

An E3 ubiquitin ligase controls DDX3X ubiquitination and stability and therefore suppresses RLR-dependent antiviral responses.
Abstract
Retinoic acid–inducible gene-I (RIG-I)–like receptors (RLRs) are major cytosolic RNA sensors and play crucial roles in initiating antiviral innate immunity. Furthermore, RLRs have been implicated in multiple autoimmune disorders. Thus, RLR activation should be tightly controlled to avoid detrimental effects. “DEAD-box RNA helicase 3, X-linked” (DDX3X) is a key adaptor in RLR signaling, but its regulatory mechanisms remain unknown. Here, we show that the E3 ubiquitin ligase RNF39 inhibits RLR pathways through mediating K48-linked ubiquitination and proteasomal degradation of DDX3X. Concordantly, Rnf39 deficiency enhances RNA virus–triggered innate immune responses and attenuates viral replication. Thus, our results uncover a previously unknown mechanism for the control of DDX3X activity and suggest RNF39 as a priming intervention target for diseases caused by aberrant RLR activation.
INTRODUCTION
Cytosolic sensing of nucleic acids through pattern recognition receptors is vital for activating innate immune responses under various physiological and pathological conditions, such as viral infections, inflammation, and cancer (1–3). Pathogen-derived RNA in the cytosol is sensed by the retinoic acid–inducible gene-I (RIG-I)–like receptors (RLRs), including RIG-I and melanoma differentiation-associated gene 5 (MDA5) (3–5). RIG-I and MDA5 sense single- or double-stranded RNA generated during viral infection and then bind to the key adaptor mitochondrial antiviral-signaling protein (MAVS; also known as IPS-1, VISA, and CARDIF) on the surface of mitochondria (5, 6). Upon activation, MAVS forms prion-like aggregates, which further activates TANK-binding kinase 1 (TBK1)/IκB kinase ε (IKKε) and interferon regulatory factor 3 (IRF3) for type I interferon (IFN) responses.
“DEAD-box RNA helicase 3, X-linked” (DDX3X) is a key adaptor involved in the RLR signaling, which facilitates RNA sensing pathway through interacting with MAVS, IKKε, and the IFN-β promoter (7–11). Following viral infection, DDX3X is recruited to MAVS and acts as a scaffold to mediate the formation of MAVS-TRAF3 complex, resulting in the enhancement of RLR activation (10, 12). Deletion of Ddx3x in the hematopoietic system reduced the synthesis of type I interferons following vesicular stomatitis virus (VSV) infection and promoted VSV replication in vitro (13). A couple of viruses, such as HIV-1 and hepatitis B virus, have evolved sophisticated mechanisms to evade the induced RLR pathway activation by targeting DDX3X (14, 15). HIV-1 blocks DDX3X-MAVS signaling to evade antiviral host defense (14), suggesting the control of DDX3X activity as a viral strategy that accelerates HIV-1 replication. However, how DDX3X activity is negatively regulated remains unknown.
Protein ubiquitination is a crucial posttranscriptional modification to provide specificity and regulate the intensity of innate immunity (16). A variety of RING domain E3 ubiquitin ligases organized in a tight cluster within the major histocompatibility complex (MHC) class I region have been implicated in the regulation of innate immunity (17–19). RING finger protein 39 (RNF39; also called HZFw), which contains a RING domain and has potential E3 ubiquitin ligase activity, is also encoded within the MHC class I region (20). Genetic variant of RNF39 is associated with a variety of viral diseases and autoimmune diseases, such as HIV, multiple sclerosis (MS), Behcet’s disease, allergic rhinitis, and systemic lupus erythematosus (SLE) (21–27). Previous genome-wide association studies have mapped to a region close to the RNF39 associated with HIV-1 disease progression (21). Plasma HIV-1 viral loads are associated with RNF39 genetic variants in patients infected with HIV-1, and RNF39 inhibits cellular HIV-1 replication (22). However, the biological function of RNF39 in antiviral responses remains unknown.
In the present study, we show that RNF39 attenuates RLR signaling by mediating K48-linked polyubiquitination and proteasomal degradation of DDX3X. Concordantly, Rnf39 deficiency enhances innate immune responses against RNA viruses. Thus, our results uncover a previously unknown mechanism for the control of DDX3X activity and suggest RNF39 as a priming target for the intervention of diseases caused by aberrant RLR activation.
RESULTS
RNF39 inhibits RLR-induced innate immune response
To determine the potential role of RNF39 in antiviral responses, we first examined its expression during viral infection in mouse primary peritoneal macrophages (PMs). VSV and Sendai virus (SeV), two types of single-stranded RNA viruses recognized by RIG-I, markedly increased RNF39 expression (fig. S1, A and B). Following viral infection, multiple immune cells, including macrophages, secrete amounts of type I IFNs to initiate host antiviral responses. RNF39 expression was considerably enhanced following IFN-β stimulation (fig. S1C). Similarly, viral infection and IFN-β stimulation also induced RNF39 expression in mouse embryonic fibroblasts (MEFs) (fig. S1, D to F). Together, these data indicate that RNF39 expression is induced during viral infection, suggesting that RNF39 is involved in host antiviral responses.
