

Abstract
One goal of microbial ecology researchers is to capture the maximum amount of information from all organisms in a sample. The recent COVID-19 pandemic, caused by the RNA virus SARS-CoV-2, has highlighted a gap in traditional DNA-based protocols, including the high-throughput methods the authors previously established as field standards. To enable simultaneous SARS-CoV-2 and microbial community profiling, the authors compared the relative performance of two total nucleic acid extraction protocols with the authors' previously benchmarked protocol. The authors included a diverse panel of environmental and host-associated sample types, including body sites commonly swabbed for COVID-19 testing. Here the authors present results comparing the cost, processing time, DNA and RNA yield, microbial community composition, limit of detection and well-to-well contamination between these protocols.
Keywords: : 16S rRNA, DNA extraction, high-throughput sequencing, limit of detection, microbial community, microbiome, RNA extraction, shotgun metagenomics, well-to-well contamination
Accession numbers
Raw sequence data were deposited at the European Nucleotide Archive (accession number ERP124610), and raw and processed data are available at Qiita (study identifier 12201). Processing and analysis code is available on GitHub (https://github.com/justinshaffer/Extraction_test_MagMAX).
METHOD SUMMARY
To allow for downstream applications involving RNA-based organisms such as SARS-CoV-2, the authors compared the two extraction protocols designed to extract DNA and RNA with the authors' previously established protocol for extracting only DNA for microbial community analyses. Across ten diverse sample types, one of the two protocols was equivalent or better than the authors' established DNA-based protocol. The authors' conclusion is based on per-sample comparisons of DNA and RNA yield, number of quality sequences generated, microbial community alpha- and beta-diversity and taxonomic composition, limit of detection and extent of well-to-well contamination.
Our growing understanding of microbial communities continues to reveal knowledge important for fostering human and environmental sustainability [1–4]. Nearly every day, new links are made between the human microbiome and human health [5–7], and the development of methods related to studying microbial communities is ever-expanding [8–10]. One major roadblock to studying microbial communities is that single methods rarely capture information from all organisms in a sample or from across diverse sample types [11–13].
The ongoing COVID-19 pandemic driven by SARS-CoV-2 has infected over 40 million human individuals and killed 1.1 million (as of 18 October 2020) [14]. Such an event represents an invaluable opportunity to study the effects of a novel pathogen on microbial interactions relevant to human hosts and other ecosystems [15–17]. Currently, the authors' protocol benchmarked for high-throughput microbiome sequencing focuses on extracting high-quality DNA from samples [18] and therefore will not capture RNA-based genomes such as that of SARS-CoV-2, which is a positive-sense, single-stranded RNA virus [19].
Here the authors aim was to identify an extraction protocol that extracts high-quality RNA while also producing DNA output and community composition comparable to the authors' previously benchmarked protocol [18]. The authors also considered technical differences regarding the detection ability [20] and extent of contamination [21–23] among protocols. The authors compared DNA and RNA yield, number of quality sequences, microbial community alpha- and beta-diversity and taxonomic composition, limit of detection (LOD) and extent of well-to-well contamination across common sample types and among three extraction protocols.
Methods
Sample collection
To compare extraction protocols, the authors collected biological materials from a broad range of human and environmental samples, focusing on types widely used in studies of microbial communities and SARS-CoV-2 detection [18,24,25]. Each unique sample was aliquoted across extraction plates for comparison of extraction efficiency among protocols. The authors included a total of 33 human skin samples, 30 human oral samples, eight built environment samples, six fecal samples, six human urine samples, six soil samples, four water samples, four fermented food samples and two tissue samples. The authors collected most sample types using wooden handle cotton swabs (Puritan, CA, USA) following the standard Earth Microbiome Project protocol [26]. To make comparisons relevant to SARS-CoV-2 detection, the authors collected additional samples, mimicking those collected from patients, using BBL CultureSwab plastic handle polyester swabs (category number 220135; BD Biosciences, NJ, USA) following the CDC's specimen collection guidelines [24,25].
The authors collected samples to allow for technical replication across three extraction protocols. Human skin samples included those from the foot, armpit, forehead and nostril interior. Foot and armpit samples were collected from three individuals by rubbing five cotton swabs simultaneously on the sole of each foot or armpit for 30 s. Forehead and nostril samples were collected from 12 individuals by rubbing two polyester swabs over the forehead for 30 s or in each nostril for 15 s each. Human oral samples included throat, saliva and oral saline rinses and the same rinses diluted in viral transport medium [27]. Throat samples were collected from 12 individuals by rubbing two polyester swabs across the pharynx for 30 s. Saliva was collected from 12 individuals using active spitting into a 50-ml centrifuge tube. Saline rinses were collected from three individuals by swishing 10 ml 0.9% saline for 30 s and spitting into a 50-ml centrifuge tube. To mimic storage in viral transport medium, 5 ml of saline rinse was mixed with 100 μl 50× viral transport medium in a 15-ml centrifuge tube. Built environment samples included floors and door handles. Floor and door handle samples were collected from two rooms using cotton swabs and two rooms using polyester swabs by rubbing nine swabs simultaneously across the surface of a 1 sq ft tile for 30 s or one entire door handle, respectively. Fecal samples included human, mouse and cat samples. Human feces were collected from two individuals using commode collectors (commode specimen collection system, Thermo Fisher Scientific, MA, USA). Mouse feces were collected from two individuals and stored in 1.5-ml microcentrifuge tubes. Cat feces were collected from two individuals and stored in plastic zip-top bags. Human urine samples included samples from female and male individuals. Urine was collected from three female and three male individuals and stored in commode collectors or 50-ml centrifuge tubes. Soil samples included tree rhizosphere and bare soil. For each type, soil was collected from two adjacent sites down to a depth of 20 cm using a sterile trowel and stored in plastic zip-top bags. Water samples included fresh- and seawater collected from two sites at the San Diego River and two sites at the Scripps Institution of Oceanography, respectively. Water was collected and stored in 50-ml centrifuge tubes. Fermented food samples included yogurt and sauerkraut samples. Two varieties of one brand of each food were purchased commercially and stored in 50-ml centrifuge tubes. Tissue samples included jejunum tissue from eight mice. Approximately 3.8 cm of the middle small intestine was removed, and any fecal matter inside was squeezed out lengthwise. Each tissue section was added to a 2-ml microcentrifuge tube containing 1 ml sterile 1× phosphate-buffered saline and approximately 40 mg sterile 1-mm silicone beads and homogenized at 6000 rpm for 1 min with a MagNA Lyser (Roche Diagnostics, CA, USA). The liquid homogenate from three intestinal sections from cohoused mice was pooled to create a single sample (one sample per cage). All samples were stored at -80°C within 3 h of collection and frozen for a maximum of 24 h before extraction. To compare LOD – defined as the number of cells required to detect a microbe in the sequence data – the authors included serial dilutions of cultures of Bacillus subtilis (Firmicutes) and Paracoccus denitrificans (Alphaproteobacteria) [20]. Input cell densities ranged from 2.0 to 9.6E7 cells for B. subtilis and 0.0 to 3.1E7 cells for P. denitrificans. To compare well-to-well contamination [23], the authors included plasmid-borne, synthetic 16S rRNA gene spike-ins (i.e., 4 ng of unique spike-in to one well of each column in the plate) [28] and at least five extraction blanks per plate.
