at 10 μg/ml suspended in ELISA coating buffer. Coated plates were incubated overnight at 4°C. Unbound Chlamydia antigen was then removed from the wells, and plates were blocked for 1 h at 37°C with 200 μl of ELISA blocking buffer (0.2% porcine gelatin in 1× PBS). Plates were then washed with ELISA wash buffer before the addition of samples (serum, vaginal washes, or fecal extracts). Following an overnight incubation, plates were washed and monoclonal alkaline phosphatase (AP)-conjugated goat anti-mouse IgG1, IgG2c, or IgA antibodies diluted in blocking buffer were added and allowed to incubate at RT for 2 h. Plates were washed again in ELISA wash buffer, and AP activity was determined as described previously (28, 29, 58). Antibody titers are expressed as log10 values of the highest serial dilution that yielded an optical density at 405 nm (OD405) that was twice that of negative controls. The presence and specificity of antibodies in sera, fecal extracts, and vaginal washes were also determined by Western blot analysis, for which 10 μg of gradient-purified Chlamydia EB protein was loaded per well. Similarly, sera, intestinal extracts, and vaginal washes were examined for the presence of C. muridarum MOMP.
Determination of C. muridarum titers in tissues.
Tissues were excised aseptically from mice at different times postinfection and placed in sterile tubes containing SPG. Tissues were then processed and Chlamydia titers determined as previously described (26, 33, 59). Titers are expressed as log10 IFU per vaginal swab, uterus, ovary/oviduct, cecum, or iliac lymph node (ILN) or per g of spleen. Chlamydia-negative samples were expanded for six passages to ensure that low titers could be detected. Negative cultures at passage 1 remained negative throughout the 6 expansions. All reported titers are from the first passage.
Evaluation of FRT pathology.
The FRTs were excised aseptically, imaged, and scored in order to evaluate the extent of pathology according to an ordinal scale as described previously (60). Hydrosalpinx severity scores are reported as average bilateral scores per mouse in all treatment groups.
In vitro C. muridarum neutralization assays.
Plasma and fecal extracts from immunized and naive mice were assayed for in vitro antibody-mediated neutralization. In order to exclude any fecal debris, fecal extracts were passed through a premoistened 0.45-μm filter. Briefly, fecal extracts or complement-inactivated sera were incubated with 1 × 103 C. muridarum IFU for 1 h at 37°C and 5% CO2 prior to infection of monolayers in 96-well plates (61). At 24 h postinfection (hpi), cells were fixed in ice-cold absolute methanol and IFU were numerated. Percentage neutralization is reported as percent reduction of IFU by a test serum compared to naive sera or fecal extracts.
In vivo C. muridarum neutralization.
To examine whether antibodies found in vaginal washes can neutralize Chlamydia in vivo, EBs were incubated on ice for 30 min with pooled vaginal washes of naive mice (unimmunized and unchallenged), PBS-PBS mice (unimmunized and challenged) or PO(L)-SC(K) mice, collected at 6 weeks postchallenge (day 84). After incubation, 10 μl of EB and vaginal wash mixture containing 1 × 106 IFU of Chlamydia was used to p.v. infect groups of 6 naive mice. Vaginal swabs were collected at 3 and 7 days postinfection (dpi), at which time tissues were collected for examining whether neutralization affected Chlamydia dissemination to the GIT. For this study, mice were housed in wire mesh cages and were fitted with neck collars in order to preclude GIT infection via grooming as described previously (33). An identical study was conducted to examine the in vivo C. muridarum neutralization at early stages of infection (days 1 to 7).
Staining and analysis of frozen tissue sections by IFM.
Excised tissues were snap-frozen in Tissue-Tek OCT compound (Sakura Finetek) on dry ice and stored at −80°C. Cryosections (5 to 7 μm thick) were fixed in 4% PFA and stained with monoclonal antibodies and actin-binding phalloidin-Alexa Fluor 350 for 1 to 2 h. All antibodies were used at a dilution of 1:100. Stained tissue sections were washed in PBS and mounted using Fluoromount-G (Southern Biotech), and images were acquired and analyzed as described previously (30, 62).
Statistical analysis.
All data were analyzed using analysis of variance (ANOVA) procedures and SAS software. Population means were separated using Student’s t test or Tukey’s multiple-comparison procedures and were declared significantly different at a P value of <0.05.
ACKNOWLEDGMENTS
We express our gratitude to Kyle Ramsey (Midwestern University, Downers Grove, IL) for providing detailed protocols for Chlamydia titer determination, purification, and genotyping. We also thank Matthew Coleman (Lawrence Livermore National Laboratories; U19AI144184) for providing MOMP antigen for Western blot analysis.