To evaluate the physiological function of RNF39, we then generated Rnf39-deficient (Rnf39−/−) mice (fig. S2, A to D). SeV- and VSV-induced IFN-β, tumor necrosis factor–α (TNF-α), and interleukin-6 (IL-6) secretion and mRNA expression were enhanced by Rnf39 deficiency (Fig. 1, A and B). To further confirm the regulatory roles of RNF39 on the RLR pathway, we designed small interfering RNAs (siRNAs) targeting Rnf39 and confirmed the knockdown efficiency in mouse PMs (fig. S2E). Rnf39 knockdown greatly enhanced VSV-induced IFN-β expression and IRF3 phosphorylation (Figs. 1E and 2C). siRNA2, which has the highest efficiency to knock down RNF39 expression, has the highest potential to increase IFN-β expression (Fig. 1E). Next, we evaluated the roles of RNF39 in human leukemia monocytic cell line THP-1 cells and mouse bone marrow–derived dendritic cells (BMDCs). RNF39 knockdown enhanced VSV-induced IFN-β expression in THP-1 cells (fig. S3A). In addition, Rnf39 deficiency enhanced RNA virus–induced IFN-β expression in BMDCs (fig. S3, B and C).
Fig. 1. RNF39 inhibits RNA-mediated innate immune response.
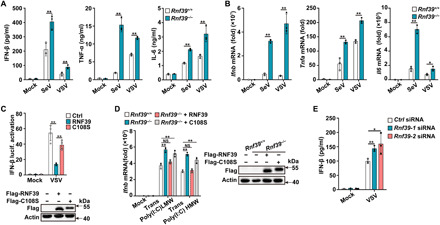
(A and B) Enzyme-linked immunosorbent assay (ELISA) (A) and reverse transcription polymerase chain reaction (RT-PCR) (B) analysis of IFN-β, TNF-α, and IL-6 expression in PMs from Rnf39+/+ or Rnf39−/− mice infected with SeV and VSV. (C) Luciferase activity analysis of IFN-β promoter activity in human embryonic kidney (HEK) 293T cells transfected with IFN-β reporter plasmid, together with empty vector control (Ctrl) plasmid, RNF39, or its mutant C108S after being infected with VSV. Plasmid expression was confirmed by immunoblot analysis (below blots). (D) RT-PCR analysis of Ifnb mRNA expression in Rnf39+/+ MEFs, Rnf39−/− MEFs, or Rnf39−/− MEFs transfected with RNF39 or its mutant C108S plasmid, followed by transfection with low–molecular weight (LMW) or high–molecular weight (HMW) polyinosinic:polycytidylic acid [poly(I:C)] for 4 hours. Plasmid expression was confirmed by immunoblot analysis (right blots). (E) ELISA analysis of IFN-β expression in mouse PMs transfected with Ctrl siRNA and Rnf39 siRNA 1 or 2 for 48 hours and then infected with VSV. All data are represented as means ± SD. Statistical significance was determined by unpaired two-tailed Student’s t tests: *P < 0.05 and **P < 0.01; NS, not significant. All experiments were repeated at a minimum of three times.
Fig. 2. RNF39 attenuates RLR-induced IRF3 activation.
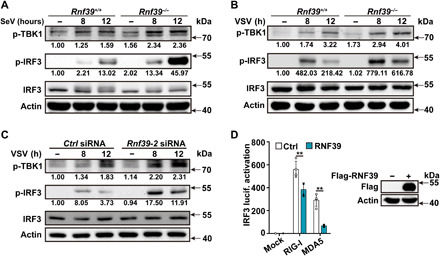
(A and B) Immunoblot analysis of p-TBK1, p-IRF3, and IRF3 in PMs from Rnf39+/+ or Rnf39−/− mice after being infected with SeV and VSV for the indicated time periods. (C) Immunoblot analysis of p-TBK1, p-IRF3, and IRF3 in mouse PMs transfected with Ctrl siRNA or Rnf39 siRNA 2 for 48 hours and then infected with VSV for the indicated time periods. (D) Luciferase activity analysis of IRF3 promoter activity in HEK293T cells transfected with IRF3 reporter plasmid and the indicated adaptor plasmids, together with RNF39 or empty vector control plasmid. Plasmid expression was confirmed by immunoblot analysis (right blots). All data are represented as means ± SD. Significance was determined by unpaired two-tailed Student’s t test: **P < 0.01. All experiments were repeated at a minimum of three times.
Next, we examined the biological effects of RNF39 on the RNA sensing pathway. RNF39 notably attenuated VSV-induced IFN-β reporter activation, while RNF39 C108S mutant (an E3 ubiquitin ligase activity–disrupted mutant, in which a Cys-to-Ser point mutation was introduced) lost the inhibitory effects on RIG-I–triggered IFN-β reporter activation (Fig. 1C). RIG-I and MDA5 selectively recognize cytoplasmic short and long double-stranded RNAs, respectively. Rnf39 deficiency greatly promoted Ifnb expression induced by transfection of both polyinosinic:polycytidylic acid [poly(I:C)] high molecular weight (HMW) and poly(I:C) low molecular weight (LMW) (Fig. 1D). In addition, in this rescue experiments, the transfection of RNF39 wild-type (WT) plasmid, but not C108S mutant, can restore the inhibitory effects on Ifnb expression (Fig. 1D). Together, these data indicate that RNF39 inhibits the RLR pathway via its E3 ubiquitin ligase activity.
IRF3 is a key transcription factor for IFN-β expression. We next examined the effects of RNF39 on IRF3 activation. Rnf39 deficiency and knockdown both enhanced RNA virus–induced IRF3 phosphorylation (Fig. 2, A to C, and fig. S3D). Furthermore, RNF39 overexpression inhibited RIG-I– and MDA5-induced IRF3 reporter activation (Fig. 2D). These results indicate that RNF39 attenuates RLR-induced IRF3 activation, leading to the inhibition of downstream signaling.