DNA and RNA extraction
The authors compared two extraction protocols that use a 96-sample magnetic bead cleanup format: the MagAttract PowerSoil DNA isolation kit (category number 27000-4-KF; Qiagen, CA, USA) and the MagMAX microbiome ultra nucleic acid isolation kit (category number A42357; Applied Biosystems, CA, USA). The authors considered that the PowerSoil kit protocol includes heating the lysis solution to 60°C when mixing with samples as well as a subsequent 20-min bead-beating step, whereas the MagMAX kit uses no heating and only a 2-min bead-beating step. Additional heating and extended bead-beating may alter the extent of cellular lysis and degradation of extracellular nucleic acids and, subsequently, microbial community composition. The authors therefore included a third protocol, a variant of the MagMAX protocol, including 60°C incubation and 20-min bead-beating steps, and refer to the three protocols as PowerSoil, MagMAX 20-min and MagMAX 2-min.
For extraction, aliquots of each sample were transferred to unique wells of a 96-well extraction plate. For samples collected with swabs, the entire swab head was broken off into the lysis plate. For liquid samples, the authors transferred 200 μl. For bulk samples, the authors used cotton swabs to collect approximately 100 mg of homogenized material and broke the entire swab head off into the lysis plate. Extractions were performed following the manufacturer's protocol, except for the modifications made to the previously described MagMAX 20-min protocol. Lysis was performed with a TissueLyser II (Qiagen). Bead clean-ups were performed with the KingFisher flex purification system (Thermo Fisher Scientific). Extracted nucleic acids were stored at -80°C prior to quantification of RNA yield, fragment length distribution and integrity as well as quantification of DNA yield and downstream sequencing.
16S rRNA gene and shotgun metagenomics sequencing
The authors prepared DNA for 16S rRNA gene and shallow shotgun metagenomics sequencing as described previously [10,29–31]. For 16S data, raw sequence files were demultiplexed using Qiita [32], and suboperational taxonomic units were generated using Deblur [33]. For shallow shotgun metagenomics data, raw sequence files were demultiplexed using BaseSpace (Illumina, CA, USA), quality-filtered using Atropos [34] and human read-depleted by alignment to human reference genome GRCh38 using bowtie2 [35]. Filtered reads were aligned to the Web of Life database [36] using Shogun [31] with default parameters and using bowtie2 as the aligner, followed by read classification with the Web of Life Toolkit App [36,37]. Raw sequence data were deposited at the European Nucleotide Archive (accession number ERP124610), and raw and processed data are available at Qiita (study identifier 12201). Processing and analysis code is available on GitHub (https://github.com/justinshaffer/Extraction_test_MagMAX).
Results & discussion
The authors found DNA yield to be similar across the three extraction protocols and note that when considering all sample types (n = 615 samples), the extraction efficiency of the PowerSoil protocol was more similar to that of the MagMAX 20-min protocol compared with MagMAX 2-min protocol (paired-sample Wilcoxon signed-rank test: PowerSoil vs MagMAX 20-min W = 10,540; p = 0.6 and PowerSoil vs MagMAX 2-min W = 81,170; p = 0.01) (Supplementary Figure 1). The authors observed a similar pattern for the number of quality-filtered 16S reads (PowerSoil vs MagMAX 20-min W = 11,482; p = 0.1 and PowerSoil vs MagMAX 2-min W = 4651; p = 2.74E-11). However, for quality- and human-filtered shotgun metagenomics reads, both MagMAX protocols varied from the PowerSoil protocol (PowerSoil vs MagMAX 20-min W = 15,873; p = 1.41E-11 and PowerSoil vs MagMAX 2-min W = 17,148; p = 2.24E-15) (Figure 1 & Supplementary Figure 2).
Figure 1. . Sequences per sample across extraction protocols and sample types.
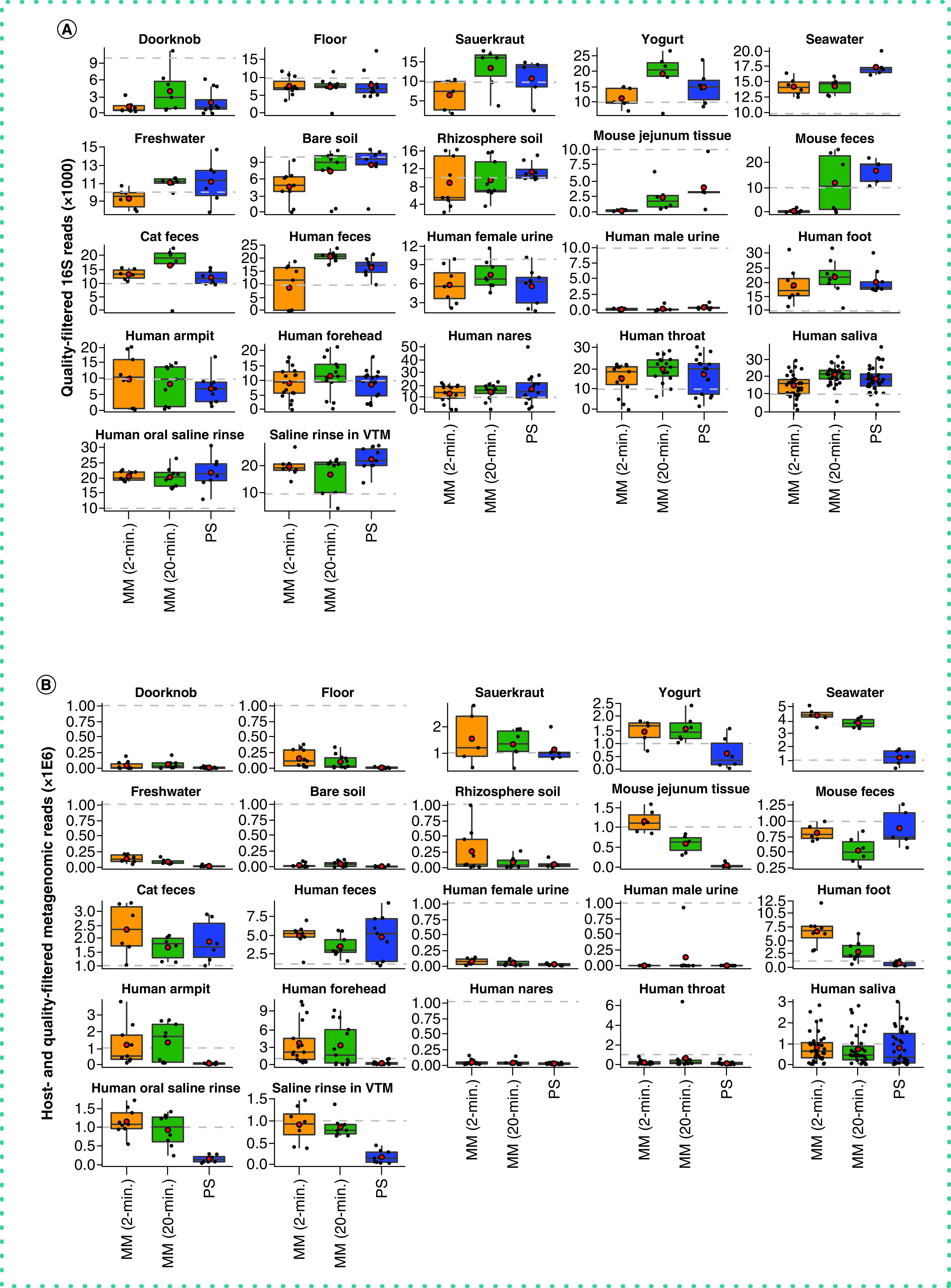
Average number of quality sequences for (A) 16S and (B) metagenomics data (n = 660 samples included). Red circles indicate means. Dashed lines indicate our expectation of (A) 10,000 from 16S and (B) 1 million reads from metagenomics, respectively, for human fecal samples. Note that additional samples included here absent from our statistical test (n = 45) include those for which technical replication across protocols was not feasible due to recommended sampling protocols (e.g., human nares, human throat), so we included biological replicates instead. Sample types missing here lacked representation by both MagMAX protocols.
