

Abstract
Neuropsychiatric phenotypes have long been known to be influenced by heritable risk factors, directly confirmed by the past decade of genetic studies which have revealed specific genetic variants enriched in disease cohorts. However, the initial hope that a small set of genes would be responsible for a given disorder proved false. The more complex reality is that a given disorder may be influenced by myriad small-effect noncoding variants, and/or by rare but severe coding variants, many de novo. Noncoding genomic sequences—for which molecular functions cannot usually be inferred—harbor a large portion of these variants, creating a substantial barrier to understanding higher-order molecular and biological systems of disease. Fortunately, novel genetic technologies—scalable oligonucleotide synthesis, RNA sequencing, and CRISPR—have opened novel avenues to experimentally identify biologically significant variants en masse. An especially versatile technique resulting from such innovations are Massively Parallel Reporter Assays (MPRAs), powerful molecular genetic tools that can be used to screen ≥thousands of untranscribed or untranslated sequences and their variants for functional effects in a single experiment. This approach, though underutilized in psychiatric genetics, has several useful features for the field. Here, we review methods for assaying putatively functional genetic variants and regions, emphasizing MPRAs and the opportunities they hold for dissection of psychiatric polygenicity. We discuss literature applying functional assays in neurogenetics, highlighting strengths, caveats, and design considerations—especially regarding disease-relevant variables (cell type, neurodevelopment, and sex), and ultimately propose applications of MPRA to both computational and experimental neurogenetics of polygenic disease risk.
Keywords: Reporter assay, noncoding variants, GWAS, UTR, Enhancer, Polygenicity
Introduction
Psychiatric diseases are genetically influenced by both heritable variation (common and rare) and non-inherited, de novo mutations. Estimated common variant (frequency ≥ 1%) influence on disease liability ranges from 10–33% for major depressive disorder (MDD) (1–3) and schizophrenia (SCZ) (1,4,5) to over 50% in autism spectrum disorders (ASD) (6,7). The remaining familial heritability of psychiatric—especially neurodevelopmental and psychotic (8,9)—diseases is largely conferred by rare variants (7). Two major hurdles have prevented variant data from illuminating disease mechanisms: the volume of variant discoveries/associations to test for functionality and causality, and imperfect methods of predicting variant consequences.
Variant-disease associations arise from correlational methodologies. Genome-wide association studies (GWAS) identify overrepresented single nucleotide polymorphisms (SNPs), tagging hundreds of mostly untranscribed, linked SNPs (7). Similarly, family studies identify thousands of proband-specific (de novo) or -enriched (rare, inherited) variants, though 0–2 per patient may be causal. However, discovery-oriented approaches alone are incapable of specifying which variants have biological function.
Predicting whether and how noncoding variants are functional is a nontrivial enterprise. The majority of GWAS loci bear indirect indication(s) of transcriptional regulatory function, including expression quantitative trait locus (eQTL) associations, chromatin accessibility, or histone marks (10–12). As others have noted, these data alone cannot define functional regulatory elements/variants (13,14). However, even within one cell type, such data are often mutually discordant: an emerging (i.e., preprinted) study examining six epigenomic datasets from K562 cells showed 49% of functional regulators did not overlap any epigenomic annotations; another 40% only overlapped one of the six (15). Similarly, MPRA of chromatin-based K562 enhancer predictions found only 30% regulated transcription (16). Unsurprisingly, these discrepancies apparently extend to disease variant interpretation: only a minority of GWAS variants (except for blood traits) overlap tissue-specific regulatory predictions (17) from histone marks (18). Such findings collectively suggest that heritable, disease tissue-specific regulatory phenomenon are both missed and mislabeled when relying solely on chromatin states.
Despite the clear excess of de novo variation in coding sequences in ASD and other neurodevelopmental disorders, and though coding variant consequences can often be predicted (e.g., nonsense mutations), this constitutes the minority of heritable risk for several psychiatric diagnoses (19). The remaining burden falls within putative transcriptional (19) and translational regulatory elements (e.g., promoters, UTRs) (20,21). ASDs provide a representative case: among 1,902 subjects, over 255,106 de novo variants were recently identified, with thousands each in upstream/promoter sequences and untranslated regions (UTRs) (22). UTRs regulate transcript stability and miRNA interactions (23); emerging work further implicates UTRs in nuclear transcript trafficking in the brain (24). The occurrence of most disease-linked variation in the least-well understood features of the genome/transcriptome thus obstructs understanding of disease biology. Collectively, these two problems necessitate high-throughput assays with functional readout for putative regulatory elements and variants. Such assays enable identification of functional variants and the biological contexts in which they act. This knowledge can shape hypotheses regarding shared mechanisms by which disparate genetic factors converge on shared phenotypic endpoints.
Here, we will primarily discuss MPRAs for high-throughput parcellation of genetic discoveries. MPRA technology pairs genomic features (e.g., each allele of a genomic sequence) to a reporter gene bearing unique, transcribed barcodes, allowing multiplexed RNA-level readout of element activity (25,26). Critically, there is substantial potential for MPRAs to identify functional variants from neuropsychiatric-associated loci. In part one, we discuss uses of MPRAs in functional identification of gene regulatory elements and variants, design/interpretation considerations for MPRA, and methods to complement/follow-up findings. In part two, we discuss potential applications of MPRA to identify mechanistic convergence across polygenic risk space.
Part 1: MPRAs for Identification of Functional Regulatory Elements and Variants
MPRAs offer a flexible framework to study elements regulating transcription (e.g., enhancers, promoters), splicing, protein translation, and post-transcriptional phenomenon. Though too numerous to review deeply here, we point readers to published and emerging applications of MPRA to splicing (27–29), RNA editing (30), and protein translation (31). MPRAs have been most broadly applied to explore and computationally model transcriptional “regulatory grammar”—how sequence features such as binding motifs, their abundance, and arrangement affect regulatory capacity (16,32–38). More recently, these approaches have been applied to characterize UTR functions in RNA stability and translation (39–43), and to identify SNPs and rare variants influencing transcription (44–50).
As shown in Figure S1A–B, a canonical ‘enhancer’ MPRA utilizes a promoter with candidate elements either upstream or in a 3’UTR (STARRseq) (51). Each element is paired with unique barcodes in the transcribed UTRs, which are sequenced as quantitative readouts. Expression—representing transcription or RNA stability—is measured as the RNA barcode counts per DNA barcode count (Figure S1E). This measure can be leveraged to define active or differentially active enhancer elements. Functional elements have been defined by either comparing to minimal-promoter only barcodes (16,34,37,38,52,53), or individual sequences against their shuffled counterparts (32,38); MPRAs have also successfully compared activity between alleles (44,45,47–50,54).
MPRAs also enable study of post-transcriptional regulatory elements. As shown in Figure 1E–G, the RNA/DNA expression metric assess UTR effects on transcript stability. Published and emerging UTR MPRAs have not yet considered human variants directly, but have distinguished functions of ASD/ID-implicated CELF proteins and related RBPs (42,55,56), and defined 3’UTR (39–41) and 5’UTR (43,57) features influencing transcript stability and translation. Both UTR and enhancer MPRAs enable assessment of disease-associated variant function across model systems (Figure 1A–C; Figure S2).
Figure 1 |. Example Allele-Differential Phenomenon in Common MPRA Approaches, and Analysis of MPRA Data.
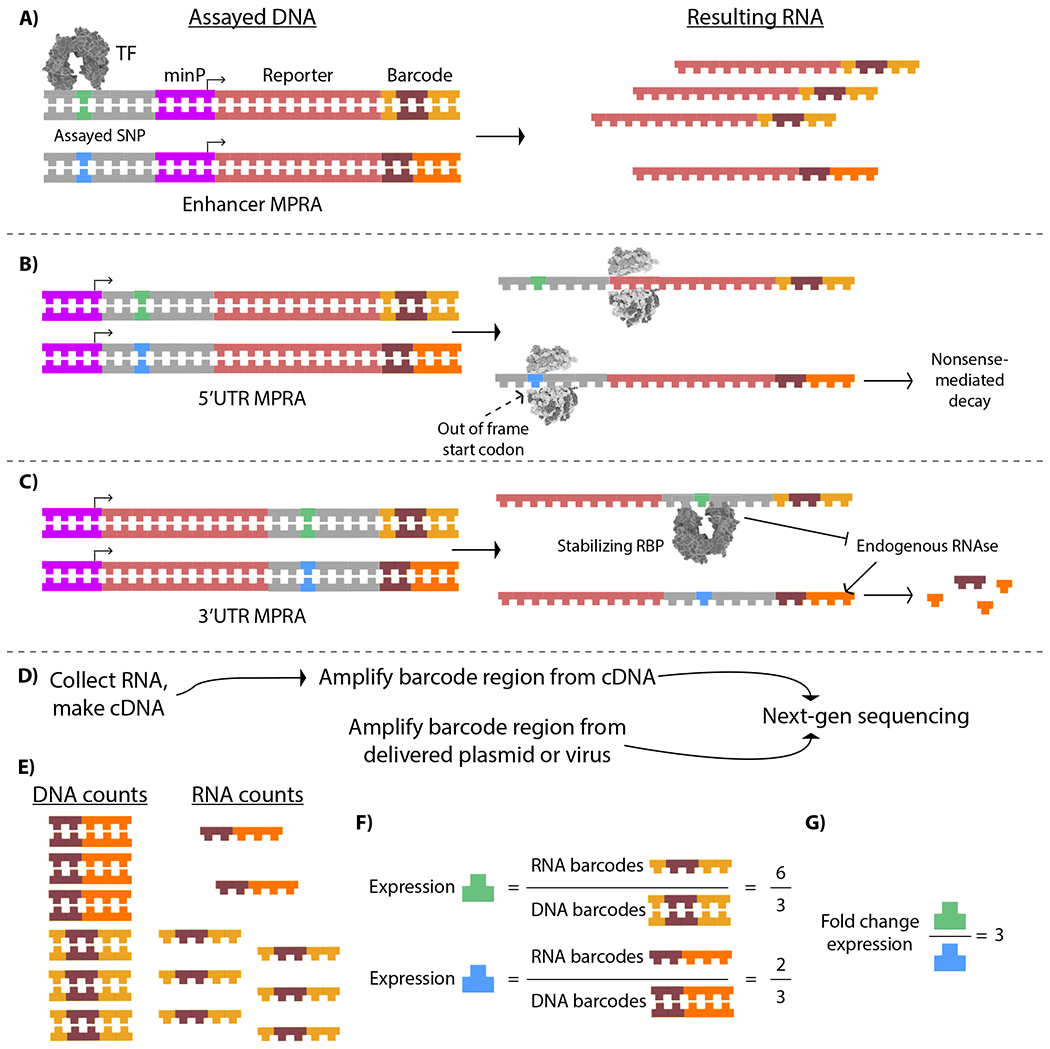
A) In a transcriptional-regulatory assay, a putative regulatory SNP may create, ablate, strengthen, or weaken a TF binding site. As a result, one allele drives more transcription (detected via its 3’UTR barcodes) per encoding DNA than the other allele. B) In a 5’UTR assay, a functional SNP may sequence features controlling translation initiation. For example, a variant allele may introduce an upstream start codon out of frame with the reporter gene, resulting in nonsense mediated decay, and thus, decreased detection of the barcodes paired to that UTR allele. C) In a 3’UTR assay, a variant may alter an RBP binding site; in this example, an RBP site specific to one allele increases the stability of the reporter transcript, and thus of the barcode paired to it. D) After transfection/transduction, RNA is collected from specimens and prepared along with DNA (often the delivered DNA, though sometimes this is recovered from the specimens as well) to generate sequencing libraries to quantify expression of the delivered elements in the RNA, compared to starting abundance in the DNA. E) Example read counts, presented visually, for the DNA and RNA barcode counts of one barcode paired to each allele. F) MPRA analysis centers on taking the ratio of RNA/DNA counts (or counts per million), represented by the sequence fragments at top left, as a measure of expression—i.e., approximating the number of transcripts generated per encoding DNA. These can be compared relative to the expression of all elements to find the strongest features (e.g., strongest enhancers and repressors, or most stabilizing and destabilizing UTR elements), or G) compared on a variant-wise basis to determine significant allelic regulatory effects.
