

Abstract
Defensins, a class of small cysteine‐rich cationic polypeptides across cellular life, are identified as antimicrobial compounds that display direct antimicrobial and immune signaling activities that are involved in the host defense. In addition to their roles in the innate immune system, accumulating studies have reported that some members of defensins are expressed and involved in some cancer cells, such as colon cancer, colorectal cancer, lung cancer and renal cell carcinomas. However, the roles of α‐Defensin 5 (DEFA5) in tumorigenesis and development remain unknown. In the present study, bioinformatics analysis and quantitative PCR results showed that the expression level of DEFA5 was dramatically downregulated in human gastric cancer. Overexpression of human DEFA5 in gastric cancer cell lines SGC7901 and BGC823 effectively diminished cell proliferation and reduced the colony forming ability. Moreover, DEFA5 overexpression induced cell cycle arrest by significantly increasing the number of G1‐phase cells. Consistently, in vivo tumor formation experiments in nude mice showed the suppression of the tumor growth by DEFA5 overexpression, suggesting an inhibitory effect of DEFA5 in gastric cancer. Mechanistically, DEFA5 directly binds to BMI1, which subsequently decreased its binding at the CDKN2a locus and upregulated the expression of 2 cyclin‐dependent kinase inhibitors, p16 and p19. Taken together, we concluded that DEFA5 showed an inhibitory effect in gastric cancer cell growth and may serve as a potential tumor suppressor in gastric cancer.
Keywords: BMI1, DEFA5, defensins, gastric cancer, tumor suppressor
DEFA5 directly binds to BMI1, which subsequently decreased its binding at the CDKN2a locus and upregulated the expression of 2 cyclin‐dependent kinase inhibitors, p16 and p19.
![]()
Abbreviations
- DEFA5
α‐Defensin 5
- TCGA
The Cancer Genome Atlas
1. INTRODUCTION
Stomach cancer, also called gastric cancer, is one of the most common cancers worldwide with very high morbidity and mortality. 1 In accordance with the global cancer statistics in 2018, gastric cancer is the fifth most frequently diagnosed neoplasms and the third leading cause of cancer death, causing around 783 000 deaths worldwide in 2018. 2 In fact, the incidence and mortality of gastric cancer are highly dependent on diet and Helicobacter pylori infection and therefore show a very high variation in different regions of the world. 1 , 3 , 4 For example, the rates of gastric cancer in Korea, Mongolia and Japan are extremely high. 1 Recently, considerable progress was made in the early detection of precancerous lesions and the multimodal therapy of gastric cancer. 1 However, an effective method for the treatment of gastric cancer is still lacking and the overall survival rate is still not promising, probably due to the lack of an understanding of the underlying molecular mechanisms. Therefore, there is an urgent need to identify novel targets and molecular markers associated with the progression of gastric cancer.
Defensins, a class of small cysteine‐rich cationic polypeptides across cellular life, are secreted by specific leukocytes and epithelial cells and identified as antimicrobial compounds, which display direct antimicrobial, antiviral toxin neutralizing and immunomodulatory properties. 5 , 6 Depending on their highly conserved disulfide bridging pattern, defensins are classified into 2 subfamilies in humans and mice, α‐ and β‐defensins. 5 , 7 Numerous studies have demonstrated the roles of defensins in innate immunity as well as adaptive immunity. 5 , 6 In addition, aberrant expression of defensins has also been reported in multiple types of cancer, such as colon cancer, lung cancer and renal cell carcinomas, suggesting a potential involvement of defensins in cancer initiation and progression. 8 , 9 For example, human β‐defensin 2 was reported to promote the proliferation of lung cancer cells through ATP‐binding cassette transporter G2. 10 β‐Defensin‐3 can be secreted in head and neck squamous cell cancer cells and induce CCR7 expression, providing migratory and pro‐survival signals to cancer cells. 11 Therefore, defensins may serve as cancer biomarkers and anti‐tumor molecules. 9 However, relatively few studies have focused on the roles of α‐defensins in cancer, although previous reports have shown that elevated plasma levels of α‐defensin 1‐3 strongly reflect clinical stages of bladder cancer, as well as colorectal cancer. 12 DEFA5 peptide is present in colon cancer, 13 , 14 ovary cancer, endometrium cancer, and lung cancer. 15
In the present study, we systematically studied the potential roles of DEFA5 in gastric cancer. First, we found that the expression level of DEFA5 was significantly reduced in gastric cancer cells. Overexpression of DEFA5 effectively reduced cell proliferation and tumor growth in vitro and in vivo. Finally, we uncovered the mechanisms that showed that the inhibitory effect of DEFA5 in cell proliferation was mediated by direct binding of DEFA5 to BMI1 and antagonizing BMI1‐mediated transcription repression of p16 and p19, leading to cell cycle arrest. This finding may provide a new strategy for the treatment of gastric cancer.
2. MATERIALS AND METHODS
2.1. Patients and tissue specimens
Gastric tumors and their adjacent normal tissue were collected from the gastric cancer surgery at the First Affiliated Hospital of Zhengzhou University, Henan, China. These specimens were classified by tumor mode metastasis. This study comprised 7 men and 5 women aged 25–56 years old, with the mean age of 43 years old. This study conformed to the ethical guidelines of the 1975 Declaration of Helsinki. The experimental protocols were approved by Zhengzhou University, with all participants signing informed consent prior to participation.