MM: MagMAX; PS: PowerSoil.
From a technical perspective, the authors' comparison of the LOD of each protocol indicates that the MagMAX 2-min protocol requires ten times the number of cells required by PowerSoil for accurate detection in mixed bacterial cultures (Table 1). This is compared with the 10,000 times required by the MagMAX 20-min protocol (Table 1). This pattern is mirrored when considering sample retention following filtering based on LOD thresholds, for which the MagMAX 2-min is better with high-biomass samples and the MagMAX 20-min with low-biomass samples. However, the authors observed an increase in well-to-well contamination in the MagMAX 20-min protocol compared with the MagMAX 2-min protocol (Figure 2). As all other parameters were consistent between the two MagMAX protocols, this indicates that mimicking lysis parameters from the PowerSoil protocol in the MagMAX protocol can have undesirable consequences. The extended bead-beating time may lead to lysate leaking into the thin ridges of the 96-well plate, which is covered by a plastic film. From this perspective, the authors favor the MagMAX 2-min protocol.
Table 1. . Limit of detection across extraction protocols.
| Extraction protocol | Sample set | Threshold (%) | LOD Gram-negative | LOD Gram-positive | LOD mixed culture | Read depth | Samples retained | Samples retained (%) |
|---|---|---|---|---|---|---|---|---|
| MagMAX 2-min | High biomass | 50 | – | – | 5.73E+02 | 362 | 76 | 80 |
| 80 | – | – | 5.73E+04 | 1392 | 69 | 73 | ||
| 90 | – | – | 5.73E+04 | 3512 | 64 | 67 | ||
| 95 | – | – | 5.73E+06 | 9144 | 46 | 48 | ||
| Low biomass | 50 | 1.60E+03 | 3.10E+03 | – | 637 | 69 | 73 | |
| 80 | 1.60E+04 | 3.10E+03 | – | 1631 | 63 | 66 | ||
| 90 | 1.60E+04 | 3.10E+03 | – | 3007 | 59 | 62 | ||
| 95 | 1.60E+05 | 3.10E+04 | – | 5526 | 52 | 55 | ||
| MagMAX 20-min | High biomass | 50 | – | – | 5.73E+05 | 8499 | 68 | 71 |
| 80 | – | – | 5.73E+07 | 14,522 | 55 | 57 | ||
| 90 | – | – | 5.73E+07 | 20,158 | 30 | 31 | ||
| 95 | – | – | 5.73E+07 | 27,541 | 3 | 3 | ||
| Low biomass | 50 | 1.60E+01 | 3.10E+01 | – | 491 | 79 | 83 | |
| 80 | 1.60E+03 | 3.10E+03 | – | 776 | 71 | 75 | ||
| 90 | 1.60E+03 | 3.10E+03 | – | 1031 | 68 | 72 | ||
| 95 | 1.60E+04 | 3.10E+03 | – | 1354 | 64 | 67 | ||
| PowerSoil | High biomass | 50 | – | – | 5.70E+01 | 1050 | 87 | 92 |
| 80 | – | – | 5.73E+07 | 14,632 | 36 | 38 | ||
| 90 | – | – | NA | 106,110 | 0 | 0 | ||
| 95 | – | – | NA | 944,308 | 0 | 0 | ||
| Low biomass | 50 | 1.60E+03 | 3.10E+00 | – | 1836 | 69 | 72 | |
| 80 | 1.60E+04 | 3.10E+02 | – | 3797 | 62 | 65 | ||
| 90 | 1.60E+05 | 3.10E+03 | – | 5997 | 57 | 59 | ||
| 95 | 1.60E+05 | 3.10E+03 | – | 9345 | 40 | 42 |
Titrations of cultured cells were used to identify the number of reads needed per sample to meet various thresholds of detection (i.e., percentage of reads mapped to expected taxa vs background contaminants). Read depths corresponding to a threshold of 50% were used for filtering samples prior to community analyses of microbial 16S data, as recommended [20]. Retention of samples following filtering based on read depth for each threshold is shown.
Gram+: Bacillus subtilis; Gram–: Paracoccus denitrificans; Mixed culture: B. subtilis and P. denitrificans; NA: Not applicable.
Figure 2. . Well-to-well contamination across extraction protocols.

Plasmids harboring synthetic 16S sequences were spiked into a single well per plate column (i.e., alternating from row C to F across columns: C1, F2, C3, F4, etc.) of each high-biomass sample plate prior to extraction. (A) The number of reads matching synthetic 16S sequences was quantified for all wells that did not receive a spike-in. Asterisks indicate significant differences between pairs of extraction protocols as determined by a Kruskal-Wallis post-hoc Dunn’s test with a Benjimini-Hochberg correction for multiple comparisons. (B) The percentage of spike-in reads among all reads per well shown as a heatmap.
*** p < 0.001.
With respect to microbial community composition, the authors found bias introduced by extraction protocol to be small compared with variation among sample types or replicates of the same sample (i.e., one to two orders of magnitude weaker in explaining beta-diversity) (Tables 1 & 2 & Supplementary Figures 3 & 4). The authors also found strong correlations in microbial community beta-diversity among samples between any two extraction protocols; however, relationships with the PowerSoil protocol were slightly stronger for MagMAX 2-min compared with MagMAX 20-min (Supplementary Table 1). The authors used principal coordinates analysis of unweighted UniFrac distances to visualize these trends and confirmed that samples clustered strongly by type and host subject and not by extraction protocol for both 16S and metagenomics data (Figure 3 & Supplementary Figures 5 & 6). Estimates of alpha-diversity were more comparable to those from PowerSoil for the MagMAX 2-min protocol (paired-sample Wilcoxon signed-rank test: PowerSoil vs MagMAX 2-min W = 5916; p = 0.0001 and PowerSoil vs MagMAX 20-min W = 7058; p = 1.53E-06) (Figure 4 & Supplementary Figure 7). Finally, the majority of genera (16S) and species (metagenomics) were shared across all three extraction protocols; however, for both datasets, the MagMAX 2-min protocol shared a greater number of exclusive taxa with the PowerSoil protocol than the MagMAX 20-min protocol did (Figure 5).
