

Abstract
The structure of the TriABC inner membrane component of the triclosan/SDS-specific efflux pump from Pseudomonas aeruginosa was determined by cryo-electron microscopy to 4.5Å resolution. The complete structure of the inner membrane transporter TriC of the Resistance-Nodulation-Division (RND) superfamily was solved including a partial structure of the fused periplasmic membrane fusion subunits, TriA and TriB. The substrate-free conformation of TriABC represents an intermediate step in efflux complex assembly prior to the engagement of the outer membrane channel. Structural analysis identified a tunnel network whose constriction impedes substrate efflux, indicating inhibition of TriABC in the unengaged state. Blind docking studies revealed binding to TriC at the same loci by substrates and bulkier non-substrates. Together with functional analyses, we propose that selective substrate translocation involves conformational gating at the tunnel narrowing that together with conformational ordering of TriA and TriB creates an engaged state capable of mediating substrate efflux.
Graphical Abstract
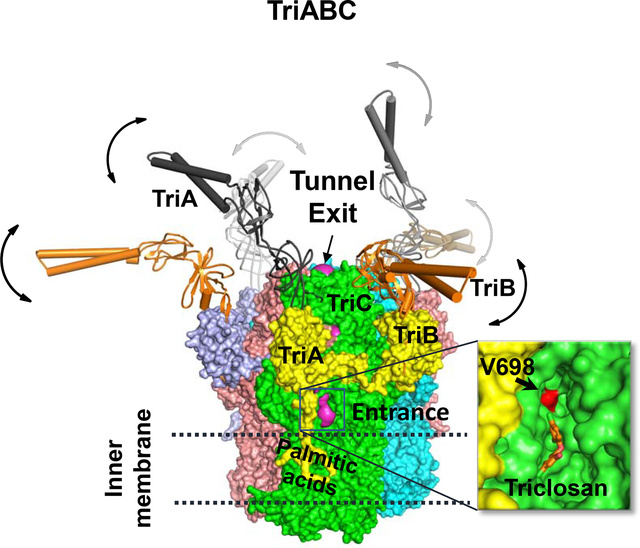
In Brief:
TriABC is a triclosan/SDS-specific efflux pump from Pseudomonas aeruginosa and linked to biocide resistance. Subunits TriA and TriB show disorder in absence of substrate. Efflux implicates a tunnel in TriC subunits, supported by functional and docking data. Tunnel narrowing by an internal gating loop situated after Val 698 inhibits efflux.
Introduction
Gram-negative bacteria are resistant to a broad range of antimicrobial agents due to a combined action of a permeability barrier of the outer membrane and active multidrug efflux (Zgurskaya et al., 2015a; Zgurskaya and Nikaido, 2000). The notoriously antibiotic resistant Pseudomonas aeruginosa species, a nosocomial human pathogen, possesses both an extended repertoire of multidrug efflux pumps and the outer membrane of extremely low permeability (De Kievit et al., 2001; Poole, 2004). Multidrug efflux pumps belonging to the RND (Resistance-Nodulation-cell Division) superfamily of transporters are at the core of the P. aeruginosa antibiotic resistance and the focus of the present study.
Among twelve RND transporters encoded on the chromosome of P. aeruginosa, TriABC-OpmH stands apart because of its unusual structural and functional features (Mima et al., 2007; Weeks et al., 2015). Although most RND-type efflux pumps are tri-partite protein complexes, TriABC-OpmH complex comprises four subunits: the inner membrane RND transporter TriC, the outer membrane channel OpmH from the Outer Membrane Factor (OMF) family and two different periplasmic membrane fusion proteins (MFPs) TriA and TriB. Together the four components form a trans-envelope efflux pump spanning the two membranes and the periplasm of P. aeruginosa cell envelope. TriABC-OpmH has a narrow substrate specificity with an antimicrobial agent triclosan, a detergent SDS, and a lipophilic stain Nile Red, as the only known substrates of this pump (Mima et al., 2007; Weeks et al., 2015). Both triclosan and SDS are also substrates of various RND efflux pumps including the well-characterized E. coli AcrAB-TolC and P. aeruginosa MexAB-OprM (Chuanchuen et al., 2001; Weeks et al., 2015).
TriC assembles as a trimer. Each protomer comprises a periplasmic porter domain and a transmembrane domain with of two large periplasmic loops and twelve transmembrane segments (TM1 to TM12) (Murakami et al., 2002). In AcrB, substrates bind to a large cavity of the porter domain and in a groove bound by TM1 and TM2 (Eicher et al., 2012; Nakashima et al., 2011; Oswald et al., 2016). The conformational changes in the porter domain are driven by the proton translocation engaging TM4 and TM10, where several highly conserved charged residues are located (Eicher et al., 2014; Seeger et al., 2009; Su et al., 2006). During active transport, the three RND protomers adopt three distinct conformations driving the substrate from the access pocket (AP) into the deep binding pocket (DBP) and then into the tunnel of the MFP and are named Loose (L), Tight (T) and Open (O), respectively (Seeger et al., 2006).
The porter domains of RND transporters are the docking sites for the assembly of the trans-envelope complexes with MFP and outer membrane proteins (Du et al., 2014; Tikhonova and Zgurskaya, 2004). Based on homology with other MFPs (Higgins et al., 2004; Mikolosko et al., 2006), the structures of TriA and TriB each comprise four domains: an α-hairpin, a lipoyl, a α-β barrel and a membrane proximal (MP) domains. Structural studies of AcrAB-TolC support a model of the trans-envelope complex, in which the MFPs form a hexameric, funnel-like structure that connects the porter domain of the RND transporter with a trimeric OMF via its α-hairpins, which are fully extended into periplasm (Daury et al., 2016; Du et al., 2014). Functional and biochemical studies showed that the complex is dynamic and its assembly is concurrent with the conformational changes in MFP needed to activate the transporter and open the channel for a substrate to cross the outer membrane (Tikhonova et al., 2009; Tikhonova et al., 2011; Zgurskaya et al., 2015a).
In the assembled trans-envelope complex, the MFP subunit forms two different interfaces, with both the RND transporter and with the OMF. These two interfaces are functionally non-equivalent and in the TriABC-OpmH complex are specific for TriA and TriB proteins (Ntreh et al., 2016; Weeks et al., 2015). The α-hairpin of TriA but not TriB is essential for recruitment of OMF, whereas the MP domain of TriB, but not TriA, is important for the activity of the efflux pump (Weeks et al., 2015). In the complex, TriA and TriB are arranged in the symmetric manner and each MFPs occupies a specific binding site in TriC and OpmH (Krishnamoorthy et al., 2016; Ntreh et al., 2016).
An outstanding question is the conformational dynamics of MFPs that drive tripartite complex formation. Structural studies have provided insight into the molecular architecture of the tripartite complex promoted by MFPs (Daury et al., 2016; Du et al., 2014), however the conformational state of MFPs in complex with the inner membrane transporter prior to the tripartite complex formation is largely unknown. This substrate-free state is important to prevent the transporter from futile efflux cycling and depletion of cell energy. The substrate free-state can engage in formation of the tripartite complex (Wang et al., 2017) and in subsequent activation of drug efflux (Tikhonova et al., 2011; Zgurskaya et al., 2015b). A model has been postulated for the substrate free complex formed by the inner membrane transporter with its cognate MFP, and later nick-named “drug sweeping” state (Symmons et al., 2015; Tikhonova et al., 2011; Yamaguchi et al., 2015; Zgurskaya et al., 2015b).
To investigate the conformation of such a “drug sweeping” state, the structure of TriC interacting with the fused TriAB dimer was determined using single particle cryo-electron microscopy. Our results provide evidence for a substrate-free open conformation of TriABC, an intermediate in the assembly of the trans-envelope efflux complex.
Results
Fusion of TriA and TriB is required to purify the TriABC transporter complex:
To examine the structural integrity of the complex, we co-expressed the His-tagged TriC transporter with TriA and TriB produced either as two separate polypeptides or as a fused TriAB dimer (Weeks et al., 2015). When overexpressed in E. coli Δ9-Pore cells lacking all TolC-dependent transporters (Krishnamoorthy et al., 2016), both TriABC variants associate with the endogenous TolC channel and confer resistance to triclosan and SDS (Weeks et al., 2015) (Table S1). Similarly, both complexes exhibited comparable efflux efficiency, when their activities were reconstituted in E. coli Δ9-Pore cells using the fluorescent membrane probe Nile Red (Fig. 1A).
Figure 1. Activities and purification of TriABC pump.
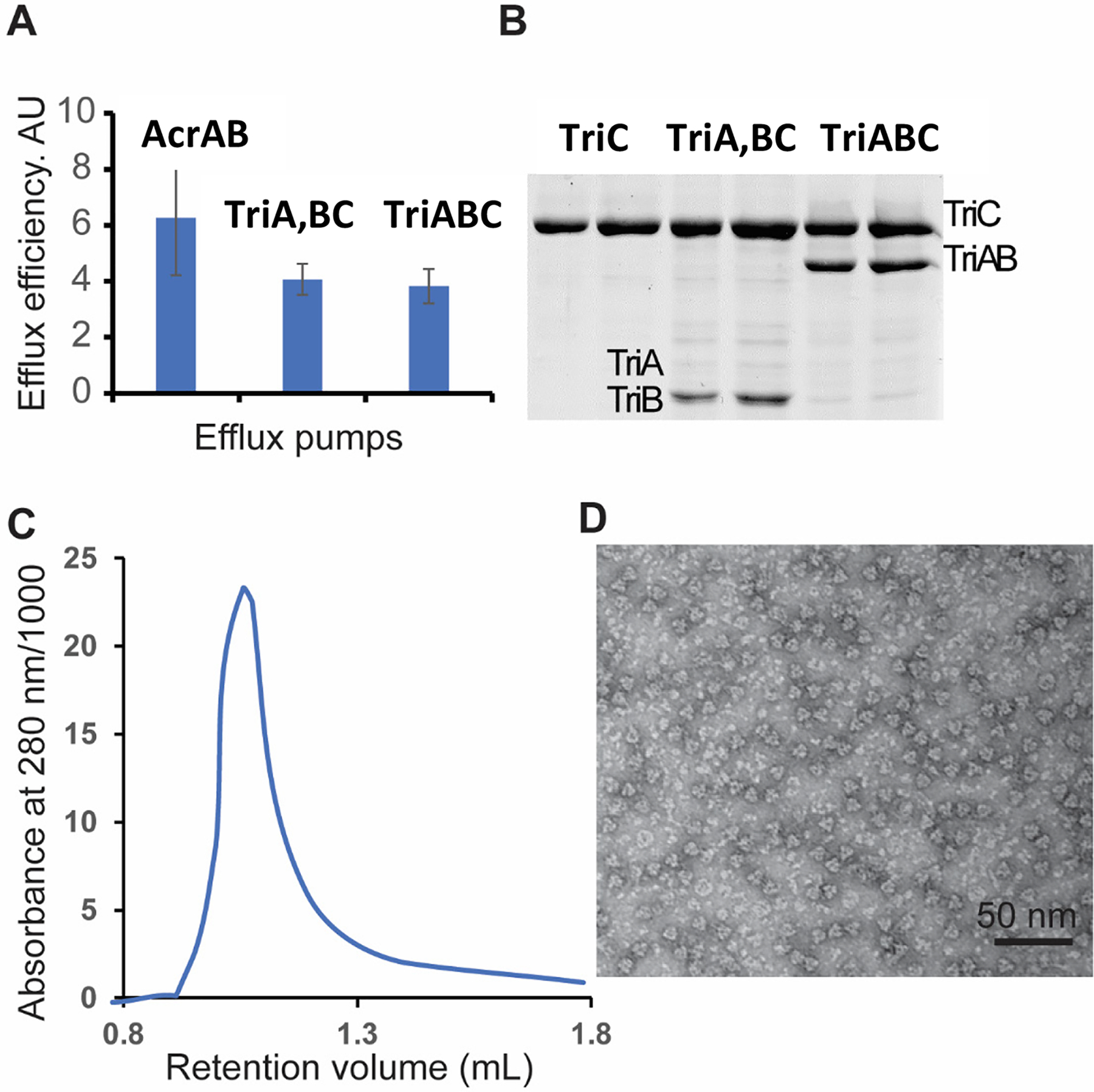
A. Intracellular accumulation of Nile Red (NR) in E. coli Δ9-Pore overproducing AcrAB and TriA,BC with TriA and TriB as separate (TriA, TriB) and TriABC with fused (TriAB) polypeptides respectively. Efflux efficiency is estimated as a ratio of NR accumulation in cells carrying an empty vector and in cells overproducing the indicated efflux pumps.
B. TriC-His and TriABC-His complexes were purified from E. coli JWW2 carrying respective plasmid constructs, separated by 10% SDS-PAGE and stained with Coomassie Brilliant Blue. The molecular masses of TriA and TriB are too close for a clear band separation. The dominant band is TriA, as seen by immunoblotting of TriABC fractions with anti-TriA and anti-TriB polyclonal antibodies in Fig. 8A–B.
C. The TriABC sample was injected onto a Superdex 200 increase 3.2/300 column (flowrate: 75 μL/min, mobile phase: 100 mM Tris/HCl, 150 mM NaCl, 0.03 % (w/v) DDM, pH 8.0, column volume 2.4 mL). The retention volume of the peak corresponded to an estimated molecular weight of 620 kDa.
D. Negative stain electron microscopy image of purified TriABC.
TriA and TriB both co-purified with TriC His-affinity tag fraction, however the purified complex was heterogeneous and contained substoichiometric amounts of TriB protein (Fig. 1B). In contrast, the fused TriAB protein formed a stable complex with TriC (TriABC thereafter). The structure of this complex was determined using single particle cryo-EM. Size exclusion chromatography yielded a single homogeneous peak that corroborated the 1:1 TriC:TriAB stoichiometry. The position of the peak fraction (~ 620 kDa) (Fig. 1C) is consistent with the expected molecular weight for a stoichiometric trimeric TriABC complex (560 kDa) surrounded by a n-Dodecyl-β-D-Maltoside (DDM) detergent corona. Homogeneity of the purified complex particles was further corroborated by negative stain electron microscopy (Fig 1D).
Different states of TriC alone and in the complex with TriAB:
Single particle analysis in negative stain EM and cryo-EM highlighted unique features of TriC alone and TriABC complex. When displayed at threshold volume of 330 Å3, the negative stain reconstructions of TriC particles showed a clearly distinguishable OMF-docking, porter and transmembrane domains (Fig. S1A–B). The differences observed in five volumes of TriC, calculated by Maximum-likelihood three-dimensional classification (ML3D), indicated that in solution isolated TriC appears conformationally flexible.
In the negative staining map of TriABC complex, densities assigned to TriAB protein are seen, surrounding the porter domain of TriC (Fig. S1C–D). The cryo-EM difference map between the 2D classes corresponding to a bottom view of TriC and TriABC complexes clearly shows the presence of six residual densities corresponding to trimeric TriAB (Fig. S1D).
Structure of the TriABC complex:
The cryo-EM analysis of TriABC delineated TriC and showed partially ordered MFP chains (Fig. 2 and S1). Without imposing symmetry, the resolution of the EM map was estimated at 6.5 Å using the gold-standard Fourier shell correlation criterion of 0.143 (Fig. S2B). The imposition of C3 symmetry significantly increased the resolution to 4.5 Å (Fig. S2C and S3). Twelve transmembrane helices were resolved in each TriC protomer. Although the 2D class averages point to weak densities that extend into the periplasm (Fig 2G), the 3D reconstruction averaged out these densities rather than consolidating them into interpretable features.
Figure 2. The structure of TriABC trimer as determined using single particle cryo-EM.