2.2. Plasmids and antibodies
Complementary DNAs (cDNA) for human BMI1 and DEFA5 were amplified from reverse‐transcribed cDNA of HEK293T cells. For recombinant expression in E coli, the cDNA of DEFA5 was cloned into a modified RSF‐Duet vector with an N‐terminal 6× His‐tag and BMI1 was inserted into a pGEX‐6p‐1 vector. cDNAs of human DEFA5 were inserted into the pCDH vector for lentivirus packaging in HEK293T cells. All plasmids were verified using DNA sequencing.
Antibodies for AKT (4691), Phospho‐AKT (4060), Erk1/2 (4695), and Phospho‐Erk1/2 (4370) were obtained from Cell Signaling Technology. Anti‐DEFA5 (ab180515), anti‐BMI1 (ab126783), anti‐p16 (ab51243), anti‐PTEN (ab267787), anti‐Spry‐2 (ab180527), anti‐E‐cadherin (ab40772) and anti‐p19 (ab102842) antibodies were obtained from Abcam. Anti‐HA (H3663) tag and anti‐β‐actin (A5441) antibody was obtained from Sigma‐Aldrich.
2.3. Cells and cell culture
The human gastric cancer cell line SGC7901 (BICR, 3111C0001CCC000236), and HEK293T cells (ATCC CRL‐11268™) were obtained from the ATCC. BGC823 cells (3111C0001CCC000062) were purchased from BMCR. SGC7901 and BGC823 cells were cultured in RPMI‐1640 medium (Gibco), and HEK293T cells were cultured in DMEM (Gibco). All cultured media were supplemented with 10% FBS (Gibco), 1% penicillin–streptomycin in a 37°C incubator with an atmosphere of 5% CO2 and 95% air.
DEFA5 overexpression was accomplished by cloning DEFA5 cDNA into the pCDH vector, and the empty vector was used as a control. The vectors were transfected into HEK293T cells for lentivirus packaging. SGC7901 cells were then infected using HEK293T cell supernatant and were selected in medium supplemented with 5 μg/mL of puromycin. BMI1 knockout was accomplished using cloning BMI1 sgRNA into PX459 vector. The vectors were transfected SGC7901 cells and SGC7901 cells were selected by 5 μg/mL puromycin. The gRNA sequence for BMI1 is: 1# 5′‐ctctcggagtctgtgcagaa‐3′ and 2# 5′‐acaaagcacacacatcaggt‐3′.
2.4. Evaluation of cell proliferation
Cell proliferation ability was determined by the Cell Count Kit 8 (Beyotime, C0038) in accordance with the instruction. Briefly, SGC7901 cells were seeded into 96‐well plates and cultured with 100 μL medium. After 24, 48, 72, and 96 h incubation, 100 μL RPMI‐1640 containing 10 μL CCK8 reagent was added to each well. Multiskan™ FC (Thermo) was used to measure the value of OD450 at each well of the plates after incubation for 1 h.
2.5. Colony formation assay
SGC7901 cells were placed in 6‐well plates (500 cells/well) with 4 mL complete medium. After 2 wk incubation, the cells were washed with cold PBS and then fixed with methanol for 30 min. Then cells were incubated in crystal violet dye. After 30 min staining, the plates were observed with a microscope and the colony number were counted.
2.6. Cell migration assay
Cell migration ability was measured using a 24‐well transwell chamber system (Costar 3422, Corning Inc). The cells were washed twice with PBS, resuspended in 100 μL serum‐free medium and placed into the upper chambers. The lower chambers were filled with 500 μL medium containing 20% FBS. After 48 h incubation, the membrane then was fixed in methanol for 30 min at room temperature, stained with crystal violet for 30 min. The images were randomly capture (×20) and the cell number in the membrane was counted to determine the cell migration ability.
2.7. Quantitative real‐time PCR
Total RNA was extracted from cells using TRIzol reagent (Invitrogen) in accordance with the manufacturer's instructions. The first strand of cDNA was synthesized from 2 μg of total RNA using random primers and reverse transcriptase (Invitrogen). Real‐time quantitative PCR analyses were performed using the CFX96 Real‐Time PCR System (Bio‐Rad). GAPDH was used as a loading control for normalization. Each sample was measured in duplicates. The primer sequences are as follows: human GAPDH: sense, 5′‐GAAGGTGAAGGTCGGAGTC‐3′; antisense, 5′‐GAAGATGGTGATGGGATTTC‐3′; DEFA5: sense, 5′‐CCTCAGGTTCTCAGGCAAGA‐3′; antisense, 5′‐TCTAGGAAGCTCAGCGAAG‐3′; p16: sense, 5′‐GAAGGTCCCTCAGACATCCC‐3′; antisense, 5′‐CAGTTGTGGCCCTGTAGGA‐3′; p19: sense, 5′‐CGCTGCAGGTCATGATGTTT‐3′; antisense, 5′‐CCTTCAGGGTGTCCAGGAAT‐3′.