Table 2. . Results from a forward, stepwise model selection of factors influencing microbial community beta-diversity.
| Data type | Distance metric | Factor | Adjusted R2 | df | AIC | F | p-value |
|---|---|---|---|---|---|---|---|
| 16S | Unweighted UniFrac | Sample type | 0.87 | 24 | -556.59 | 172.97 | 0.0002 |
| Host identity | 0.01 | 30 | -583.89 | 2.85 | 0.0002 | ||
| Extraction protocol | 0.001 | 2 | -588.47 | 3.92 | 0.004 | ||
| Weighted UniFrac | Sample type | 0.76 | 24 | -165.42 | 79.55 | 0.0002 | |
| Host identity | 0.06 | 30 | -320.67 | 7.83 | 0.0002 | ||
| Extraction protocol | 0.001 | 2 | -323.72 | 3.21 | 0.02 | ||
| Jaccard | Sample type | 0.89 | 24 | -651.49 | 206.18 | 0.0002 | |
| Host identity | 0.02 | 30 | -756.85 | 5.76 | 0.0002 | ||
| Extraction protocol | 0.001 | 2 | -762.48 | 4.40 | 0.0008 | ||
| RPCA | Sample type | 0.86 | 24 | -495.50 | 154.16 | 0.0002 | |
| Host identity | 0.03 | 30 | -619.04 | 6.49 | 0.0002 | ||
| Extraction protocol | 0.001 | 2 | -625.14 | 4.61 | 0.0002 | ||
| Metagenomics | Unweighted UniFrac | Sample type | 0.93 | 26 | -958.24 | 317.60 | 0.0002 |
| Host identity | 0.01 | 31 | -1062.60 | 5.57 | 0.0002 | ||
| Extraction protocol | 0.001 | 2 | -1067.53 | 4.08 | 0.0006 | ||
| Weighted UniFrac | Sample type | 0.87 | 26 | -602.92 | 173.32 | 0.0002 | |
| Host identity | 0.02 | 31 | -676.11 | 4.42 | 0.0002 | ||
| Extraction protocol | 0.003 | 2 | -693.97 | 10.09 | 0.0002 | ||
| Jaccard | Sample type | 0.94 | 26 | -1084.87 | 391.42 | 0.0002 | |
| Host identity | 0.01 | 31 | -1217.42 | 6.67 | 0.0002 | ||
| RPCA | Sample type | 0.85 | 26 | -496.04 | 143.29 | 0.0002 | |
| Host identity | 0.03 | 31 | -620.86 | 6.36 | 0.0002 | ||
| Extraction protocol | 0.005 | 2 | -645.41 | 13.24 | 0.0002 |
Values are based on permutation tests of variation explained by redundancy analysis, done separately for four unique metrics for both 16S and metagenomics data. The full model included bead-beating time (i.e., 2 vs 20 min), sample biomass (i.e., high vs low biomass), sample type, host subject identity and extraction protocol (i.e., MagMAX 2-min, MagMAX 20-min, PowerSoil) as model variables. The 16S data were rarefied, as noted for Figure 3. Metagenomics data were rarefied to 17,000 host- and quality-filtered reads per sample or had samples with fewer than 17,000 reads excluded when using RPCA distances (n = 647 samples). Rarefaction depths were selected to maintain at least 75% samples from both high- and low-biomass datasets.
AIC: Akaike information criterion; df: degrees of freedom; RPCA: Robust principal component analysis.
Figure 3. . Beta-diversity among extraction protocols and sample types.
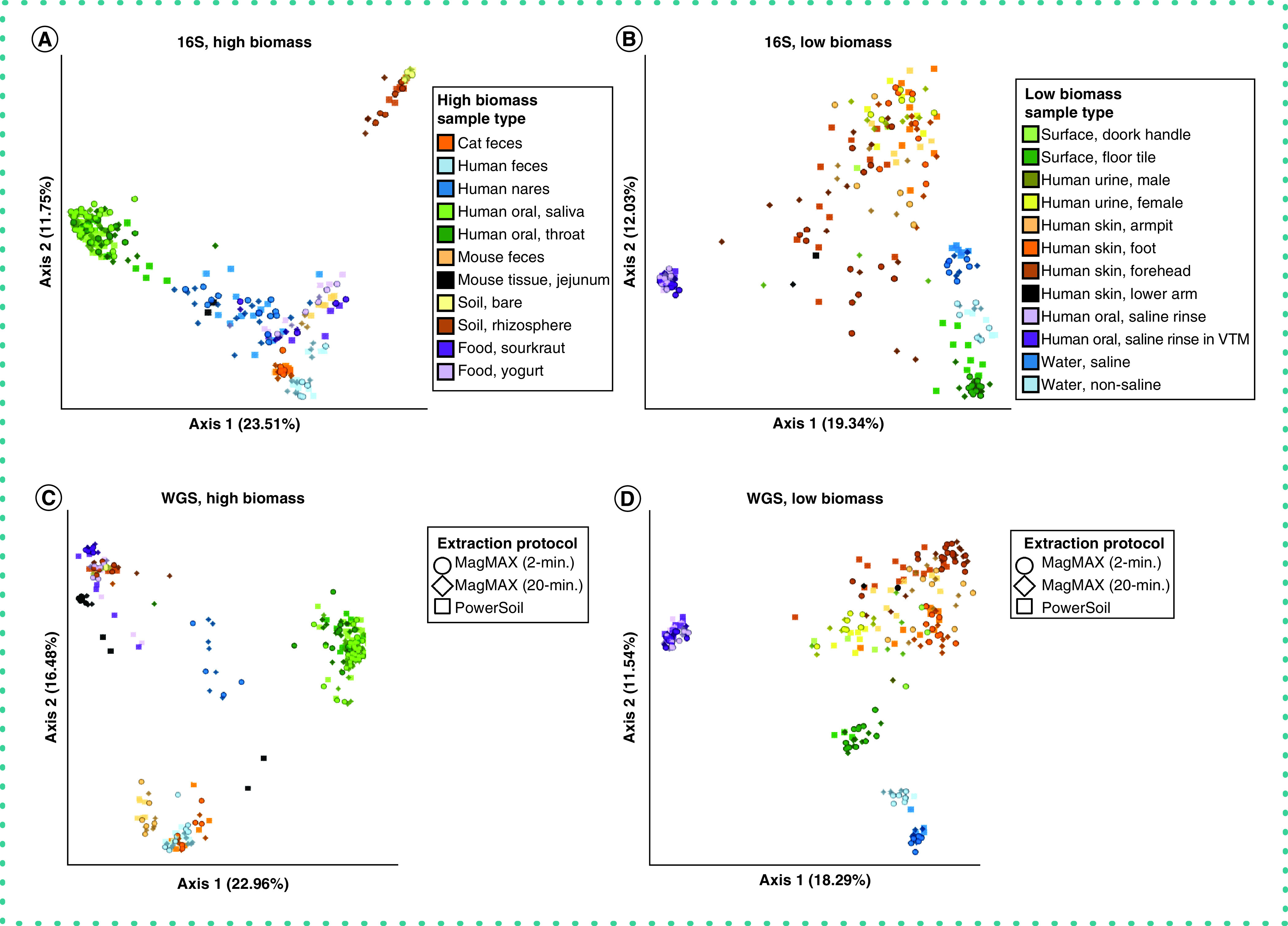
Principal coordinates analysis (PCoA) plots showing unweighted UniFrac distances based on 16S data for (A) high biomass samples and (B) low biomass samples, and shotgun metagenomics data for (C) high biomass samples and (D) low biomass samples. Colors indicate sample types and shapes indicate extraction protocols. Mock community and control blanks were excluded for clarity. 16S data were rarefied to 5,000 quality-filtered reads per sample for both high- and low-biomass samples (n = 611 samples). Metagenomics data were rarefied to 35,000 host- and quality-filtered reads per high-biomass sample (n = 287 samples), and to 20,000 reads per low-biomass sample (n = 242 samples). When using RPCA distances rather than using rarefied data, we excluded samples with fewer reads than the rarefaction depth for that dataset. Rarefaction depths were selected to maintain at least 75% samples from both high- and low-biomass datasets.