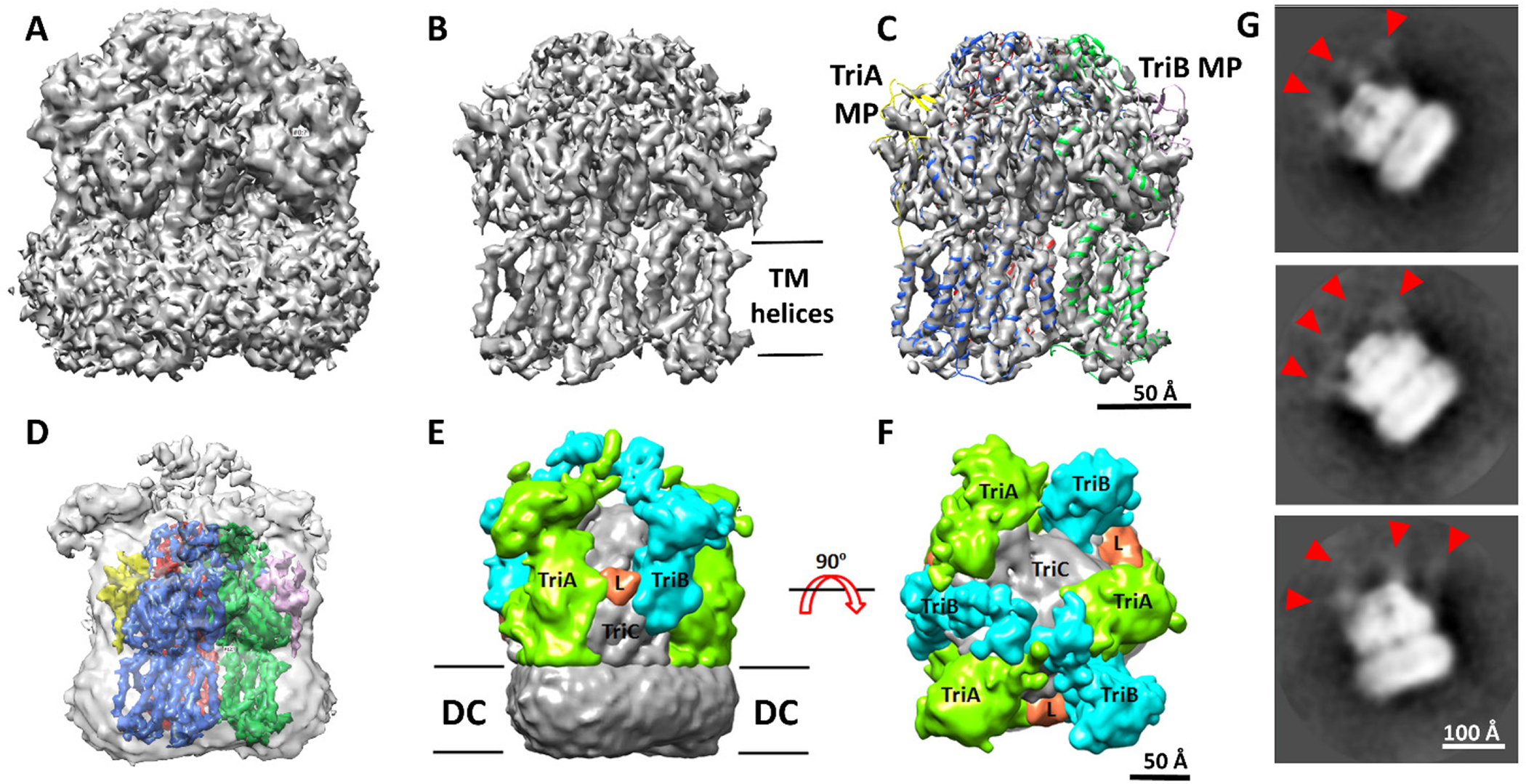
A-C. Isosurface representation of the cryo-EM map calculated imposing C3 symmetry is displayed at low (1.97, ~ 2.0σ) and high (4.35, ~ 4.3σ) contour levels in panels A and B respectively. In panel C, the model built and refined is shown as a cartoon representation superposed with the C3 symmetry map shown in panel B, with red, green and blue corresponding to the three TriC subunits, yellow and pink for the TriA MP and TriB MP domains, respectively (see also movie M1).
D. Confidence map of the C1 reconstruction calculated as described in (Beckers et al., 2019) and shown at the confidence level of 99.9% as a transparent surface representation with TriABC model superposed.
E-F. Composite map highlighting densities corresponding to TriA and TriB. The colored densities were derived from the difference map calculated between the C1 cryo EM map and a density map generated from the TriC model and low-pass-filtered to the resolution of the C1 cryo EM map (6.5Å). The difference map was then segmented using Segger (Pintilie et al., 2010) implemented in Chimera (Pettersen et al., 2004). Densities attributed to TriA, TriB and the linker L are shown colored in lime, cyan and orange respectively.
G. Examples of 2D class averages. Red arrowheads point to densities corresponding to the putative MFPs extending into the periplasm.
Structure of TriC transporter:
The cryo-EM map was next used for model building. The entire amino acid sequence for the TriC subunit was successfully fitted into the C3 cryo-EM map from an ab initio model (Table S2) predicted by the I-TASSER server (Xu and Zhang, 2010; Yang et al., 2015; Zhang and Skolnick, 2004, 2005). The TriC structure assembles as a 120 Å long and 100 Å wide homotrimer (Fig. 2 and 3) and can be decomposed figuratively into a TM domain and a periplasmic ‘headpiece’, comprising porter and OMF-docking or funnel domains (Murakami et al., 2002). Each protomer comprises 12 transmembrane helices (TM1–TM12). A large periplasmic domain is created by two extensive periplasmic loops connecting TM1 with TM2 and TM7 with TM8, respectively. This periplasmic domain can be divided into six sub-domains: the periplasmic N- (PN1 and PN2) and C- (PC1 and PC2) terminal regions and docking N-terminal (DN) and C-terminal (DC) regions (Fig. 3A). Sub-domains PN1, PN2, PC1 and PC2 form the pore domain, with PN1 making up the central pore and stabilizing the trimeric organization. Sub-domains DN and DC contribute to form the docking domain. The trimeric TriC structure suggests that sub-domains PN2, PC1 and PC2 are located at the outermost core of the periplasmic domain, facing the periplasm. Sub-domains PC1 and PC2 also form an external cleft while an adjacent second external cleft is formed between sub-domains PC1 and PN2 in the TriABC structure.
Figure 3. The structural model of TriABC interpreted from the C3 cryo-EM map.
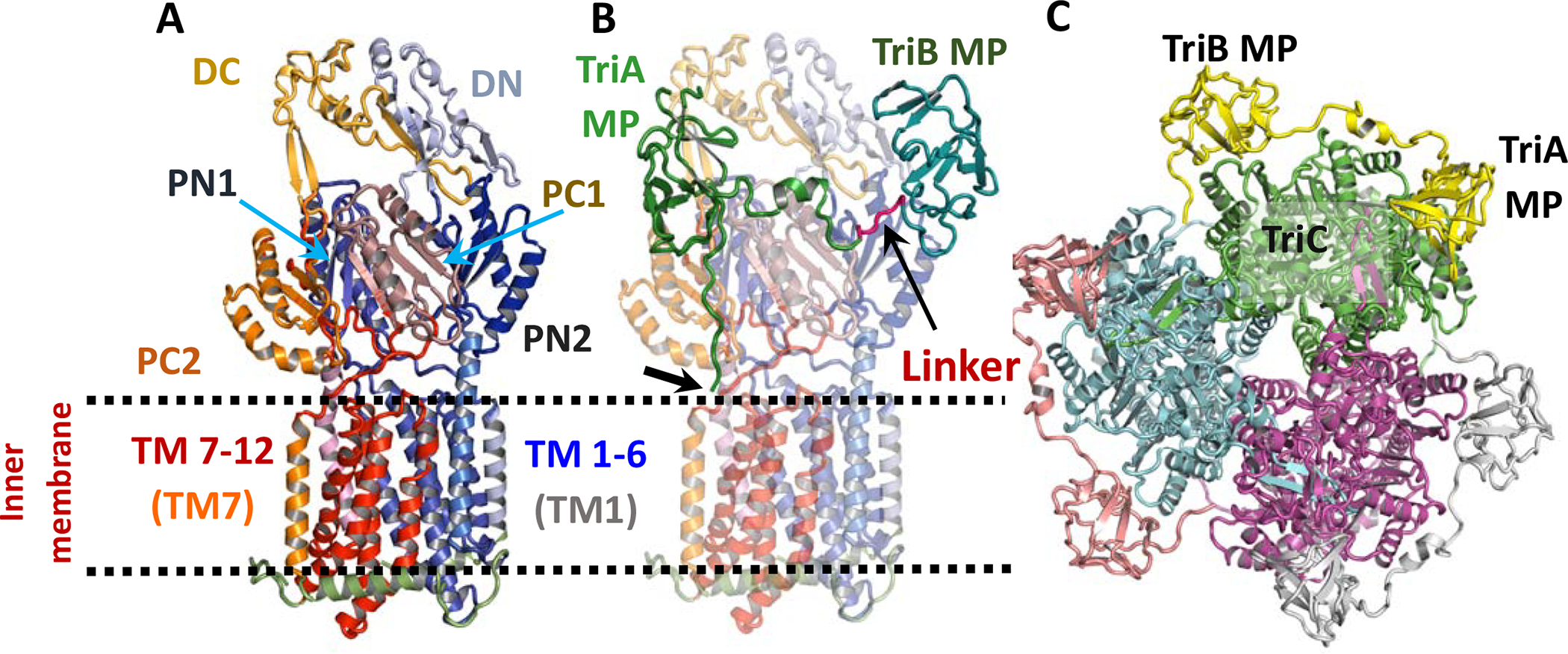
A. Ribbon diagram of a TriC protomer. The N-terminal fold comprises the helical transmembrane regions TM1-TM6 (coloured blue) and a large periplasmic domain between TM1 (grey-blue) and TM2 (slate) while C-terminal region is related to the N-terminal region by a pseudo two-fold rotation in the membrane plane and comprises the helical transmembrane regions TM7-TM12 and a large periplasmic domain between TM7 (tv-orange) and TM8 (light-pink). Subdomains PN1 (K48 to D138), PN2 (V143 to Q186 and F278 to N327), DN (E187 to L277), PC1 (P557 to L655), PC2 (I665 to W701 and P804 to G846), DC (N702 to V797) as well as TM1 (M1 to N36) and TM7 (R524 to F556) are labelled.
B. Ribbon diagram of the TriAB protomer viewed as in panel A. The TriC protomer has been rendered semi-transparent. The TriA MP domain is shown connected to the TriB MP domain via a linker region shown in red. The thick arrow points to the tripalmitoylated N-terminus of TriAB.
C. Ribbon diagram of the TriABC trimer viewed perpendicular to the membrane plane from the periplasmic space. Each subunit of TriABC is shown with different colour.
Substrate binding sites:
Comparison of the structures of AcrB and TriC (Fig. 4A) highlighted several differences between the two transporters. In AcrB, the access (proximal, AP) and deep binding (distal, DBP) pockets (Nakashima et al., 2011) are separated by a switch loop of 8 residues that is bounded by conserved residues Gly-614 and Gln-622, with Phe-617 creating a partition between the two sites and restricting the channel connecting them (Eicher et al., 2012; Nakashima et al., 2011; Zgurskaya et al., 2015a). The conformation of this loop, initially described as critical for binding and efflux of such antibiotics as doxorubicin and erythromycin (Eicher et al., 2012; Nakashima et al., 2011), was recently shown to be important solely for export activity (Ababou and Koronakis, 2016). Furthermore, it is the switch loop dynamics that mediates substrate export activity (Muller et al., 2017). In TriC, the equivalent loop is much longer consisting of 15 residues, bounded by conserved residues Gly-598 and Gln-613, and penetrates further into the proximal binding site (Fig. 4B–C), and thus lining the putative proximal binding site. The tip of the loop contains residues Phe-604, Tyr-605, Leu-606 in TriC while in AcrB, the tip of the loop has residues Phe-615, Gly-616, Phe-617. A conserved Arg-603 (Arg-620 in AcrB) is present in the loop and is oriented facing into the TriC interior.
Fig. 4. Structural comparison of the TriC and AcrB protomers.
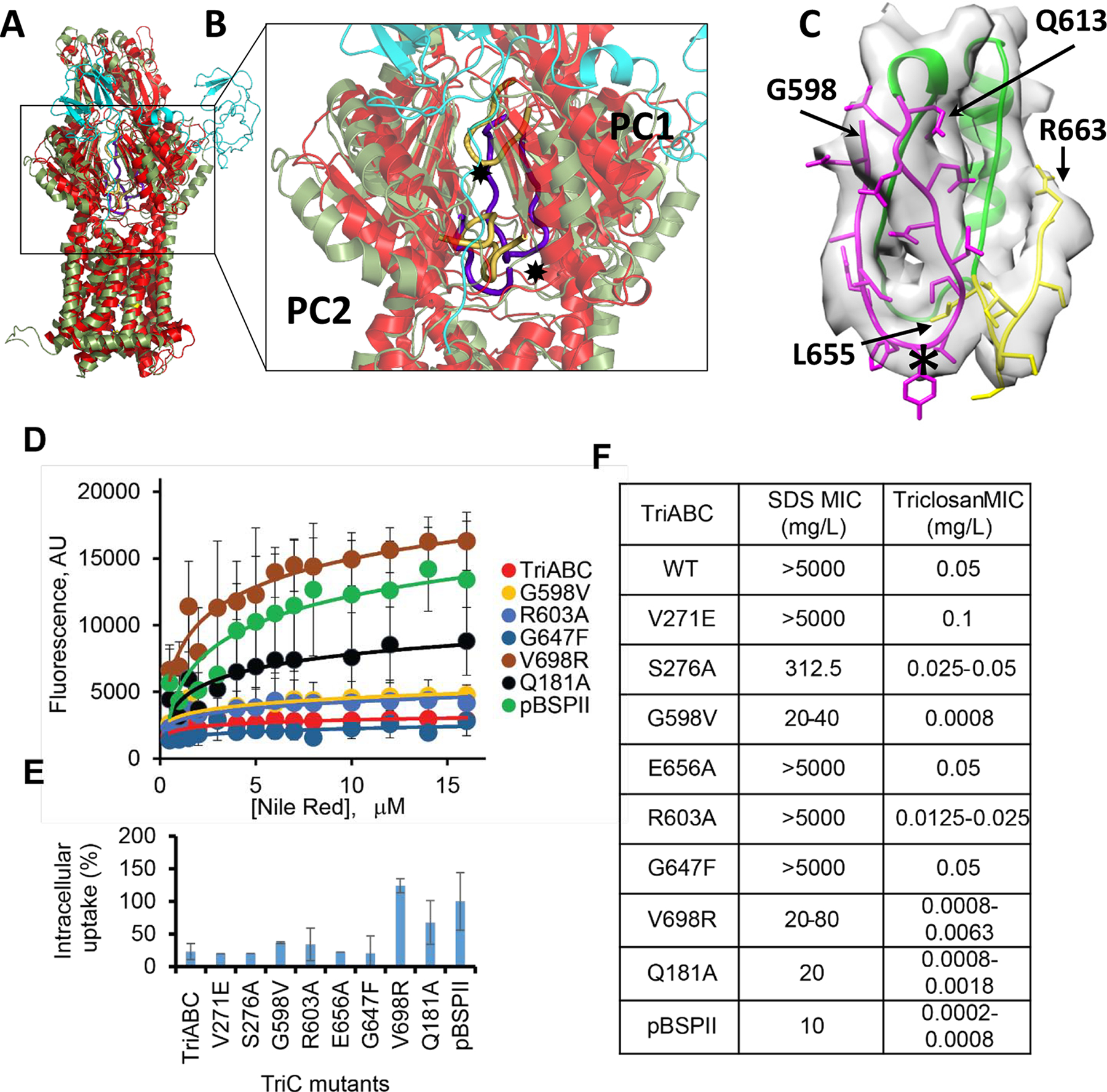
A. Ribbon diagram of protomers of TriC and AcrB viewed in the membrane plane. A TriC subunit of TriABC (green) is superimposed onto a “substrate-free” protomer (extrusion subunit) of AcrB (red). The corresponding TriAB subunit is shown in cyan.
B. Front view of the periplasmic clefts formed by sub-domains PC1, PN1, PC2, and PN2 of TriC and AcrB shown as semi-transparent objects. PN1 and PN2 lie behind PC2 and PC1, respectively, and are not labelled. Residues 614–622 in AcrB form a flexible loop (a thick yellow trace) that gates multidrug binding within the periplasmic cleft. The loop apex (✸) contains two Phe residues, 615 and 617, and are thought to gate drug extrusion in AcrB. TriC has an equivalent yet longer loop, (a thick purple trace), consisting of residues 598–614 that penetrate the cleft. The loop apex (✸) contains hydrophobic residues Phe-604, Tyr-605, Leu-606. Noteworthy also is the loop, residues 667–680 in AcrB (thick yellow), and residues 655–663 in TriC (thick purple), that form the basement of the periplasmic cleft. This loop is shorter in TriC allowing the ‘gating’ loop in TriC to penetrate deeper into the periplasmic cleft.