Rnf39 deficiency enhances anti-RNA viral responses in vitro and in vivo
Next, we investigated the function of RNF39 in antiviral immunity. Rnf39 deficiency promoted VSV infection– and SeV infection–induced signal transducer and activator of transcription 1 (STAT1) phosphorylation and subsequent interferon-stimulated genes (ISGs) expression, including Ifit1, Ifit2, Isg15, Mx1, and Rantes (regulated upon activation of normal “T cell” expressed and secreted) in macrophages (Fig. 3, A to C, and fig. S4). VSV replication was notably attenuated by Rnf39 deficiency (Fig. 3, D and E). To preclude the possibility that RNF39 functions downstream IFN-β, we examined IFN-β–triggered signaling. Rnf39 deficiency had no effects on IFN-β–induced STAT1 phosphorylation and ISG expression (Fig. 3, F and G). Together, these data indicate that RNF39 inhibits cellular anti-RNA viral immune responses and promotes RNA viral replication.
Fig. 3. Rnf39 deficiency enhances anti-RNA viral responses.
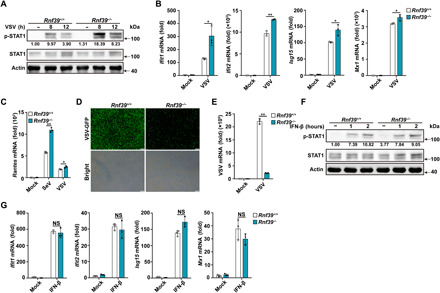
(A and F) Immunoblot analysis of p-STAT1 and STAT1 in Rnf39+/+ or Rnf39−/− PMs infected with VSV [1 multiplicity of infection (MOI)] (A) or stimulated with IFN-β (20 ng/ml) (F) for the indicated time periods. (B and G) RT-PCR analysis of ISG mRNA expression in Rn39f +/+ or Rnf39−/− PMs infected with VSV (B) or stimulated with IFN-β (G). (C) RT-PCR analysis of Rantes mRNA expression in PMs from Rnf39+/+ or Rnf39−/− mice infected with SeV or VSV. (D and E) Microscopy (D) and RT-PCR (E) analysis of VSV replication in Rnf39+/+ or Rnf39−/− PMs infected with VSV-GFP (1 MOI) for 12 hours. Scale bars, 100 μm. All data are represented as means ± SD. Significance was determined by unpaired two-tailed Student’s t test: *P < 0.05 and **P < 0.01. All experiments were repeated at a minimum of three times.
We next investigated the physiological and pathological relevance of the regulatory effects of RNF39 on RLR signaling in the context of viral infection in vivo. Following VSV infection, Rnf39-deficient mice produced notably more IFN-β, TNF-α, and IL-6 in sera than WT mice (Fig. 4A). Concordantly, the VSV viral burden was notably decreased in lung and spleen of Rnf39-deficient mice (Fig. 4B). Less infiltration of inflammatory cells was observed in the lungs of Rnf39-deficient mice (Fig. 4C). Moreover, Rnf39-deficient mice were more resistant in survival assays upon VSV infection (Fig. 4D). Collectively, these data indicate that Rnf39 deficiency enhances RNA-dependent antiviral responses and suppresses RNA viral replication.
Fig. 4. Rnf39 deficiency enhances innate immune responses against RNA viral infection in vivo.
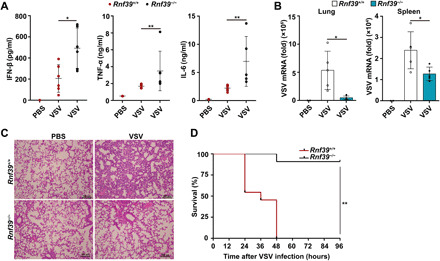
(A) ELISA analysis of IFN-β, TNF-α, and IL-6 production in serum from Rnf39+/+ or Rnf39−/− mice infected with VSV by intraperitoneal injection [phosphate-buffered saline (PBS), n = 2; VSV, n = 6 per condition]. (B) RT-PCR analysis of VSV replication in lung and spleen from Rnf39+/+ or Rnf39−/− mice infected with VSV by intraperitoneal injection (PBS, n = 2; VSV, n = 5 per condition). (C) Hematoxylin and eosin staining of lung tissue sections from Rnf39+/+ or Rnf39−/− mice infected with VSV by intraperitoneal injection (PBS, n = 4; VSV, n = 4 per condition). Scale bars, 100 μm. (D) Survival of Rnf39+/+ or Rnf39−/− mice infected with VSV by intraperitoneal injection (n = 11 per condition). The data are represented as means ± SD. Significance was determined by unpaired two-tailed Student’s t test: *P < 0.05 and **P < 0.01. The data of TNF-α and IL-6 in (C) are represented as medians ± SD. Significance was determined by nonparametric tests: *P < 0.05 and **P < 0.01. All experiments were repeated at a minimum of three times.
RNF39 targets DDX3X
Multiple Ring finger proteins have been implicated in promoting the protein degradation of their respective substrates via E3 ubiquitin ligase activity (16, 18). To clarify its molecular targets, we first examined whether RNF39 regulated the expression of a variety of molecules involved in RLR signaling. RNF39 greatly inhibited DDX3X protein expression but had no effects on the expression of RIG-I, MAVS, TRAF3, and TBK1 (fig. S5A). Moreover, Rnf39 knockdown substantially enhanced DDX3X protein level (fig. S5B). Together, these data indicate that DDX3X may be a target of RNF39.
