

Abstract
Post-transcriptional modifications of pre-mRNAs expand the diversity of proteomes in higher eukaryotes. In the brain, these modifications diversify the functional output of many critical neuronal signal molecules. In this study, we identified a brain-specific A-to-I RNA editing that changed glutamine to arginine (Q/R) at exon 20 and an alternative splicing of exon 4 in Tmem63b, which encodes a ubiquitously expressed osmosensitive cation channel. The channel isoforms lacking exon 4 occurred in ∼80% of Tmem63b mRNAs in the brain but were not detected in other tissues, suggesting a brain-specific splicing. We found that the Q/R editing was catalyzed by Adar2 (Adarb1) and required an editing site complementary sequence located in the proximal 5′ end of intron 20. Moreover, the Q/R editing was almost exclusively identified in the splicing isoform lacking exon 4, indicating a coupling between the editing and the splicing. Elimination of the Q/R editing in brain-specific Adar2 knockout mice did not affect the splicing efficiency of exon 4. Furthermore, transfection with the splicing isoform containing exon 4 suppressed the Q/R editing in primary cultured cerebellar granule neurons. Thus, our study revealed a coupling between an RNA editing and a distant alternative splicing in the Tmem63b pre-mRNA, in which the splicing plays a dominant role. Finally, physiological analysis showed that the splicing and the editing coordinately regulate Ca2+ permeability and osmosensitivity of channel proteins, which may contribute to their functions in the brain.
Keywords: Tmem63b, A-to-I RNA editing, alternative splicing, brain-specific, mechanosensitive, osmosensitive, mechanotransduction, RNA editing, osmotic swelling, ion channel
RNA editing is a post-transcriptional modification of pre-mRNAs that can introduce codon changes in mature mRNAs. The A (adenosine)-to-I (inosine) deamination in pre-mRNAs is the most abundant RNA editing in mammals (1). Because inosine in mRNA is interpreted as guanosine (G) during translation (2), the A-to-I editing often leads to changes in amino acid sequence. The A-to-I editing occurs in important signal molecules, including α-amino-3-hydroxy-5-methyl-4-isoxazolepropionic acid receptor GluA2 subunit (3), kainate receptor GluK1 and GluK2 subunits (4), Kv1.1 α subunit, and 5-HT2C receptors (5, 6), which are known to regulate neuronal development, circuit formation, neuronal degeneration, and synaptic transmission (7, 8, 9, 10). The A-to-I editing is catalyzed by Adar (adenosine deaminases acting on RNA) enzymes, which recognize the dsRNA hairpin structure formed by the editing region and the editing site complementary sequence (ECS) and deaminate the targeted adenosine to inosine (11). In mammals, Adar1 (Adar) and Adar2 (Adarb1) catalyze deamination, whereas Adar3 (Adarb2) has no enzyme activity (1).
Alternative splicing is another type of post-transcriptional modification of pre-mRNAs in eukaryotes (12). Whereas more than 95% of genes undergo alternative splicing in human, around 63% do so in mouse (13, 14). Aberrant alternative splicing is widely observed in various diseases including Mediterranean anemia (15), Alzheimer's disease (16, 17), spinal muscular atrophy (18), ALS (19, 20), and cancers (21). Intriguingly, the A-to-I RNA editing in exons and the alternative splicing of nearby introns are often coupled. For instance, the R/G editing at exon 13 of Gria2 regulates alternative splicing of exon 14 (flop) and exon 15 (flip) (22, 23, 24). The splicing efficiency also regulates nearby upstream editing when it is guided by intronic ECS (25, 26).