MPRAs Identify Functional Elements in Specific Cellular Contexts
Perhaps the most exciting—if underappreciated—property of MPRAs is the ability to assay elements using disease-relevant cells and conditions. Functional elements are defined by each cell type’s unique milieu of expressed TFs, chromatin modifiers, miRNAs, and RBPs, which mediate regulatory element activity. The breadth of published and emerging tissue/cell type differences in gene expression (58,59), chromatin marks (18,60) and chromatin interactions (61–63) all illustrate the magnitude of these regulatory differences. The importance of cell type was illustrated by an MPRA of the same elements in U87 glioblastoma and neural progenitor cells (NPCs): the most active enhancers in each cell type contained entirely different motifs and sequence features (37). Recent (64,65) and emerging (66) approaches have identified highly cell type-specific brain enhancers using adeno-associated viral (AAV) vectors alongside traditional (e.g., immunofluorescent) readouts. Moreover, a novel, AAV-based MPRA (i.e., using barcodes) identified novel functional enhancers for somatostatin interneurons (67). Aside from these examples, MPRAs in neural cells have been limited to date. Several early MPRAs utilized explanted retina to explore influences of TF binding sites and their arrangements on activity (38,48,53,68). One novel study, relevant to functional contexts (discussed below), assayed mouse neuron enhancers for activity changes following KCl depolarization (69). Other studies include an MPRA characterizing temporal patterns of cis-regulatory element activity across seven timepoints in human NPC differentiation into neurons (70). This delineation of regulatory element function illustrates the power of regulatory assays to reveal timepoints and cellular states wherein gene regulation—especially for neurodevelopmental disorders—may exert its causal effects.
In vivo regulatory assays—including in the brain—have more recently been demonstrated, generally at smaller scales than in vitro MPRAs. Osterwalder, et. al (71) singly or multiply knocked out putative limb development enhancers in mice, illustrating enhancer redundancy—that is, limb development disruption only with perturbation of multiple elements. McClymont et. al (72) identified 2,000 candidate embryonic mouse enhancers in purified midbrain dopamine neurons, and validated the developmental and regional specificity of a subset using transgenic reporter mice. The scale of these assays has been expanded by groundbreaking implementation of MPRA in the brain in vivo (48,67) to query functional effects in native cell contexts.
This transition to in vivo MPRA is beneficial because, while cell type overwhelmingly influences regulatory assays, additional conditions may equally alter outcomes (Figure 2A–C). Age, sex, pharmacology, and environment (e.g., stress)—all can shape gene expression. For example, MPRAs have identified elements responsive to hormonal contexts such as steroid-responsive glucocorticoid receptor binding (73). Altogether, MPRAs enable identification of functional regulatory elements across varied internal and external environments.
Figure 2 |. Regulatory assays are influenced by a range of conditions, from environment to sequence context.
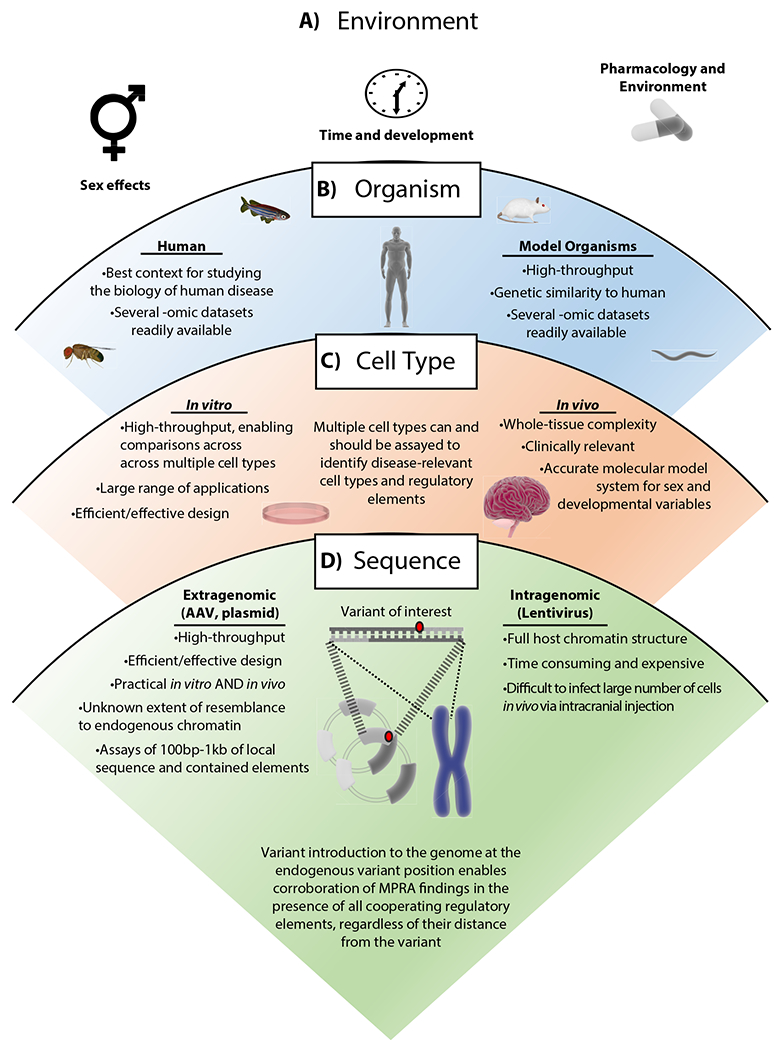
The range of conditions that influence regulatory assays (from top to bottom) starts when considering A) the environment, e.g., sex, time, and pharmacology. These parameters have the potential to affect various –omic profiles in a given system. B) The next level of consideration is the organism, which can include human-derived tissue or one of the many model organisms. Human genomic context is ideal for studying the biology of human disease – though a comparatively limited scope of techniques for human-derived tissues exists. C) Next, one should consider the selected cell type(s) and whether to assay in vitro or in vivo. Each of these provides a unique set of benefits, and one approach can be used to validate findings from the other (45,74). In the case of modeling the brain and psychiatric genetic variants, cell type-specific/enriched MPRAs in vivo would constitute the highest-fidelity model of variant effects by accounting for regulatory effects of endogenously interacting cell types. D) Lastly, the sequence context will be influenced by the delivery method, which results in transcription from extragenomic or intragenomic MPRA DNAs. In either case, only limited length of sequence surrounding a feature of interest is preserved (e.g., in ~120bp tiles of genomic sequence in custom oligonucleotide cloning, or ≤ 1kb in clone-and-capture methods), preventing assessment of any interactive effects from elements further away. (A recent study suggests that size of a tile negatively correlates with reproducibility of expression driven compared to that driven by ~120bp tiles, emphasizing the importance of this consideration (93)). While AAV-transduced episomes gain histones (105) and chromosome-like nucleosome spacing (106), it is unknown whether gene-regulatory histone marks on these episomes mirror those of endogenous regulatory chromatin. For these reasons of both local sequence context and chromatin context, we suggest corroboration of MPRA findings in native genomic settings, by, for example, introducing the variant to the genome of a cell line using CRISPR methods.