Next, we examined the interaction between RNF39 and DDX3X. RNF39 was cotransfected with DDX3X, TBK1, TRAF3, IRF3, MDA5, RIG-I, or MAVS in human embryonic kidney (HEK) 293T cells. RNF39 coprecipitated with DDX3X (Fig. 5A and fig. S5E). Furthermore, RNF39 interacted with DDX3X in SeV-infected mouse PMs (Fig. 5B and fig. S5F). Consistently, confocal analysis showed the colocalization between RNF39 and DDX3X in MEFs (Fig. 5C). In vitro binding assays demonstrated that RNF39 could directly interact with DDX3X (Fig. 5D). Together, these data suggest that RNF39 directly interacts with DDX3X.
Fig. 5. RNF39 targets DDX3X.
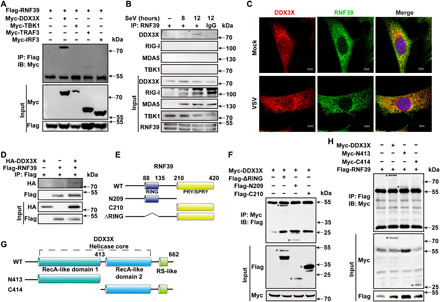
(A) Coimmunoprecipitation of RNF39 with the indicated adaptors from HEK293T cells transfected with Flag-RNF39 and the indicated Myc-tagged plasmids. IP, immunoprecipitation; IB, immunoblot. (B) Coimmunoprecipitation of endogenous RNF39 with endogenous DDX3X from mouse PMs infected with SeV for the indicated time periods. (C) Colocalization between RNF39 and DDX3X in MEFs was examined by confocal microscopy infected with VSV. Scale bars, 10 μm. (D) HA-DDX3X was obtained by in vitro transcription and translation. Interaction between RNF39 and DDX3X was assayed by mixing recombinant protein of human RING finger protein 39 (RNF39) transcript and DDX3X-HA together, followed by immunoprecipitation with Flag antibody and immunoblot analysis with HA antibody. (E) Schematic diagram of RNF39 and its truncation mutants. RING, RING domain; PRY/SPRY, PRY/SPRY domain. (F) Flag-tagged RNF39 mutants and Myc-DDX3X were individually transfected into HEK293T cells. The cell lysates were immunoprecipitated with an anti-Myc antibody and then immunoblotted with the indicated antibodies. The specific bands were marked by asterisk. (G) Schematic diagram of DDX3X and its truncation mutants. RecA-1, RecA-like domains-1; RecA-2, RecA-like domains-2; RS, arginine-serine-rich domain. (H) Myc-tagged DDX3X or its mutants and Flag-RNF39 were individually transfected into HEK293T cells. The cell lysates were immunoprecipitated with Flag antibody and then immunoblotted with the indicated antibodies. The specific bands were marked by asterisk. All experiments were repeated at a minimum of three times.
RNF39 contains an N-terminal RING finger domain and a C-terminal B30.2/SPRY domain (Fig. 5E). To search for the domain of RNF39 that is responsible for the interaction with DDX3X, a series of RNF39 truncated mutants were constructed (Fig. 5E). DDX3X was coprecipitated with RNF39 RING domain deletion mutant (ΔRING) and N209, but not with C210 (Fig. 5F). These results indicated that RNF39 interacted with DDX3X via its N-terminal region. DDX3X contains two RecA-like domains and a C-terminal arginine-serine–rich (RS)–like domain (Fig. 5G). Coimmunoprecipitation experiments with DDX3X truncated mutants showed that the N-terminal recombinase A (RecA)–like domain 1 of DDX3X is required for its interaction with RNF39 (Fig. 5H).
RNF39 promotes DDX3X degradation in proteasome
We next investigated whether RNF39 could promote the protein degradation of DDX3X. Rnf39 deficiency increased DDX3X protein expression in both resting and viral-infected mouse PMs (Fig. 6A), with no influence on Ddx3x mRNA expression (Fig. 6B). Consistently, RNF39 knockdown enhanced DDX3X protein expression in THP-1 (fig. S5C), and Rnf39 deficiency enhanced DDX3X protein expression in BMDCs (fig. S5D). RNF39 overexpression dose-dependently inhibited DDX3X expression in HEK293T cells (Fig. 6C), while other MHC locus–encoded E3 ubiquitin ligases including tripartite motif (TRIM)13, TRIM31, and TRIM40 had no effects on DDX3X expression (fig. S6). Furthermore, Rnf39 deficiency notably inhibited the protein degradation of DDX3X in cycloheximide (CHX) chase experiments (Fig. 6, D and E). RNF39-mediated inhibition of DDX3X expression could be reversed by the proteasome inhibitor MG132, but not by lysosome/autophagy inhibitors chloroquine and 3-methyladenine (3-MA) (Fig. 6F). In addition, the RNF39 point mutation (C108S) lost the ability to promote DDX3X degradation (Fig. 6G). Collectively, these data indicate that RNF39 promotes DDX3X protein degradation in proteasome via its E3 ubiquitin ligase activity.
Fig. 6. RNF39 promotes proteasomal degradation of DDX3X.