2.8. Cell cycle assay
The distribution of the cell cycle was evaluated using a cell cycle analysis kit (Beyotime, C1052) in accordance with the instruction. Briefly, SGC7901 cells (2*105 cells/well) were placed in 6‐well plates. After 24 h, the cells were collected using trypsin digestion and centrifugation. After washing with cold PBS, cells were fixed in 70% methanol at 4°C overnight. Then, the cells were stained with a propidium iodide (PI) that containing 10 mL of RNase A at 37°C for 30 min. The samples were analyzed using a FACS flow cytometer (BD C6) and at least 10 000 cells were collected per sample. The data were analyzed using FlowJo 10.0 software (TreeStar).
2.9. Cell apoptosis assay
Single‐cell suspension was collected by trypsinization and then were washed with PBS twice, followed by staining with PI and FITC‐ANNEXIN‐V for 30 min at dark. Cell apoptosis was then analyzed with FACS flow cytometer (BD C6) and analyzed using FlowJo 10.0 software (TreeStar).
2.10. Immunoblotting and immunoprecipitation
After washing with cold PBS, the cells were lysed in a lysis buffer (20 mmol/L Tris, pH 7.5, 150 mmol/L NaCl, 5 mmol/L EDTA, 0.5% NP‐40, 10% glycerol, protease inhibitor cocktail [Roche]). Protein G beads coupled with 2 μg BMI1 antibody were incubated with cell lysates at 4°C for 4 h and then the beads were washed 4 times with lysis buffer. Proteins bound to the beads were separated using SDS‐PAGE and immunoblotted with anti‐BMI1 and DEFA5 antibody. The SGC7901 cells WT, BMI1−/− and DEFA5 overexpression cells were washed twice with cold PBS and collected in lysis buffer. Proteins were separated using SDS‐PAGE and immunoblotted with anti‐AKT, p‐AKT, Erk1/2, p‐Erk1/2, p16, p19, β‐actin, BMI1 and DEFA5 antibodies.
2.11. Recombinant proteins and in vitro pull‐down assay
Recombinant BMI1 and DEFA5 were expressed in E coli that were cultured in Luria‐Bertani medium at 37°C with 100 mg/L ampicillin or kanamycin. Cells were collected by centrifugation at 4000 g for 15 min, and then the pellets were resuspended in lysis buffer (20 mmol/L Tris at pH 8.0, 400 mmol/L NaCl). For DEFA5 purification, the lysate was separated by centrifugation at 22000 g for 30 min, and the recovered supernatant was applied to a Ni‐NTA affinity column (Qiagen), followed by intensive washing. Recombinant protein was eluted from the Ni‐NTA affinity column using elution buffer. For GST‐BMI1, the supernatant was incubated with Glutathione Sepharose 4B resin (GE Healthcare) at 4°C for 1 h. After incubation, the resin was washed with lysis buffer. The recombinant protein was eluted with 20 mmol/L GSH and further purified by gel filtration with a Superdex200 column.
Glutathione Sepharose 4B resin beads coupled with 2 μg GST‐BMI1 fusion proteins were incubated with His‐tag DEFA5 for 4 h at 4°C. After washing 4 times with PBS buffer, proteins bound to the beads were separated using SDS‐PAGE and immunoblotted with anti‐His and anti‐GST antibodies.
2.12. Chromatin immunoprecipitation assay
ChIP assay was performed as described previously{Lv, 2010 #53} Primers used to amplify P16 and P19 promoter were P16.S: CCCATTTTCCTATCTGC and P16.A: CTAGTTCAAAGGATTCC, P19.S: CTCCTGCAGCACGCGAGGTTC (from −200 to −180 bp) and P19.A: GACTTCGGCAGCTGCTCACACC (from +7 to +28). Primers used to amplify the non‐responsive region were TCTACCTCACACCCCTGACC (from −1182 to −1161 bp) and CTGGGCAGAGATTTCCAGAC (from −1384 to −1363 bp).
2.13. Statistical analysis
The data were expressed as mean ± standard deviation. T test and a one‐way ANOVA was used for the statistical analysis, which was performed with SPSS 17.0 statistical software. P < .05 was represented as being statistically significant.
3. RESULTS
3.1. DEFA5 is downregulated in gastric cancer
To investigate the potential role of DEFA5 in tumorigenesis and development, we first analyzed the expression level of DEFA5 in gastric cancer from the clinically annotated genomic database, TCGA, and found that the transcripts of DEFA5 in gastric cancer samples was dramatically decreased compared with the normal samples (Figure 1A,B and Supplemental Figure 2). To confirm the downregulation of DEFA5 in gastric cancer, quantitative real‐time PCR was performed to determine the mRNA expression of DEFA5 in the tumor tissue from gastric cancer patients. Paired tumor and adjacent normal tissue were collected from the gastric cancer surgery. As shown in Figure 1C, the mRNA expression of DEFA5 was significantly downregulated in gastric cancer tissue. To further confirm the DEFA5‐expressing cells within gastric cancer tissues, we analyze the single‐cell sequencing data (GSE134520) and the expression of DEFA5 was observed in immune cells, stromal cells, as well as malignant cells (Figure 1D and Supplemental Figure 1). However, most of the cells did not express DEFA5. Considering the roles of DEFA5 in pathogen infection, we next checked the expression status in samples with Helicobacter pylori. Compared with uninfected patients, expression of DEFA5 was significantly increased upon Helicobacter pylori infection (Figure 1E).