MPRAs, assay context, and functional variants
MPRAs can be designed not only to identify functional elements, but to assay and compare genetic variants in contexts known—or predicted—to mediate disease. As transcriptomic and epigenomic studies highlight an enormous role for cell type, it is unsurprising that this influence extends to regulatory variants. For example, variants exert cell type-dependent effects on chromatin structure even within a neurodevelopmental lineage: an emerging study discovered chromatin accessibility QTLs in human NPCs and neurons, with ~80% of QTL SNPs being specific to one of the cell types (74). Cell type roles in putatively functional variation are also implicated by GWAS SNPs enrichment in tissue-specific eQTLs (59,75), neural cell type-specific chromatin interactions (62), and eQTLs that evade detection in bulk brain (i.e., multi-cell type) tissue but are evident in purified populations like dentate granule neurons (76). MPRA has likewise demonstrated the essentiality of cell type in defining functional variants: the Critical Assessment of Genome Interpretation 5 (CAGI5) consortium performed an MPRA on saturation-mutagenized human enhancers and disease-associated promoters in numerous cell lines, challenging analysts to computationally predict functionality and effect size for held-out variants. The most predictive annotations for a given cell line were often from the same cells across several top-performing analyses (77). Thus, experimental study of putative disease-associated variants requires firm hypotheses on where (tissue/cell type), when (development/differentiation), and how elements are expressed/active and biologically relevant. Careful consideration needs to be given to the appropriate cellular context when designing assays for psychiatric genetics: key variant-interacting TFs and RBPs expressed in neurons may not be present in convenient cell lines (e.g., K562), potentially rendering functional neural elements/variants apparently silent.
Despite their potential, few MPRAs have examined disease-associated variants while considering both cell type and –omic predictions. Tewhey, et. al (49) screened 30,000 eQTL SNPs from human lymphoblastoid cell lines (LCLs) using MPRA in LCLs, maintaining the discovery context in their assay. Over 3,400 active regulatory sequences were identified, including 850 activity-modulating variants (24%), consistent with functional (expression-modulating) SNP associations tagging linked, non-functional SNPs, akin to GWAS. Illustrating MPRA’s sensitivity, significant allelic differences in activity were detectable at effect sizes <2-fold. Emerging work by Choi, et. al (78) prioritized over 800 SNPs—guided by fine-mapping and epigenomics—from 16 melanoma GWAS loci, to assay for transcriptional-regulatory activity in cultured melanocytes. Candidate variants with concordant eQTL signal in independent melanocyte data were further investigated, ultimately enabling experimental demonstration of biophysical (TF binding), molecular (target gene expression), cellular (growth rate), and in vivo (melanoma rate in transgenic zebrafish) variant mechanisms. Finally, a recent MPRA of autoimmune GWAS loci yielded replicable findings across 12 donor lines of CD4+ T-cells, which were discordant with the more easily accessible—but leukemic—Jurkat cell line (79). These experiments exquisitely illustrate MPRA’s capacity for sensitivity, context specificity, and high discovery rates, especially when integrating both association data and multi-omic annotations.
As with functional element assays, functional variant assays have recently moved in vivo, again including the brain. Kvon, et. al (80) utilized a novel knock-in system to generate transgenic mice expressing a LacZ reporter expressed under putatively regulatory elements containing rare, polydactyly-associated variants; subsequent LacZ staining clarified which variants were functional based on alterations of limb bud LacZ patterns. Excitingly, a small-scale MPRA has recently emerged using in vivo tissue: after prioritizing a single SNP from a bipolar disorder GWAS locus using epigenomic annotations, the two alleles of this sequence region were paired to 20 barcodes each and electroporated into embryonic mouse brains to confirm variant function (48).
Limitations and Design Considerations
With the powerful opportunities of MPRA come limitations. A major caveat lies in gathering candidate variants to assay. For example, a prominent and functionally characterized schizophrenia GWAS locus in the major histocompatibility complex (MHC) region (4)—containing hundreds of linked SNPs—turns out to mark heritable copy number variations in the complement C4a gene (81); assaying only SNPs from this locus would not reveal the primary causal variant. Likewise, an MDD-associated SNP tags the absence of a transposon with regulatory effects on a noncoding RNA (82). In other words, MPRA’s usefulness is contingent on investigation (and size—see below) of sequences to be assayed.
Further considerations include appropriateness of biological ‘contexts’ (Figure 2). At the level of ‘sequence context,’ MPRAs generally use multiplex oligonucleotide synthesis to custom-design sequences and variations by the thousands. However, such approaches are size-limited to ~300bp, which precludes assay of large or spaced regulatory sequences. Oligonucleotide synthesis also is error-prone; tagging each element with multiple barcodes safeguards against error-driven false-positives. Bulk capture-and-clone strategies circumvent these issues by utilizing larger, genomic DNA fragments directly (47,83–85) at the expense of precision assay design. Finally, element positioning can substantially influence results and replicability. While STARR-seq is favorable for one-step cloning (putative enhancers doubling as 3’UTR barcodes), emerging works illustrate that enhancer-like sequence placement in 3’UTRs yields results which cluster separately from other MPRA designs testing the same sequences (86), and that such sequence placement can exert RNA stability effects that, without correction, may confound interpretation (87).
Reporter gene features are also important in regulatory assays. Previous enhancer MPRAs have demonstrated replicability by testing the same elements with a second promoter, with element activities highly correlated between the two (45,88,89). However, these cross-promoter correlations (Pearson r 0.7–0.8) have been weaker than often reported for biological replicates in MPRA (r>0.9). Promoter choice thus can influence assay results, via, for example, absence—or species differences in—promoter elements a cis-regulator requires. Likewise, UTR regulatory elements may be sensitive to the stoichiometry of transcripts and RBPs or miRNAs in the cell; excess transcript production by a strong promoter could potentially render effects of interacting regulators undetectable. In brief, rigorous MPRAs or follow-up assays should use both a minimal promoter and either a strong exogenous (e.g., CMV) or a genomic promoter from the pertinent cell/tissue type (e.g., a constitutively expressed, neuron-specific gene).
Importantly, the ability to test candidate sequences in their endogenous locus is not a feature of MPRAs. Thus, ‘genomic context’—that is, episomal (AAV, plasmid) vs. genome-integrated (lentiviral) approaches—require consideration. Emerging comparisons find these approaches correlate well (86), though certain applications may require a specific approach (e.g. MPRA of chromatin conformation (90)). The comparative throughput for a fixed number of cells is greater for plasmid transfection—thousands of plasmids per neural cell in vitro (91) compared to viral transduction (≤ tens of sequences/cell). These limitations and alternative methods are further considered in Table 1.
Table 1 |. Strengths and Limitations of Functional-Regulatory Assays in Terms of Throughput and Sequence and Cellular Contexts.
Method family: An umbrella term covering multiple adaptations of an assay. Technique: The particular adaptation of the family’s assay. “CRISPR editing” signifies precision replacement of an endogenous genomic sequence with a desired sequence (as opposed to CRISPR mutagenesis). Largest target/sequence assayable: Largest sequence currently supported by viral vectors (AAV assays), pooled oligonucleotide synthesis (MPRA), or guide RNAs (CRISPR), respectively. Simultaneous throughput for variants/perturbations per sample: Based on literature cited in the text and in this table, the largest range of elements that have been simultaneously targeted or tested within experimental replicates for the technique. Can assay cellular phenotypes? For CRISPR assays, the phenotype is generally cell division or death, measured by enrichment/depletion of genome-integrated sgRNAs in a cell culture relative to the pool of sgRNAs originally transfected. Genome-integrated? Does/can the assay utilize genome-integrated sequences (i.e., lentiviral delivery)? Assays at the endogenous genomic sequence? Whether the assay tests the putative regulatory sequence in its standard location in the genome (rather than testing a short piece of genomic sequence elsewhere in the genome or outside of the genome entirely). Can identify target gene of endogenous transcriptional regulator? Whether the assay allows for pairing of a functional element to its target gene(s). This is effectively contingent on testing the element in its endogenous genomic position.
| Method Family | Method | Can assay variants (e.g. SNPs) for function? | Can assay elements (e.g., TFBS, enhancers) for function? | Largest sequence / target | Simultaneous throughput for variants / perturbations per sample | Can assay cellular phenotype? | Genome-integrated? | Assays at the endogenous genomic sequence? | Demonstrated in model organism CNS in vivo? | Demonstrated in human primary or iPSC-derived neural stem cells, NPCs or neurons? | Can identify target gene of endogenous cis-regulator? |
|---|---|---|---|---|---|---|---|---|---|---|---|
| MPRA | Multiplex (Barcoded) AAV Transcriptional Regulator Assays | Yes | Yes | 3–5 kb | 100s-1000s | No | No | No | Yes (67) | No | No |
| Enhancer MPRA and STARR-seq | ~150–200 (custom oligos); ~700 (capture-and-clone) | 10,000–10^6 | Often not (exception: lentivirus random or targeted integration) | Yes (48) | Yes (48,95,97) | No | |||||
| 3’UTR MPRA/PTRE-seq | Not demonstrated but see above | No | N/A | ||||||||
| 5’UTR MPRA | “ | No | N/A | ||||||||
| RNA Splicing MPRA | “ | No | N/A | ||||||||
| Protein Translation MPRA | “ | No | N/A | ||||||||
| CRISPR Regulatory Disruption Assays | CRISPRi | No | Yes | ~50 bp | •Max demonstrated in CNS in vivo: 5 targets (2 sgRNAs each) •Max demonstrated in neural cell types in vitro: ~12,000 |
Yes | Yes | Yes | Yes (136) | Yes (107,137,138) | Yes |
| CRISPRa | •Max demonstrated in CNS in vivo: 10 targets (5 sgRNAs each) •Max demonstrated in neural cell types in vitro: 3 |
Yes | Yes | Yes (139–141) | Yes (113,138,142,143) | Yes | |||||
| CRISPR Mutagenesis of regulatory elements | •Max demonstrated in neural cell types in vitro: 26,000 targets (2 sgRNAs per target) | Yes | Yes | No | Yes (104) | For an a priori defined gene (144,145) | |||||
| CRISPRi-FlowFISH | ~900 | Not demonstrated | Yes | No | No | Yes | |||||
| Low/single throughput | CRISPR Editing | Yes | Yes | Several kb | 1–2 | Yes | Yes | Yes | Yes | Yes | Yes |
| AAV Transcriptional Regulator Assays with traditional readouts (fluorescence, LacZ, etc.) | 3–5 kb | 1 | No | No | No | Yes (esp. tacitly via cell-type targeted optogenetic, chemogenetic, and circuit-labeling techniques, as in (99)) | Yes (99) | No | |||
| Luciferase Reporter Assay | 3–5 kb | 1 | No | No | No | Yes (146) | Yes | No |
Other considerations include determining an appropriate ‘cellular’ and ‘organismal’ context (Figure 2B–C). Common strategies for choosing cellular contexts include using pathology (e.g., substantia nigra in Parkinson’s disease), expression patterns of disease-associated genes (e.g., cortical excitatory neurons in SCZ (92)), or GWAS-eQTL overlaps (e.g., neurogenic niches of mid-fetal brain in ASD and SCZ (93)). Cell type prioritization is further covered elsewhere in this Special Issue (94).