This work was supported by grants to V.K. (R21AI133062) from the National Institutes of Health and to D.R. (DMR-1757954) from the National Science Foundation.
REFERENCES
- 1.WHO. 2019. Sexually transmitted infections (STIs). https://www.who.int/en/news-room/fact-sheets/detail/sexually-transmitted-infections-(stis). Accessed 16 June 2019.
- 2.CDC. 2018. Sexually transmitted disease surveillance 2018. https://www.cdc.gov/std/stats18/default.htm.
- 3.Brunham RC, Gottlieb SL, Paavonen J. 2015. Pelvic inflammatory disease. N Engl J Med 372:2039–2048. doi: 10.1056/NEJMra1411426. [DOI] [PubMed] [Google Scholar]
- 4.Soper DE, Brockwell NJ, Dalton HP. 1992. Microbial etiology of urban emergency department acute salpingitis: treatment with ofloxacin. Am J Obstet Gynecol 167:653–660. doi: 10.1016/s0002-9378(11)91566-x. [DOI] [PubMed] [Google Scholar]
- 5.Haggerty CL, Gottlieb SL, Taylor BD, Low N, Xu F, Ness RB. 2010. Risk of sequelae after Chlamydia trachomatis genital infection in women. J Infect Dis 201(Suppl 2):S134–S155. doi: 10.1086/652395. [DOI] [PubMed] [Google Scholar]
- 6.Madeleine MM, Anttila T, Schwartz SM, Saikku P, Leinonen M, Carter JJ, Wurscher M, Johnson LG, Galloway DA, Daling JR. 2007. Risk of cervical cancer associated with Chlamydia trachomatis antibodies by histology, HPV type and HPV cofactors. Int J Cancer 120:650–655. doi: 10.1002/ijc.22325. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Schust DJ, Ibana JA, Buckner LR, Ficarra M, Sugimoto J, Amedee AM, Quayle AJ. 2012. Potential mechanisms for increased HIV-1 transmission across the endocervical epithelium during C. trachomatis infection. Curr HIV Res 10:218–227. doi: 10.2174/157016212800618093. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Schiller JT, Castellsague X, Garland SM. 2012. A review of clinical trials of human papillomavirus prophylactic vaccines. Vaccine 30(Suppl 5):F123–F138. doi: 10.1016/j.vaccine.2012.04.108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Landers DV, Erlich K, Sung M, Schachter J. 1991. Role of L3T4-bearing T-cell populations in experimental murine chlamydial salpingitis. Infect Immun 59:3774–3777. doi: 10.1128/IAI.59.10.3774-3777.1991. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Morrison RP, Feilzer K, Tumas DB. 1995. Gene knockout mice establish a primary protective role for major histocompatibility complex class II-restricted responses in Chlamydia trachomatis genital tract infection. Infect Immun 63:4661–4668. doi: 10.1128/IAI.63.12.4661-4668.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Morrison SG, Morrison RP. 2001. Resolution of secondary Chlamydia trachomatis genital tract infection in immune mice with depletion of both CD4+ and CD8+ T cells. Infect Immun 69:2643–2649. doi: 10.1128/IAI.69.4.2643-2649.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Su H, Caldwell HD. 1995. CD4+ T cells play a significant role in adoptive immunity to Chlamydia trachomatis infection of the mouse genital tract. Infect Immun 63:3302–3308. doi: 10.1128/IAI.63.9.3302-3308.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Morrison SG, Su H, Caldwell HD, Morrison RP. 2000. Immunity to murine Chlamydia trachomatis genital tract reinfection involves B cells and CD4(+) T cells but not CD8(+) T cells. Infect Immun 68:6979–6987. doi: 10.1128/iai.68.12.6979-6987.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Morrison SG, Morrison RP. 2005. A predominant role for antibody in acquired immunity to chlamydial genital tract reinfection. J Immunol 175:7536–7542. doi: 10.4049/jimmunol.175.11.7536. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Morrison SG, Morrison RP. 2005. The protective effect of antibody in immunity to murine chlamydial genital tract reinfection is independent of immunoglobulin A. Infect Immun 73:6183–6186. doi: 10.1128/IAI.73.9.6183-6186.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Barron AL, Rank RG, Moses EB. 1984. Immune response in mice infected in the genital tract with mouse pneumonitis agent (Chlamydia trachomatis biovar). Infect Immun 44:82–85. doi: 10.1128/IAI.44.1.82-85.1984. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Batteiger BE, Rank RG. 1987. Analysis of the humoral immune response to chlamydial genital infection in guinea pigs. Infect Immun 55:1767–1773. doi: 10.1128/IAI.55.8.1767-1773.1987. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Rank RG, Barron AL. 1983. Humoral immune response in acquired immunity to chlamydial genital infection of female guinea pigs. Infect Immun 39:463–465. doi: 10.1128/IAI.39.1.463-465.1983. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Rank RG, Batteiger BE. 1989. Protective role of serum antibody in immunity to chlamydial genital infection. Infect Immun 57:299–301. doi: 10.1128/IAI.57.1.299-301.1989. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Su H, Feilzer K, Caldwell HD, Morrison RP. 1997. Chlamydia trachomatis genital tract infection of antibody-deficient gene knockout mice. Infect Immun 65:1993–1999. doi: 10.1128/IAI.65.6.1993-1999.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Ramsey KH, Soderberg LS, Rank RG. 1988. Resolution of chlamydial genital infection in B-cell-deficient mice and immunity to reinfection. Infect Immun 56:1320–1325. doi: 10.1128/IAI.56.5.1320-1325.1988. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Brunham RC, Kuo CC, Cles L, Holmes KK. 1983. Correlation of host immune response with quantitative recovery of Chlamydia trachomatis from the human endocervix. Infect Immun 39:1491–1494. doi: 10.1128/IAI.39.3.1491-1494.1983. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.de la Maza LM, Pal S, Khamesipour A, Peterson EM. 1994. Intravaginal inoculation of mice with the Chlamydia trachomatis mouse pneumonitis biovar results in infertility. Infect Immun 62:2094–2097. doi: 10.1128/IAI.62.5.2094-2097.1994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Shah AA, Schripsema JH, Imtiaz MT, Sigar IM, Kasimos J, Matos PG, Inouye S, Ramsey KH. 2005. Histopathologic changes related to fibrotic oviduct occlusion after genital tract infection of mice with Chlamydia muridarum. Sex Transm Dis 32:49–56. doi: 10.1097/01.olq.0000148299.14513.11. [DOI] [PubMed] [Google Scholar]
- 25.Rank RG, Yeruva L. 2014. Hidden in plain sight: chlamydial gastrointestinal infection and its relevance to persistence in human genital infection. Infect Immun 82:1362–1371. doi: 10.1128/IAI.01244-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Yeruva L, Spencer N, Bowlin AK, Wang Y, Rank RG. 2013. Chlamydial infection of the gastrointestinal tract: a reservoir for persistent infection. Pathog Dis 68:88–95. doi: 10.1111/2049-632X.12052. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Wang L, Zhu C, Zhang T, Tian Q, Zhang N, Morrison S, Morrison R, Xue M, Zhong G. 2018. Nonpathogenic colonization with Chlamydia in the gastrointestinal tract as oral vaccination for inducing transmucosal protection. Infect Immun 86:e00630-17. doi: 10.1128/IAI.00630-17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Howe SE, Konjufca VH. 2014. Protein-coated nanoparticles are internalized by the epithelial cells of the female reproductive tract and induce systemic and mucosal immune responses. PLoS One 9:e114601. doi: 10.1371/journal.pone.0114601. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Howe SE, Konjufca VH. 2015. Per-oral immunization with antigen-conjugated nanoparticles followed by sub-cutaneous boosting immunization induces long-lasting mucosal and systemic antibody responses in mice. PLoS One 10:e0118067. doi: 10.1371/journal.pone.0118067. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Howe SE, Sowa G, Konjufca V. 2016. Systemic and mucosal antibody responses to soluble and nanoparticle-conjugated antigens administered intranasally. Antibodies 5:20. doi: 10.3390/antib5040020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Gratrix J, Singh AE, Bergman J, Egan C, Plitt SS, McGinnis J, Bell CA, Drews SJ, Read R. 2015. Evidence for increased Chlamydia case finding after the introduction of rectal screening among women attending 2 Canadian sexually transmitted infection clinics. Clin Infect Dis 60:398–404. doi: 10.1093/cid/ciu831. [DOI] [PubMed] [Google Scholar]
- 32.Jones RB, Rabinovitch RA, Katz BP, Batteiger BE, Quinn TS, Terho P, Lapworth MA. 1985. Chlamydia trachomatis in the pharynx and rectum of heterosexual patients at risk for genital infection. Ann Intern Med 102:757–762. doi: 10.7326/0003-4819-102-6-757. [DOI] [PubMed] [Google Scholar]