FIGURE 1.
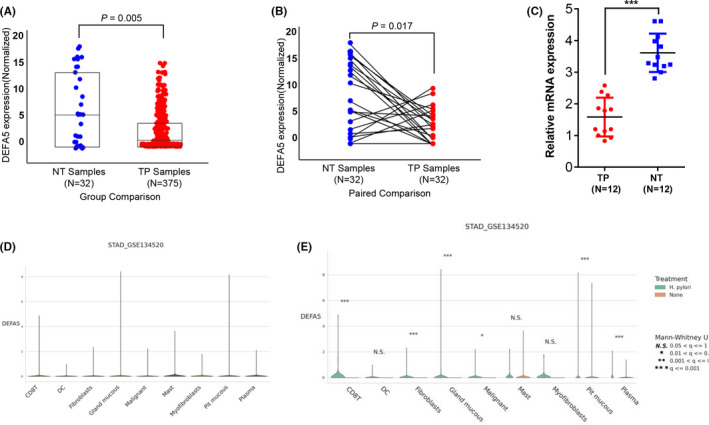
DEFA5 is downregulated in gastric cancer. A, B, RNA sequencing data showing the expression level of DEFA5 in gastric cancer from TCGA database. Data from 32 normal and 375 cancer samples in gastric cancer. C, Quantitative real‐time PCR experiment showing the mRNA expression of DEFA5 in the tumor tissue from gastric cancer patients. n = 12. **P < .01 vs normal control group. D, E, expression of DEFA5 in single‐cell dataset (GSE134520) were analyzed using webtool: https://www.tcpaportal.org/tcpa/index.html.
3.2. DEFA5 overexpression suppresses cell proliferation in vitro
To understand the roles of DEFA5 in the development of gastric cancer, we attempted to overexpress DEFA5 in gastric cancer cells. First, gastric cancer cell lines SGC7901 and BGC823 that stably expresses DEFA5 was constructed by lentivirus infection and puromycin selection. The mRNA and protein expression of DEFA5 was verified using real‐time PCR and western blot, respectively. As shown in Figure 2A,B, DEFA5 was overexpressed in SGC7901 and BGC823 cells compared with control vector. Interestingly, DEFA5 overexpression significantly inhibited the cell proliferation of gastric cancer cells in a time‐dependent manner (Figure 2C). Consistently, in the colony forming assay, the colony number of gastric cancer cells that stably expresses DEFA5 were much lower compared with those of the control cells (Figure 2F,G), suggesting that DEFA5 overexpression impaired the colony forming ability of gastric cancer cells. However, the transwell migration assay results showed that the cell migration and invasion ability, determined by the numbers of cells that migrated through the membranes after 24 h in the assay, was unchanged after DEFA5 overexpression in gastric cancer cells (Figure 2D,E).
FIGURE 2.
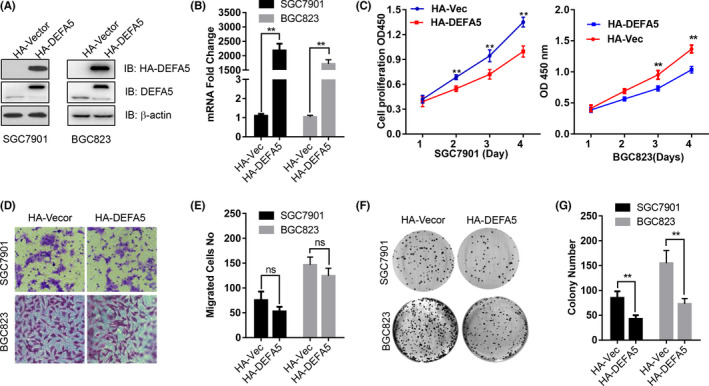
DEFA5 overexpression suppresses cell proliferation in vitro. A, B, Western blot and real‐time PCR results showing DEFA5 was overexpressed in SGC7901 cells and BGC823. C, OD450 value indicating the capacity of cell proliferation. D, E, Representative images and the numbers of cells that migrated through the membranes after 24 h. **, P < .01 vs HA‐Vec group. Data were presented as mean ± SD. The results were reproducible in 3 independent experiments. F, G, Representative images and the calculated results of the colony forming assay showing the colony forming ability of SGC7901 and BGC823 cells after DEFA5 overexpression. Data were presented as mean ± SD. The results were reproducible in 3 independent experiments
3.3. DEFA5 overexpression inhibits tumor growth in vivo
To further determine the effect of DEFA5 on the growth of gastric cancer cells in vivo, single‐cell suspensions from SGC7901 cells were injected subcutaneously into the nude mice. At 6‐wk after inoculation in nude mice, the size of subcutaneous tumors was measured to determine the cancer cell growth rate in vivo. We found that the mean tumor volume of DEFA5 overexpression group was 325 ± 86 mm3, this was much smaller compared with that of the control group (865 ± 124 mm3) (Figure 3), suggesting that DEFA5 overexpression significantly inhibited the growth of gastric cancer cells in vivo. Altogether, the above results demonstrated an inhibitory effect of DEFA5 in cell proliferation and tumor growth in gastric cancer.