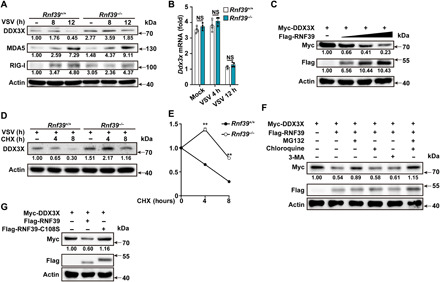
(A and B) Immunoblot analysis of extracts (A) or RT-PCR analysis (B) of mouse PMs from Rnf39+/+ or Rnf39−/− mice infected with VSV for the indicated time points. (C) Immunoblot analysis of extracts from HEK293T cells transfected with Myc-DDX3X and increasing amount of Flag-RNF39 (0, 0.5, 1, or 2 μg) expression plasmids. (D and E) Immunoblot analysis of extracts (D) from Rnf39+/+ or Rnf39−/− mouse PMs infected with VSV for 4 hours and then treated with cycloheximide (CHX) for various times. DDX3X expression level was quantitated by measuring band intensities using “ImageJ” software (E). The values were normalized to actin. (F) Immunoblot analysis of extracts from HEK293T cells transfected with Myc-DDX3X and Flag-RNF39 expression plasmid and then treated with MG132, chloroquine, or 3-MA for 4 hours. (G) Immunoblot analysis of extracts from HEK293T cells transfected with Myc-DDX3X, together with Flag-tagged RNF39 or RNF39 C108S expression plasmids. All data are represented as means ± SD. Significance was determined by unpaired two-tailed Student’s t test: **P < 0.01. All experiments were repeated at a minimum of three times.
RNF39 selectively promotes K48-linked ubiquitination of DDX3X
Protein ubiquitination is a key step in the ubiquitin-proteasome degradation pathway. We thereby examined the effects of RNF39 on DDX3X ubiquitination. Polyubiquitination of DDX3X was substantially increased in the presence of RNF39 expression plasmid in HEK293T cells (Fig. 7A). The RNF39 C108S mutant lost the ability to enhance the polyubiquitination of DDX3X (Fig. 7A). Given that DDX3X or RNF39 perhaps form a complex with other nonspecific proteins, we performed a two-step immunoprecipitation assay (Re-IP) to reduce the presence of nonspecific associated proteins, and similar results were observed (Fig. 7B). To dissect the polyubiquitin chain linkage on DDX3X catalyzed by RNF39, a panel of ubiquitin mutants that contains arginine substitutions of all its lysine (K) residues except the one as indicated was used in the transfection assays. DDX3X polyubiquitination could be detected in the presence of K48 or K63 plasmid, but not with other mutants (Fig. 7C). Moreover, RNF39 markedly enhanced K48-linked polyubiquitination of DDX3X, with no effects on K63-linked polyubiquitination of DDX3X (Fig. 7D). Collectively, these data indicate that RNF39 selectively promotes K48-linked polyubiquitination of DDX3X.
Fig. 7. RNF39 promotes K48-linked ubiquitination of DDX3X.
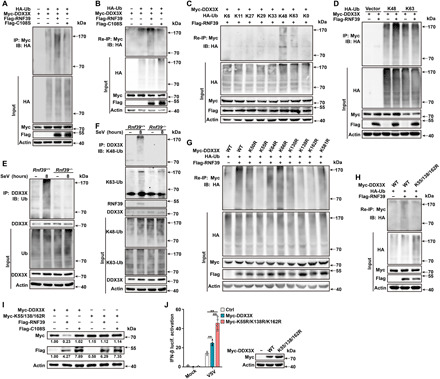
(A) Lysates from HEK293T cells transiently cotransfected with HA-Ub, Myc-DDX3X along with RNF39-Flag or RNF39 C108S-Flag, were subjected to immunoprecipitation with Myc antibody followed by immunoblot analysis with HA antibody. (B) Lysates from HEK293T cells transiently cotransfected with HA-Ub, Myc-DDX3X along with RNF39-Flag or RNF39 C108S-Flag, were subjected to immunoprecipitation with Myc antibody. The immunoprecipitates were denatured and re-immunoprecipitated with Myc antibody (two-step immunoprecipitation, Re-IP) and then analyzed by immunoblot analysis. (C) Re-IP analysis lysates from HEK293T cells transiently cotransfected with HA-Ub (WT and its mutants), Flag-RNF39, and Myc-DDX3X. (D) Immunoprecipitation analysis lysates from HEK293T cells transiently cotransfected with K48-Ub or K63-Ub mutant, Flag-RNF39, and Myc-DDX3X. (E and F) Lysates from Rnf39+/+ or Rnf39−/− mouse PMs infected with SeV for 8 hours were immunoprecipitated with DDX3X antibody, followed by immunoblot analysis with indicated antibodies. (G and H) Re-IP analysis lysates from HEK293T cells transiently cotransfected with HA-Ub, Flag-RNF39, along with Myc-DDX3X (WT and its point mutants). (I) Immunoblot analysis of extracts from HEK293T cells transfected with Myc-DDX3X or Myc-DDX3X K55/138/162R mutant, together with Flag-tagged RNF39 or RNF39 C108S expression plasmid. (J) Luciferase activity analysis of IFN-β promoter activity in HEK293T cells transfected with IFN-β reporter plasmid, together with empty vector control plasmid, Myc-DDX3X, or Myc-DDX3X K55/138/162R mutant after being infected with VSV. Plasmid expression in the HEK293T cells was confirmed by immunoblot analysis. All data are represented as means ± SD. Significance was determined by unpaired two-tailed Student’s t test: **P < 0.01. All experiments were repeated at a minimum of three times.
Under physiological conditions, the endogenous DDX3X was observed to be robustly ubiquitinated upon viral infection in mouse PMs and MEFs (Fig. 7E and fig. S7A). However, DDX3X polyubiquitination was almost completely abolished in both Rnf39-deficient and Rnf39 siRNA–transfected cells (Fig. 7E and fig. S7B). In addition, Rnf39 deficiency selectively inhibited K48-linked polyubiquitination of DDX3X, whereas K63-linked polyubiquitination of DDX3X was not affected (Fig. 7F). Collectively, these data firmly establish that RNF39 acts as an E3 ubiquitin ligase and directly catalyzes the K48-linked polyubiquitination of DDX3X.