A notable opportunity is utilization of MPRAs in human iPSC-derived neural cell types, which offer the ability to conduct cell type-specific assays in a human genetic context. Very recent (70) and emerging (48,95) MPRAs are proof-of-principle for this approach, supporting advancement to assaying variants in the setting of iPSC derivates. Moreover, while cell type-specific MPRAs have been restricted to in vitro settings, where reproducing tissue physiology (e.g., inter-cell type interactions, hormones, stress) is difficult, barcoded multiplex AAV regulatory assays (67) indicate in vivo, cell type-specific MRPA is possible. Nonetheless, negative MPRA results should be interpreted cautiously; absence of function in one context may not extend to all contexts.
Statistical considerations in MPRA include appropriate library size (number of elements and paired barcodes) for the cell type to be tested. Generally, library size should be downsized for rarer, hard-to-maintain, or hard-to-transfect/transduce cell types to ensure robust barcode recovery and measurement. MPRAs have tested ~107 sequences simultaneously in easily transfected cancer cell lines (84,85), though in physiological cell types, like NPCs, this capacity is 104-105 (37,69,96), with emerging work approaching 106 (95,97). Library size is further constrained by element-per-cell (i.e., lentiviral) approaches. In other words, the fidelity of the model system and the MPRA library size it can support are generally anticorrelated. A consensus on the depth of barcodes-per-element is, to date, absent, ranging from 1 (STARR-seq (51)) to several hundred in previous (70) and emerging (97,98) work, with highly correlated replicate measurements across this range. Tewhey, et. al estimated that statistical benefits for small-effect transcriptional-regulatory variants accrue by 5 barcodes, and asymptote around 25–50 (49); another finds > 10 barcodes consistently yield inter-replicate r>0.97 in several cell types (86). Whether these guidelines apply to UTR assays remains unclear. Overall, MPRA power guidelines would benefit substantially from deep assessment by modelers and statisticians.
Finally, given a finite number of elements that can be simultaneously assayed, one can choose whether to prioritize candidate variants using epigenomic data, or simply include all linked SNPs (Figure S2). An assay’s ‘hit rate’ may be improved by prioritizing variants with indirect evidence of function, with the caveats of relying on epigenomic data discussed previously. However, foregoing such prioritization enables analysis of how well such features actually predict measured expression. Thus, the decision of prioritization must balance the value of ‘hits’ vs. identifying functionally predictive indirect measures (epigenetics) for the target cell type or disease.
Complementary methods in high-throughput study of DNA and RNA regulatory elements
There are a variety of other approaches that complement MPRAs (Table 1). Of course, lower throughput enhancer assays allow screening of the same elements or variants across a variety of contexts, even in vivo. Whether conducted using AAV (e.g., (99)), or transgenesis (e.g. (100)), these should remain gold standard approaches for validation and deep characterization of small numbers of elements and variants, including those identified by MPRAs.
A primary limitation of MPRA is the inability to test regulators in their endogenous genomic position and sequence context. Sequence-specific targeting using CRISPR/Cas9 has enabled several additional techniques for probing molecular and cellular effects of regulatory variation, with the caveat that, unlike MPRA, these techniques do not currently allow for the multiplexed study of post-transcriptional/translational regulatory variants. Nonetheless, these techniques enable study of putative disease gene roles in gene expression networks and cellular phenotypes. Perturb-seq (101) combines genewise perturbation by CRISPR with single-cell RNA-seq to identify gene sets dysregulated by loss of function of each candidate gene. These have, for example, been used to discover co-transcribed gene networks involved in neuronal remodeling (102) and for in vivo assessment of genes harboring de novo loss of function mutations in ASDs (103). Likewise, CRISPR screens can be used to define functional elements influencing selectable traits (e.g., proliferation), as in an emerging study perturbing both genes and cis-regulatory elements to define their roles in human neural stem cell proliferation (104). Finally, CRISPR editing has been used in vitro to assess single-transcript noncoding variant effects by comparing allelic RNA and genomic DNA abundances in edited cultures (105), a potential means of single-variant validation/follow-up of UTR MPRA findings. To our knowledge, such assays have not been conducted at a genome-wide scale in psychiatric disease, but have been used to identify genes that alter expression of the Parkinson’s-associated PARKIN (106).
Cis-regulatory MPRAs cannot identify the endogenous target gene(s) of functional elements. Fortunately, CRISPR-derived methods using a mutagenically-‘dead’ Cas9 (dCas9) conjugated to a transcriptional activator or repressor allow targeted potentiation or repression of endogenous genomic regulatory elements (CRISPRa and CRISPRi, respectively) to assess altered gene expression and other outcomes. These technologies are already online in state-of-the-art human neuroscience models: a recent CRISPRi study knocked down over 2000 genes by targeting their promoters in iPSC-derived excitatory neurons, defining their context-specific roles in their survival, differentiation, and proliferation—including gene effects altered by co-culture with astrocytes (107). Emerging work has further leveraged CRISPRi’s cell type specificity to study ASD-associated gene knockdown effects in an etiologically relevant cellular context (NPCs) (108). A recently introduced extension of CRISPRi (‘CRISPRi-FlowFISH’) targets intergenic regulators, identifying their target gene by concurrent fluorescent in situ hybridization against genes from the same chromodomain. Fluorescence-intensity sorting into bins and subsequent RNA-seq can then pair regulators (via guide RNA sequence) and target genes (altered FISH signal in a guide RNA’s presence) (109). While this assay was performed in K562 cells, it is not hard to envision its extension to neural cell types in vitro or in vivo. Altogether, CRISPR-based follow-up of MPRA candidates to define target genes and verify of genomic activity of regulators/variants will be key to developing insights in psychiatric genomics.
Part 2: MPRAs as an avenue to dissect multiallelic and polygenic mechanisms of neuropsychiatric traits
While MPRAs cannot intrinsically scale up to functional demonstration of cell-, tissue-, or behavior-level phenotypes, they have the potential to provide key information to guide molecular hypotheses for how these higher-order phenotypes emerge from large sets of regulators and/or their target genes. We focus here on examination of variants in trans space—that is, defining shared and recurrent features among MPRA-nominated functional variants across the genome that may collectively underlie large portions of polygenic disease risk. A brief examination of functional SNP interactions within linkage blocks can be found in the Supplemental Text.
The utility of MPRAs in identifying commonalities from variants across the genome
The most vexing question that remains after individual functional variant mechanisms are elucidated is how variants collectively contribute to phenotypic risk. MPRAs provide several ways to begin addressing this question: 1) identifying regulatory features shared by across several functional risk variants; 2) identifying functional modules enriched for variant-impacted genes; 3) providing functional annotations to variants for computational genomic approaches; and finally, 4) by enabling study of variant-by-environment interactions by performing MPRA across conditions.
Firstly, MPRA experiments running the gamut from basic regulatory genomics to human traits and variation have defined ‘regulatory grammars’ of assayed contexts. Identification of functional variants in the MPRA setting enables similar establishment of the ‘regulatory grammar’ of a trait or disease. Functional variants identified by MPRA across several UTRs may feature a specific RBP’s binding site, for example, or could be used to deliberately define functional activity of a disease-associated miRNA, like miR-137 (4). Likewise, variants associated with a trait could be more likely to fall in particular TF binding sites or be enriched in cell type-specific marks of genomic regulation. Evidence of this convergence is seen in de novo variants associated with ASD: several distinct variants disrupt binding sites for a single TF, NFIX (110). Similarly, putative gene targets of schizophrenia-associated variants are also putative—biases aside (111)—Fmrp targets (112). MPRA has also identified such regulatory convergence by, for example, intersecting identified functional SNPs with TF ChIP-seq datasets in pertinent cell types to discover recurrently disrupted TF binding sites (98). Assays of downstream consequences of variation also confirm biological convergence across association loci. A four-element-target CRISPRi/a assay revealed that schizophrenia risk genes act synergistically via shared influence on synaptic activity, and concurrent alteration of expression of all four genes results in a cellular transcriptome more accurately reflective of postmortem schizophrenia brain tissue (113). For both rare and common variants, identifying common regulators among risk genes provides information which can refine predictions of disease-related cell types based on TF, RBP, or epigenomic mark expression.
Secondly, genes and gene networks affected by statistically associated variation are often predicted using MAGMA (114), which in essence scores genes based on proximity to an associated variant and its linkage partners. Resulting gene sets are subjected to analyses such as Gene Ontology enrichment or are examined for enrichment in WGCNA (coexpression) networks from candidate tissue types to identify pathways and mechanisms on which these genes converge. While its use is ubiquitous in genomic studies, standard MAGMA gene association statistics for psychiatric disorders only modestly correlate to those from a tissue-specific, chromatin configuration-aware modification of MAGMA (115), suggesting that biological hypotheses from MAGMA gene sets may miss disease-associated genes in brain. Being able to refine implicated genes by functional validation using—or in follow-up to—MPRA will help to benchmark such approaches and refine prediction convergence with ‘truly’ dysregulated candidate genes.