FIGURE 3.
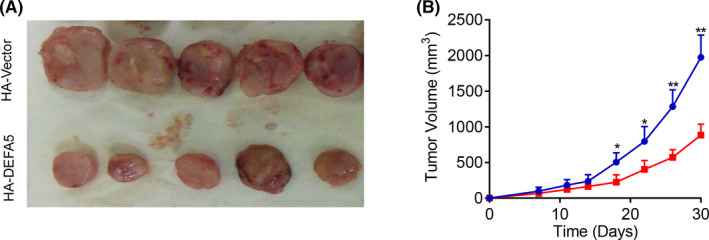
DEFA5 overexpression inhibits tumor growth in vivo. A, Typical image of the subcutaneous tumors in nude mice 6 wk after inoculation. B, The size of subcutaneous tumors was measured to determine the cancer cell growth rate. *P < .05, **P < .01 vs HA‐Vector group. Data are presented as the mean ± SD of 5 individual mice (n = 5)
3.4. DEFA5 overexpression induces cell cycle arrest by increasing the number of G1‐phase cells
To explore the potential mechanisms underlying the inhibitory effect of DEFA5 in cell proliferation, PI staining and flow cytometry analysis were performed to evaluate whether DEFA5 overexpression affected the cell cycle profile in SGC7901 and BGC823 cells. As shown in Figure 4A,B, compared with the control group, DEFA5 overexpression in gastric cancer cells dramatically increased the number of G1‐phase cells, but significantly decreased G2/M‐phase cells, suggesting that DEFA5 overexpression induced cell cycle arrest to suppress the cell proliferation in gastric cancer cells. Furthermore, western blot results showed that DEFA5 overexpression significantly decreased the phosphorylation levels of cell proliferation‐related marker proteins AKT and ERK, but the total protein levels of AKT and ERK did not change (Figure 4C,D), indicating that the inhibitory effect of DEFA5 in cell proliferation was partly resulted from the inhibition of AKT and ERK signaling pathway. Consistently, the expression of PTEN was increased upon DEFA5 overexpression, which partially explained the dephosphorylation of AKT.
FIGURE 4.

DEFA5 overexpression induces cell cycle arrest by increasing the number of S‐phase cells. A, Representative samples of propidium iodide staining and flow cytometry analysis showing the cells those in S‐phase, G0/G1‐phase and G2/M‐phase; B, The collective data showing the percentage of G0/G1‐phase cells, S‐phase cells and G2/M‐phase cells. C, D, Western blot results showing the phosphorylation levels of cell proliferation‐related marker proteins AKT and ERK. *P < .05, **P < .01 vs HA‐Vec group. Data are presented as mean ± SD. The results were reproducible in 3 independent experiments
3.5. DEFA5 interacts with BMI1
To further figure out the mechanism how DEFA5 induced cell cycle arrest in gastric cancer cells, co‐immunoprecipitation (Co‐IP) was performed to determine the direct interaction of DEFA5 with BMI1, which is highly expressed in tumor cells, and is related to tumor pathogenesis, development, invasion, and prognosis. 16 As shown in Figure 5A, DEFA5 can strongly bind to BMI1. To rule out the possibility of non‐specific binding that caused by antibodies, we purified both proteins with different tags in E coli and performed the in vitro glutathione S‐transferase (GST) pull‐down assay to confirm the interaction between DEFA5 and BMI1. Consistent with the Co‐IP experiments, His‐tag‐DEFA5 detected by His antibody can be pulled down by GST‐tag‐BMI1 (Figure 5B), indicating the specific interaction between DEFA5 and BMI1. Subsequently, we constructed BMI1 into the N‐terminal fragment (Flag‐BMI1‐N) and the C‐terminal fragment (Flag‐BMI1‐C), to check the specific region for DEFA5 binding, and the result demonstrated that DEFA5 binds to BMI1 at the C‐terminal region (Figure 5C).
FIGURE 5.