C. Switch loop region of TriC showing the fit to the density map of residues 598–613 (purple) that penetrate the cleft, residues 655–663 (yellow) that form the basement of the periplasmic cleft, and a cartoon trace of the intervening residues 615–654. The switch loop apex (✸) is also shown. Map densities are shown as greyish isosurfaces using a 3σ cutoff (in UCSF Chimera). The switch loop region was rotated slightly clockwise about the vertical axis with respect to panel B.
D. Concentration dependence curves of NR accumulation in E. coli Δ9-Pore cells producing TriABC and its mutant TriC variants. Error bars, SE (N=3).
E. Intracellular concentration of NR expressed as a percent of NR accumulation in cells carrying empty pBSPII vector. Cells were incubated in the presence of 12 mM NR for 5 min and fluorescence intensity measured.
F. Susceptibilities to triclosan and SDS of E. coli D9 cells producing TriABC and its TriC mutant variants (N=3).
To test whether the residues lining the putative binding pockets of TriC impact activity, we introduced amino acid substitutions vicinal to the putative proximal access (AP) site and the deep distal binding (DBP) pocket (Fig. 4F) and analyzed the ability of the expressed mutants (Fig. S4) to confer resistance to triclosan and SDS and to expel Nile Red from E. coli (Fig. 4D–F). Substitutions, Glu181Ala, Gly598Val, and Val698Arg, had a significant effect on substrate specificity and activity of TriC. The conserved Gly598Val substitution at the start of the putative switch loop in TriC led to a reduction of Nile Red efflux by TriABC and obliterated the TriABC-dependent resistance to SDS and triclosan. The Gln181Ala and Val698Arg variants resulted in a more than 8-fold decrease in the MIC (minimal inhibitory concentration) of triclosan, a decrease in the ability to expel Nile Red, and a concurrent the loss of protection against SDS. The Arg603Ala substitution had a mild effect on TriABC activity with the partial loss in the ability to confer triclosan resistance (2 to 4 fold MIC decrease in comparison to WT) and was matched by a partial loss of Nile Red efflux (Fig. 4D–E). Finally, the Ser276Ala variant was active in efflux of Nile Red and displayed resistance to triclosan but could only partially complement the SDS susceptibility of efflux-deficient Δ9 cells. Thus, TriABC appears to have substrate binding sites consistent with the binding pockets of AcrB. These trends in substrate specificities indicate that the mutations in TriC impact similarly the efflux of Nile Red and the protection against triclosan and SDS and suggesting that the efflux mechanism for these substrates are broadly similar (Fig. 4D–F).
TriC Tunnel Network:
Substrates can reach the postulated binding sites of TriC through a tunnel network identical in each TriC subunit. The continuous tunnel network was specific to the periplasmic domain of TriC and extends from the inner membrane (labelled ENTRANCE) to the apical center of the trimeric complex (labelled EXIT) corresponding to a 190 Å long trajectory (Fig. 5A). The tunnel network features a bottleneck region with diameter of 2.2 Å, less than the smallest circular cross-section diameters of SDS, triclosan and Nile Red. Tunnel networks with larger bottleneck diameters could not be found. The network entrance is at the outer leaflet of the inner membrane and leads to the access (AP) binding loci, identified from superposition of TriC with AcrB (Fig 5B, Video M2). The tunnel merges with the identical tunnels from the other 2 subunits in the apical center of the complex. Large antibiotics such as erythromycin or rifampicin attach in the AP site of AcrB (Eicher et al., 2012; Nakashima et al., 2011), and are then transferred into the distal deep pocket (DBP) (Fig 5B) during L→T conformational transition. In the TriABC structure, the tunnel network links the DBP site with the AP site and includes the bottleneck region of ~ 10Å long (Fig. 5C).
Fig. 5. Intramolecular ligand-accessible channels in the TriABC trimer.
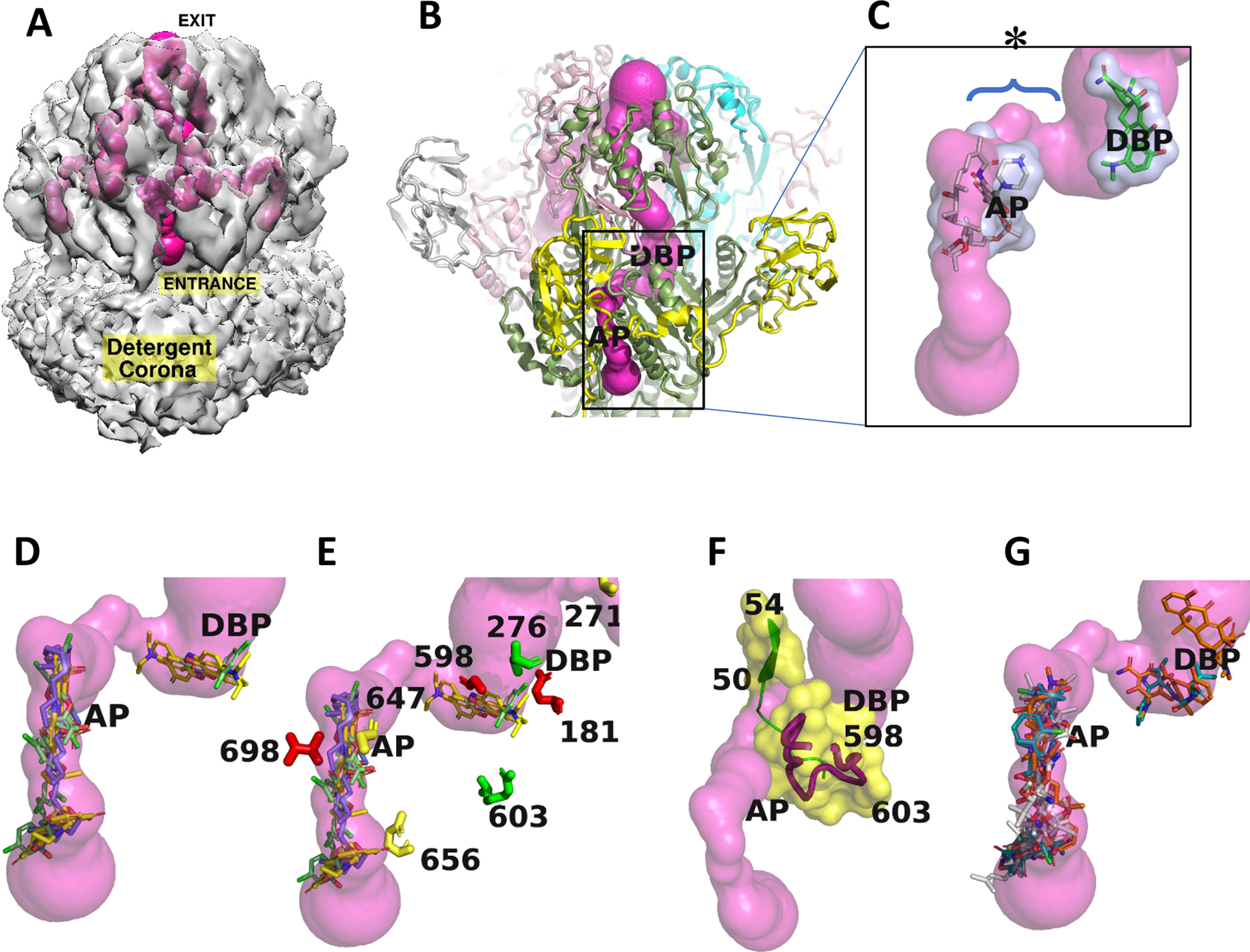
Trimeric tunnel network is depicted as solid surfaces colored (hot pink), using the structure of a TriABC protomer.
A. The tunnel network is shown embedded in the C3 cryoEM density (2σ), and has an overall length of approximately 190 Å in each TriC subunit. The entrance to the tunnel is shown labelled for the TriC subunit facing the viewer and commences at the interface of TriC with the outer leaflet of the inner membrane with egress at the funnel like exit formed by the docking domains of the TriC protomers. The complete tunnel network of TriABC including that in the adjacent subunits is shown by making the C3 cryo-EM density semi-transparent.
B. Superposition of AcrB (Nakashima et al, 2011) onto the TriC protomer positions in TriC the AcrB ligand binding sites referred to as the proximal access pocket (AP) and distal binding pocket (DBP). The tunnel narrows to 2.2Å diameter in the region between the AP and DBP sites.
C. Close-up of rifampicin (grey) and minocycline (green) bound in AcrB (Nakashima et al, 2011) at the AP and DBP loci, respectively, and shown as stick representations surrounded by transparent van der Waals surfaces, positioned at the putative homologous binding loci in TriC, and coincide with the tunnel network. The labels AP and DBP were placed the center of mass of the respective antibiotic. The bottleneck region is identified by an * and its extent by a brace.
D. Docking calculations using BSP-SLIM (Lee and Zhang, 2012) showing the 5 highest scoring poses for each substrate: SDS, triclosan, and Nile Red, as stick representations in a TriABC protomer, their respective carbon atoms are colored slate, green, and yellow.
E. Close-up of the region between AP and DBP (labels slightly displaced for clarity) showing the position of the mutated residues that are listed in Fig. 5. Coloured in red are the residues that upon mutation severely modified extrusion activity, in green partially impacted activity, while the residues whose mutation did not impact extrusion activity are coloured in yellow.
F. Tunnel closeup showing secondary structure and associated van der Waals surfaces adjacent to the tunnel bottleneck (2.2Å) between the AP and DBP sites. The bottleneck is bounded by β strand (50–54) on one side and the gating loop (598–614) on the other side, Cα traces are shown in green and red respectively. Position of mutated residues are labelled in the gating loop.
G. Docking calculations using BSP-SLIM showing the 5 highest scoring poses for antibiotics tetracycline, cloxacillin, and novobiocin, that are not extruded by TriABC, as stick representations. Their respective carbon atoms are colored brown, green, and white.
To assess whether the surfaces of the tunnel network are conducive to substrate binding, blind docking calculations were performed with SDS, triclosan, and Nile Red for one TriABC protomer using the BSP-SLIM server (Lee and Zhang, 2012). The calculations identified two binding clusters, one vicinal to the cavity entrance of the tunnel network and extending to the AP site and the other clustering at the DBP site (Fig. 5D). All poses overlapped with the surface of the tunnel network at the AP and DBP loci reinforcing that the tunnel network linking these two sites is the conduit for substrate efflux.
Three substitutions Glu656Val (Glu673 in AcrB), Gly647Phe (Phe664 in AcrB) and Val271Glu (Gly271 in AcrB) did not alter the specificity or activity of TriABC and their location does not overlap with the tunnel networks. In contrast, the TriC variant with substitution Val698Arg (Arg715 in AcrB) was inactive in MIC measurements and Nile Red uptake assays. Val698 is located near the interface with TriA (see below ‘Partially ordered TriAB subunit’) as such the introduction of the positive charge may affect interactions between TriC and TriA. Arg698 is also adjacent to the AP site (Fig. 5E) and a long Arg side chain pointing into tunnel could create steric hindrance to substrate translocation. Equally, Gln-181 points into the DBP site (Fig. 5E) and loss of the Gln-181 carboxamide side chain in Gln→Ala mutation could reduce stabilizing interactions with substrate (Fig 5E), thereby affecting their efflux. Similarly, the loss of the hydroxyl group in Ser276Ala would abrogate potential hydrogen bonding interaction with TriABC substrates (Fig 5E) and compromise translocation. Thus, the TriC transporter likely contains two putative substrate binding pockets with Val698 and Gln181 mediating substrate stabilization.
Two of the TriC variants, Gly598Val and Arg603Ala which also hinder efflux in TriABC, are located on the switch loop in a region between the AP and the DBP sites and (Fig. 5F). Intriguingly, the tunnel bottleneck region is defined by the β strand (50–54, PN1) and loop PC1 residues 608–614 in the switch loop (Fig. 5F). The β strand (50–54, PN1) forms the interior strand of a four stranded antiparallel β sheet and its extensive hydrogen bonding interactions would limit conformational changes. Switch loop dynamics could however alleviate the bottleneck constriction and enable substrate translocation between the AP and the DBP sites consistent with the interpretation that switch loop variants Gly598Val and Arg603Ala inhibit efflux in TriABC.
To gain insight into the reduced specificity of TriABC substrates, blind docking calculations were also performed with tetracycline, cloxacillin, and novobiocin (Fig. 5G), antibiotics, that are not extruded by TriABC (Weeks et al., 2015) The inability by TriABC to efflux the bulkier antibiotics even though their binding loci are homologous to those of the TriABC substrates (AP and DBP sites, Fig. 5G) implicates switch loop dynamics as the underlying mechanism for the selective efflux in TriABC.
Proton-Relay Network:
Drug efflux by RND-type exporters is driven by a proton motive force (PMF) (Zgurskaya and Nikaido, 1999). Four essential charged residues, Asp407, Asp408, Lys940, and Arg971, in addition to Thr978, have been identified in AcrB as well as other RND transporters to form a transmembrane proton-relay network (Eicher et al., 2014; Su et al., 2006; Takatsuka and Nikaido, 2006; Yue et al., 2017). In the structure of the TriABC transporter, the same five conserved residues superimpose onto Asp405, Asp406, Arg925, Arg955 and Thr962 (Fig. 6). Asp405 and Asp406 (TM4) and Arg925 (TM10) can form ion pairs, while Thr962 faces Asp406 similarly to the homologous residues in AcrB (Murakami et al., 2002). The TM4 and TM10 helices of TriC are located at the center of the 12 α-helix bundle and Arg955 on TM11 is located near the cytoplasmic surface of the membrane. Notable is the replacement of the lysine residue at position 940 in AcrB with that of an arginine residue, Arg925, in TriC. Because of the pKa of 13.8 of arginine, the Arg guanidinium side chain remains charged even in lipid bilayers (Fitch et al., 2015; Thibado et al., 2016). This is in agreement with the molecular dynamical simulations indicating that Lys940 in AcrB remains protonated throughout the transport cycle (Eicher et al., 2014; Yue et al., 2017) and supports the putative role of this charged residue in the proton transport cycle of RND transporters.
Figure 6. Charged residues in the central transmembrane domains of TriC and AcrB.
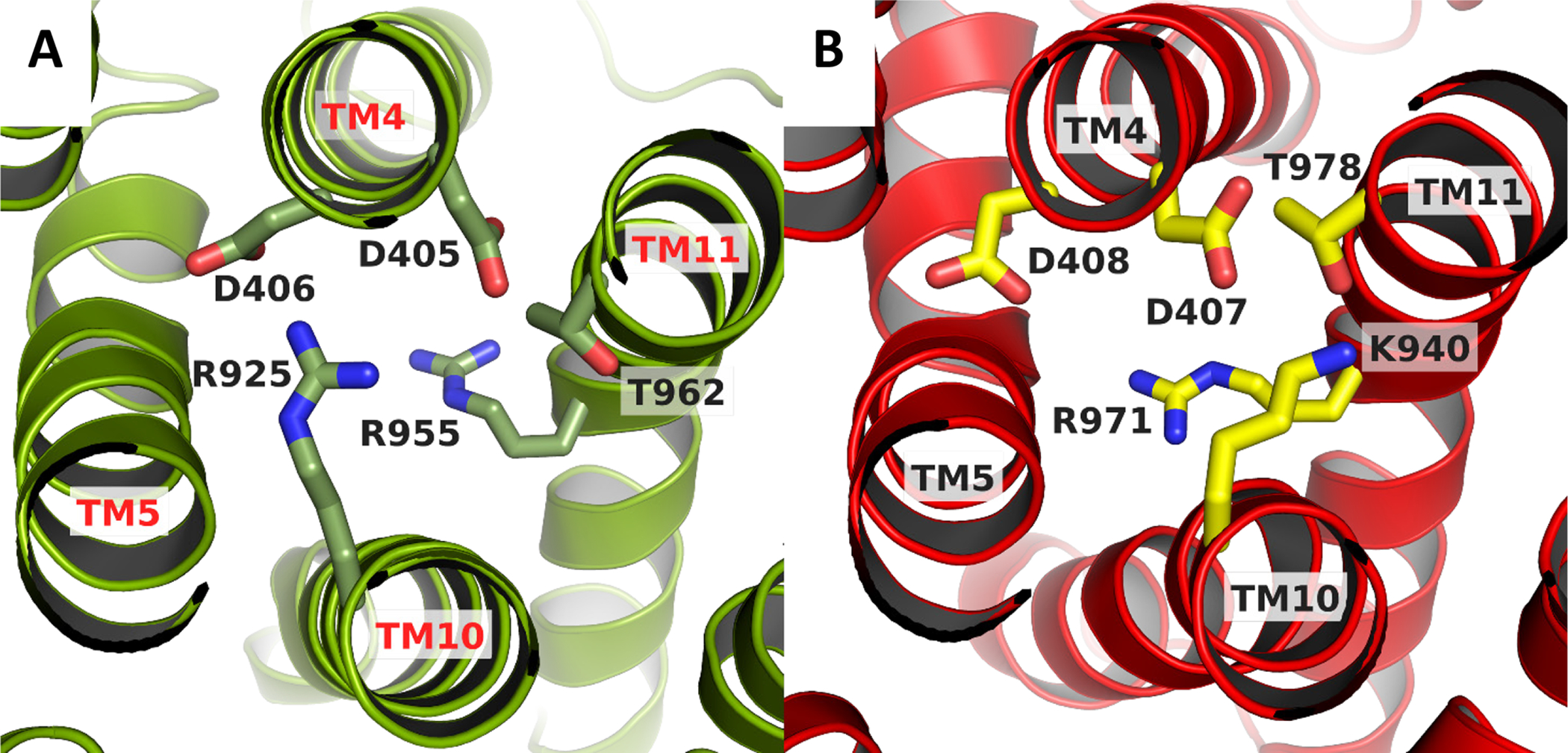
AcrB substrate free protomer was superimposed onto TriC protomer using the Super tool in PyMol (Schrödinger).