- 33.Howe SE, Shillova N, Konjufca V. 2019. Dissemination of Chlamydia from the reproductive tract to the gastro-intestinal tract occurs in stages and relies on Chlamydia transport by host cells. PLoS Pathog 15:e1008207. doi: 10.1371/journal.ppat.1008207. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Pal S, Fielder TJ, Peterson EM, de la Maza LM. 1994. Protection against infertility in a BALB/c mouse salpingitis model by intranasal immunization with the mouse pneumonitis biovar of Chlamydia trachomatis. Infect Immun 62:3354–3362. doi: 10.1128/IAI.62.8.3354-3362.1994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Cotter TW, Ramsey KH, Miranpuri GS, Poulsen CE, Byrne GI. 1997. Dissemination of Chlamydia trachomatis chronic genital tract infection in gamma interferon gene knockout mice. Infect Immun 65:2145–2152. doi: 10.1128/IAI.65.6.2145-2152.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Howe SE, Lickteig DJ, Plunkett KN, Ryerse JS, Konjufca V. 2014. The uptake of soluble and particulate antigens by epithelial cells in the mouse small intestine. PLoS One 9:e86656. doi: 10.1371/journal.pone.0086656. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.He W, Felderman M, Evans AC, Geng J, Homan D, Bourguet F, Fischer NO, Li YP, Lam KS, Noy A, Xing L, Cheng RH, Rasley A, Blanchette CD, Kamrud K, Wang N, Gouvis H, Peterson TC, Hubby B, Coleman MA. 2017. Cell-free production of a functional oligomeric form of a Chlamydia major outer-membrane protein (MOMP) for vaccine development. J Biol Chem 292:15121–15132. doi: 10.1074/jbc.M117.784561. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Eisinger RW, Erbelding E, Fauci AS. 9 September 2019. Refocusing research on sexually transmitted infections. J Infect Dis doi: 10.1093/infdis/jiz442. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Tifrea DF, Pal S, Popot JL, Cocco MJ, de la Maza LM. 2014. Increased immunoaccessibility of MOMP epitopes in a vaccine formulated with amphipols may account for the very robust protection elicited against a vaginal challenge with Chlamydia muridarum. J Immunol 192:5201–5213. doi: 10.4049/jimmunol.1303392. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Carmichael JR, Pal S, Tifrea D, de la Maza LM. 2011. Induction of protection against vaginal shedding and infertility by a recombinant Chlamydia vaccine. Vaccine 29:5276–5283. doi: 10.1016/j.vaccine.2011.05.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Mutsch M, Zhou W, Rhodes P, Bopp M, Chen RT, Linder T, Spyr C, Steffen R. 2004. Use of the inactivated intranasal influenza vaccine and the risk of Bell’s palsy in Switzerland. N Engl J Med 350:896–903. doi: 10.1056/NEJMoa030595. [DOI] [PubMed] [Google Scholar]
- 42.Jiang JN, Liu GC, Kickhoefer VA, Rome LH, Li LX, McSorley SJ, Kelly KA. 2017. A protective vaccine against Chlamydia genital infection using vault nanoparticles without an added adjuvant. Vaccines 5:3. doi: 10.3390/vaccines5010003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Zhu C, Lin H, Tang L, Chen J, Wu Y, Zhong G. 2018. Oral Chlamydia vaccination induces transmucosal protection in the airway. Vaccine 36:2061–2068. doi: 10.1016/j.vaccine.2018.03.015. [DOI] [PubMed] [Google Scholar]
- 44.Darville T, Andrews CW, Jr, Laffoon KK, Shymasani W, Kishen LR, Rank RG. 1997. Mouse strain-dependent variation in the course and outcome of chlamydial genital tract infection is associated with differences in host response. Infect Immun 65:3065–3073. doi: 10.1128/IAI.65.8.3065-3073.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Zhang Q, Huang Y, Gong S, Yang Z, Sun X, Schenken R, Zhong G. 2015. In vivo and ex vivo imaging reveals a long-lasting chlamydial infection in the mouse gastrointestinal tract following genital tract inoculation. Infect Immun 83:3568–3577. doi: 10.1128/IAI.00673-15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Vlcek KR, Li W, Manam S, Zanotti B, Nicholson BJ, Ramsey KH, Murthy AK. 2016. The contribution of Chlamydia-specific CD8(+) T cells to upper genital tract pathology. Immunol Cell Biol 94:208–212. doi: 10.1038/icb.2015.74. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Zhong G. 2018. Chlamydia spreading from the genital tract to the gastrointestinal tract—a two-hit hypothesis. Trends Microbiol 26:611–623. doi: 10.1016/j.tim.2017.12.