Thirdly, epigenomic data alone is not comprehensively predictive of active regulators. However, well-informed analyses of human genetic findings rely heavily on such annotations to convert associations into biological hypotheses. Critically, these epigenomic data—unlike MPRA data—can be collected from postmortem human tissue. MPRAs focused on neuropsychiatric disorder associated variation stand to benefit from high-information datasets by aiding variant prioritization for assay inclusion. Several recent datasets on synthetic UTRs (39,43), RNA binding proteins (116,117), and postmortem human brain multi-omics (60,118–127) are worth noting for readers investigating disease-associated variation. Integrative computational analyses have brought these datasets together predict functional variation in SCZ, bipolar disorder, and ASDs (128,129). These constitute high-priority candidates for experimental validation by MPRA. Furthermore, emerging work reveals a symbiotic relationship developing between epigenomics and functional assays: functional element/variant information from MPRA has been used alongside epigenomic annotations to improve machine learning predictions of functional variants (130). Predictions from these refined algorithms are another low-hanging fruit for candidates to assay by MPRA; those results could then constitute new training data. Such refinement of epigenomic data interpretation coupled with functionally-demonstrated regulatory variation would mutually benefit one another and myriad downstream analyses, such as variant enrichment in genomic features and disease gene identification. For example, TWAS (131) and Predixcan (132) intersect gene expression QTLs (eQTL) with trait-associated variants to predict expression differences between cases and controls, thus identifying dysregulated gene sets. MPRA data can disentangle which eQTL SNPs are truly functional from those associated only due to LD, which could thus refine variant-gene pairings used in these analyses. Altogether, MPRA can serve to refine both epigenomic and genic definitions of truly causal disease features.
Finally, the context-specificity of MPRA represents a newfound ability to assess variant effects on gene regulation en masse under different biological and environmental contexts, including with in vivo models. While issues of convergent disease effects across genes and regulators are indeed complex, environmental effects—perhaps most canonically, stress—on these regulators are questions at the forefront of understanding polygenic risk in neuropsychiatric disorders. Pharmacologic variables have been successfully tested in MPRA, namely in the identification of glucocorticoid-responsive (73) and p53-responsive (133) regulatory elements. MPRAs could further be layered with concurrent gene perturbations (e.g., knockdown of a putative regulator), or cell culture conditions for in vitro identification of variant-environment interactions, exemplified by MPRA identification of neuronal activity-dependent enhancers (69). As mouse and human brain cell types and their gene expression patterns are largely (though not entirely) conserved both in development (134) and adulthood (135), the extension of MPRAs to the in vivo context will enable study of broader endogenous and exogenous disease-associated factors, such as sex or stress. Identifying variants with environment-dependent functions would be a start toward identifying convergent molecular mechanisms behind conditional disease risk in disorders such as MDD.
Conclusion
MPRA presents unique opportunities to dissect polygenicity of psychiatric disorders via simultaneous identification of functional variants across identified risk space. Beyond the primary benefits of identifying ‘true positive’ functional variants in specific biological and environmental contexts, MPRAs stand to rapidly broaden, deepen, and refine hypotheses and mechanisms of both noncoding disease risk and of gene-regulatory architecture itself.
Supplementary Material
ACKNOWLEDGEMENTS
This work was funded by NIMH grants 1F30MH1116654 and 1R01MH116999, and Simons Foundation 571009. We would like to thank Sergej Djuranovic, Ph.D., Barak Cohen, Ph.D., and Cohen lab alumni Dana King, Ph.D., and Brett Maricque, Ph.D. for their collaboration and guidance designing, adapting, and analyzing MPRAs applied to neuropsychiatry. We would also like to thank Idoya Lahortiga, Ph.D. and Luk Cox, Ph.D. curators of Somersault1824 (https://www.Somersault1824.com), for their open-access, Creative Commons BY-NC-SA 4.0 licensed libraries of high-quality biomedicine graphics (especially those from Graphite Life Explorer, ePMV, and Eyewire), adapted for figures in this review. Finally, we would like to thank Lexi Harris for assistance with figure design, and Mike Vasek and Tony Fischer for their assistance in editing this manuscript.This work was simultaneously submitted for peer review and as a preprint on Biorxiv (doi: 10.1101/2020.02.02.931337).
Footnotes
CONFLICT OF INTEREST
No authors declare a conflict of interest.
References
- 1.Consortium C-DG of the PG, Lee SH, Ripke S, Neale BM, Faraone SV, Purcell SM, et al. (2013): Genetic relationship between five psychiatric disorders estimated from genome-wide SNPs. Nat Genet 45: 984–994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Bulik-Sullivan B, Finucane HK, Anttila V, Gusev A, Day FR, Loh P-R, et al. (2015): An atlas of genetic correlations across human diseases and traits. Nat Genet 47: 1236–41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Wray NR, Ripke S, Mattheisen M, Trzaskowski M, Byrne EM, Abdellaoui A, et al. (2018): Genome-wide association analyses identify 44 risk variants and refine the genetic architecture of major depression. Nat Genet 50: 668–681. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Ripke S, Neale BM, Corvin A, Walters JTR, Farh K-H, Holmans PA, et al. (2014): Biological insights from 108 schizophrenia-associated genetic loci. Nature 511: 421–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Purcell SM, Wray NR, Stone JL, Visscher PM, O’Donovan MC, Sullivan PF, et al. (2009): Common polygenic variation contributes to risk of schizophrenia and bipolar disorder. Nature 460: 748–52. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Gaugler T, Klei L, Sanders SJ, Bodea CA, Goldberg AP, Lee AB, et al. (2014): Most genetic risk for autism resides with common variation. Nat Genet 46: 881–5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Pettersson E, Lichtenstein P, Larsson H, Song J, PGC ADDWG of the iPSYCH-B-PC Autism Spectrum Disorder Working Group of the iPSYCH-Broad-PGC Consortium, Bipolar Disorder Working Group of the PGC, Eating Disorder Working Group of the PGC, Major Depressive Disorder Working Group of the PGC, Obsessive Compulsive Disorders and Tourette Syndrome Working Group of the PGC, Schizophrenia CLOZUK, Substance Use Disorder Working Group of the, Agrawal A, et al. (2018): Genetic influences on eight psychiatric disorders based on family data of 4 408 646 full and half-siblings, and genetic data of 333 748 cases and controls. Psychol Med 49: 1–8.30409236 [Google Scholar]
- 8.Ryan NM, Lihm J, Kramer M, McCarthy S, Morris SW, Arnau-Soler A, et al. (2018): DNA sequence-level analyses reveal potential phenotypic modifiers in a large family with psychiatric disorders. Mol Psychiatr 23: 2254–2265. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Howrigan DP, Rose SA, Samocha KE, Fromer M, Cerrato F, Chen WJ, et al. (2020): Exome sequencing in schizophrenia-affected parent-offspring trios reveals risk conferred by protein-coding de novo mutations. Nat Neurosci 1–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Gusev A, Lee SH, Trynka G, Finucane H, Vilhjálmsson BJ, Xu H, et al. (2014): Partitioning heritability of regulatory and cell-type-specific variants across 11 common diseases. Am J Hum Genet 95: 535–52. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Gamazon ER, Segrè AV, Bunt M van de, Wen X, Xi HS, Hormozdiari F, et al. (2018): Using an atlas of gene regulation across 44 human tissues to inform complex disease- and trait-associated variation. Nat Genet 50: 956–967. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Schaub MA, Boyle AP, Kundaje A, Batzoglou S, Snyder M (2012): Linking disease associations with regulatory information in the human genome. Genome Res 22: 1748–1759. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Doolittle WF (2013): Is junk DNA bunk? A critique of ENCODE. P Natl Acad Sci Usa 110: 5294–300. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Eddy SR (2013): The ENCODE project: missteps overshadowing a success. Curr Biology Cb 23: R259–61. [DOI] [PubMed] [Google Scholar]
- 15.Benton ML, Talipineni SC, Kostka D, Capra JA (2019): Genome-wide enhancer annotations differ significantly in genomic distribution, evolution, and function. Bmc Genomics 20: 511. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Kwasnieski JC, Fiore C, Chaudhari HG, Cohen BA (2014): High-throughput functional testing of ENCODE segmentation predictions. Genome Res 24: 1595–1602. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Brown AA, Viñuela A, Delaneau O, Spector TD, Small KS, Dermitzakis ET (2017): Predicting causal variants affecting expression by using whole-genome sequencing and RNA-seq from multiple human tissues. Nat Genet 49: 1747–1751. [DOI] [PubMed] [Google Scholar]
- 18.Kundaje A, Meuleman W, Ernst J, Bilenky M, Yen A, Heravi-Moussavi A, et al. (2015): Integrative analysis of 111 reference human epigenomes. Nature 518: 317–30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.An J-Y, Lin K, Zhu L, Werling DM, Dong S, Brand H, et al. (2018): Genome-wide de novo risk score implicates promoter variation in autism spectrum disorder. Science 362: eaat6576. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Mao F, Wang L, Zhao X, Li Z, Xiao L, Rao RC, et al. (2019): Post-transcriptionally impaired de novo mutations contribute to the genetic etiology of four neuropsychiatric disorders. Biorxiv 175844. [Google Scholar]