A. The spatial arrangement in TriC is isostructural with that of AcrB, shown in panel B. The pertinent residues are shown as sticksThe view is looking down from the periplasmic side. The figure was prepared using PyMol (Schrödinger).
Partially ordered TriAB subunit:
A region of residual cryo-EM density was present on each TriC protomer (Fig. 7A). This region comprised two compact densities facing into the periplasm, one density interacting tightly with each TriC protomer while the second density exhibited a reduced interaction with each TriC protomer. An additional strand to the 12 transmembrane helices of TriC was observed extending from the PC2 region into the detergent micelle (Fig. 7A). This strand partially occludes the periplasmic cleft formed by PC1 and PC2 sub-domains of each TriC protomer and terminated in a well-defined region of density. A palmitic acid in the form of S-(2,3-bis(palmitoyloxy)propyl)-N-palmitoylcysteine (Sankaran and Wu, 1994) could be fitted into this later density (Fig. 7B), in agreement with the previously described lipid modification present in the mature form at the TriA N-terminus (Weeks et al., 2015). The strand of residual EM density then extending from the lipid modification acid was interpreted as TriA residues 1–12.
Figure 7. Partial structure of TriAB.
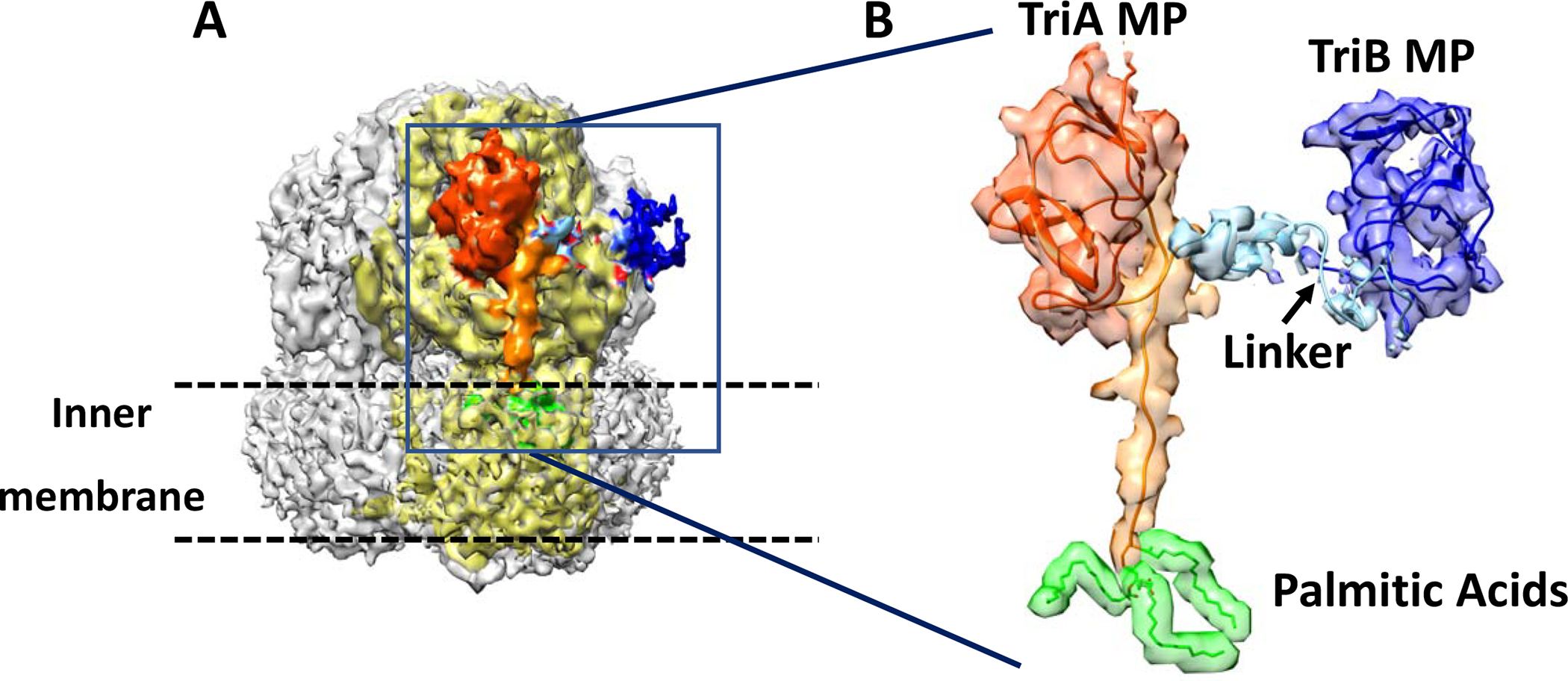
A. Isosurface representation the cryoEM C3 map (contoured at 2σ) with densities associated with refined partial model of TriAB colored. TriA and TriB MP domains and their connecting junction are shown in orange/red-orange, dark blue and light blue, respectively. The TriC protomer interacting with TriAB is shown in light-yellow. The green density was interpreted as tripalmitoylated modification of Cys-1 of TriAB and is embedded in the detergent micelle.
B. Showing agreement of the refined partial model of TriAB with the cryoEM C3 density map. The TriB MP domain, the connecting linker, and the three palmitic acids at Cys-1 are shown at 1σ density level instead of 2σ.
The sequence corresponding to residues 12–24 and 260–333 of the predicted fold of the TriA MP domain (Table S2) was readily fitted into the globular residual density interacting tightly with TriC. The strongest residual density adjacent to TriA MP domain in the C1 difference map (Fig. 2E and 2F), was next used to extend the interpretation of the cryoEM density to include the TriA C-terminal residues (334–352). The second compact globular density was interpreted as the TriB MP domain (residues 367–375 and 621–695; Table S2). Linker (353–356) and TriB MP N-terminal sequence (357–366) were modeled as coiled linear strands into the map connecting the 2 globular densities. Model was improved and validated as described in method section.
The TriA N-terminal region is stabilized by a number of polar interactions with the porter subdomains PC1 and PC2 of the periplasmic cleft of TriC, while the interaction surface of the TriA MP domain with DC and PC2 sub-domains is largely polar as well as involving several hydrophobic patches. The TriB MP domain interacts through a single locus with the PN2 subdomain in TriC. The residues in the two TriC loops (151–157, 315–321), each connecting a helix to strand structural motif in PN2, make a limited number of hydrophobic contacts and hydrogen bonds with surface residues of the TriB MP domain that are clustered about residues 371, 626, and 677. The weaker interaction of the TriB MP domain is reflected by the 2-fold greater average B factor as compared to the TriA MP domain consistent with greater positional displacement and hence a weaker cryo-EM density and lower associated CC value reported by real_space_refinement in Phenix (Afonine et al., 2018). The polypeptide sequence corresponding to the remaining polypeptide domains of TriA and TriB could not be associated with map density in the EM maps. However, the weak densities noted in the 2D class averages (Fig 2G) are not inconsistent with traces of the polypeptide sequences for the missing MFP domains that would extend into the periplasm. Conformational disorder by these MFP domains would obliterate their densities in the 3D reconstruction.
MFP conformational transitions:
Proteolysis in vitro and in vivo was used next to probe conformational changes in TriA and TriB by examining their interactions with TriC and the OMF TolC. Trypsin digests of the purified TriABC complexes followed by immunoblotting with anti-TriA antibodies showed that TriA as a part of the fused TriAB construct and as a separate polypeptide is cleaved by trypsin at the same sites (~40 kDa Fig. 8A). N-terminal sequencing showed that stable 27 and 30 kDa tryptic fragments (Fig. S5) are generated by the cleavage at residues, K134 and R106, respectively. These residues are located at the base of the α-helical hairpin. TriB and its tryptic fragments could be readily seen as a part of the fused TriAB dimer in the TriABC complex, but not when TriA and TriB were expressed as two separate polypeptide chains (A, B Fig. 8B) due to substochiometric purification issues (Fig.1 and S4).
Figure 8. In vivo and in vitro proteolytic profiles of TriA and TriB.
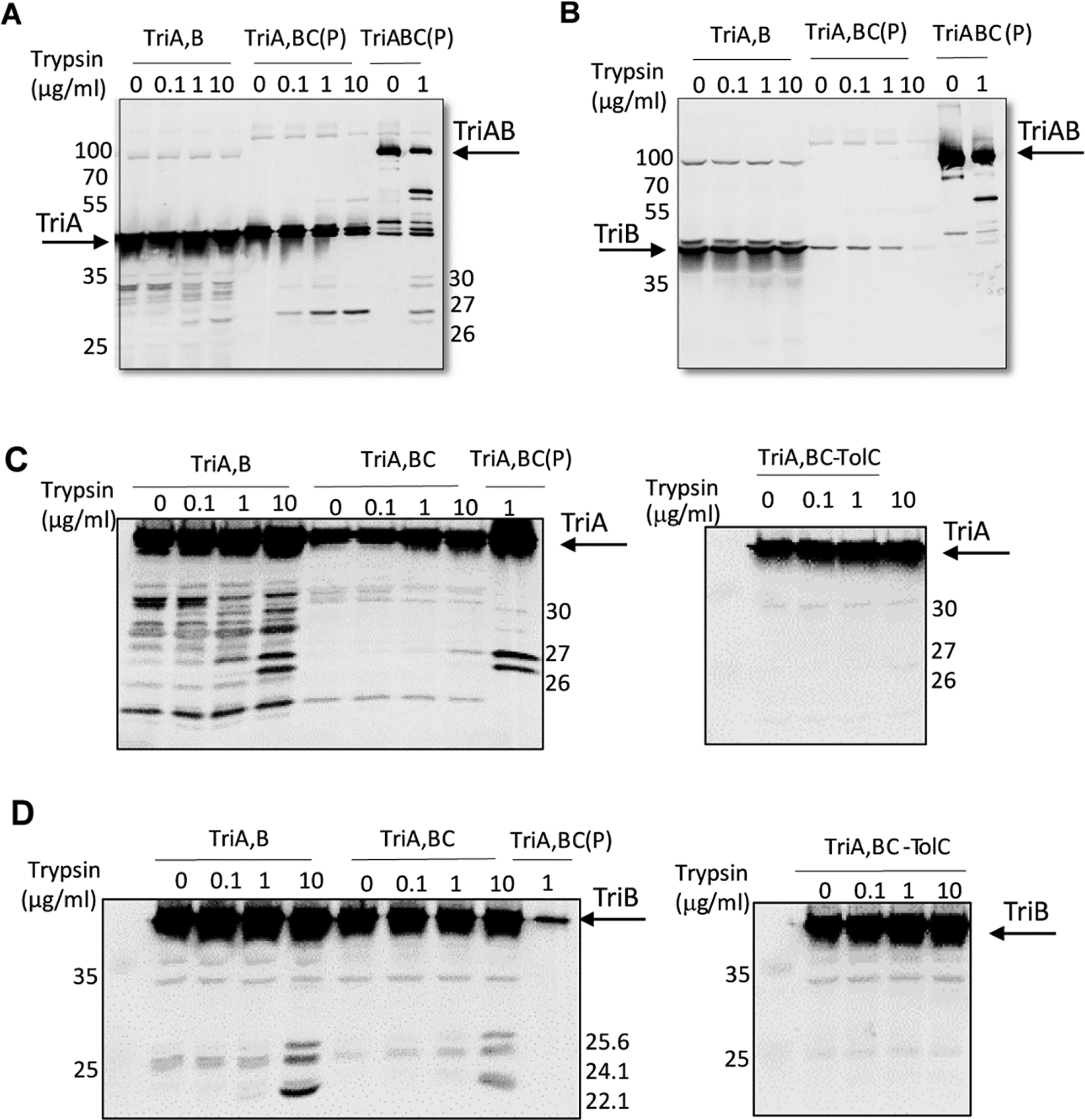
A. Anti-TriA immunoblotting analysis of purified (P) TriABC complexes with TriA and TriB as separate (TriA,B) and fused (TriAB) polypeptides. Complexes treated with indicated concentrations of trypsin. The trypsin-treated whole E. coli GKCW102(pTriA,B) cells producing TriA and TriB were boiled in SDS sample buffer and immunoblotted along with the purified complexes.
B. The same as in A but immunoblotting with anti-TriB antibodies was carried out.
C. Whole E. coli GKCW102(pTriA,B), GKCW102(pTriA,BC) and GKCW104 (pTriA,BC) cells producing TriA and TriB as separate polypeptides, alone or along with TriC, were treated with indicated concentrations of trypsin and TriA fragments visualized by immunoblotting with anti-TriA antibodies. Purified TriA,BC(P) complex treated with trypsin was loaded as a control.
D. The same as in C but immunoblotting was done with anti-TriB antibody. Results representative of at least two independent experiments are shown.
The in vivo proteolysis followed by immunoblotting of E. coli cells carrying plasmid bearing TriA and TriB showed different sensitivity to proteases despite similar expression of the two MFPs (Fig. 8C–D). In the periplasm, TriA was found to be proteolytically labile and cleaved by intracellular proteases, whereas externally added trypsin generated additional 30, 26 and 27 kD proteolytic fragments identical to those in the tryptic profile of the purified TriABC construct (Fig. 8C, left panel). Surprisingly, TriA was stable and resistant to trypsin when co-expressed with TriB and TriC (Fig. 8C, left panel). In contrast, the periplasmic TriB is proteolytically stable but can be cleaved by exogenously added trypsin (Fig. 8D, left panel). From comparison with the tryptic digest of the purified TriB (Fig. S5), these fragments according to N-terminal sequencing of tryptic fragments of the purified TriB are the cleavage products at R126 (25. 6kD fragment), R140 (24.1 kD fragment) and K152 (22.8 kD), all located in the α-helical hairpin of TriB. No significant differences in proteolytic profiles of TriB were detected in the presence or absence of TriC and the protein was cleaved by trypsin into stable fragments (Fig. 8D, left panel). In contrast, both TriA and TriB were protected in complex with TriC from the cleavage by proteases in the presence of TolC (Fig. 8C and 8D, right panels). The differential proteolysis of the α-helical hairpin domains of the MFPs indicates that the MFPs undergo conformational transitions upon interaction with specific cognate partners that modifies their accessibilities to proteolysis. Thus, TriA is structurally dynamic and is stabilized by interactions with TriC, whereas the structure of TriB is not sensitive to the presence of transporter. The recruitment of OMF into the complex stabilizes and protects both MFP subunits from proteases.