Figure 4. . Alpha-diversity across extraction protocols and sample types.
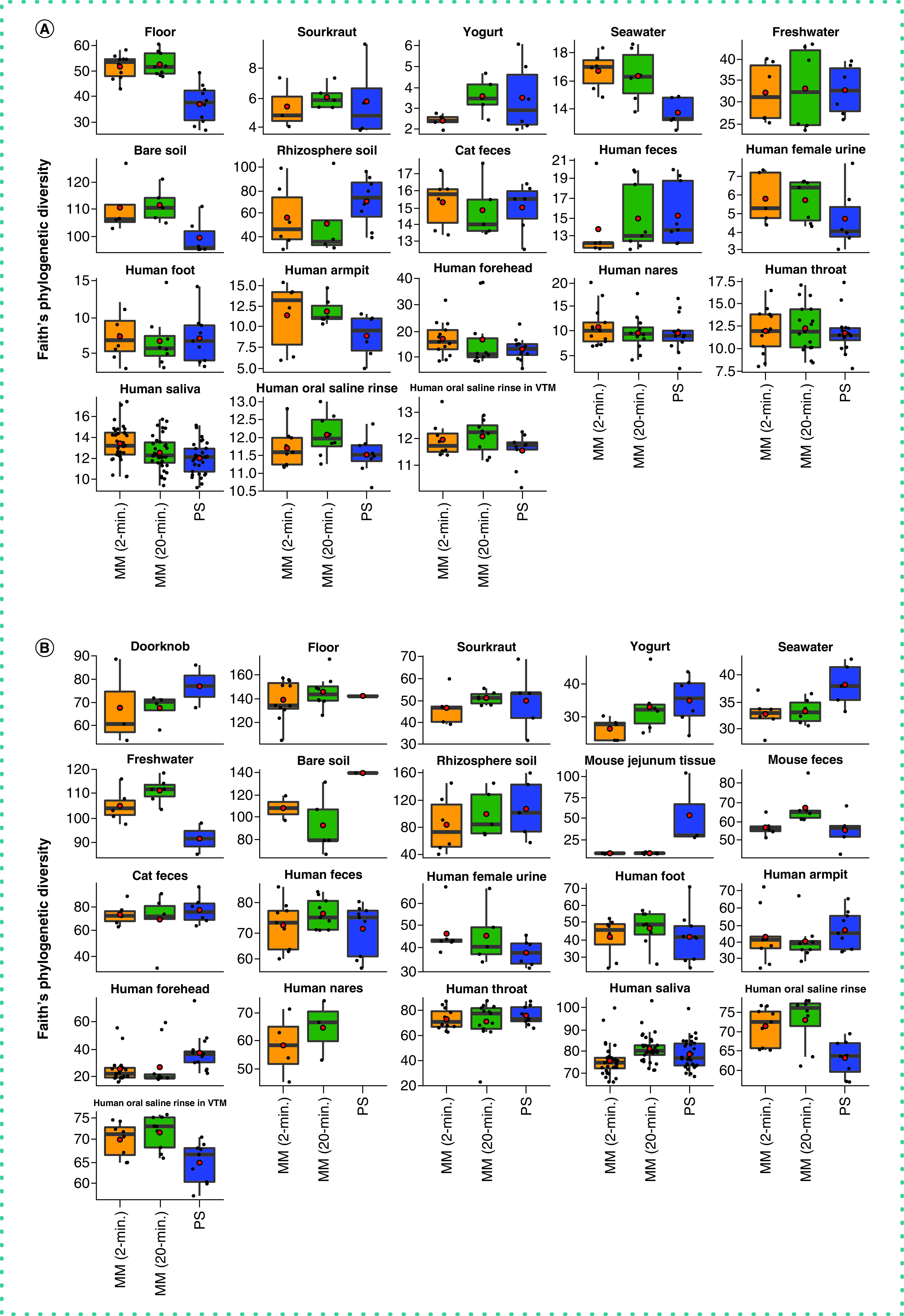
Faith’s Phylogenetic Diversity among the three extraction protocols based on (A) 16S and (B) metagenomics data. Red circles indicate means. Data were rarefied as noted for Figure 3.
MM: MagMAX; PS: PowerSoil.
Figure 5. . Taxonomic bias among extraction protocols.