- 21.Turner TN, Eichler EE (2018): The Role of De Novo Noncoding Regulatory Mutations in Neurodevelopmental Disorders. Trends Neurosci 42: 115–127. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Satterstrom FK, Kosmicki JA, Wang J, Breen MS, Rubeis SD, An J-Y, et al. (2020): Large-Scale Exome Sequencing Study Implicates Both Developmental and Functional Changes in the Neurobiology of Autism. Cell. 10.1016/j.cell.2019.12.036 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Mayr C (2017): Regulation by 3′-Untranslated Regions. Annu Rev Genet 51: 171–194. [DOI] [PubMed] [Google Scholar]
- 24.Price AJ, Hwang T, Tao R, Burke EE, Rajpurohit A, Shin JH, et al. (2019): Characterizing the nuclear and cytoplasmic transcriptomes in developing and mature human cortex uncovers new insight into psychiatric disease gene regulation. Genome Res 30: 1–11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Kinney JB, Murugan A, Callan CG, Cox EC (2010): Using deep sequencing to characterize the biophysical mechanism of a transcriptional regulatory sequence. P Natl Acad Sci Usa 107: 9158–63. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Patwardhan RP, Lee C, Litvin O, Young DL, Pe’er D, Shendure J (2009): High-resolution analysis of DNA regulatory elements by synthetic saturation mutagenesis. Nat Biotechnol 27: 1173–1175. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Baeza-Centurion P, Miñana B, Schmiedel JM, Valcárcel J, Lehner B (2019): Combinatorial Genetics Reveals a Scaling Law for the Effects of Mutations on Splicing. Cell 176: 549–563.e23. [DOI] [PubMed] [Google Scholar]
- 28.Rosenberg AB, Patwardhan RP, Shendure J, Seelig G (2015): Learning the sequence determinants of alternative splicing from millions of random sequences. Cell 163: 698–711. [DOI] [PubMed] [Google Scholar]
- 29.Wong MS, Kinney JB, Krainer AR (2018): Quantitative Activity Profile and Context Dependence of All Human 5’ Splice Sites. Mol Cell 71: 1012–1026.e3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Safra M, Nir R, Farouq D, Slutzkin IV, Schwartz S (2017): TRUB1 is the predominant pseudouridine synthase acting on mammalian mRNA via a predictable and conserved code. Genome Res 27: 393–406. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Matreyek KA, Starita LM, Stephany JJ, Martin B, Chiasson MA, Gray VE, et al. (2018): Multiplex assessment of protein variant abundance by massively parallel sequencing. Nat Genet 50: 874–882. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Grossman SR, Zhang X, Wang L, Engreitz J, Melnikov A, Rogov P, et al. (2017): Systematic dissection of genomic features determining transcription factor binding and enhancer function. Proc National Acad Sci 114: E1291–E1300. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Gisselbrecht SS, Barrera LA, Porsch M, Aboukhalil A, Estep PW, Vedenko A, et al. (2013): Highly parallel assays of tissue-specific enhancers in whole Drosophila embryos. Nat Methods 10: 774–780. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.King DM, Maricque BB, Cohen BA (2018): Synthetic and genomic regulatory elements reveal aspects of cis regulatory grammar in Mouse Embryonic Stem Cells. Biorxiv 398107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Fiore C, Cohen BA (2016): Interactions between pluripotency factors specify cis-regulation in embryonic stem cells. Genome Res 26: 778–86. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Levo M, Avnit-Sagi T, Lotan-Pompan M, Kalma Y, Weinberger A, Yakhini Z, Segal E (2017): Systematic Investigation of Transcription Factor Activity in the Context of Chromatin Using Massively Parallel Binding and Expression Assays. Mol Cell 65: 604–617.e6. [DOI] [PubMed] [Google Scholar]
- 37.Maricque BB, Dougherty JD, Cohen BA (2016): A genome-integrated massively parallel reporter assay reveals DNA sequence determinants of cis -regulatory activity in neural cells. Nucleic Acids Res 45: gkw942. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.White MA, Myers CA, Corbo JC, Cohen BA (2013): Massively parallel in vivo enhancer assay reveals that highly local features determine the cis-regulatory function of ChIP-seq peaks. Proc National Acad Sci 110: 11952–11957. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Cottrell KA, Chaudhari HG, Cohen BA, Djuranovic S (2018): PTRE-seq reveals mechanism and interactions of RNA binding proteins and miRNAs. Nat Commun 9: 301. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Litterman AJ, Kageyama R, Tonqueze OL, Zhao W, Gagnon JD, Goodarzi H, et al. (2019): A massively parallel 3′ UTR reporter assay reveals relationships between nucleotide content, sequence conservation, and mRNA destabilization. Genome Res 29: 896–906. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Rabani M, Pieper L, Chew G-L, Schier AF (2018): A Massively Parallel Reporter Assay of 3′ UTR Sequences Identifies In Vivo Rules for mRNA Degradation. Mol Cell 70: 565. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Rieger MA, King DM, Cohen BA, Dougherty JD (2018): CLIP-Seq and massively parallel functional analysis of the CELF6 RNA binding protein reveals a role in destabilizing synaptic gene mRNAs through interaction with 3’UTR elements in vivo. Biorxiv 401604. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Sample PJ, Wang B, Reid DW, Presnyak V, McFadyen IJ, Morris DR, Seelig G (2019): Human 5′ UTR design and variant effect prediction from a massively parallel translation assay. Nat Biotechnol 37: 803–809. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Klein JC, Keith A, Rice SJ, Shepherd C, Agarwal V, Loughlin J, Shendure J (2018): Functional Testing of Thousands of Osteoarthritis-Associated Variants for Regulatory Activity. Biorxiv 379727. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Castaldi PJ, Guo F, Qiao D, Du F, Naing ZZC, Li Y, et al. (2019): Identification of Functional Variants in the FAM13A Chronic Obstructive Pulmonary Disease Genome-Wide Association Study Locus by Massively Parallel Reporter Assays. Am J Resp Crit Care 199: 52–61. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Myint L, Avramopoulos DG, Goff LA, Hansen KD (2018): Linear models enable powerful differential activity analysis in massively parallel reporter assays. Biorxiv 196394. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Shen SQ, Myers CA, Hughes AEO, Byrne LC, Flannery JG, Corbo JC (2015): Massively parallel cis -regulatory analysis in the mammalian central nervous system. Genome Res 26: 238–255. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Shen SQ, Kim-Han JS, Cheng L, Xu D, Gokcumen O, Hughes AEO, et al. (2019): A candidate causal variant underlying both higher intelligence and increased risk of bipolar disorder. Biorxiv 580258. [Google Scholar]
- 49.Tewhey R, Kotliar D, Park DS, Liu B, Winnicki S, Reilly SK, et al. (2016): Direct Identification of Hundreds of Expression-Modulating Variants using a Multiplexed Reporter Assay. Cell 165: 1519–1529. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Ulirsch JC, Nandakumar SK, Wang L, Giani FC, Zhang X, Rogov P, et al. (2016): Systematic Functional Dissection of Common Genetic Variation Affecting Red Blood Cell Traits. Cell 165: 1530–1545. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Arnold CD, Gerlach D, Stelzer C, Boryn LM, Rath M, Stark A (2013): Genome-Wide Quantitative Enhancer Activity Maps Identified by STARR-seq. Science 339: 1074–1077. [DOI] [PubMed] [Google Scholar]
- 52.Mogno I, Kwasnieski JC, Cohen BA (2013): Massively parallel synthetic promoter assays reveal the in vivo effects of binding site variants. Genome Res 23: 1908–1915. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Kwasnieski JC, Mogno I, Myers CA, Corbo JC, Cohen BA (2012): Complex effects of nucleotide variants in a mammalian cis-regulatory element. P Natl Acad Sci Usa 109: 19498–503. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Myint L, Wang R, Boukas L, Hansen KD, Goff LA, Avramopoulos D (2019): A screen of 1,049 schizophrenia and 30 Alzheimer’s-associated variants for regulatory potential. Biorxiv 447557. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Dougherty JD, Maloney SE, Wozniak DF, Rieger MA, Sonnenblick L, Coppola G, et al. (2013): The Disruption of Celf6, a Gene Identified by Translational Profiling of Serotonergic Neurons, Results in Autism-Related Behaviors. J Neurosci 33: 2732–2753. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Wagnon JL, Briese M, Sun W, Mahaffey CL, Curk T, Rot G, et al. (2012): CELF4 regulates translation and local abundance of a vast set of mRNAs, including genes associated with regulation of synaptic function. Plos Genet 8: e1003067. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Jia L, Mao Y, Ji Q, Dersh D, Yewdell JW, Qian S-B (2020): Decoding mRNA translatability and stability from 5′UTR. bioRxiv 2020.03.13.990887. [DOI] [PubMed] [Google Scholar]
- 58.(DGT) FC and the RP and C, Forrest ARR, Kawaji H, Rehli M, Baillie JK, Hoon MJL de, et al. (2014): A promoter-level mammalian expression atlas. Nature 507: 462–70. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Aguet F, Brown AA, Castel SE, Davis JR, He Y, Jo B, et al. (2017): Genetic effects on gene expression across human tissues. Nature 550: 204–213. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Dunham I, Kundaje A, Aldred SF, Collins PJ, Davis CA, Doyle F, et al. (2012): An integrated encyclopedia of DNA elements in the human genome. Nature 489: 57–74. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Song M, Pebworth M-P, Yang X, Abnousi A, Fan C, Wen J, et al. (2020): 3D Epigenomic Characterization Reveals Insights Into Gene Regulation and Lineage Specification During Corticogenesis. Biorxiv 2020.02.24.963652. [Google Scholar]