DEFA5 interacts with BMI1 and suppresses its recruitment to the promoter region of CDKN2a. A, Co‐IP experiment showing the direct interaction between DEFA5 and BMI1. B, The specific interaction between DEFA5 and BMI1 was confirmed by in vitro GST pull‐down assay. C, Co‐IP experiment indicating the interaction between HA‐DEFA5 and full‐length BMI1 or different truncated molecules. D, E, ChIP assay combined with quantitative PCR showing the recruitment of BMI1 to the promoter region of CDKN2a was significantly reduced by DEFA5 overexpression. D, Representative images. E, The promoter region of CDKN2a that pulled down by BMI1 antibody. F, Dual‐luciferase assay demonstrating the promoter activity of CDKN2a. (G, H, I) the protein and mRNA expression levels of BIM‐1, p16 and p19. **P < .01 vs HA‐Vec group. Data were presented as mean ± SD. The results were reproducible in 3 independent experiments
3.6. DEFA5 binds to BMI1 and suppresses its recruitment to the promoter region of CDKN2a
BMI1 is reported to serve as the transcription repression of the CDKN2a locus that encodes 2 proteins, p16 and p19, both of which act as tumor suppressors by regulating the cell cycle. 17 Therefore, we hypothesized here that DEFA5 overexpression may recruit and bind with BMI1, which subsequently decreased the recruitment of BMI1 to the promoter region of CDKN2a locus and upregulated the expression of 2 cyclin‐dependent kinase inhibitors, p16 and p19. To test this hypothesis, chromatin immunoprecipitation (ChIP) assay combined with quantitative PCR were performed to test the interaction of BMI1 and CDKN2a promoter. We found that the recruitment of BMI1 to the promoter region of CDKN2a was significantly reduced by DEFA5 overexpression in gastric cancer cells (Figure 5D,E). Therefore, the promoter activity of CDKN2a was dramatically increased by DEFA5 overexpression detected by dual‐luciferase assay (Figure 5F). Consistently, western blot analysis showed that, compared with control cells, DEFA5 overexpression increased the mRNA and protein expression levels of both p16 and p19 in gastric cancer cells (Figure 5G‐I). Notably, BMI1 expression downregulated DEFA5 overexpression, although the underlying mechanism is still unknown.
3.7. BMI1 knockout mimics the inhibitory effect of DEFA5 by increasing p16 and p19 expression
Based on the above findings, DEFA5 exerted its inhibitory effect in cell proliferation by antagonizing BMI1‐mediated inhibition of CDKN2a promoter activity. Therefore, knocking out BMI1 may mimic the inhibitory effect of DEFA5. First, we constructed BMI1 knockout cells by introducing sgRNA targeting BMI1 coding region and the Cas9 endonuclease. Western blot analysis results showed that no BMI1 was expressed in SGC7901 cells (Figure 6A). Expectedly, cell proliferation was significantly suppressed by knocking out BMI1 (Figure 6C), agreeing well with the effect of DEFA5 overexpression. Interestingly, BMI1 knockout itself significantly elevated the mRNA and protein expression levels of p16 and p19 (Figure 6A,B). However, DEFA5 overexpression failed to further increase both mRNA and protein levels of p16 and p19 in BMI1 knockout gastric cancer cells (Figure 6A,B). These results indicated that DEFA5 exerted its inhibitory effect in the cell proliferation by antagonizing BMI1‐mediated inhibition of p16 and p19 expression.
FIGURE 6.

BMI1 knockout mimics the inhibitory effect of DEFA5 by increasing p16 and p19 expression. A, B, The protein expression levels of p16 and p19 after BMI1 knockout and/or DEFA5 overexpression. C, The capacity of cell proliferation after BMI1 knockout. **P < .01 vs HA‐Vec group. Data were presented as mean ± SD. The results were reproducible in 3 independent experiments
4. DISCUSSION
Gastric cancer is the most common malignant tumor of the digestive tract. 1 , 2 At present, clinical treatment mainly relies on the standard treatment of surgical resection combined with radiotherapy and chemotherapy, and the clinical effect is not satisfactory. 1 Therefore, new biomarkers and therapeutic targets for gastric cancer are urgently needed. In the present study, for the first time we demonstrated an inhibitory effect of DEFA5 in the cell growth of gastric cancer by elevating the expression of 2 cyclin‐dependent kinase inhibitors, p16 and p19, to induce cell cycle arrest. Therefore, DEFA5 may serve as a potential tumor suppressor in gastric cancer.
In addition to its antimicrobial activity, defensins are widely involved in the innate immunity of pathogens and have been reported to be closely related to tumorigenesis and tumor immunity. 5 , 7 , 9 , 18 DEFA5 belongs to the α‐defensins subfamily and was originally reported to be produced by and secreted from Paneth cells, secretory epithelial cells in the small intestine. 19 In Paneth cells, DEFA5 is primarily secreted as a precursor molecule, and matured into a functional form with antimicrobial activity through a proteolytic processing by Paneth cell‐derived trypsin. 19 In mice, DEFA5 is a major antimicrobial peptide in the intestine and has been estimated to reach ~250 μg/mL in the lumen and ~100 mg/mL in the crypts. 19 , 20 , 21 , 22 Previous reports have shown that in DEFA5 transgenic mice, the luminal bacterial composition was changed, and the number of segmented filamentous bacteria was decreased, indicating that DEFA5 plays a critical role for gut microbiome homeostasis. 23 DEFA5 is also expressed in epithelial cells of the urinary and reproductive tracts, and contributes to the innate immune protection from pathogens. 24 , 25 In addition, human DEFA5 was reported to be a candidate biomarker to delineate inflammatory bowel disease, 26 and plays an important role in the anti‐inflammatory responses. 27 , 28
So far, the association of DEFA5 with cancer has been reported, although the role of DEFA5 in tumorigenesis is still unknown. Some studies have demonstrated that DEFA5 peptide is present in colon cancer, 13 , 14 ovary cancer, endometrium cancer, and lung cancer. 15 In 2013, Inada et al reported for the first time that DEFA5 may inhibit the growth of esophageal squamous carcinoma cells by down‐regulating the expression of E‐cadherin, suggesting that DEFA5 may function as a tumor suppressor. 29 Here, decreased expression of DEFA5 was observed in the tumor tissues from gastric cancer patients, and provides a hint that DEFA5 may play a role in the process of gastric cancer. Both in vitro and in vivo experiments indicated that DEFA5 overexpression inhibited cell proliferation and tumor growth by halting cell cycle at G1/S phase. However, the ability of cell migration and invasion was not affected. In addition, DEFA5 overexpression dramatically reduced the phosphorylation levels of AKT and ERK, suggesting that the AKT/ERK signaling pathway that mediates cell proliferation and development was impaired, but the mechanism is still unknown and needs to be further explored.