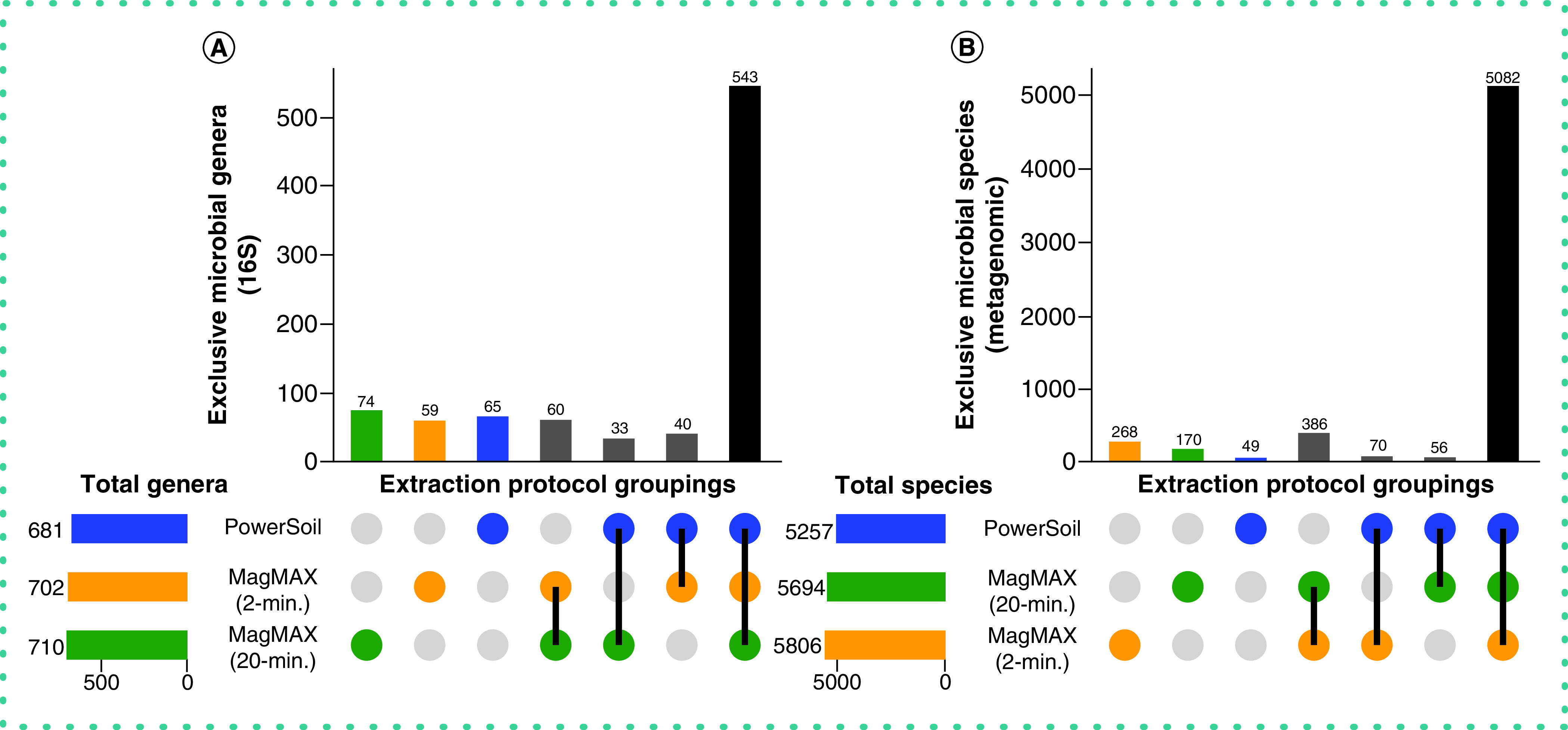
Upset plots showing (A) genera for 16S data and (B) species for metagenomics data, highlighting taxa shared among extraction protocols. Data were rarefied as noted for Figure 3.
Together, these results highlight that, despite variation in DNA yield, sequence read counts and LOD of microbial cells among extraction protocols, differences in microbial taxonomic and community composition resulting from the different methods were minor for both 16S and metagenomics microbial sequence data. However, between the two MagMAX protocols, the authors note that for beta-diversity, alpha-diversity and taxonomic composition, the MagMAX 2-min protocol generated results more comparable to the PowerSoil protocol.
Importantly, whereas RNA yield was comparable between the two MagMAX protocols (Figure 6A), the authors observed a higher quality of extracted RNA using the MagMAX 2-min versus MagMAX 20-min protocol (Figure 6B & C). In addition to reduced well-to-well contamination from a shorter bead-beating time during lysis for the MagMAX 2-min versus MagMAX 20-min protocol, the lack of incubation of the lysis buffer resulted in relatively high-quality RNA produced with the former compared with the latter (Figure 6).
Figure 6. . RNA output across extraction protocols.
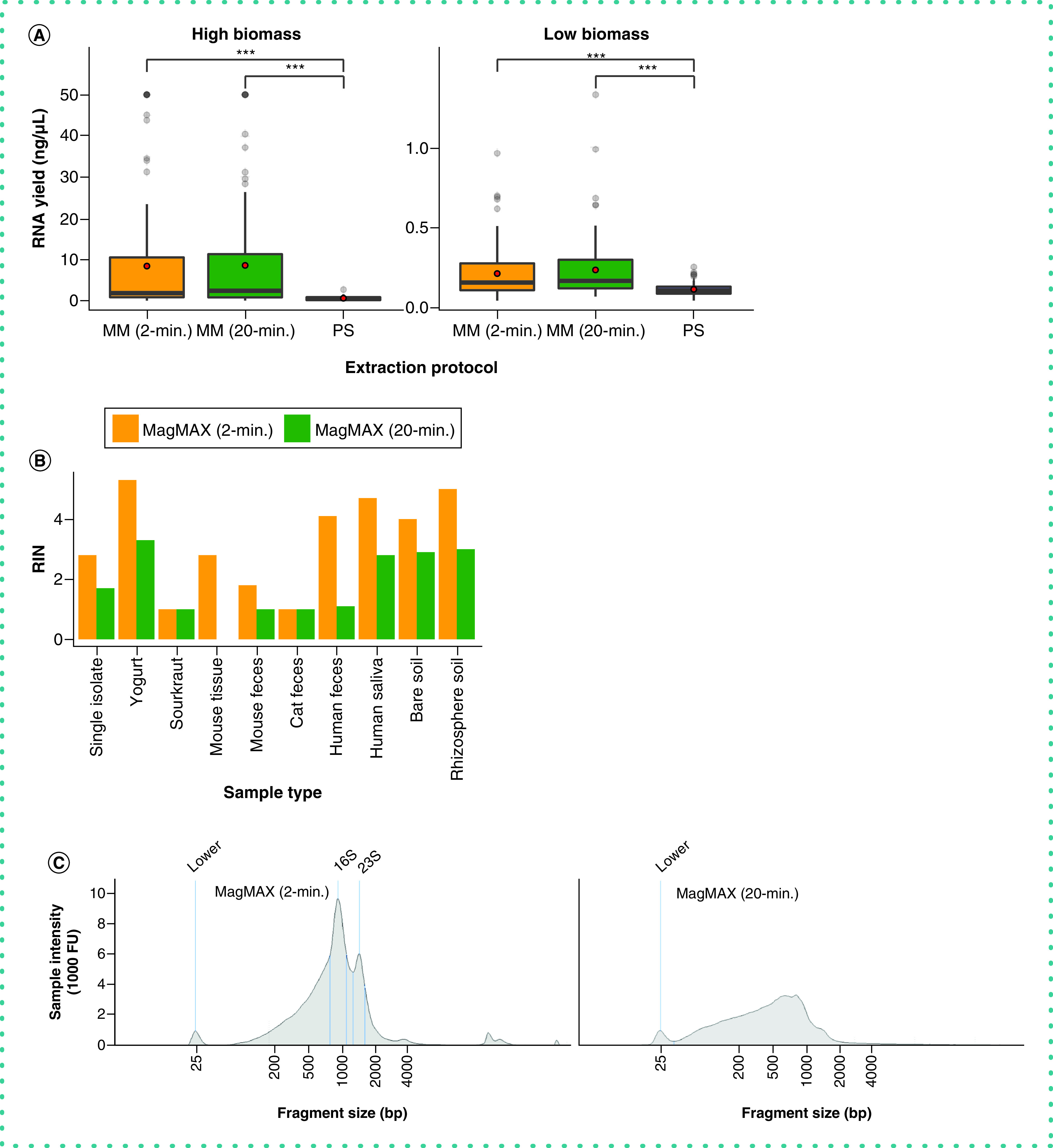
(A) RNA yield quantified using the Qubit RNA assay. Red circles indicate means. Asterisks indicate significant differences between pairs of extraction protocols as determined by paired-sample Wilcoxon signed-rank tests; ***p < 2.2E-16. Values at 50 ng/µl are at the upper limit of detection for the Qubit assay, and may underestimate actual yields for those samples. (B) RNA Integrity Number (RIN) across a subset of samples for the MagMAX extraction protocols, estimated using the TapeStation high-sensitivity (HS) RNA assay. PowerSoil extracts were excluded from the assay due to poor RNA yield, however we note that this may be to our exclusion of the RNAse step available in that protocol. (C) RNA fragment length distribution estimated using the TapeStation HS RNA assay for one human fecal sample. The distribution for the MagMAX (2-min) is on the left and that for the MagMAX (20-min) on the right. The positive control marker at 25-bp is annotated. Peaks corresponding to expected lengths for 16S and 23S rRNA are annotated for the 2-min. protocol and are missing from output from the 2-min. one.