Discussion
The ordered molecular architecture of TriABC was determined by single particle cryo-EM and corresponded to a complete trace of the polypeptide backbone of the TriC protomer and a trace limited to the respective MP domains of the fused TriAB protomer (Fig. 2). The structure of TriABC exhibits three-fold symmetry, with three protomers of TriC and TriAB per complex, consistent with the molecular weight estimated by size exclusion chromatography. The agreement of the refined model with all EM densities (Video M1) corroborates that most if not all ordered map density features present in the cryo-EM map has been accounted for. The increase in resolution upon imposing C3 symmetry, from 6.5Å to 4.5Å, enhanced the signal to noise ratio and corroborating the three-fold symmetry of the TriABC transporter.
The polypeptide fold of TriC is homologous to that of AcrB ((Murakami et al., 2002; Seeger et al., 2006)), CusA (Su et al., 2011), ZneA (Pak et al., 2013), and MtrD (Bolla et al., 2014). The TriC protomer has greatest structural similarity with CusA used to model TriC (RMSD Cα = 3.38 Å), followed with the Open/Extrusion subunits of AcrB (RMSD Cα = 3.57 Å) (Fig. 4A). A notable difference is the structure and position of the ‘switch’ loop of TriC in Fig. 4B–C. The homologous switch loop is flexible in AcrB and MexB (Sennhauser et al., 2009) and gates multidrug binding within the periplasmic cleft (Eicher et al., 2012; Nakashima et al., 2011). A conformational change of the switch loop enables the translocation of large antibiotics from the AP site to the DBP site in AcrB. This loop is considerably longer in TriC and extends deeper into the periplasmic cleft. In both TriC and AcrB, the loop apex is hydrophobic consistent with loop penetration into a largely hydrophobic binding pocket. Concomitantly, the complementary loop (residues 655–663 in TriC) which forms the floor of the periplasmic cleft (Fig. 4B–C), is much shorter than its homologous in AcrB (residues 667–680), allowing for deeper penetration into the periplasmic cleft by the ‘switch loop’. In AcrB, the conformational changes of the floor and switch loops occur concurrently. In TriC, the loss in activities upon mutation of residue Gly598 → Val at the loop hinge and of Arg603 →Ala at the loop median (Fig. 5F) suggests that this longer ‘switch’ loop is indeed implicated in drug binding and/or mediates drug extrusion. The variant Val598 would rigidify hinge motion and directly or indirectly interfere with binding at the AP site. The variant Ala603 would change the amphipathic character of the median loop, due to the loss of the Arg side chain, which would affect hydrophobic and charged interactions with TM2 of subdomain PN2. The considerable differences in chemical nature and shape of the TriABC efflux substrates would result in differential interactions and ensuing dynamics due to the side chains of the variants thereby creating differential reductions in drug efflux (Fig. 4D–F).
PMF mechanism:
TriABC represents the first structure of an RND transporter from the HAE-1 protein family (Tseng et al., 1999) that has the central residue, Lys940 in AcrB, essential for proton translocation, replaced by an arginine, Arg925. Evolutionarily, lysine is a much-preferred basic residue compared to arginine among HAE-RND family members (Takatsuka and Nikaido, 2006). The isostructural substitution of Arg925 in TriC (Fig. 6), offers mechanistic insight into the proton transport cycle of RND transporters. Because of its high intrinsic pKa of 13.3 (Li et al., 2008), it is expected that the arginine would always be positively charged, even when buried in a highly hydrophobic environment such as the interior of a protein or in the middle of a lipid bilayer (Li et al., 2008; Yoo and Cui, 2008). The energetic penalty to deprotonate it at pH 7 is much costlier than to deprotonate a lysine residue whose pKa is near 10.4 and can be completely deprotonated at physiological pH (Isom et al., 2011). Of note is the property of the long and flexible side chain of arginine that enables the guanidinium group to interact with polar atoms and donate up to five hydrogen bonds (Harms et al., 2011). However, the lipid bilayer, including local instabilities leading to water penetration (Eicher et al., 2014), as well as interactions with polar head groups preclude the existence of strictly hydrophobic micro-environments to keep the arginine side chain in a deprotonated state.
The very similar drug efflux activities by E. coli cells expressing AcrAB and TriABC transporters (Fig. 1 and Table S1) and the identical spatial arrangement of isostructural charged residues in the central region of the transmembrane domain (Fig. 6) suggest that both transporters use similar proton-relay mechanisms. From a mechanistic perspective, the presence of a charged arginine residue in TriC throughout the proton transport cycle corroborates the finding from dynamical simulations that Lys940 in AcrB remains protonated throughout the PMF cycle (Eicher et al., 2014). A positively charged residue enhances or ensures by electrostatic stabilization an acidic population of the neighboring aspartate residues. Aspartate residues when buried in the nonpolar hydrophobic core of a folded protein or a lipid bilayer interior can undergo substantial pKa shifts exceeding a value of 10 (Isom et al., 2010). Such a high pKa would inhibit proton transfers due to a negligible population of negatively charged carboxylates to act as proton acceptors. Indeed, the isosteric mutations of Asp407Asn and Asp408Asn in AcrB renders the transporter incapable of mediating proton translocation and drug efflux (Eicher et al., 2014). Proton transport in HAE1-RND transporters thus depends on maintaining an adequate pKa for the aspartate residues in the central transmembrane region to ensure sufficient populations of both charged and neutral states in order to mediate proton transport by Asp408.
Tunnel network:
Superposition of TriC with ligand bound subunits in (RMSD Cα = 4.55 Å) identified two ligand binding loci, the AP and the DBP site, that overlap with the tunnel network (Fig 5C). The blind docking calculations corroborate interaction with TriC at the AP or the DBP loci by each pose of the TriABC substrates: SDS, triclosan and Nile Red (Fig. 5D). The multisite binding at each loci is not inconsistent with a binding mechanism involving multidrug recognition as has been proposed for AcrB (Kobylka et al., 2020; Yamaguchi et al., 2015). Furthermore, the bipartite poses for each substrate overlapped with tunnel surfaces and concurred with the interpretation of the mutagenic data (Fig 5E) further argues in favor of two binding loci. The bottleneck of ~ 10 Å long linking the two sites in TriC (Fig. 5C) would diminish the likelihood of the TriABC complex, in absence of OMF, of extruding antibiotics and biocides into the periplasmic space instead of the extracellular milieu. Accordingly, the symmetric trimeric structure of the TriABC complex represents a conformer that is functionally incompetent for drug extrusion.
Translocation from the AP site to the DBP site based on the structural data necessitates a conformational fluctuation to open a sufficiently large channel enabling translocation between the two sites. From the DBP site, substrates would then be readily extruded without any significant steric hindrances in the funnel tunnel. The entrances to the tunnel network are of sufficient dimensions to accommodate the substrates (Fig. 5A, 5D) and thus the symmetric TriABC structure is primed for uptake of lipophilic substrates intercalated into the cytoplasmic membrane. Indeed, all three known substrates of TriC would preferentially intercalate into the inner membrane based on their estimated logP values: Nile Red – 4.39; Triclosan – 5.53; SDS – 3.86 (Tetko et al., 2005).
The extent of the conformational fluctuations that enlarge the tunnel bottleneck to facilitate access to the DBP site from the AP site must however be restricted. TriABC is not known to efflux bulkier antibiotics such as tetracycline, cloxacillin, and novobiocin (Weeks et al., 2015) yet blind docking calculations with these antibiotics nevertheless show binding at the AP and DBP loci (Fig. 5G). The inability by TriABC to efflux these bulkier antibiotics eve006E though their binding loci are homologous to those of the TriABC substrates suggests that switch loop dynamics may act as a gating mechanism for selective efflux. Smaller conformational fluctuations of the almost two-fold longer switch loop, which is deeply imbedded in the TriC tertiary structure (Fig 4A, 4B) and has more polar and charged side chains, could restrict translocation of bulkier and longer substrates through the bottleneck region (Fig 5F). This interpretation is in agreement with studies that showed the influence of loop integrity and rigidifications on efflux activity and substrate extrusion (Ababou and Koronakis, 2016) (Muller et al., 2017).
TriAB dynamics:
Previous studies showed non-equivalent functions of TriA and TriB in the complex assembly and efflux (Ntreh et al., 2016; Weeks et al., 2015). This study reveals structural difference in the interactions of the two MFPs with the TriC transporter and a dynamic role by the TriAB membrane fusion protein in the complex. TriA but not TriB forms a stable complex with TriC as seen from the co-purification experiments and the cryo-EM structure (Fig. 1 and 2). Importantly, TriA but not TriB is essential for recruitment of TolC/OpmH (Ntreh et al., 2016; Weeks et al., 2015), the result suggesting that TriA and other MFP subunits that occupy the same site on the transporter serve to snare OMFs. The α-hairpin, the lipoyl, and the α-β barrel constituent sub-domains of TriA, not well-defined in the cryoEM map, are likely bent away from the TolC docking region in the bipartite complex. Of note, the high flexibility between subdomains of AcrA (Hazel et al., 2019; Ip et al., 2003; Mikolosko et al., 2006) would accommodate bent conformations. The susceptibility of the α-helical hairpin region of TriA to protease action in the TriABC complex (Fig 8A) indicates that the TriA α-helical hairpin region is not protected upon bipartite complex formation and lends further support to conformational mobility by the MFP TriA in the TriABC complex.
Although both TriA and TriB undergo conformational changes upon TriABC-TolC complex formation that is manifested by their complete resistance to trypsin action in the tripartite complex (Fig. 8C and 8D, right panel), TriB does not undergo an apparent conformational change similar to TriA upon TriABC complex formation, as its resistance to trypsin action does not change whether it is a separate peptide or fused to TriA (Fig. 8C and 8D, left panel). Such behavior is entirely consistent with TriA, mediating formation of the tripartite complex via its interaction with TriC. The tighter interaction with TriC may reduce the conformational search space of TriA, and accelerate complex formation with the OMF. As TriB association with TriC is weaker, it is likely that interactions between the OMF and the TriAC complex stabilize conformational changes in TriB to fully engage it into the assembled complex. The reduced interaction of TriB with TriC is consistent with the observed weaker cryoEM density for the TriB MP domain (Fig. 2 and Video M1). Site-directed mutagenesis of the MP domain of TriB showed that this MFP subunit but not TriA is essential for the stimulation of the transporter (Weeks et al., 2015) by allowing TriB to form critical interactions that stabilize the open conformation of OMF (Ntreh et al., 2016). Hence, conformational changes in both MFPs drive the recruitment of OMF, by TriA to facilitate the full engagement of TriB into the complex, which activates the transporter and concomitantly stabilizes the open conformation of OMF.
The absence of EM density for the α-helical hairpins of TriAB underpins conformational flexibility in the TriABC complex; the ensemble of conformations would be averaged out by single particle image reconstruction. Multiple conformational states for TriAB in the TriABC complex would provide a decided intrinsic advantage compared to a restricted number of discrete conformations by increasing the configurational entropy which in turn enhances the thermodynamic stability of the TriABC complex. The observed cryo-EM complex thus typifies the default molecular architecture for the TriABC complex in absence of OMF. The determined structure further represents an open dynamic state of the TriABC efflux pump, which precedes the recruitment of the outer membrane factor.
STAR★Methods
RESOURCE AVAILABILITY
Lead Contact
Further information and requests for resources and reagents should be directed to and will be fulfilled by the Lead Contact: Jurgen Sygusch (jurgen.sygusch@umontreal.ca)
Materials Availability
The plasmids and bacterial strains generated in this study are available upon request.
Data and Code Availability
The TriABC trimer cryo-EM maps reconstructed using 35,520 best particles from 1080 micrographs are deposited in the EMDB under accession number 21361 for the 6.5Å C1 map and under accession number 21363 for the 4.5Å C3 symmetry map. The C3 symmetry negative staining map has been deposited in the EMDB under accession number 21362.
Raw frames, particles stacks, resulting reconstructions, and associated metadata for the cryo-EM data set have also been deposited into the EMPIAR database: 10128.
The TriABC atomic coordinates (PDB ID: 6VEJ) have been deposited in the Protein Data Bank, Research Collaboratory for Structural Bioinformatics, Rutgers University, New Brunswick, NJ (http://www.rcsb.org/).
EXPERIMENTAL MODEL and SUBJECT DETAILS
Recombinant proteins were produced in E. coli cells as outlined in Method Details.
METHOD DETAILS
Bacterial strains
The functionality of TriABC and its variants were first tested by their ability to complement the triclosan/SDS hypersusceptibility phenotype of acrAB-deficient E. coli JWW2 strain (Δ(araD-araB)567 Δ(rhaD-rhaB)568 ΔlacZ4787 (::rrnB-3) hsdR514 rph-1 ΔompT-scar ΔacrAB::Kmr) (Weeks et al., 2015). This strain was also used for protein purification. The Nile Red uptake experiments were carried out in the hyperporinated E. coli Δ9-Pore (GKCW107); proteolysis was done in GKCW102 and GKCW104 (Krishnamoorthy et al., 2016). The Δ9-Pore cells overproduce a large Pore that enables intracellular accumulation of various compounds including trypsin and the fluorescent probe Nile Red (Krishnamoorthy et al., 2016).
Culture conditions for in vitro system
All cells were grown in Luria-Bertani (LB) broth (tryptone 10 g/L, yeast extract 5 g/L and NaCl 5g/L) with appropriate selection markers as needed, at 37°C with shaking at 200 rpm. For uptake experiments, at OD600 ~0.3, L-arabinose (up to 0.2%) was added to induce the expression of the Pore and cells were further incubated until OD600 reached 1.0.
Minimal Inhibitory Concentrations (MICs)
MICs of antimicrobial agents were measured using the two-fold dilution technique in 96-well microtitre plates (Tikhonova et al., 2002).
Site-directed mutagenesis
Point mutations were introduced using a QuikChange Lightning site-directed mutagenesis kit and primers designed per manufacturer’s guidelines (Agilent Technologies). Plasmids were purified using a GeneJet Kit (Fisher Scientific) and sequenced at the Oklahoma Medical Research Foundation Sequencing Facility to confirm the introduction of the desired substitutions.
Protein expression
E. coli cultures were grown overnight in LB medium, supplemented with 100 μg ml−1 ampicillin as needed, and subcultured 1:100 in fresh LB for 6 hours. Cells were collected via centrifugation at 3,220 × g for 20 min and stored at −20°C. Cells were resuspended in 1 ml lysis buffer (10 mM Tris-HCl pH 7.5, 5 mM EDTA, 100 μg ml−1 lysozyme) and incubated on ice for 1 hour, then subjected to sonication. Unbroken cells were collected, and membranes were pelleted by centrifugation at >100,000 × g for 1 hour. Membrane pellets were then resuspended in 100 μl of 10 mM Tris-HCl, pH 7.5, 100 mM NaCl, and 1 mM phenylmethylsulfonyl fluoride (PMSF) and relative protein amounts quantified using the Bio-Rad Protein Assay before SDS-PAGE and immunoblotting. Polyclonal anti-TriA, anti-TriB and anti-TriC antibodies were reported before (Weeks et al., 2015). For anti-TriC immunoblots, 5 μg of membrane proteins were resolved on SDS-polyacrylamide gels and transferred onto PVDF membranes. Anti-TriC antibodies have previously been shown to also recognize TriAB (Weeks et al., 2015). Proteins were detected using standard immunoblotting protocols with anti-TriC primary antibodies produced in rabbit, combined with secondary anti-rabbit-alkaline phosphatase conjugated antibodies (Sigma Aldrich) or anti-Penta His monoclonal mouse primary antibodies (Pierce), combined with secondary anti-mouse-alkaline phosphatase conjugated antibodies (Sigma Aldrich). Membranes were developed using 5-bromo-4-chloro-3-indoyl phosphate–Nitro Blue Tetrazolium (Pierce).