The BMI1 gene is one of the core members of the PcG family and plays an important role in the development of bone, hematopoietic and nerve development in embryonic mammals. 16 , 30 In addition, BMI1 is highly expressed in tumor cells, and is related to tumor pathogenesis, development, invasion, and prognosis. 31 Considering the strong interaction between DEFA5 and BMI1 shown in this study, we provide evidence to demonstrate that DEFA5 regulated the cell cycle and proliferation of gastric cancer cells through the BMI1 gene. It has been reported that BMI1 promotes cell transformation and tumor formation by transcriptional regulation of multiple downstream pathways including the cell cycle, DNA damage repair, EMT, and apoptosis. 16 One of the most studied and validated BMI1 targets is the CDKN2a locus, which is ubiquitously expressed in many tissues and encodes 2 cyclin‐dependent kinase inhibitors, p16Ink4a and p19 Arf. 17 Here, we found that DEFA5 when overexpressed can bind to BMI1 and decrease its recruitment to the promoter region of CDKN2a, and which subsequently elevated the transcriptional activity of CDKN2a and increased expression levels of p16 and p19, finally leading to cell cycle arrest of gastric cancer cells. The BMI1 knockout mimics the roles of DEFA5 overexpression in cell proliferation of gastric cancer cells. In other words, DEFA5 overexpression revived BMI1‐mediated transcriptional repression of the CDKN2a locus through direct interaction, suggesting its potential as a drug for gastric cancer treatment.
In conclusion, our findings demonstrated that DEFA5 might serve as a tumor suppressor in gastric cancer. Importantly, we provided a novel regulatory mechanism of α‐defensins in gastric cancer, by which DEFA5 inhibited BMI1 transcription activity and increased the expression of p16 and p19 to suppress cell proliferation. We hope that our findings could provide a new insight toward the treatment of gastric cancer, although DEFA5 transgenic and knockout mice are needed to further emphasize the roles of DEFA5 in vivo in gastric cancer.
DISCLOSURE
The authors have declared no conflict of interest.
Supporting information
Figs S1‐S3
Wu Z, Ding Z, Cheng B, Cui Z. The inhibitory effect of human DEFA5 in growth of gastric cancer by targeting BMI1. Cancer Sci. 2021;112:1075–1083. 10.1111/cas.14827
REFERENCES
- 1. Rawla P, Barsouk A. Epidemiology of gastric cancer: global trends, risk factors and prevention. Prz Gastroenterol. 2019;14:26‐38. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Bray F, Ferlay J, Soerjomataram I, et al. GLOBOCAN estimates of incidence and mortality worldwide for 36 cancers in 185 countries. CA Cancer J Clin. 2018;68(2018):394‐424. [DOI] [PubMed] [Google Scholar]
- 3. Karimi P, Islami F, Anandasabapathy S, Freedman ND, Kamangar F. Gastric cancer: descriptive epidemiology, risk factors, screening, and prevention. Cancer Epidemiol Biomarkers Prev. 2014;23:700‐713. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Lunet N, Barros H. Helicobacter pylori infection and gastric cancer: facing the enigmas. Int J Cancer. 2003;106:953‐960. [DOI] [PubMed] [Google Scholar]
- 5. Ganz T. Defensins: antimicrobial peptides of innate immunity. Nat Rev Immunol. 2003;3:710‐720. [DOI] [PubMed] [Google Scholar]
- 6. Raj PA, Dentino AR. Current status of defensins and their role in innate and adaptive immunity. FEMS Microbiol Lett. 2002;206:9‐18. [DOI] [PubMed] [Google Scholar]
- 7. Lehrer RI, Lu W. alpha‐Defensins in human innate immunity. Immunol Rev. 2012;245:84‐112. [DOI] [PubMed] [Google Scholar]
- 8. Ghosh SK, McCormick TS, Weinberg A. Human beta defensins and cancer: contradictions and common ground. Front Oncol. 2019;9:341. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Droin N, Hendra JB, Ducoroy P, Solary E. Human defensins as cancer biomarkers and antitumour molecules. J Proteomics. 2009;72:918‐927. [DOI] [PubMed] [Google Scholar]
- 10. Cun Gao WY, Tian H, Li L, Li S, Si L. Human beta‐defensin 2 promotes the proliferation of lung cancer cells through ATP‐binding cassette transporter G2. Int J Clin Exp Pathol. 2016;9:5944‐5949. [Google Scholar]