MM: MagMAX; PS: PowerSoil.
Conclusion
We conclude that the MagMAX 2-min extraction protocol is comparable to our established PowerSoil protocol with respect to characterizing microbial community composition and therefore should allow for comparisons such as meta-analysis across 16S and metagenomics data produced using both protocols and downstream methods similar to those used here. In addition to extracting both DNA and RNA, the more rapid processing time (i.e., approximately 2 h faster than PowerSoil per 96 samples), use of fewer consumables (i.e., approximately 70% of plastics) and lower cost (i.e., $5.56 vs $5.65 per sample) highlight the MagMAX 2-min protocol as a comparable and efficient alternative to the PowerSoil protocol that also allows for downstream applications using RNA.
Future perspective
Future optimization of molecular methods for microbial community analyses should focus on increasing representation of all microbes in a sample as well as diverse sample types, including those used here. Achieving these goals will allow for more widely adopted use of the same methods. As no single study can be completely comprehensive, making advances that allow us to better compare across studies, particularly past studies, is an important step [38]. Alongside the development of computational methods that bioinformatically reduce experimental variation, continuing to explore new molecular methods for capturing important ecological interactions will support our growing understanding of microbial communities.
Executive summary.
Established protocols were compared for DNA extraction with two alternative protocols that also extract RNA.
The authors included a diverse panel of sample types, ranging from host-associated to environmental.
Controls were included for detecting well-to-well contamination and LOD of microbial cells.
The authors observed sample type-specific differences in DNA extraction efficiency among three extraction protocols.
Both new protocols were similar with respect to RNA extraction efficiency but varied in RNA quality.
Sample type and host identity were stronger drivers of microbial community beta-diversity compared with the extraction protocol used.
A protocol was identified that generates both DNA and RNA and produces data that are highly similar to their established protocol with respect to microbial community alpha-diversity, beta-diversity and taxonomic composition.
The similarity between the optimal protocol and the authors' existing protocol will allow for meta-analyses across both with negligible technical bias.
Supplementary Material
Acknowledgments
The authors thank K Dao, R Moranchel, C Nguyen, C Morris and R Simpson for providing samples and J DeReus for assistance with data processing.
Footnotes
Supplementary data
To view the supplementary data that accompany this paper please visit the journal website at: www.future-science.com/doi/suppl/10.2144/btn-2020-0153
Author contributions
JP Shaffer, C Marotz, P Belda-Ferre, RA Salido, CS Carpenter, LS Zaramela, JJ Minich, G Humphrey, AD Swafford, S Miller-Montgomery and R Knight designed the study; JP Shaffer, C Marotz, P Belda-Ferre, RA Salido, CS Carpenter, L Zaramela, M Bryant and G Humphrey provided samples; JP Shaffer, C Marotz, CS Carpenter and S Fraraccio performed extractions; RA Salido, M Bryant, K Sanders and G Humphrey performed quality control and sequencing; JP Shaffer, C Marotz, P Belda-Ferre, C Martino, S Wandro, M Estaki and RA Salido performed data analyses; and JP Shaffer wrote the manuscript, with contributions from all authors.
Financial & competing interests disclosure
JP Shaffer was supported by NIH San Diego Institutional Research and Academic Career Development Award (5K12GM068524-17) and the United States Department of Agriculture - National Institute of Food and Agriculture (USDA-NIFA) (2019-67013-29137). C Martino was supported by NIH (1RF1-AG058942-01) and Semiconductor Research Corporation and Defense Advanced Research Projects Agency (SRC/DARPA) (GI18518). P Belda-Ferre was supported by National Science Foundation - Center for Aerosol Impacts on Chemistry of the Environment and Crohn's & Colitis Foundation Award (CCFA) (675191). G Ackermann was supported by NIH (R01HL140976, R01DK102932, R01HL134887) and Department of Defense (W81XWH-17-1-0589). G Humphrey was supported by NIH (U19AG063744, U01 AI124316), Office of Naval Research (ONR) (N00014-15-1-2809) and the Emerald Foundation (3022). R Knight was supported by NIH (1RF1-AG058942-01, 1DP1AT010885, R01HL140976, R01DK102932, R01HL134887), USDA-NIFA (2019-67013-29137), SRC/DARPA (GI18518), CCFA (675191), ONR (N00014-15-1-2809) and the Emerald Foundation (3022). The authors have no other relevant affiliations or financial involvement with any organization or entity with a financial interest in or financial conflict with the subject matter or materials discussed in the manuscript apart from those disclosed.
No writing assistance was utilized in the production of this manuscript.
Ethical conduct of research
The authors state that they have obtained appropriate institutional review board approval; the human subject work conducted here has been approved through University of California, San Diego IRB#150275. In addition, informed consent has been obtained from the participants involved.