Next, we tried to identify the ubiquitinated lysine sites of DDX3X. DDX3X contains 32 lysine residues. First, we predicted the possible ubiquitination sites by the UbPred program (28). Eight lysine (K) residues were predicted to be possible ubiquitination sites, and they are conserved in human and mouse (table S2). Then, we replaced each of the eight DDX3X lysine (K) residues individually with arginine (R) to create K50R, K55R, K64R, K66R, K130R, K138R, K162R, and K581 mutants of DDX3X, respectively. Re-IP assay revealed that only K55R, K138R, and K162R partly blocked the ubiquitination of DDX3X mediated by RNF39 (Fig. 7G). We then constructed the DDX3X K55/K138/K162R mutant, which harbors three mutations. RNF39-mediated ubiquitination and protein degradation of DDX3X were completely abolished in DDX3X K55/K138/K162R mutant (Fig. 7, H and I). In addition, compared to DDX3X WT, the K55/K138/K162R mutant notably enhanced VSV-induced IFN-β activation (Fig. 7J). Thus, these results suggest that K55, K138, and K162 of DDX3X are essential residues for its ubiquitination and degradation.
DISCUSSION
MHC locus consists of a tightly linked set of genes encoding many proteins involved in immune responses. A variety of nonclassical genes in MHC locus–encoded molecules are implicated in immune regulation, such as human leucocyte antigen (HLA)-E and HLA-G in human. A couple of E3 ubiquitin ligase genes are located in the MHC class I region and have important regulatory roles in antiviral immune responses, inflammation, and autoimmunity, such as TRIM31 and TRIM38 (17–19). RNF39 is also encoded within the MHC class I region, which implies its potential roles in immune regulation. In the present study, we show that RNF39 selectively targets DDX3X, promotes its degradation via the ubiquitin-proteasome pathway, and suppresses RLR signaling (fig. S6). Viral infection induced RNF39 expression to achieve the fine-tune regulation of host antiviral responses. RNA viruses impair the RLR-mediated signaling pathway by targeting RNF39, which indicates the complexity of the interaction between hosts and viruses.
DDX3X is a multifunctional protein involved in mRNA metabolism, cell cycle control, and the formation of stress granules, apoptosis, tumorigenesis, and viral infection (11, 29–31). Recently, DDX3X has been identified as a live-or-die checkpoint in stressed cells by driving NLRP3 inflammasome and stress granule assembly (31). Furthermore, DDX3X is considered as a valuable target for developing broad-spectrum antiviral agents (32, 33). Thus, control of DDX3X expression via RNF39 will be a prime target for designing therapeutic strategies for multiple diseases.
Self-nucleic acid–driven type I IFN production and inflammation play crucial or contributing roles in the initiation of autoinflammatory and autoimmune diseases. For example, MDA5 gain-of-function mutants could cause a variety of autoimmune disorders, such as type 1 diabetes and SLE (34, 35). Genetic variant of RNF39 is associated with autoimmune diseases, including SLE and MS (23–27). Deficiency of RNF39 promotes RLR activation and enhances type I IFN production. Thus, our data further imply the potential roles of RNF39 in autoimmunity and suggest RNF39 as a priming target for the intervention of autoimmune diseases caused by excessive RLR activation.
In summary, we identified RNF39 as a suppressor of anti-RNA viral innate immunity by targeting the adaptor DDX3X. Because of the fundamental and complicated functions of RLR, fine-tuning of the nucleic acid–sensing pathway is critical for the viral disease resistance and maintenance of immune homeostasis. Our research uncovers a previously unknown regulatory mechanism of RLR pathway under physiological conditions. Furthermore, control of DDX3X expression by RNF39 will be a priming therapeutic strategy for the intervention of diseases with aberrant activation of innate immune responses.
MATERIALS AND METHODS
Study design
The aims of the study were to characterize the regulatory mechanisms of DDX3X in the RLR signaling. The experiments and assessment of outcomes were performed in a blinded fashion. Descriptions of experimental replicate are found in the figure legends and/or subsections of Materials and Methods.
Mice
Rnf39-deficient mice on C57BL/6 background were generated by CRISPR-Cas9–mediated genome engineering. C57BL/6 mice were from Vital River Laboratory Animal Technology Co. (Beijing, China). All animal experiments were undertaken in accordance with the National Institute of Health Guide for the Care and Use of Laboratory Animals, with the approval of the Scientific Investigation Board of School of Basic Medical Science, Shandong University, Jinan, Shandong Province, China.
Statistical analysis
Statistical significance between groups was determined by unpaired two-tailed Student’s t test and nonparametric tests. Statistical significance in examining survival among Rnf39+/+ and Rnf39−/− mice was performed via the Kaplan-Meier survival by GraphPad Prism 6.0. Values of P < 0.05 were considered to be statistically significant.
Acknowledgments
Funding: This work was supported by grants from the National Natural Science Foundation of China (31870866, 81622030, 81901609, and 81861130369) and the National Key Research and Developmental Program of China (2017YFC1001100). W.Z. is a Newton Advanced Fellow awarded by the Academy of Medical Sciences (NAF\R1\180232). Author contributions: W.Z. conceived the study, designed the experiments, and provided overall direction. W.W., M.J., C.Z., Z.Y., H.S., and Y.Q. did the experiments. W.W., M.J., and W.Z. analyzed the data and wrote the paper. Competing interests: The authors declare that they have no competing interests. Data and materials availability: All data needed to evaluate the conclusions in the paper are present in the paper or the Supplementary Materials. Additional data related to this paper may be requested from the authors.