Protein purification
E. coli JWW2 competent cells were co-transformed with pBAD33-TriAxBC and pBAD-CHis constructs (Weeks et al., 2015), plated on selective media supplemented with ampicillin and chloramphenicol, and grown overnight at 37°C. The TriABC complex is purified by the C-terminal His-tag of TriC. For this purpose cells were harvested by centrifugation at 3,200 × g for 20 mins at 4°C, then the pellets were resuspended in 20 mM Tris-HCl pH 7.5, 5 mM MgCl2, 1 mM phenylmethylsulfonyl fluoride (PMSF), and 0.05 mg/ml DNAse and incubated on ice for 30 mins. The cells were lysed thrice for 30–45 sec using an ultrasonicator (Branson). Unbroken cells were removed by low speed centrifugation at 3,220 × g for 30 mins. Cell membranes were isolated by high speed centrifugation at 100,000 × g for 1 hr at 4°C, then washed for 1.5 hours at 100,000 × g in 10 mM Tris-HCl pH 7.5, 100 mM NaCl, and 1 mM PMSF. Membrane pellets were then re-suspended in 3 ml solubilization buffer (20 mM Tris-HCl pH 7.5, 150 mM NaCl, 1 mM PMSF, 5 mM imidazole, 2% DDM) and incubated overnight on a rotator. Insoluble fractions were collected via centrifugation at 100,000 × g for 1 hour, and the solubilized membrane fractions were loaded onto a 1 ml Cu2+ charged HisBind Resin (EMD Millipore) column equilibrated with Buffer A (50 mM Tris-HCl pH 7.5, 150 mM NaCl, 1 mM PMSF, 0.03% DDM) with 5 mM imidazole. Flow-through was re-applied to the column to ensure complete binding, and the column subsequently washed with 10 column volumes of Buffer A each containing 5 mM, 20 mM and 40 mM imidazole. Protein was eluted using 3.5 column volumes of Buffer A containing 400 mM imidazole. Fractions were then analyzed by SDS-PAGE to determine the amount of protein and combined for gel filtration and ultracentrifugation. The authenticity of TriABC was confirmed by cutting-out the bands corresponding to the purified fractions of TriABC, namely for TriC and TriAB of the SDS-PAGE gel, followed by in-gel digestion with trypsin, and mass spectrometry analysis. For gel filtration chromatography, protein samples were loaded onto a size exclusion column (Superdex 200 GE 3.2/300) equilibrated in eluting buffer (100 mM Tris HCl (pH 8.0), 150 mM NaCl, 1 mM PMSF, 0.03% (w/v) DDM) using a GE micro ӒKTA system. Protein concentrations were determined from OD280 measurements using a GE NanoPhotometer. The final TriABC protein yield is approximately 200 μg per litre of E. coli culture. The fraction corresponding to TriABC after size exclusion chromatography was then used for cryo-EM.
Proteolysis
For in vitro proteolysis experiments, TriA, TriB and TriABC complex (1–5 μM final concentration each) were incubated with increasing concentrations of trypsin for 30 min at 37°C. Reactions were terminated by adding SDS sample buffer and boiling for 5 min. Samples were analyzed by 12% and 16% SDS-PAGE and protein fragments visualized by silver staining or by immunoblotting with corresponding polyclonal rabbit antibodies. For N-terminal sequencing, the fragments of interest were transferred onto PVDF membrane, stained with Coomassie Brilliant Blue R and excised. Peptide sequences were determined at the Iowa State University Proteomics facility.
For in vivo proteolysis experiments, 5 ml of exponentially growing cells (OD600 ~ 0.5–0.6) were harvested and washed twice with buffer containing 20 mM Tris-HCl (pH 7.5), 100 mM NaCl. The cells were resuspended in 200 μl of buffer containing 20 mM Tris-HCl (pH 7.5), 5 mM EDTA, 20% sucrose. Digestion reactions were set up by adding increasing concentration of trypsin to 36 μl of concentrated cells (2.7 × 107) and were incubated at 37°C for 60 min. Reactions were terminated by cooling on ice for 5–10 min then adding SDS sample buffer and boiling for 5 min. The whole cell lysates were resolved on 12% SDS-PAGE. TriA and TriB proteins and their proteolytic fragments were visualized by immunoblotting with polyclonal anti-TriA and anti-TriB antibodies. At least two independent experiments were carried out for each strain.
Negative stain EM microscopy
Purified TriC and TriABC samples (each 5 μL diluted to ~50 μg/mL) were applied onto negatively glow-discharged carbon-coated grids (400 mesh, copper grid) for 1 min. Excess liquid was removed by blotting with filter paper. Freshly prepared 1.5% uranyl formate (pH 5.0) was added (5 μl) for 1 min and then blotted dry. Digital micrographs were collected using a FEI Tecnai G2 F20 microscope operated at 200 kV and equipped with a Gatan Ultrascan 4k × 4k Digital CCD Camera. The images were recorded at defocus between 1.0–2.0 μm at a magnification of 134,010X at the camera and a pixel size of 1.12 Å. 11,393 protein particles were boxed using e2boxer from the EMAN2 software suite (Tang et al., 2007). Initial reference maps were generated de novo using PRIME (Elmlund et al., 2013) and 3D refinement was performed using RELION-1.3 (Scheres, 2012).
Cryo-EM data acquisition
Graphene oxide dispersion in H2O (Sigma) was diluted to 0.2 mg.ml−1. C-flat CF-2/2–2C holey grids were glow discharged for 1 min and 3 μl of the graphene suspension was added to the grids for 1 min. Grids were subsequently blotted briefly using Whatman No1 filter paper and washed three times on 20 μl drops of H2O (twice on the graphene side and once on the reverse side) (Pantelic et al., 2012). Grids were then used for plunge-freezing without further treatment. 3 μL of purified TriABC was deposited on C-flat CF-2/2–2C holey grids previously treated with graphene oxide and plunge-frozen in liquid ethane cooled by liquid nitrogen.
Grids were imaged using a Titan Krios transmission electron microscope (FEI Company) operated at 300 kV, with the specimen maintained at liquid nitrogen. A total of 3483 movie frames were recorded on a FEI Falcon II camera at a magnification of 128,771 X at the camera and a pixel size of 1.08 Å. The total dose per image was ~50 e-/Å2 split into 7 frames with frames 1 to 5 having a dose of 6 e-/Å2 and frames 6 and 7, 10 e-/Å2. Data was typically collected with a defocus range between −1.6 and −3.0 μm.
EM Data Processing and Image Analysis
Movie frames were first aligned using Unblur (Grant and Grigorieff, 2015) and averaged and the contrast transfer function parameters were determined using CTFFIND4 (Rohou and Grigorieff, 2015). All micrographs were inspected manually to discard the micrographs having no graphene oxide support and thus, no particles. Protein particles were boxed using e2boxer from the EMAN2 software suite (Tang et al., 2007). After extraction, images of the particles were binned twice for a final box size of 100×100 pixels and a resulting pixel size of 2.17 Å at specimen level.
A total of 138,282 candidate particle images were analyzed using RELION-1.3 (Scheres, 2012). First, a 2D classification was performed using 8 starting seeds (Fig. S2A). 3 classes were selected by visual inspection showing different orientations of TriABC to yield 35,520 particle images, which were used for all further data processing. Particles were 3D aligned using a negative staining map previously calculated and low pass filtered at 60 Å with a C3 symmetry imposed. Particles were polished and used for new 3D refinements both with C3 and C1 symmetry imposed. A mask of outer diameter of 50 pixels was kept during the C1 refinement. This EM-map was used against all data set particles for a final refinement with no symmetry imposed.
Map resolutions were computed using gold standard FSC (Fig. S2B–C). Further image analysis with 3D classifications did not improved map resolution.
TriABC structure determination
The structure of TriABC was determined in two stages. First a model for the 3D structure of TriC was prepared from the primary amino acid sequence (Yang et al., 2015; Zhang and Skolnick, 2004, 2005) using the I-TASSER server (Xu and Zhang, 2010; Yang et al., 2015; Zhang and Skolnick, 2004, 2005) and the top structural homolog, CusA, was chosen based on the highest TM-score (0.96) (Table S2). The EM structure exhibited a deep central vestibule with distinct crevasses that afforded facile orientation of the TriC structural model. The fit of the modeled TriC trimer to the cryo-EM map was then further improved using iterative real space refinement in Phenix (Adams et al., 2010; Afonine et al., 2018) and model rebuilding with Coot (Emsley et al., 2010) yielding a high map correlation coefficient (CC) of 0.84. To better visualize side chains, the cryoEM maps were oversampled at a frequency of 1.4× along each axis. The oversampled map was used for refinement and model building. Bulky side chains such as tryptophan residues in the entire polypeptide sequence of TriC were unambiguously associated with EM density using a 3σ cutoff (Fig. S6). Comparison of the refined TriC subunit with the conformation of the CusA subunit spanning the inner membrane yielded a RMSD of 3.38 Å for Cα atoms. A similar RMSD value of 3.57Å was obtained when comparing the TriC protomer with the substrate ‘free’ B (Open/Extrusion) subunit of AcrB and that of the MexB subunit F, RMSD of 3.66 Å.
The identification of the residual density corresponding to the TriA N-terminal sequence exploited that the TriA N-terminus is lipidated (Weeks et al., 2015) and would thus be situated proximal to the outer leaflet of the inner membrane. A linear strand of residual density terminating at the detergent micelle and clearly visible in the cryo-EM map could be interpreted as residues 1–12 of the mature TriA sequence while palmitic acid covalently bound to the TriA N-terminus Cys-1 in the form of S-(2,3-bis(palmitoyloxy)propyl)-N-palmitoyl-cysteine (Sankaran and Wu, 1994) could be fitted into an adjacent region of density in the detergent micelle (Fig. 7B).
The identification of the compact EM density adjacent to the N-terminal residue 12 of the TriA MP domain was then made using a modelling tool in Coot (jiggle-fit) (Emsley et al., 2010), which uniquely oriented the TriA MP C-terminal domain predicted by I-TASSER (Table S2) into the C3 cryo-EM density. The structure of the TriA sequence fold was modelled based on the subunit A of ZneB MFP from C. metallidurans (De Angelis et al., 2010) predicted by the I-TASSER server (Yang et al., 2015) with a high TM-score (0.90) (Table S2). Separately fitting the α-hairpin, lipoyl, and αβ-barrel sub-domains of the modelled TriA into the outstanding compact residual density did not provide a satisfactory fit whereby map density could be associated with contiguous polypeptide strands. In ZneB, AcrA, and CusB MFP structures, the MP C-terminal region is found proximal to its N-terminal region (Mikolosko et al., 2006; Su et al., 2009) that further corroborated our interpretation of the density. We next turned to the C1 difference map shown in panel E and F of Figure 2 to extend the interpretation of the TriA MP C-terminal sequence (334–353) by fitting it into the strongest residual uninterpreted density feature adjacent to TriA MP domain. A long tube-like density starting at Ala-333 was modelled as a helical structure for residues 338–347 and a linear strand for residues 348–352. The final modelling fitted residues 1–24 and 260–352 of the TriA MP C-terminal region to the map density with a map correlation coefficient (CC) of 0.75 upon real_space_refinement in Phenix (Adams et al., 2010; Afonine et al., 2018). Using the Super alignment feature in PyMol (Schrödinger), the fold of the fitted residues when superimposed against the MP domain of the ZneB structure yielded a very low RMSD = 1.55 Å for the Cα atoms indicating excellent modelling by I-TASSER of the Cα fold for TriA MP domain sequence.
The linker sequence (353–356) and the TriB MP N-terminal sequence (357–366; using TriAB sequence numbering for TriB) could be next fitted into the difference map density, corresponding to region L in Fig. 2E and 2F, as a coiled linear strand which terminated at the second compact globular region of density. The structure of the fold for the TriB domain sequence used for fitting into the compact globular region of density was modelled based on the subunit M of MexB MFP from P. aeruginosa (De Angelis et al., 2010) and was predicted by the I-TASSER (Yang et al., 2015) with high TM-score (0.86) (Table S2). The fitting to the compact globular density by the Coot jiggle-fit tool of the predicted fold for the TriB MP domain (367–375, 621–695; using TriAB sequence numbering for TriB) resulted in a unique orientation placing residue 367 of the TriB MP domain vicinal to the previous residue 366 of the TriB MP N-terminal sequence. Separately fitting models of the α-hairpin, lipoyl, and αβ-barrel sub-domains of modelled TriB into the compact residual density did not provide a satisfactory fit whereby map density could be associated with contiguous polypeptide strands. The final modelling of residues 367–375 and 621–695 of TriAB MP C-terminal region to the map density yielded a map correlation coefficient (CC) of 0.55 upon real_space_refinement in Phenix (Adams et al., 2010; Afonine et al., 2018). Using the Super alignment feature in PyMol (Schrödinger), the fold of the fitted residues when superimposed against the MP domain of the M subunit of the MexA structure yielded an RMSD = 3.10 Å for Cα atoms and indicating very good modelling by I-TASSER of the TriB MP domain fold.
The TriABC structure consisting of TriC and the TriAB MP domain trimer was then further improved by iterative real space refinement against the C3 cryo-EM map (Afonine et al., 2018). To expedite the fitting to the C3 cryo-EM map, the TriABC model was refined using a molecular dynamics flexible fitting simulation coupled to real-space refinement prior to the last stages of refinement (Kidmose et al., 2019). Table 1 summarizes the resultant refinement showing a high overall CC = 0.80 and few outliers < 1%. A three-dimensional perspective of the model fits to the C3 and C1 cryo-EM maps as well as to the C3 negative staining map is shown in Video M1. The refined structure explained virtually all observed density in the C3 cryo-EM map, except for the detergent corona which was not modelled due to its apparent disordered structure.
Table 1.
Summary of the real-space refinement by Phenix (release 3594) of the TriABC atomic model against the C3 cryo-EM map.
| Metric | |
|---|---|
| Resolution of cryo-EM map | 4.35 Å |
| Map CC a | 0.800 |
| rmsd (bonds): | 0.004 Å |
| rmsd (angles): | 0.783° |
| All-atom clash score b | 19.55 |
| Ramachandran plot c | |
| outliers: | 0.16 % |
| allowed: | 8.61 % |
| favored: | 91.23 % |
| Rotamer outliers: | 0.49 % |
| C-beta deviations: | 0 |
Correlation coefficient calculated by Phenix between map density and atomic coordinates of model
calculated as 1000 × (number of bad overlaps) / (number of atoms)
Peptide link settings used Ramachandran restraints in Phenix. Ramachandran restraints were assigned a weight of 14.0 that was used throughout the real space refinement. Additionally, the Apply peptide plane was turned on in Phenix.
To assess overfitting, we refined the atomic model against half-map1 and determined the map to model FSC curve. At FSC = 0.143, the resolution was 4.5 Å. We then calculated the FSC curve for the same model (without re-refinement) against half-map2 and obtained a resolution of 4.5 Å. The two FSC curves were virtually identical (not shown) suggesting little, if any, overfitting. Finally, the FSC curve for the refined model to the full map yielded a resolution of 4.5 Å at FSC = 0.143 consistent with the resolution obtained from the two half-maps.
Tunnel network in each TriC subunit was identified using MOLEonline (Pravda et al., 2018). Figures 2, 5A and 7 were prepared using Chimera (Pettersen et al., 2004). Structure figures 3, 4A–B, 5B-G and 6 were prepared with PyMol (Schrödinger).
QUANTIFICATION and STATISTICAL ANALYSIS
Data Collection and Final Refinement
Data was collected on a Titan Krios equipped with a FEI Falcon II camera at the Facility for Electron Microscopy Research at McGill University. 3483 movie frames were recorded at a magnification of 128,771 X and a pixel size of 1.08 Å. The total dose per image was ~50 e-/Å2 split into 7 frames with frames 1 to 5 having a dose of 6 e-/Å2 and frames 6 and 7, 10 e-/Å2.
Final refinement statistics are summarized in Table 1.
ADDITIONAL RESOURCES
This study has not generated new reagents or contributed to a new website/forum.
Supplementary Material
Video M1, Related to Figure 2, 3 and 7. Fit of the refined TriABC structure to the C3 and C1 cryo-EM maps and the C3 negative stain map.
Video M1. This video provides a three-dimensional perspective of the fit of the refined TriABC structure to the C3 and C1 cryo-EM maps and the C3 negative stain map. The atomic models of the trimeric TriC subunits (green, blue, turquoise) and of the partial structural model for the trimeric TriAB subunits (red, light green, yellow) are shown in cartoon style. For ease of viewing, the various subunits comprising a single TriABC protomer are identified as well as the DDM detergent corona. Furthermore, views from the periplasm and cytoplasm are annotated for the cartoon model and associated densities. The tripalmitoylated N-terminus is annotated in magenta. A putative DDM molecule placed in the central cavity and associated with a TriC subunit near its interface with an adjacent TriC subunit has carbon atoms colored yellow and oxygen atoms in red. Transparency of the map densities have been varied to facilitate viewing of the fit of the TriABC model (Cα trace) into the various maps. Various map density levels used are annotated corresponding to highest density level set to 4 σ, the lowest set to 1 σ and an intermediate density level displayed at 2 σ.