Open access
This work is licensed under the Attribution-NonCommercial-NoDerivatives 4.0 Unported License. To view a copy of this license, visit http://creativecommons.org/licenses/by-nc-nd/4.0/
References
Papers of special note have been highlighted as: • of interest; •• of considerable interest
- 1.Kelly CR, Ihunnah C, Fischer M et al. Fecal microbiota transplant for treatment of Clostridium difficile infection in immunocompromised patients. Am. J. Gastroenterol. 109(7), 1065–1071 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Bokulich NA, Chung J, Battaglia T et al. Antibiotics, birth mode, and diet shape microbiome maturation during early life. Sci. Transl. Med. 8(343), 343ra82 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Panke-Buisse K, Poole AC, Goodrich JK, Ley RE, Kao-Kniffin J. Selection on soil microbiomes reveals reproducible impacts on plant function. ISME J. 9, 980–989 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Bell TH, Stefani FOP, Abram K et al. A diverse soil microbiome degrades more crude oil than specialized bacterial assemblages obtained in culture. Appl. Environ. Microbiol. 82(18), 5530–5541 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Dominguez-Bello MG, Godoy-Vitorino F, Knight R, Blaser MJ. Role of the microbiome in human development. Gut 68, 1108–1114 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Poore GD, Kopylova E, Zhu Q et al. Microbiome analysis of blood and tissues suggest cancer diagnostic approach. Nature 579, 567–574 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Taylor BC, Lejzerowicz F, Poirel M et al. Consumption of fermented foods is associated with systematic differences in gut microbiome and metabolome. mSystems 5(2), e00901-19 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Knight R, Vrbanac A, Taylor BC et al. Best practices for analysing microbiomes. Nat. Rev. Microbiol. 16, 410–422 (2018). [DOI] [PubMed] [Google Scholar]
- 9.Bolyen E, Rideout JR, Dillon MR et al. Reproducible, interactive, scalable, and extensible microbiome data science using QIIME2. Nat. Biotechnol. 37, 852–857 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Sanders JG, Nurk S, Salido RA et al. Optimizing sequencing protocols for leaderboard metagenomics by combining long and short reads. Genome Biol. 20(1), 226 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Song SJ, Amir A, Metcalf JL et al. Preservation methods differ in fecal microbiome stability, affecting suitability for field studies. mSystems 1(3), e00021-16 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]; •• Highlights the importance of considering how samples are maintained prior to processing for analysis of microbial communities.
- 12.Parada AE, Needham DM, Fuhrman JA. Every base matters: assessing small subunit rRNA primers for marine microbiomes with mock communities, time series and global field samples. Environ. Microbiol. 18(5), 1403–1414 (2016). [DOI] [PubMed] [Google Scholar]
- 13.Bjerre RD, Hugerth LW, Boulund F, Seifert FM, Johansen JD, Engstrand L. Effects of sampling strategy and DNA extraction on human skin microbiome investigations. Sci. Rep. 9(1), 17287 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]; • Important for understanding the influence of technical factors on analysis of microbial communities.
- 14.WHO. Coronavirus disease (COVID-19) weekly situation report, October 18, 2020. www.who.int/docs/default-source/coronaviruse/situation-reports/20200928-weekly-epi-update.pdf?sfvrsn=9e354665_6
- 15.Domingues CPF, Rebelo JS, Dionsio F, Botelho A, Nogueira T. The social distancing imposed to contain COVID-19 can affect our microbiome: a double-edged sword in human health. mSphere 5(5), e00716-20 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Hughes S, Troise O, Donaldson H, Mughal N, Moore LSP. Bacterial and fungal coinfection among hospitalized patients with COVID-19: a retrospective cohort study in a UK secondary-care setting. Clin. Microbiol. Infect. 26, 1395–1399 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]; • Highlights the importance of understanding interactions of SARS coronavirus 2 (SARS-CoV-2) with other illnesses caused by bacteria and fungi.
- 17.Zuo T, Zhang F, Lui G et al. Alterations in gut microbiota of patients with COVID-19 during time of hospitalization. Gastroenterol. 159, 944–955 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Marotz C, Amir A, Humphrey G, Gaffney J, Gogul G, Knight R. DNA extraction for streamlined metagenomics of diverse environmental samples. BioTechniques 62(6), 290–293 (2017). [DOI] [PubMed] [Google Scholar]; •• Important for understanding differences in DNA extraction methods, particularly different bead clean-up methods, for downstream analysis of microbial communities.
- 19.Khailany RA, Safdar M, Ozaslan M. Genomic characterization of a novel SARS-CoV-2. Gene Rep. 19, 100682 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Minich JJ, Zhu Q, Janssen S et al. KatharoSeq enables high-throughput microbiome analysis from low-biomass samples. mSystems 3(3), e00218-17 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]; •• Important for understanding differences in DNA extraction methods for regular or high- versus low-biomass sample types for analysis of microbial communities.
- 21.Salter SJ, Cox MJ, Turek EM et al. Reagent and laboratory contamination can critically impact sequence-based microbiome analysis. BMC Biol. 12, 87 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Eisenhofer R, Minich JJ, Marotz C, Cooper A, Knight R, Weyrich LS. Contamination in low microbial biomass microbiome studies: issues and recommendations. Trends Microbiol. 27(2), 105–117 (2019). [DOI] [PubMed] [Google Scholar]
- 23.Minich JJ, Sanders JG, Amir A, Humphrey G, Gilbert JA, Knight R. Quantifying and understanding well-to-well contamination in microbiome research. mSystems 4(4), e00186-19 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Sehulster LM, Chinn RYW, Arduino MJ et al. Guidelines for environmental infection control in health-care facilities. In: Recommendations from CDC and the Healthcare Infection Control Practices Advisory Committee (HICPAC). American Society for Healthcare Engineering/American Hospital Association, IL, USA: (2004). [Google Scholar]
- 25.CDC. Specimen collection guidelines. 1–8 (2020). www.cdc.gov/urdo/downloads/SpecCollectionGuidelines.pdf
- 26.Thompson LR, Sanders JG, McDonald D et al. A communal catalogue reveals Earth's multiscale microbial diversity. Nature 551, 457–463 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.CDC. Preparation of viral transport medium. SOP#: DSR-052-02. 1–8 (2020). www.cdc.gov/coronavirus/2019-ncov/downloads/Viral-Transport-Medium.pdf
- 28.Tourlousse DM, Yoshiike S, Ohashi A, Matsukura S, Noda N, Sekiguchi Y. Synthetic spike-in standards for high-throughput 16S rRNA gene amplicon sequencing. Nucleic Acids Res. 45(4), e23 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Caporaso JG, Lauber CL, Walters WA et al. Ultra-high-throughput microbial community analysis on the Illumina HiSeq and MiSeq platforms. ISME J. 6, 1621–1624 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Minich JJ, Humphrey G, Benitez RAS et al. High-throughput miniaturized 16S rRNA amplicon library preparation reduces costs while preserving microbiome integrity. mSystems 3(6), e00166-18 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Hillmann B, Al-Ghalith GA, Shields-Cutler RR et al. Evaluating the information content of shallow shotgun metagenomics. mSystems 3(6), e00069-18 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Gonzalez A, Navas-Molina JA, Kosciolek T et al. Qiita: rapid, web-enabled microbiome meta-analysis. Nat. Methods 15, 796–798 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Amir A, McDonald D, Navas-Molina JA et al. Deblur rapidly resolves single-nucleotide community sequence patterns. mSystems 2(2), e00191-16 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Didion JP, Martin M, Collins FS. Atropos: specific, sensitive, and speedy trimming of sequencing reads. PeerJ 5, e3720 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Langmead B, Salzberg SL. Fast gapped-read alignment with Bowtie 2. Nat. Methods 9(4), 357–359 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Zhu Q, Mai U, Pfeiffer W et al. Phylogenomics of 10,575 genomes reveals evolutionary proximity between domains Bacteria and Archaea. Nat. Commun. 10(1), 5477 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Zhu Q. Web of Life Toolkit App. https://github.com/qiyunzhu/woltka
- 38.Greathouse KL, Sinha R, Vogtmann E. DNA extraction for human microbiome studies: the issue of standardization. Genome Biol. 20(1), 212 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.