SUPPLEMENTARY MATERIALS
Supplementary material for this article is available at http://advances.sciencemag.org/cgi/content/full/7/10/eabe5877/DC1
REFERENCES AND NOTES
- 1.Ablasser A., Hur S., Regulation of cGAS- and RLR-mediated immunity to nucleic acids. Nat. Immunol. 21, 17–29 (2020). [DOI] [PubMed] [Google Scholar]
- 2.Rehwinkel J., Gack M. U., RIG-I-like receptors: Their regulation and roles in RNA sensing. Nat. Rev. Immunol. 20, 537–551 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Tan X., Sun L., Chen J., Chen Z. J., Detection of microbial infections through innate immune sensing of nucleic acids. Annu. Rev. Microbiol. 72, 447–478 (2018). [DOI] [PubMed] [Google Scholar]
- 4.Takeuchi O., Akira S., Pattern recognition receptors and inflammation. Cell 140, 805–820 (2010). [DOI] [PubMed] [Google Scholar]
- 5.Chow K. T., Gale M. Jr., Loo Y.-M., RIG-I and other RNA sensors in antiviral immunity. Annu. Rev. Immunol. 36, 667–694 (2018). [DOI] [PubMed] [Google Scholar]
- 6.Zevini A., Olagnier D., Hiscott J., Crosstalk between cytoplasmic RIG-I and STING sensing pathways. Trends Immunol. 38, 194–205 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Oshiumi H., Ikeda M., Matsumoto M., Watanabe A., Takeuchi O., Akira S., Kato N., Shimotohno K., Seya T., Hepatitis C virus core protein abrogates the DDX3 function that enhances IPS-1-mediated IFN–beta induction. PLOS ONE 5, e14258 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Soulat D., Bürckstümmer T., Westermayer S., Goncalves A., Bauch A., Stefanovic A., Hantschel O., Bennett K. L., Decker T., Superti-Furga G., The DEAD-box helicase DDX3X is a critical component of the TANK-binding kinase 1-dependent innate immune response. EMBO J. 27, 2135–2146 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Gu L., Fullam A., Brennan R., Schröder M., Human DEAD box helicase 3 couples IκB kinase ε to interferon regulatory factor 3 activation. Mol. Cell. Biol. 33, 2004–2015 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Gu L., Fullam A., McCormack N., Höhn Y., Schröder M., DDX3 directly regulates TRAF3 ubiquitination and acts as a scaffold to co-ordinate assembly of signalling complexes downstream from MAVS. Biochem. J. 474, 571–587 (2017). [DOI] [PubMed] [Google Scholar]
- 11.Ariumi Y., Multiple functions of DDX3 RNA helicase in gene regulation, tumorigenesis, and viral infection. Front. Genet. 5, 423 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Oshiumi H., Sakai K., Matsumoto M., Seya T., DEAD/H BOX 3 (DDX3) helicase binds the RIG-I adaptor IPS-1 to up-regulate IFN-beta-inducing potential. Eur. J. Immunol. 40, 940–948 (2010). [DOI] [PubMed] [Google Scholar]
- 13.Szappanos D., Tschismarov R., Perlot T., Westermayer S., Fischer K., Platanitis E., Kallinger F., Novatchkova M., Lassnig C., Müller M., Sexl V., Bennett K. L., Foong-Sobis M., Penninger J. M., Decker T., The RNA helicase DDX3X is an essential mediator of innate antimicrobial immunity. PLOS Pathog. 14, e1007397 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Gringhuis S. I., Hertoghs N., Kaptein T. M., Zijlstra-Willems E. M., Sarrami-Forooshani R., Sprokholt J. K., van Teijlingen N. H., Kootstra N. A., Booiman T., van Dort K. A., Ribeiro C. M. S., Drewniak A., Geijtenbeek T. B. H., HIV-1 blocks the signaling adaptor MAVS to evade antiviral host defense after sensing of abortive HIV-1 RNA by the host helicase DDX3. Nat. Immunol. 18, 225–235 (2017). [DOI] [PubMed] [Google Scholar]
- 15.Wang H., Ryu W. S., Hepatitis B virus polymerase blocks pattern recognition receptor signaling via interaction with DDX3: Implications for immune evasion. PLOS Pathog. 6, e1000986 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Heaton S. M., Borg N. A., Dixit V. M., Ubiquitin in the activation and attenuation of innate antiviral immunity. J. Exp. Med. 213, 1–13 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Liu B., Zhang M., Chu H., Zhang H., Wu H., Song G., Wang P., Zhao K., Hou J., Wang X., Zhang L., Gao C., The ubiquitin E3 ligase TRIM31 promotes aggregation and activation of the signaling adaptor MAVS through Lys63-linked polyubiquitination. Nat. Immunol. 18, 214–224 (2017). [DOI] [PubMed] [Google Scholar]
- 18.Song H., Liu B., Huai W., Yu Z., Wang W., Zhao J., Han L., Jiang G., Zhang L., Gao C., Zhao W., The E3 ubiquitin ligase TRIM31 attenuates NLRP3 inflammasome activation by promoting proteasomal degradation of NLRP3. Nat. Commun. 7, 13727 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Hu M. M., Yang Q., Xie X. Q., Liao C. Y., Lin H., Liu T. T., Yin L., Shu H. B., Sumoylation promotes the stability of the DNA sensor cGAS and the adaptor STING to regulate the kinetics of response to DNA virus. Immunity 45, 555–569 (2016). [DOI] [PubMed] [Google Scholar]
- 20.Meyer M., Gaudieri S., Rhodes D. A., Trowsdale J., Cluster of TRIM genes in the human MHC class I region sharing the B30.2 domain. Tissue Antigens 61, 63–71 (2003). [DOI] [PubMed] [Google Scholar]