The same atomic model of TriABC was used to fit to each map. In the part one of the movie, the fit is shown for the TriABC atomic model that was refined against the C3 cryo-EM maps, while the second part shows the same refined TriABC model and fitted into the C1 cryo-EM map density without any additional refinement, while the last part shows the same fit of TriABC but to the C3 negative staining map. The TriC quaternary structure completely fitted into all density maps at 2 σ. The structure of the TriA MP domains of all three TriAB subunits fitted both the C3 and C1 cryo-EM density maps at 2 σ, while two TriB MP domains of the TriAB subunits, colored green and red, fitted best into the C1 cryo-EM map at 1 σ. The linker junction of TriAB in the subunit that is colored green, provided the best fit in the C1 cryo-EM map. There is good overall fit by the partial TriAB model into the knob like features of the C3 negative staining map at 1 σ. Movie was made using Chimera 1.14 (Pettersen et al., 2004). The DDM detergent corona which becomes visible at 2.3σ in the C3 cryo-EM map was not modeled.
Video M2, Related to Figure 5. Efflux tunnels in trimeric TriABC.
Video M2. This video shows the efflux tunnels (pink), calculated by MOLEonline (Pravda et al., 2018), using the refined atomic model of trimeric TriABC. Coloring scheme and annotation is the same as presented in Video M1. The efflux tunnels have well defined three-fold symmetry consistent with the C3 cryo-EM density. Each tunnel entrance is at the edge of the detergent corona while egress occurs at the trimeric interface made by the docking domains of the TriC subunits. Transparency of the map density has been varied to facilitate viewing of the efflux tunnels. Map levels were annotated as in Video M1. Notably, the antibiotics, rifampicin (RFP) and minocycline (MIY), found in the structure of AcrB (PDB: 3AOD) at the access pocket (AP) and distal binding pocket (DBP), respectively, are shown as stick representations surrounded by transparent van der Waals surfaces. The positions of these antibiotics in TriC were derived by superposition of appropriate AcrB subunits onto the same TriC subunit (RMS = 4.6Å Cα atoms), using the Super tool in PyMol (Schrödinger). The putative homologous antibiotic binding loci in TriC coincided with the tunnel network. Movie was made using Chimera 1.14 (Pettersen et al., 2004).
Table S1, Related to Figure 1A. Antimicrobial susceptibility of E. coli Δ9-Pore cells expressing AcrAB and TriABC efflux complexes.
Table S2, Related to Figures 2, 3 and 7. I-TASSER homology modelling of TriC, TriA, and TriB polypeptide folds.
KEY RESOURCES TABLE
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Antibodies | ||
| Anti-TriC antibodies | (Weeks et al., 2015) | N/A |
| Anti-Rabbit IgG (whole molecule) -Alkaline Phosphatase antibody | Sigma Aldrich | Cat#A3687 |
| Penta-His Tag Monoclonal Antibody | ThermoFisher Scientific | Cat#P-21315 |
| Anti-Mouse IgG (whole molecule) −Alkaline Phosphatase antibody | Sigma Aldrich | Cat#A9316 |
| Polyclonal anti-TriA and anti-TriB antibodies | (Weeks et al., 2015) | N/A |
| Bacterial and Virus Strains | ||
| E. coli JWW2 Δ(araD-araB)567 Δ(rhaD-rhaB)568 ΔlacZ4787 (::rrnB-3) hsdR514 rph-1 ΔompT-scar ΔacrAB::Kmr | (Weeks et al., 2015) | N/A |
| E. coli GKCW107 Δ(argF-lac)169 λ− IN(rrnD-rrnE)1 rpsL179(strR) ΔacrB ΔacrD ΔacrEF::spc ΔemrB ΔemrY ΔentS::cam ΔmacB ΔmdtC ΔmdtF attTn7::mini-Tn7T-Tpr -araC-ParaBAD-fhuAΔCΔ4L | (Krishnamoorthy et al., 2016) | N/A |
| GKCW102 (BW25113 attTn7::mini-Tn7T-Kmrr -araC-ParaBAD- fhuAΔCΔ4L) | (Krishnamoorthy et al., 2016) | N/A |
| GKCW104 (BW25113 ΔtolC-ygiBC (attTn7::mini-Tn7T-Kmr -araC-ParaBAD- fhuAΔCΔ4L) | (Krishnamoorthy et al., 2016) | N/A |
| Chemicals, Peptides, and Recombinant Proteins | ||
| Nile Red | Sigma Aldrich | Cat#19123, CAS: 7385-67-3 |
| 5-bromo-4-chloro-3-indoyl phosphate–Nitro Blue Tetrazolium | ThermoFisher Scientific | Cat#34040 and Cat#34035 |
| IPTG (Isopropyl-beta-D-thiogalactoside) | GoldBio | Cat#I2481C, CAS: 367-93-1 |
| L-arabinose | Sigma Aldrich | Cat#A91906, CAS: 5328-37-0 |
| Triclosan | Sigma Aldrich | CAS: 3380-34-5 |
| SDS (sodium dodecyl sulfate) | Sigma Aldrich | CAS: 151-21-3 |
| Pentapeptide GNSRG | (Weeks et al., 2015) | N/A |
| HisBind Resin | EMD Millipore | Cat#69670–5 |
| Critical Commercial Assays | ||
| QuikChange Lightning site-directed mutagenesis kit | Agilent Technologies | Cat#210519 |
| GeneJet Plasmid Miniprep Kit | Fisher Scientific | Cat#K0503 |
| Recombinant DNA | ||
| pBAD33-TriAxBC | (Weeks et al., 2015) | N/A |
| pBAD-CHis | (Weeks et al., 2015) | N/A |
| Deposited Data | ||
| TriABC trimer C3 cryo-EM map | This work | EMDB-21363 |
| TriABC trimer C1 cryo-EM map | This work | EMDB-21361 |
| TriABC trimer C3 negative stain map | This work | EMDB-21362 |
| Raw frames, particles stacks, resulting reconstructions, and associated metadata | This work | EMPIAR-10128 |
| TriABC atomic coordinates | This work | PDB: 6VEJ |
| AcrB | (Murakami et al., 2002; Seeger et al., 2006) | PDB:4DX5 |
| AcrB | (Nakashima et al., 2011) | PDB: 3AOD |
| MexB | (Sennhauser et al., 2009) | PDB: 3W9I |
| ZneA | (Pak et al., 2013) | PDB: 4K0E |
| MtrD | (Bolla et al., 2014) | PDB: 4MT1 |
| CusA | (Su et al., 2011) | PDB: 3NE5 |
| Software and Algorithms | ||
| EMAN2 software suite | (Tang et al., 2007) | https://cryoem.bcm.edu/cryoem/downloads/view_eman2_versions |
| PRIME | (Elmlund et al., 2013) | https://simplecryoem.com/SIMPLE3.0/download.html |
| RELION-1.3 | (Scheres, 2012) | https://www3.mrc-lmb.cam.ac.uk/relion/index.php/Download_%26_install |
| Unblur | (Grant and Grigorieff, 2015) | https://grigoriefflab.umassmed.edu/unblur_summovie |
| CTFFIND4 | (Rohou and Grigorieff, 2015) | https://grigoriefflab.umassmed.edu/ctffind4 |
| Monores | (Vilas et al., 2018) | http://scipion.i2pc.es/download_form |
| I-TASSER server | (Xu and Zhang, 2010); (Yang et al., 2015) (Zhang and Skolnick, 2004, 2005) |
https://zhanglab.ccmb.med.umich.edu/I-TASSER/ |
| Phenix.real.space.refine | (Afonine et al., 2018) | https://www.phenix-online.org/ |
| Coot | (Emsley et al., 2010) | https://github.com/bernhardcl/coot/releases |
| PyMol | The PyMOL Molecular Graphics System, Version 2.0 | https://pymol.org |
| Segger | (Pintilie et al., 2010) | https://cryoem.slac.stanford.edu/ncmi/resources/software/segger |
| MOLEonline | (Pravda et al., 2018) | https://mole.upol.cz/online/ |
| UCSF Chimera | (Pettersen et al., 2004) | https://www.cgl.ucsf.edu/chimera/download.html |
| BSP-SLIM | (Lee and Zhang, 2012) | https://zhanglab.ccmb.med.umich.edu/BSP-SLIM/ |
| Other | ||
| Exclusion column (Superdex 200 GE 3.2/300) | GE Healthcare Bio-Sciences | Cat#28990946 |
Significance.
P. aeruginosa is an important human pathogen, which is extremely resistant to antibiotics and biocides. TriABC and other efflux transporters play a critical role in this resistance. Although a significant body of structural and functional data has been accumulated in recent years, we have no understanding of steps leading to the assembly and activation of these drug efflux pumps. This study presents the first insight into the state of an efflux complex prior to the engagement of the outer membrane channel. This inactive state would be important to prevent the depletion of cell energy, for initial substrate recognition and activation of drug efflux. The results of this study will facilitate the design of inhibitors preventing the activation of efflux pumps.
Highlights:
CryoEM structure of substrate free TriABC at 4.5 Å resolution.
Periplasmic membrane fusion subunits are partially ordered.
An arginine controls the pKa of surrounding aspartate residues to donate protons.
Loop motion gates substrate tunnel opening, controlling substrate specificity.
ACKNOWLEDGEMENTS
This work was supported by National Institutes of Health/ National Institute of Allergy and Infectious Diseases Grants AI052293 (to H.I.Z.), by the Natural Sciences and Engineering Research Council of Canada RGPIN-2014-04798 (to I.R.), and RGPIN-2016-04898 (to J.S.), and by the University of Melbourne STMM Funds (to I.R.), by GEPROM-FRQS Intramural funding (to J.S., A.Y., L.F. and I.R.). J.R. was a MITACS Research Intern. The content is solely the responsibility of the authors and does not necessarily represent the official views of the NIH. The Canadian Foundation for Innovation provided infrastructure and equipment necessary for these studies. The Facility for Electron Microscopy Research at McGill University (FEMR) provided access to electron microscopes. Proteomics analyses were performed by the Center for Advanced Proteomics Analyses at the Université de Montreal, a Node of the Canadian Genomic Innovation Network that is supported by the Canadian government through Genome Canada. This work was facilitated by computing resources from CLUMEQ by Compute/Calcul Canada.
We thank Mohsen Kazemi for the local resolution calculations.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Conflict of Interest
The authors declare that they have no conflict of interest.
Supplemental Information
Document S1. Figures S1–S6 and Tables S1–S2.
REFERENCES
- Ababou A, and Koronakis V (2016). Structures of Gate Loop Variants of the AcrB Drug Efflux Pump Bound by Erythromycin Substrate. PLoS One 11, e0159154. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Adams PD, Afonine PV, Bunkoczi G, Chen VB, Davis IW, Echols N, Headd JJ, Hung LW, Kapral GJ, Grosse-Kunstleve RW, et al. (2010). PHENIX: a comprehensive Python-based system for macromolecular structure solution. Acta Crystallogr D Biol Crystallogr 66, 213–221. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Afonine PV, Poon BK, Read RJ, Sobolev OV, Terwilliger TC, Urzhumtsev A, and Adams PD (2018). Real-space refinement in PHENIX for cryo-EM and crystallography. Acta Crystallogr D Struct Biol 74, 531–544. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Beckers M, Jakobi AJ, and Sachse C (2019). Thresholding of cryo-EM density maps by false discovery rate control. IUCrJ 6, 18–33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bolla JR, Su CC, Do SV, Radhakrishnan A, Kumar N, Long F, Chou TH, Delmar JA, Lei HT, Rajashankar KR, et al. (2014). Crystal structure of the Neisseria gonorrhoeae MtrD inner membrane multidrug efflux pump. PLoS One 9, e97903. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chuanchuen R, Beinlich K, Hoang TT, Becher A, Karkhoff-Schweizer RR, and Schweizer HP (2001). Cross-resistance between triclosan and antibiotics in Pseudomonas aeruginosa is mediated by multidrug efflux pumps: Exposure of a susceptible mutant strain to triclosan selects nfxB mutants overexpressing MexCD-OprJ. Antimicrobial Agents and Chemotherapy 45, 428–432. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Daury L, Orange F, Taveau JC, Verchere A, Monlezun L, Gounou C, Marreddy RK, Picard M, Broutin I, Pos KM, et al. (2016). Tripartite assembly of RND multidrug efflux pumps. Nat Commun 7, 10731. [DOI] [PMC free article] [PubMed] [Google Scholar]
- De Angelis F, Lee JK, O’Connell JD 3rd, Miercke LJ, Verschueren KH, Srinivasan V, Bauvois C, Govaerts C, Robbins RA, Ruysschaert JM, et al. (2010). Metal-induced conformational changes in ZneB suggest an active role of membrane fusion proteins in efflux resistance systems. Proc Natl Acad Sci U S A 107, 11038–11043. [DOI] [PMC free article] [PubMed] [Google Scholar]
- De Kievit TR, Parkins MD, Gillis RJ, Srikumar R, Ceri H, Poole K, Iglewski BH, and Storey DG (2001). Multidrug efflux pumps: expression patterns and contribution to antibiotic resistance in Pseudomonas aeruginosa biofilms. Antimicrob Agents Chemother 45, 1761–1770. [DOI] [PMC free article] [PubMed] [Google Scholar]
- de la Rosa-Trevin JM, Quintana A, Del Cano L, Zaldivar A, Foche I, Gutierrez J, Gomez-Blanco J, Burguet-Castell J, Cuenca-Alba J, Abrishami V, et al. (2016). Scipion: A software framework toward integration, reproducibility and validation in 3D electron microscopy. J Struct Biol 195, 93–99. [DOI] [PubMed] [Google Scholar]
- Du D, Wang Z, James NR, Voss JE, Klimont E, Ohene-Agyei T, Venter H, Chiu W, and Luisi BF (2014). Structure of the AcrAB-TolC multidrug efflux pump. Nature 509, 512–515. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eicher T, Cha HJ, Seeger MA, Brandstatter L, El-Delik J, Bohnert JA, Kern WV, Verrey F, Grutter MG, Diederichs K, et al. (2012). Transport of drugs by the multidrug transporter AcrB involves an access and a deep binding pocket that are separated by a switch-loop. Proc Natl Acad Sci U S A 109, 5687–5692. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eicher T, Seeger MA, Anselmi C, Zhou W, Brandstatter L, Verrey F, Diederichs K, Faraldo-Gomez JD, and Pos KM (2014). Coupling of remote alternating-access transport mechanisms for protons and substrates in the multidrug efflux pump AcrB. Elife 3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Elmlund H, Elmlund D, and Bengio S (2013). PRIME: probabilistic initial 3D model generation for single-particle cryo-electron microscopy. Structure 21, 1299–1306. [DOI] [PubMed] [Google Scholar]
- Emsley P, Lohkamp B, Scott WG, and Cowtan K (2010). Features and development of Coot. Acta Crystallogr D Biol Crystallogr 66, 486–501. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fitch CA, Platzer G, Okon M, Garcia-Moreno BE, and McIntosh LP (2015). Arginine: Its pKa value revisited. Protein Sci 24, 752–761. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grant T, and Grigorieff N (2015). Measuring the optimal exposure for single particle cryo-EM using a 2.6 A reconstruction of rotavirus VP6. Elife 4, e06980. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hazel AJ, Abdali N, Leus IV, Parks JM, Smith JC, Zgurskaya HI, and Gumbart JC (2019). Conformational Dynamics of AcrA Govern Multidrug Efflux Pump Assembly. ACS Infect Dis 5, 1926–1935. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Higgins MK, Bokma E, Koronakis E, Hughes C, and Koronakis V (2004). Structure of the periplasmic component of a bacterial drug efflux pump. Proc Natl Acad Sci U S A 101, 9994–9999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hohn M, Tang G, Goodyear G, Baldwin PR, Huang Z, Penczek PA, Yang C, Glaeser RM, Adams PD, and Ludtke SJ (2007). SPARX, a new environment for Cryo-EM image processing. J Struct Biol 157, 47–55. [DOI] [PubMed] [Google Scholar]
- Ip H, Stratton K, Zgurskaya H, and Liu J (2003). pH-induced conformational changes of AcrA, the membrane fusion protein of Escherichia coli multidrug efflux system. The Journal of biological chemistry 278, 50474–50482. [DOI] [PubMed] [Google Scholar]
- Isom DG, Castaneda CA, Cannon BR, and Garcia-Moreno B (2011). Large shifts in pKa values of lysine residues buried inside a protein. Proc Natl Acad Sci U S A 108, 5260–5265. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Isom DG, Castaneda CA, Cannon BR, Velu PD, and Garcia-Moreno EB (2010). Charges in the hydrophobic interior of proteins. Proc Natl Acad Sci U S A 107, 16096–16100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kidmose RT, Juhl J, Nissen P, Boesen T, Karlsen JL, and Pedersen BP (2019). Namdinator - automatic molecular dynamics flexible fitting of structural models into cryo-EM and crystallography experimental maps. IUCrJ 6, 526–531. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kobylka J, Kuth MS, Müller RT, Geertsma ER, and Pos KM (2020). AcrB: a mean, keen, drug efflux machine. Annals of the New York Academy of Sciences 1459, 38–68. [DOI] [PubMed] [Google Scholar]