- 62.Song M, Yang X, Ren X, Maliskova L, Li B, Jones IR, et al. (2019): Mapping cis-regulatory chromatin contacts in neural cells links neuropsychiatric disorder risk variants to target genes. Nat Genet 51: 1252–1262. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Nott A, Holtman IR, Coufal NG, Schlachetzki JCM, Yu M, Hu R, et al. (2019): Brain cell type-specific enhancer-promoter interactome maps and disease-risk association. Sci New York N Y 366: 1134–1139. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Blankvoort S, Witter MP, Noonan J, Cotney J, Kentros C (2018): Marked Diversity of Unique Cortical Enhancers Enables Neuron-Specific Tools by Enhancer-Driven Gene Expression. Curr Biol 28: 2103–2114.e5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Nair RR, Blankvoort S, Lagartos MJ, Kentros C (2020): Enhancer-Driven Gene Expression (EDGE) Enables the Generation of Viral Vectors Specific to Neuronal Subtypes. Iscience 23: 100888. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Graybuck LT, Daigle T, Sedeño-Cortés A, Walker M, Kalmbach B, Lenz GH, et al. (2020): Enhancer viruses and a transgenic platform for combinatorial cell subclass-specific labeling. Biorxiv 525014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Hrvatin S, Tzeng CP, Nagy MA, Stroud H, Koutsioumpa C, Wilcox OF, et al. (2019): A scalable platform for the development of cell-type-specific viral drivers. Elife 8: e48089. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Hughes AEO, Myers CA, Corbo JC (2018): A massively parallel reporter assay reveals context-dependent activity of homeodomain binding sites in vivo. Genome Res 28: 1520–1531. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Nguyen TA, Jones RD, Snavely AR, Pfenning AR, Kirchner R, Hemberg M, Gray JM (2016): High-throughput functional comparison of promoter and enhancer activities. Genome Res 26: 1023–33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Inoue F, Kreimer A, Ashuach T, Ahituv N, Yosef N (2019): Identification and Massively Parallel Characterization of Regulatory Elements Driving Neural Induction. Cell Stem Cell. 10.1016/j.stem.2019.09.010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Osterwalder M, Barozzi I, Tissières V, Fukuda-Yuzawa Y, Mannion BJ, Afzal SY, et al. (2018): Enhancer redundancy provides phenotypic robustness in mammalian development. Nature 554: 239–243. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.McClymont SA, Hook PW, Soto AI, Reed X, Law WD, Kerans SJ, et al. (2018): Parkinson-Associated SNCA Enhancer Variants Revealed by Open Chromatin in Mouse Dopamine Neurons. Am J Hum Genet 103: 874–892. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Vockley CM, D’Ippolito AM, McDowell IC, Majoros WH, Safi A, Song L, et al. (2016): Direct GR Binding Sites Potentiate Clusters of TF Binding across the Human Genome. Cell 166: 1269–1281.e19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Liang D, Elwell AL, Aygün N, Lafferty MJ, Krupa O, Cheek KE, et al. (2020): Cell-type specific effects of genetic variation on chromatin accessibility during human neuronal differentiation. Biorxiv 2020.01.13.904862. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Kopp N, Climer S, Dougherty JD (2015): Moving from capstones toward cornerstones: successes and challenges in applying systems biology to identify mechanisms of autism spectrum disorders. Frontiers Genetics 6: 301. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Jaffe AE, Hoeppner DJ, Saito T, Blanpain L, Ukaigwe J, Burke EE, et al. (2020): Profiling gene expression in the human dentate gyrus granule cell layer reveals insights into schizophrenia and its genetic risk. Nat Neurosci 23: 510–519. [DOI] [PubMed] [Google Scholar]
- 77.Shigaki D, Adato O, Adhikar AN, Dong S, Hawkins-Hooker A, Inoue F, et al. (2019): Integration of Multiple Epigenomic Marks Improves Prediction of Variant Impact in Saturation Mutagenesis Reporter Assay. Hum Mutat 40: 1280–1291. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Choi J, Zhang T, Vu A, Ablain J, Makowski MM, Colli LM, et al. (2019): Massively parallel reporter assays combined with cell-type specific eQTL informed multiple melanoma loci and identified a pleiotropic function of HIV-1 restriction gene, MX2 , in melanoma promotion. Biorxiv 625400. [Google Scholar]
- 79.Bourges C, Groff AF, Burren OS, Gerhardinger C, Mattioli K, Hutchinson A, et al. (2020): Resolving mechanisms of immune-mediated disease in primary CD4 T cells. Embo Mol Med 12: e12112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Kvon EZ, Zhu Y, Kelman G, Novak CS, Plajzer-Frick I, Kato M, et al. (2020): Comprehensive In Vivo Interrogation Reveals Phenotypic Impact of Human Enhancer Variants. Cell 180: 1262–1271.e15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Consortium SWG of the PG, Sekar A, Bialas AR, Rivera H de, Davis A, Hammond TR, et al. (2016): Schizophrenia risk from complex variation of complement component 4. Nature 530: 177–183. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Liu W, Li W, Cai X, Yang Z, Li H, Su X, et al. (2020): Identification of a functional human-unique 351-bp Alu insertion polymorphism associated with major depressive disorder in the 1p31.1 GWAS risk loci. Neuropsychopharmacol Official Publ Am Coll Neuropsychopharmacol 1–11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Vockley CM, Guo C, Majoros WH, Nodzenski M, Scholtens DM, Hayes MG, et al. (2015): Massively parallel quantification of the regulatory effects of noncoding genetic variation in a human cohort. Genome Res 25: 1206–1214. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Wang X, He L, Goggin SM, Saadat A, Wang L, Sinnott-Armstrong N, et al. (2018): High-resolution genome-wide functional dissection of transcriptional regulatory regions and nucleotides in human. Nat Commun 9: 5380. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Arensbergen J van, Pagie L, FitzPatrick VD, Haas M de, Baltissen MP, Comoglio F, et al. (2019): High-throughput identification of human SNPs affecting regulatory element activity. Nat Genet 51: 1160–1169. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Klein J, Agarwal V, Inoue F, Keith A, Martin B, Kircher M, et al. (2019): A systematic evaluation of the design, orientation, and sequence context dependencies of massively parallel reporter assays. Biorxiv 2: 576405. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Lee D, Shi M, Moran J, Wall M, Zhang J, Liu J, et al. (2019): STARRPeaker: Uniform processing and accurate identification of STARR-seq active regions. Biorxiv 694869. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Liu Y, Yu S, Dhiman VK, Brunetti T, Eckart H, White KP (2017): Functional assessment of human enhancer activities using whole-genome STARR-sequencing. Genome Biol 18: 219. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Ernst J, Melnikov A, Zhang X, Wang L, Rogov P, Mikkelsen TS, Kellis M (2016): Genome-scale high-resolution mapping of activating and repressive nucleotides in regulatory regions. Nat Biotechnol 34: 1180–1190. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Maricque BB, Chaudhari HG, Cohen BA (2018): A massively parallel reporter assay dissects the influence of chromatin structure on cis-regulatory activity. Nat Biotechnol 37: 90–95. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Madeira C, Rodrigues CAV, Reis MSC, Ferreira FFCG, Correia RESM, Diogo MM, Cabral JMS (2013): Nonviral Gene Delivery to Neural Stem Cells with Minicircles by Microporation. Biomacromolecules 14: 1379–1387. [DOI] [PubMed] [Google Scholar]
- 92.Skene NG, Bryois J, Bakken TE, Breen G, Crowley JJ, Gaspar HA, et al. (2018): Genetic identification of brain cell types underlying schizophrenia. Nat Genet 50: 825–833. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Walker RL, Ramaswami G, Hartl C, Mancuso N, Gandal MJ, Torre-Ubieta L de la, et al. (2019): Genetic Control of Expression and Splicing in Developing Human Brain Informs Disease Mechanisms. Cell 179: 750–771.e22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Uffelmann E, Posthuma D (2020): Emerging Methods and Resources for Biological Interrogation of Neuropsychiatric Polygenic-Signal. Biol Psychiatry 88: XXX–XXX. [DOI] [PubMed] [Google Scholar]
- 95.Uebbing S, Gockley J, Reilly SK, Kocher AA, Geller E, Gandotra N, et al. (2019): Massively parallel discovery of human-specific substitutions that alter neurodevelopmental enhancer activity. Biorxiv 865519. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Doan RN, Bae B-I, Cubelos B, Chang C, Hossain AA, Al-Saad S, et al. (2016): Mutations in Human Accelerated Regions Disrupt Cognition and Social Behavior. Cell 167: 341–354.e12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Ryu H, Inoue F, Whalen S, Williams A, Kircher M, Martin B, et al. (2018): Massively parallel dissection of human accelerated regions in human and chimpanzee neural progenitors. Biorxiv 256313. [Google Scholar]
- 98.Lu X, Chen X, Forney C, Donmez O, Miller D, Parameswaran S, et al. (2020): Genome-wide discovery of SLE genetic risk variant allelic enhancer activity. Biorxiv 2020.01.20.906701. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Dimidschstein J, Chen Q, Tremblay R, Rogers SL, Saldi G-A, Guo L, et al. (2016): A viral strategy for targeting and manipulating interneurons across vertebrate species. Nat Neurosci 19: 1743–1749. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Dickel DE, Barozzi I, Zhu Y, Fukuda-Yuzawa Y, Osterwalder M, Mannion BJ, et al. (2016): Genome-wide compendium and functional assessment of in vivo heart enhancers. Nat Commun 7: 12923. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Dixit A, Parnas O, Li B, Chen J, Fulco CP, Jerby-Arnon L, et al. (2016): Perturb-Seq: Dissecting Molecular Circuits with Scalable Single-Cell RNA Profiling of Pooled Genetic Screens. Cell 167: 1853–1866.e17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Alyagor I, Berkun V, Keren-Shaul H, Marmor-Kollet N, David E, Mayseless O, et al. (2018): Combining Developmental and Perturbation-Seq Uncovers Transcriptional Modules Orchestrating Neuronal Remodeling. Dev Cell 47: 38–52.e6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Jin X, Simmons SK, Guo AX, Shetty AS, Ko M, Nguyen L, et al. (2019): In vivo Perturb-Seq reveals neuronal and glial abnormalities associated with Autism risk genes. Biorxiv 791525. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Geller E, Gockley J, Emera D, Uebbing S, Cotney J, Noonan JP (2019): Massively parallel disruption of enhancers active during human corticogenesis. bioRxiv 852673. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Brandt M, Gokden A, Ziosi M, Lappalainen T (2019): A polyclonal allelic expression assay for detecting regulatory effects of transcript variants. Biorxiv 794081. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Potting C, Crochemore C, Moretti F, Nigsch F, Schmidt I, Manneville C, et al. (2017): Genome-wide CRISPR screen for PARKIN regulators reveals transcriptional repression as a determinant of mitophagy. P Natl Acad Sci Usa 115: E180–E189. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.Tian R, Gachechiladze MA, Ludwig CH, Laurie MT, Hong JY, Nathaniel D, et al. (2019): CRISPR Interference-Based Platform for Multimodal Genetic Screens in Human iPSC-Derived Neurons. Neuron 104: 239–255.e12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Lalli MA, Avey D, Dougherty JD, Milbrandt J, Mitra RD (2019): Multiplexed single-cell autism modeling reveals convergent mechanisms altering neuronal differentiation. Biorxiv 862680. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109.Fulco CP, Nasser J, Jones TR, Munson G, Bergman DT, Subramanian V, et al. (2019): Activity-by-contact model of enhancer-promoter regulation from thousands of CRISPR perturbations. Nat Genet 51: 1664–1669. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110.Amiri A, Coppola G, Scuderi S, Wu F, Roychowdhury T, Liu F, et al. (2018): Transcriptome and epigenome landscape of human cortical development modeled in organoids. Sci New York N Y 362: eaat6720. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111.Ouwenga RL, Dougherty J (2015): Fmrp targets or not: long, highly brain-expressed genes tend to be implicated in autism and brain disorders. Mol Autism 6: 16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112.Pardiñas AF, Holmans P, Pocklington AJ, Escott-Price V, Ripke S, Carrera N, et al. (2018): Common schizophrenia alleles are enriched in mutation-intolerant genes and in regions under strong background selection. Nat Genet 50: 381–389. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113.Schrode N, Ho S-M, Yamamuro K, Dobbyn A, Huckins L, Matos MR, et al. (2019): Synergistic effects of common schizophrenia risk variants. Nat Genet 51: 1475–1485. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114.Leeuw CA de, Neale BM, Heskes T, Posthuma D (2016): The statistical properties of gene-set analysis. Nat Rev Genet 17: 353–364. [DOI] [PubMed] [Google Scholar]
- 115.Sey NYA, Hu B, Mah W, Fauni H, McAfee JC, Rajarajan P, et al. (2020): A computational tool (H-MAGMA) for improved prediction of brain-disorder risk genes by incorporating brain chromatin interaction profiles. Nat Neurosci 1–11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116.Nostrand ELV, Freese P, Pratt GA, Wang X, Wei X, Blue SM, et al. (2018): A Large-Scale Binding and Functional Map of Human RNA Binding Proteins. Biorxiv 179648. [Google Scholar]
- 117.Nostrand ELV, Pratt GA, Yee BA, Wheeler E, Blue SM, Mueller J, et al. (2019): Principles of RNA processing from analysis of enhanced CLIP maps for 150 RNA binding proteins. Biorxiv 807008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118.Gandal MJ, Haney JR, Parikshak NN, Leppa V, Ramaswami G, Hartl C, et al. (2018): Shared molecular neuropathology across major psychiatric disorders parallels polygenic overlap. Sci New York N Y 359: 693–697. [Google Scholar]
- 119.Wang D, Liu S, Warrell J, Won H, Shi X, Navarro FCP, et al. (2018): Comprehensive functional genomic resource and integrative model for the human brain. Science 362: eaat8464. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 120.Consortium TB, Jaffe AE, Straub RE, Shin JH, Tao R, Gao Y, et al. (2018): Developmental and genetic regulation of the human cortex transcriptome illuminate schizophrenia pathogenesis. Nat Neurosci 21: 1117–1125. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 121.Consortium TGte, Ardlie KG, Deluca DS, Segre AV, Sullivan TJ, Young TR, et al. (2015): The Genotype-Tissue Expression (GTEx) pilot analysis: Multitissue gene regulation in humans. Science 348: 648–660. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 122.Fromer M, Roussos P, Sieberts SK, Johnson JS, Kavanagh DH, Perumal TM, et al. (2016): Gene expression elucidates functional impact of polygenic risk for schizophrenia. Nat Neurosci 19: 1442–1453. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 123.Kim-Hellmuth S, Aguet F, Oliva M, Muñoz-Aguirre M, Wucher V, Kasela S, et al. (2019): Cell type specific genetic regulation of gene expression across human tissues. Biorxiv 806117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 124.Lonsdale J, Thomas J, Salvatore M, Phillips R, Lo E, Shad S, et al. (2013): The Genotype-Tissue Expression (GTEx) project. Nat Genet 45: 580–5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 125.Moore JE, Pratt H, Purcaro M, Weng Z (2019): A curated benchmark of enhancer-gene interactions for evaluating enhancer-target gene prediction methods. Biorxiv 745844. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 126.Gandal MJ, Zhang P, Hadjimichael E, Walker RL, Chen C, Liu S, et al. (2018): Transcriptome-wide isoform-level dysregulation in ASD, schizophrenia, and bipolar disorder. Sci New York N Y 362: eaat8127. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 127.Hoffman GE, Bendl J, Voloudakis G, Montgomery KS, Sloofman L, Wang Y-C, et al. (2019): CommonMind Consortium provides transcriptomic and epigenomic data for Schizophrenia and Bipolar Disorder. Sci Data 6: 180. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 128.Li M, Santpere G, Kawasawa YI, Evgrafov OV, Gulden FO, Pochareddy S, et al. (2018): Integrative functional genomic analysis of human brain development and neuropsychiatric risks. Sci New York N Y 362: eaat7615. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 129.Zhou J, Park CY, Theesfeld CL, Wong AK, Yuan Y, Scheckel C, et al. (2019): Whole-genome deep-learning analysis identifies contribution of noncoding mutations to autism risk. Nat Genet 51: 973–980. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 130.Li Y, Shi AH, Tewhey R, Sabeti PC, Ernst J, Kellis M (2017): Genome-wide regulatory model from MPRA data predicts functional regions, eQTLs, and GWAS hits. Biorxiv 110171. [Google Scholar]
- 131.Gusev A, Ko A, Shi H, Bhatia G, Chung W, Penninx BWJH, et al. (2016): Integrative approaches for large-scale transcriptome-wide association studies. Nat Genet 48: 245–52. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 132.Gte Consortium, Barbeira AN Dickinson SP, Bonazzola R, Zheng J, Wheeler HE, et al. (2018): Exploring the phenotypic consequences of tissue specific gene expression variation inferred from GWAS summary statistics. Nat Commun 9: 1825. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 133.Catizone AN, Uzunbas GK, Celadova P, Kuang S, Bose D, Sammons MA (2020): Locally acting transcription factors regulate p53-dependent cis-regulatory element activity. Nucleic Acids Res. 10.1093/nar/gkaa147 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 134.Loo L, Simon JM, Xing L, McCoy ES, Niehaus JK, Guo J, et al. (2019): Single-cell transcriptomic analysis of mouse neocortical development. Nat Commun 10: 134. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 135.Hodge RD, Bakken TE, Miller JA, Smith KA, Barkan ER, Graybuck LT, et al. (2019): Conserved cell types with divergent features in human versus mouse cortex. Nature 573: 61–68. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 136.Zheng Y, Shen W, Zhang J, Yang B, Liu Y-N, Qi H, et al. (2018): CRISPR interference-based specific and efficient gene inactivation in the brain. Nat Neurosci 21: 447–454. [DOI] [PubMed] [Google Scholar]
- 137.Heman-Ackah SM, Bassett AR, Wood MJA (2016): Precision Modulation of Neurodegenerative Disease-Related Gene Expression in Human iPSC-Derived Neurons. Sci Rep-uk 6: 28420. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 138.Ho S-M, Hartley BJ, Flaherty E, Rajarajan P, Abdelaal R, Obiorah I, et al. (2017): Evaluating Synthetic Activation and Repression of Neuropsychiatric-Related Genes in hiPSC-Derived NPCs, Neurons, and Astrocytes. Stem Cell Rep 9: 615–628. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 139.Colasante G, Qiu Y, Massimino L, Berardino CD, Cornford JH, Snowball A, et al. (2020): In vivo CRISPRa decreases seizures and rescues cognitive deficits in a rodent model of epilepsy. Brain J Neurology 143: 891–905. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 140.Zhou H, Liu J, Zhou C, Gao N, Rao Z, Li H, et al. (2018): In vivo simultaneous transcriptional activation of multiple genes in the brain using CRISPR–dCas9-activator transgenic mice. Nat Neurosci 21: 440–446. [DOI] [PubMed] [Google Scholar]
- 141.Lau C-H, Ho JW-T, Lo PK, Tin C (2019): Targeted Transgene Activation in the Brain Tissue by Systemic Delivery of Engineered AAV1 Expressing CRISPRa. Mol Ther Nucleic Acids 16: 637–649. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 142.Rajarajan P, Borrman T, Liao W, Schrode N, Flaherty E, Casiño C, et al. (2018): Neuron-specific signatures in the chromosomal connectome associated with schizophrenia risk. Science 362: eaat4311. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 143.Won H, Huang J, Opland CK, Hartl CL, Geschwind DH (2019): Human evolved regulatory elements modulate genes involved in cortical expansion and neurodevelopmental disease susceptibility. Nat Commun 10: 2396. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 144.Diao Y, Li B, Meng Z, Jung I, Lee AY, Dixon J, et al. (2016): A new class of temporarily phenotypic enhancers identified by CRISPR/Cas9-mediated genetic screening. Genome Res 26: 397–405. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 145.Rajagopal N, Srinivasan S, Kooshesh K, Guo Y, Edwards MD, Banerjee B, et al. (2016): High-throughput mapping of regulatory DNA. Nat Biotechnol 34: 167–174. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 146.Yasuda K, Cline C, Lin YS, Scheib R, Ganguly S, Thirumaran RK, et al. (2015): In Vivo Imaging of Human MDR1 Transcription in the Brain and Spine of MDR1-Luciferase Reporter Mice. Drug Metabolism Dispos Biological Fate Chem 43: 1646–54. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.