- 21.Fellay J., Shianna K. V., Ge D., Colombo S., Ledergerber B., Weale M., Zhang K., Gumbs C., Castagna A., Cossarizza A., Cozzi-Lepri A., de Luca A., Easterbrook P., Francioli P., Mallal S., Martinez-Picado J., Miro J. M., Obel N., Smith J. P., Wyniger J., Descombes P., Antonarakis S. E., Letvin N. L., McMichael A. J., Haynes B. F., Telenti A., Goldstein D. B., A whole-genome association study of major determinants for host control of HIV-1. Science 317, 944–947 (2007). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Lin Y. J., Chen C. Y., Jeang K. T., Liu X., Wang J. H., Hung C. H., Tsang H., Lin T. H., Liao C. C., Huang S. M., Lin C. W., Ho M. W., Chien W. K., Chen J. H., Ho T. J., Tsai F. J., Ring finger protein 39 genetic variants associate with HIV-1 plasma viral loads and its replication in cell culture. Cell Biosci. 4, 40 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Kurata R., Nakaoka H., Tajima A., Hosomichi K., Shiina T., Meguro A., Mizuki N., Ohono S., Inoue I., Inoko H., TRIM39 and RNF39 are associated with Behçet’s disease independently of HLA-B∗51 and -A∗26. Biochem. Biophys. Res. Commun. 401, 533–537 (2010). [DOI] [PubMed] [Google Scholar]
- 24.Lu Y., Cheng Y., Yan W., Nardini C., Exploring the molecular causes of hepatitis B virus vaccination response: An approach with epigenomic and transcriptomic data. BMC Med. Genomics 7, 12 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Maltby V. E., Lea R. A., Sanders K. A., White N., Benton M. C., Scott R. J., Lechner-Scott J., Differential methylation at MHC in CD4+ T cells is associated with multiple sclerosis independently of HLA-DRB1. Clin. Epigenetics 9, 71 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Morin A., Laviolette M., Pastinen T., Boulet L. P., Laprise C., Combining omics data to identify genes associated with allergic rhinitis. Clin. Epigenetics 9, 3 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Renauer P., Coit P., Jeffries M. A., Merrill J. T., McCune W. J., Maksimowicz-McKinnon K., Sawalha A. H., DNA methylation patterns in naïve CD4+ T cells identify epigenetic susceptibility loci for malar rash and discoid rash in systemic lupus erythematosus. Lupus Sci. Med. 2, e000101 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Radivojac P., Vacic V., Haynes C., Cocklin R. R., Mohan A., Heyen J. W., Goebl M. G., Iakoucheva L. M., Identification, analysis, and prediction of protein ubiquitination sites. Proteins 78, 365–380 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Valiente-Echeverría F., Hermoso M. A., Soto-Rifo R., RNA helicase DDX3: At the crossroad of viral replication and antiviral immunity. Rev. Med. Virol. 25, 286–299 (2015). [DOI] [PubMed] [Google Scholar]
- 30.Hondele M., Sachdev R., Heinrich S., Wang J., Vallotton P., Fontoura B. M. A., Weis K., DEAD-box ATPases are global regulators of phase-separated organelles. Nature 573, 144–148 (2019). [DOI] [PubMed] [Google Scholar]
- 31.Samir P., Kesavardhana S., Patmore D. M., Gingras S., Malireddi R. K. S., Karki R., Guy C. S., Briard B., Place D. E., Bhattacharya A., Sharma B. R., Nourse A., King S. V., Pitre A., Burton A. R., Pelletier S., Gilbertson R. J., Kanneganti T. D., DDX3X acts as a live-or-die checkpoint in stressed cells by regulating NLRP3 inflammasome. Nature 573, 590–594 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Brai A., Fazi R., Tintori C., Zamperini C., Bugli F., Sanguinetti M., Stigliano E., Esté J., Badia R., Franco S., Martinez M. A., Martinez J. P., Meyerhans A., Saladini F., Zazzi M., Garbelli A., Maga G., Botta M., Human DDX3 protein is a valuable target to develop broad spectrum antiviral agents. Proc. Natl. Acad. Sci. U.S.A. 113, 5388–5393 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Schröder M., Human DEAD-box protein 3 has multiple functions in gene regulation and cell cycle control and is a prime target for viral manipulation. Biochem. Pharmacol. 79, 297–306 (2010). [DOI] [PubMed] [Google Scholar]
- 34.Barrat F. J., Elkon K. B., Fitzgerald K. A., Importance of nucleic acid recognition in inflammation and autoimmunity. Annu. Rev. Med. 67, 323–336 (2016). [DOI] [PubMed] [Google Scholar]
- 35.Dias Junior J. A. G., Sampaio N. G., Rehwinkel J. A., A balancing act: MDA5 in antiviral immunity and autoinflammation. Trends Microbiol. 27, 75–85 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Yu Z., Song H., Jia M., Zhang J., Wang W., Li Q., Zhang L., Zhao W., USP1-UAF1 deubiquitinase complex stabilizes TBK1 and enhances antiviral responses. J. Exp. Med. 214, 3553–3563 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Jia M., Qin D., Zhao C., Chai L., Yu Z., Wang W., Tong L., Lv L., Wang Y., Rehwinkel J., Yu J., Zhao W., Redox homeostasis maintained by GPX4 facilitates STING activation. Nat. Immunol. 21, 727–735 (2020). [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary material for this article is available at http://advances.sciencemag.org/cgi/content/full/7/10/eabe5877/DC1