- Krishnamoorthy G, Wolloscheck D, Weeks JW, Croft C, Rybenkov VV, and Zgurskaya HI (2016). Breaking the Permeability Barrier of Escherichia coli by Controlled Hyperporination of the Outer Membrane. Antimicrob Agents Chemother 60, 7372–7381. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee HS, and Zhang Y (2012). BSP-SLIM: a blind low-resolution ligand-protein docking approach using predicted protein structures. Proteins 80, 93–110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mikolosko J, Bobyk K, Zgurskaya HI, and Ghosh P (2006). Conformational flexibility in the multidrug efflux system protein AcrA. Structure 14, 577–587. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mima T, Joshi S, Gomez-Escalada M, and Schweizer HP (2007). Identification and characterization of TriABC-OpmH, a triclosan efflux pump of Pseudomonas aeruginosa requiring two membrane fusion proteins. J Bacteriol 189, 7600–7609. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Muller RT, Travers T, Cha HJ, Phillips JL, Gnanakaran S, and Pos KM (2017). Switch Loop Flexibility Affects Substrate Transport of the AcrB Efflux Pump. J Mol Biol 429, 3863–3874. [DOI] [PubMed] [Google Scholar]
- Murakami S, Nakashima R, Yamashita E, and Yamaguchi A (2002). Crystal structure of bacterial multidrug efflux transporter AcrB. Nature 419, 587–593. [DOI] [PubMed] [Google Scholar]
- Nakashima R, Sakurai K, Yamasaki S, Nishino K, and Yamaguchi A (2011). Structures of the multidrug exporter AcrB reveal a proximal multisite drug-binding pocket. Nature 480, 565–569. [DOI] [PubMed] [Google Scholar]
- Ntreh AT, Weeks JW, Nickels LM, and Zgurskaya HI (2016). Opening the Channel: the Two Functional Interfaces of Pseudomonas aeruginosa OpmH with the Triclosan Efflux Pump TriABC. J Bacteriol 198, 3176–3185. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Oswald C, Tam HK, and Pos KM (2016). Transport of lipophilic carboxylates is mediated by transmembrane helix 2 in multidrug transporter AcrB. Nat Commun 7, 13819. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pak JE, Ekendé EN, Kifle EG, O’Connell JD 3rd, De Angelis F, Tessema MB, Derfoufi K-M, Robles-Colmenares Y, Robbins RA, Goormaghtigh E, et al. (2013). Structures of intermediate transport states of ZneA, a Zn(II)/proton antiporter. Proceedings of the National Academy of Sciences of the United States of America 110, 18484–18489. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pantelic RS, Meyer JC, Kaiser U, and Stahlberg H (2012). The application of graphene as a sample support in transmission electron microscopy. Solid State Communications 152, 1375–1382. [Google Scholar]
- Pettersen EF, Goddard TD, Huang CC, Couch GS, Greenblatt DM, Meng EC, and Ferrin TE (2004). UCSF Chimera--a visualization system for exploratory research and analysis. J Comput Chem 25, 1605–1612. [DOI] [PubMed] [Google Scholar]
- Pintilie GD, Zhang J, Goddard TD, Chiu W, and Gossard DC (2010). Quantitative analysis of cryo-EM density map segmentation by watershed and scale-space filtering, and fitting of structures by alignment to regions. J Struct Biol 170, 427–438. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Poole K (2004). Efflux-mediated multiresistance in Gram-negative bacteria. Clin Microbiol Infect 10, 12–26. [DOI] [PubMed] [Google Scholar]
- Pravda L, Sehnal D, Tousek D, Navratilova V, Bazgier V, Berka K, Svobodova Varekova R, Koca J, and Otyepka M (2018). MOLEonline: a web-based tool for analyzing channels, tunnels and pores (2018 update). Nucleic Acids Res 46, W368–W373. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rohou A, and Grigorieff N (2015). CTFFIND4: Fast and accurate defocus estimation from electron micrographs. J Struct Biol 192, 216–221. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sankaran K, and Wu HC (1994). Lipid modification of bacterial prolipoprotein. Transfer of diacylglyceryl moiety from phosphatidylglycerol. The Journal of biological chemistry 269, 19701–19706. [PubMed] [Google Scholar]
- Scheres SH (2012). RELION: implementation of a Bayesian approach to cryo-EM structure determination. J Struct Biol 180, 519–530. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schneider CA, Rasband WS, and Eliceiri KW (2012). NIH Image to ImageJ: 25 years of image analysis. Nat Methods 9, 671–675. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schrödinger L The PyMOL Molecular Graphics System.
- Seeger MA, Schiefner A, Eicher T, Verrey F, Diederichs K, and Pos KM (2006). Structural asymmetry of AcrB trimer suggests a peristaltic pump mechanism. Science 313, 1295–1298. [DOI] [PubMed] [Google Scholar]
- Seeger MA, von Ballmoos C, Verrey F, and Pos KM (2009). Crucial role of Asp408 in the proton translocation pathway of multidrug transporter AcrB: evidence from site-directed mutagenesis and carbodiimide labeling. Biochemistry 48, 5801–5812. [DOI] [PubMed] [Google Scholar]
- Sennhauser G, Bukowska MA, Briand C, and Grutter MG (2009). Crystal structure of the multidrug exporter MexB from Pseudomonas aeruginosa. J Mol Biol 389, 134–145. [DOI] [PubMed] [Google Scholar]
- Su CC, Li M, Gu R, Takatsuka Y, McDermott G, Nikaido H, and Yu EW (2006). Conformation of the AcrB multidrug efflux pump in mutants of the putative proton relay pathway. J Bacteriol 188, 7290–7296. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Su CC, Long F, Zimmermann MT, Rajashankar KR, Jernigan RL, and Yu EW (2011). Crystal structure of the CusBA heavy-metal efflux complex of Escherichia coli. Nature 470, 558–562. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Su CC, Yang F, Long F, Reyon D, Routh MD, Kuo DW, Mokhtari AK, Van Ornam JD, Rabe KL, Hoy JA, et al. (2009). Crystal structure of the membrane fusion protein CusB from Escherichia coli. J Mol Biol 393, 342–355. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Symmons MF, Marshall RL, and Bavro VN (2015). Architecture and roles of periplasmic adaptor proteins in tripartite e ffl ux assemblies. Front Microbiol 6, 513. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Takatsuka Y, and Nikaido H (2006). Threonine-978 in the transmembrane segment of the multidrug efflux pump AcrB of Escherichia coli is crucial for drug transport as a probable component of the proton relay network. J Bacteriol 188, 7284–7289. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tang G, Peng L, Baldwin PR, Mann DS, Jiang W, Rees I, and Ludtke SJ (2007). EMAN2: an extensible image processing suite for electron microscopy. J Struct Biol 157, 38–46. [DOI] [PubMed] [Google Scholar]
- Tetko IV, Gasteiger J, Todeschini R, Mauri A, Livingstone D, Ertl P, Palyulin VA, Radchenko EV, Zefirov NS, Makarenko AS, et al. (2005). Virtual computational chemistry laboratory--design and description. J Comput Aided Mol Des 19, 453–463. [DOI] [PubMed] [Google Scholar]
- Thibado JK, Martfeld AN, Greathouse DV, and Koeppe RE 2nd. (2016). Influence of High pH and Cholesterol on Single Arginine-Containing Transmembrane Peptide Helices. Biochemistry 55, 6337–6343. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tikhonova EB, Dastidar V, Rybenkov VV, and Zgurskaya HI (2009). Kinetic control of TolC recruitment by multidrug efflux complexes. Proc Natl Acad Sci U S A 106, 16416–16421. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tikhonova EB, Wang Q, and Zgurskaya HI (2002). Chimeric analysis of the multicomponent multidrug efflux transporters from gram-negative bacteria. J Bacteriol 184, 6499–6507. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tikhonova EB, Yamada Y, and Zgurskaya HI (2011). Sequential mechanism of assembly of multidrug efflux pump AcrAB-TolC. Chem Biol 18, 454–463. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tikhonova EB, and Zgurskaya HI (2004). AcrA, AcrB, and TolC of Escherichia coli Form a Stable Intermembrane Multidrug Efflux Complex. The Journal of biological chemistry 279, 32116–32124. [DOI] [PubMed] [Google Scholar]
- Tseng TT, Gratwick KS, Kollman J, Park D, Nies DH, Goffeau A, and Saier MH Jr. (1999). The RND permease superfamily: an ancient, ubiquitous and diverse family that includes human disease and development proteins. J Mol Microbiol Biotechnol 1, 107–125. [PubMed] [Google Scholar]
- Vilas JL, Gomez-Blanco J, Conesa P, Melero R, Miguel de la Rosa-Trevin J, Oton J, Cuenca J, Marabini R, Carazo JM, Vargas J, et al. (2018). MonoRes: Automatic and Accurate Estimation of Local Resolution for Electron Microscopy Maps. Structure 26, 337–344 e334. [DOI] [PubMed] [Google Scholar]
- Wang Z, Fan G, Hryc CF, Blaza JN, Serysheva II, Schmid MF, Chiu W, Luisi BF, and Du D (2017). An allosteric transport mechanism for the AcrAB-TolC multidrug efflux pump. Elife 6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weeks JW, Nickels LM, Ntreh AT, and Zgurskaya HI (2015). Non-equivalent roles of two periplasmic subunits in the function and assembly of triclosan pump TriABC from Pseudomonas aeruginosa. Mol Microbiol 98, 343–356. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xu J, and Zhang Y (2010). How significant is a protein structure similarity with TM-score = 0.5? Bioinformatics 26, 889–895. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yamaguchi A, Nakashima R, and Sakurai K (2015). Structural basis of RND-type multidrug exporters. Front Microbiol 6, 327. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang J, Yan R, Roy A, Xu D, Poisson J, and Zhang Y (2015). The I-TASSER Suite: protein structure and function prediction. Nat Methods 12, 7–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yue Z, Chen W, Zgurskaya HI, and Shen J (2017). Constant pH Molecular Dynamics Reveals How Proton Release Drives the Conformational Transition of a Transmembrane Efflux Pump. J Chem Theory Comput 13, 6405–6414. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zgurskaya HI, Lopez CA, and Gnanakaran S (2015a). Permeability Barrier of Gram-Negative Cell Envelopes and Approaches To Bypass It. ACS Infect Dis 1, 512–522. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zgurskaya HI, and Nikaido H (1999). Bypassing the periplasm: reconstitution of the AcrAB multidrug efflux pump of Escherichia coli. Proc Natl Acad Sci U S A 96, 7190–7195. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zgurskaya HI, and Nikaido H (2000). Multidrug resistance mechanisms: drug efflux across two membranes. Mol Microbiol 37, 219–225. [DOI] [PubMed] [Google Scholar]
- Zgurskaya HI, Weeks JW, Ntreh AT, Nickels LM, and Wolloscheck D (2015b). Mechanism of coupling drug transport reactions located in two different membranes. Front Microbiol 6, 100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang Y, and Skolnick J (2004). Scoring function for automated assessment of protein structure template quality. Proteins 57, 702–710. [DOI] [PubMed] [Google Scholar]
- Zhang Y, and Skolnick J (2005). TM-align: a protein structure alignment algorithm based on the TM-score. Nucleic Acids Res 33, 2302–2309. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Video M1, Related to Figure 2, 3 and 7. Fit of the refined TriABC structure to the C3 and C1 cryo-EM maps and the C3 negative stain map.
Video M1. This video provides a three-dimensional perspective of the fit of the refined TriABC structure to the C3 and C1 cryo-EM maps and the C3 negative stain map. The atomic models of the trimeric TriC subunits (green, blue, turquoise) and of the partial structural model for the trimeric TriAB subunits (red, light green, yellow) are shown in cartoon style. For ease of viewing, the various subunits comprising a single TriABC protomer are identified as well as the DDM detergent corona. Furthermore, views from the periplasm and cytoplasm are annotated for the cartoon model and associated densities. The tripalmitoylated N-terminus is annotated in magenta. A putative DDM molecule placed in the central cavity and associated with a TriC subunit near its interface with an adjacent TriC subunit has carbon atoms colored yellow and oxygen atoms in red. Transparency of the map densities have been varied to facilitate viewing of the fit of the TriABC model (Cα trace) into the various maps. Various map density levels used are annotated corresponding to highest density level set to 4 σ, the lowest set to 1 σ and an intermediate density level displayed at 2 σ.
The same atomic model of TriABC was used to fit to each map. In the part one of the movie, the fit is shown for the TriABC atomic model that was refined against the C3 cryo-EM maps, while the second part shows the same refined TriABC model and fitted into the C1 cryo-EM map density without any additional refinement, while the last part shows the same fit of TriABC but to the C3 negative staining map. The TriC quaternary structure completely fitted into all density maps at 2 σ. The structure of the TriA MP domains of all three TriAB subunits fitted both the C3 and C1 cryo-EM density maps at 2 σ, while two TriB MP domains of the TriAB subunits, colored green and red, fitted best into the C1 cryo-EM map at 1 σ. The linker junction of TriAB in the subunit that is colored green, provided the best fit in the C1 cryo-EM map. There is good overall fit by the partial TriAB model into the knob like features of the C3 negative staining map at 1 σ. Movie was made using Chimera 1.14 (Pettersen et al., 2004). The DDM detergent corona which becomes visible at 2.3σ in the C3 cryo-EM map was not modeled.
Video M2, Related to Figure 5. Efflux tunnels in trimeric TriABC.
Video M2. This video shows the efflux tunnels (pink), calculated by MOLEonline (Pravda et al., 2018), using the refined atomic model of trimeric TriABC. Coloring scheme and annotation is the same as presented in Video M1. The efflux tunnels have well defined three-fold symmetry consistent with the C3 cryo-EM density. Each tunnel entrance is at the edge of the detergent corona while egress occurs at the trimeric interface made by the docking domains of the TriC subunits. Transparency of the map density has been varied to facilitate viewing of the efflux tunnels. Map levels were annotated as in Video M1. Notably, the antibiotics, rifampicin (RFP) and minocycline (MIY), found in the structure of AcrB (PDB: 3AOD) at the access pocket (AP) and distal binding pocket (DBP), respectively, are shown as stick representations surrounded by transparent van der Waals surfaces. The positions of these antibiotics in TriC were derived by superposition of appropriate AcrB subunits onto the same TriC subunit (RMS = 4.6Å Cα atoms), using the Super tool in PyMol (Schrödinger). The putative homologous antibiotic binding loci in TriC coincided with the tunnel network. Movie was made using Chimera 1.14 (Pettersen et al., 2004).
Table S1, Related to Figure 1A. Antimicrobial susceptibility of E. coli Δ9-Pore cells expressing AcrAB and TriABC efflux complexes.
Table S2, Related to Figures 2, 3 and 7. I-TASSER homology modelling of TriC, TriA, and TriB polypeptide folds.
Data Availability Statement
The TriABC trimer cryo-EM maps reconstructed using 35,520 best particles from 1080 micrographs are deposited in the EMDB under accession number 21361 for the 6.5Å C1 map and under accession number 21363 for the 4.5Å C3 symmetry map. The C3 symmetry negative staining map has been deposited in the EMDB under accession number 21362.
Raw frames, particles stacks, resulting reconstructions, and associated metadata for the cryo-EM data set have also been deposited into the EMPIAR database: 10128.
The TriABC atomic coordinates (PDB ID: 6VEJ) have been deposited in the Protein Data Bank, Research Collaboratory for Structural Bioinformatics, Rutgers University, New Brunswick, NJ (http://www.rcsb.org/).