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Stary G, Olive A, Radovic-Moreno AF, Gondek D, Alvarez D, Basto PA, Perro M, Vrbanac VD, Tager AM, Shi JJ, Yethon JA, Farokhzad OC, Langer R, Starnbach MN, von Andrian UH. 2015. A mucosal vaccine against Chlamydia trachomatis generates two waves of protective memory T cells. Science 348:aaa8205. doi: 10.1126/science.aaa8205. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Li LX, McSorley SJ. 2013. B cells enhance antigen-specific CD4 T cell priming and prevent bacteria dissemination following Chlamydia muridarum genital tract infection. PLoS Pathog 9:e1003707. doi: 10.1371/journal.ppat.1003707. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Malaviarachchi PA, Mercado MAB, McSorley SJ, Li LX. 2020. Antibody, but not B-cell-dependent antigen presentation, plays an essential role in preventing Chlamydia systemic dissemination in mice. Eur J Immunol 50:676–684. doi: 10.1002/eji.201948391. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Jenkins MC, Stevens L, O'Brien C, Parker C, Miska K, Konjufca V. 2018. Incorporation of a recombinant Eimeria maxima IMP1 antigen into nanoparticles confers protective immunity against E. Maxima challenge infection. Vaccine 36:1126–1131. doi: 10.1016/j.vaccine.2017.11.014. [DOI] [PubMed] [Google Scholar]
- 52.Armitage CW, O’Meara CP, Beagley KW. 2017. Chlamydial infection enhances expression of the polymeric immunoglobulin receptor (pIgR) and transcytosis of IgA. Am J Reprod Immunol 77:e12611. doi: 10.1111/aji.12611. [DOI] [PubMed] [Google Scholar]
- 53.Morrison SG, Farris CM, Sturdevant GL, Whitmire WM, Morrison RP. 2011. Murine Chlamydia trachomatis genital infection is unaltered by depletion of CD4+ T cells and diminished adaptive immunity. J Infect Dis 203:1120–1128. doi: 10.1093/infdis/jiq176. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Ramsey KH, Newhall WJt, Rank RG. 1989. Humoral immune response to chlamydial genital infection of mice with the agent of mouse pneumonitis. Infect Immun 57:2441–2446. doi: 10.1128/IAI.57.8.2441-2446.1989. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.National Research Council. 2011. Guide for the care and use of laboratory animals, 8th ed. National Academies Press, Washington, DC. [Google Scholar]
- 56.Batteiger BE, Newhall WJ, V, Jones RB. 1985. Differences in outer membrane proteins of the lymphogranuloma venereum and trachoma biovars of Chlamydia trachomatis. Infect Immun 50:488–494. doi: 10.1128/IAI.50.2.488-494.1985. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Newhall WJ, Batteiger B, Jones RB. 1982. Analysis of the human serological response to proteins of Chlamydia trachomatis. Infect Immun 38:1181–1189. doi: 10.1128/IAI.38.3.1181-1189.1982. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Tifrea DF, Ralli-Jain P, Pal S, de la Maza LM. 2013. Vaccination with the recombinant major outer membrane protein elicits antibodies to the constant domains and induces cross-serovar protection against intranasal challenge with Chlamydia trachomatis. Infect Immun 81:1741–1750. doi: 10.1128/IAI.00734-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Scidmore MA. 2005. Cultivation and laboratory maintenance of Chlamydia trachomatis. Curr Protoc Microbiol Chapter 11:Unit 11A.1. [DOI] [PubMed] [Google Scholar]
- 60.Conrad TA, Gong S, Yang Z, Matulich P, Keck J, Beltrami N, Chen C, Zhou Z, Dai J, Zhong G. 2016. The chromosome-encoded hypothetical protein TC0668 is an upper genital tract pathogenicity factor of Chlamydia muridarum. Infect Immun 84:467–479. doi: 10.1128/IAI.01171-15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.O’Meara CP, Armitage CW, Harvie MC, Timms P, Lycke NY, Beagley KW. 2013. Immunization with a MOMP-based vaccine protects mice against a pulmonary Chlamydia challenge and identifies a disconnection between infection and pathology. PLoS One 8:e61962. doi: 10.1371/journal.pone.0061962. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Rosche KL, Aljasham AT, Kipfer JN, Piatkowski BT, Konjufca V. 2015. Infection with Salmonella enterica serovar Typhimurium leads to increased proportions of F4/80+ red pulp macrophages and decreased proportions of B and T lymphocytes in the spleen. PLoS One 10:e0130092. doi: 10.1371/journal.pone.0130092. [DOI] [PMC free article] [PubMed] [Google Scholar]