- 11. Mburu YK, Abe K, Ferris LK, Sarkar SN, Ferris RL. Human beta‐defensin 3 promotes NF‐kappaB‐mediated CCR7 expression and anti‐apoptotic signals in squamous cell carcinoma of the head and neck. Carcinogenesis. 2011;32:168‐174. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Kemik O, Kemik AS, Sumer A, Begenik H, Purisa S, Tuzun S. Human neutrophil peptides 1, 2 and 3 (HNP 1–3): elevated serum levels in colorectal cancer and novel marker of lymphatic and hepatic metastasis. Hum Exp Toxicol. 2013;32:167‐171. [DOI] [PubMed] [Google Scholar]
- 13. Lisitsyn NA, Bukurova YA, Nikitina IG, Krasnov GS, Sykulev Y, Beresten SF. Enteric alpha defensins in norm and pathology. Ann Clin Microbiol Antimicrob. 2012;11:1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Bousserouel S, Lamy V, Gosse F, Lobstein A, Marescaux J, Raul F. Early modulation of gene expression used as a biomarker for chemoprevention in a preclinical model of colon carcinogenesis. Pathol Int. 2011;61:80‐87. [DOI] [PubMed] [Google Scholar]
- 15. Vragniau C, Hubner JM, Beidler P, et al. Studies on the interaction of tumor‐derived HD5 alpha defensins with adenoviruses and implications for oncolytic adenovirus therapy. J Virol. 2017;91:e02030‐16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Bhattacharya R, Mustafi SB, Street M, Dey A, Dwivedi SK. Bmi‐1: At the crossroads of physiological and pathological biology. Genes Dis. 2015;2:225‐239. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Meng S, Luo M, Sun H, et al. Identification and characterization of Bmi‐1‐responding element within the human p16 promoter. J Biol Chem. 2010;285:33219‐33229. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Bevins CL, Salzman NH. Paneth cells, antimicrobial peptides and maintenance of intestinal homeostasis. Nat Rev Microbiol. 2011;9:356‐368. [DOI] [PubMed] [Google Scholar]
- 19. Ghosh D, Porter E, Shen B, et al. Paneth cell trypsin is the processing enzyme for human defensin‐5. Nat Immunol. 2002;3:583‐590. [DOI] [PubMed] [Google Scholar]
- 20. Ayabe T, Satchell DP, Wilson CL, Parks WC, Selsted ME, Ouellette AJ. Secretion of microbicidal alpha‐defensins by intestinal Paneth cells in response to bacteria. Nat Immunol. 2000;1:113‐118. [DOI] [PubMed] [Google Scholar]
- 21. Ayabe T, Satchell DP, Pesendorfer P, et al. Activation of Paneth cell alpha‐defensins in mouse small intestine. J Biol Chem. 2002;277:5219‐5228. [DOI] [PubMed] [Google Scholar]
- 22. Salzman NH, Ghosh D, Huttner KM, Paterson Y, Bevins CL. Protection against enteric salmonellosis in transgenic mice expressing a human intestinal defensin. Nature. 2003;422:522‐526. [DOI] [PubMed] [Google Scholar]
- 23. Salzman NH, Hung K, Haribhai D, et al. Enteric defensins are essential regulators of intestinal microbial ecology. Nat Immunol. 2010;11:76‐83. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Spencer JD, Hains DS, Porter E, et al. Human alpha defensin 5 expression in the human kidney and urinary tract. PLoS One. 2012;7:e31712. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Svinarich DM, Wolf NA, Gomez R, Gonik B, Romero R. Detection of human defensin 5 in reproductive tissues. Am J Obstet Gynecol. 1997;176:470‐475. [DOI] [PubMed] [Google Scholar]
- 26. Williams AD, Korolkova OY, Sakwe AM, et al. Human alpha defensin 5 is a candidate biomarker to delineate inflammatory bowel disease. PLoS One. 2017;12:e0179710. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Wang C, Shen M, Zhang N, et al. Reduction impairs the antibacterial activity but benefits the LPS neutralization ability of human enteric defensin 5. Sci Rep. 2016;6:22875. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Sugi Y, Takahashi K, Kurihara K, et al. alpha‐Defensin 5 gene expression is regulated by gut microbial metabolites. Biosci Biotechnol Biochem. 2017;81:242‐248. [DOI] [PubMed] [Google Scholar]
- 29. Nomura Y, Tanabe H, Moriichi K, et al. Reduction of E‐cadherin by human defensin‐5 in esophageal squamous cells. Biochem Biophys Res Commun. 2013;439:71‐77. [DOI] [PubMed] [Google Scholar]
- 30. Park IK, Morrison SJ, Clarke MF. Bmi1, stem cells, and senescence regulation. J Clin Invest. 2004;113:175‐179. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Siddique HR, Saleem M. Role of BMI1, a stem cell factor, in cancer recurrence and chemoresistance: preclinical and clinical evidences. Stem Cells. 2012;30:372‐378. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Figs S1‐S3