We have recently found that Tmem63b serves as an osmosensor in the inner ear and is required for survival of outer hair cells and hearing (27). To expand our understanding of Tmem63b functions in other systems, we cloned Tmem63b mRNA from mouse brain in the current study. We identified four isoforms of Tmem63b resulting from an A-to-I RNA editing that changes glutamine to arginine at exon 20 and an alternative splicing of exon 4. The editing was almost exclusively detected in the spliced isoform lacking exon 4, suggesting a linkage between the two post-transcriptional events. Using Adar2 cKO mice and cultured cerebellar granule neurons (CGNs), we found that the isoform containing exon 4 suppressed the Q/R editing efficiency in a cis manner. Functional analysis demonstrated that the splicing and the editing coordinately regulated hypoosmolarity-induced Ca2+ influx. Together, these results reveal a long-distance coupling between alternative RNA splicing and RNA editing in Tmem63b, in which the splicing plays a dominant role. These post-transcriptional modifications may enable the osmosensitive Tmem63b channel to play diversified roles in the brain.
Results
Q/R editing at exon 20 and alternative splicing of exon 4 in Tmem63b cDNAs
Recently, the osmosensitive cation channels in TMEM63 family have attracted research interests (27, 28, 29). When analyzing Tmem63b cDNAs from mouse brain tissues, we obtained one cDNA sequence that was different from the documented mRNA sequence NM_198167 in the NCBI database (Fig. 1A). The nucleotide c.1856 in this cDNA was guanosine, instead of the published adenine at the same site in exon 20. This finding suggested an A-to-I RNA editing that results in the substitution of glutamine codon CAG to arginine codon CGG at position 619 of the protein sequence (Fig. 1B). In addition, exon 4 was spliced out in the cDNA (Fig. 1C). Thus, the Tmem63b transcript we obtained was a new isoform with two post-transcriptional modifications, i.e. A-to-I editing at c.1856 in exon 20 and alternative splicing of exon 4.
Figure 1.

Q/R editing site at exon 20 and alternative splicing of exon 4 in Tmem63b pre-mRNA.A, domain structure of the mouse Tmem63b protein. The transmembrane domains (M0 to M10) are depicted in blue with linking regions in light blue. The A-to-I editing (red line) is at the intracellular linker adjacent to M7. The alternative splicing exon 4 (green block) encodes amino acids in the first intracellular loop. SP, signal peptide. Scale bar, the size of the line diagram. B, schematic representation of Q/R editing site at exon 20. Lower-left shows the sequence of the Q/R site cloned from mouse brain cDNA. Lower-right shows the cloned sequence (CLONE-1) compared with documented Tmem63b mRNA sequence NM_198167. The corresponding amino acids are shown above and under the nucleotide sequences. C, schematic representation of Tmem63b exons 3–5. The comparison between cloned sequence (CLONE-1) and documented Tmem63b sequence is shown below. The corresponding amino acids are shown above and under the nucleotide sequences.
Q/R editing of Tmem63b varies with brain regions and ages
The conversion of A to G at the 1856 site creates an endonuclease site for BsaWI (Fig. 2A). Specific primer pairs were designed to amplify sequences containing the editing site from mouse genomic DNAs (311 bp) and brain cDNAs (327 bp) (Fig. 2A). The majority of PCR products amplified from brain cDNAs of adult mice were digested by BsaWI and showed two additional bands (208 bp and 119 bp), whereas the PCR products amplified from mouse genomic DNAs were not recognized by BsaWI (Fig. 2B). BsaWI failed to digest Tmem63b cDNA fragments from tissues other than brain, including lung, kidney, heart, liver, and spleen (Fig. 2E), suggesting that this editing is brain-specific. In addition, the editing efficiency varied in brain sub-regions, from ∼40% in the hypothalamus to ∼60% in other brain regions in adult mice (Fig. 2D and Fig. S1, A and C). To investigate whether such editing was affected during the development, we examined the Q/R editing in P7 and embryonic (E12.5 and E18.5) brains and observed lower editing efficiency as compared with the adult brains (Fig. 2C and Fig. S1, B and D). There was virtually no editing in the brains of E12.5 mice, likely because of low expression level of Adar enzymes at this stage (1). Results from different brain regions at P7 were similarly lower than those in the adult brain (Fig. 2D and Fig. S1, A and C).
Figure 2.
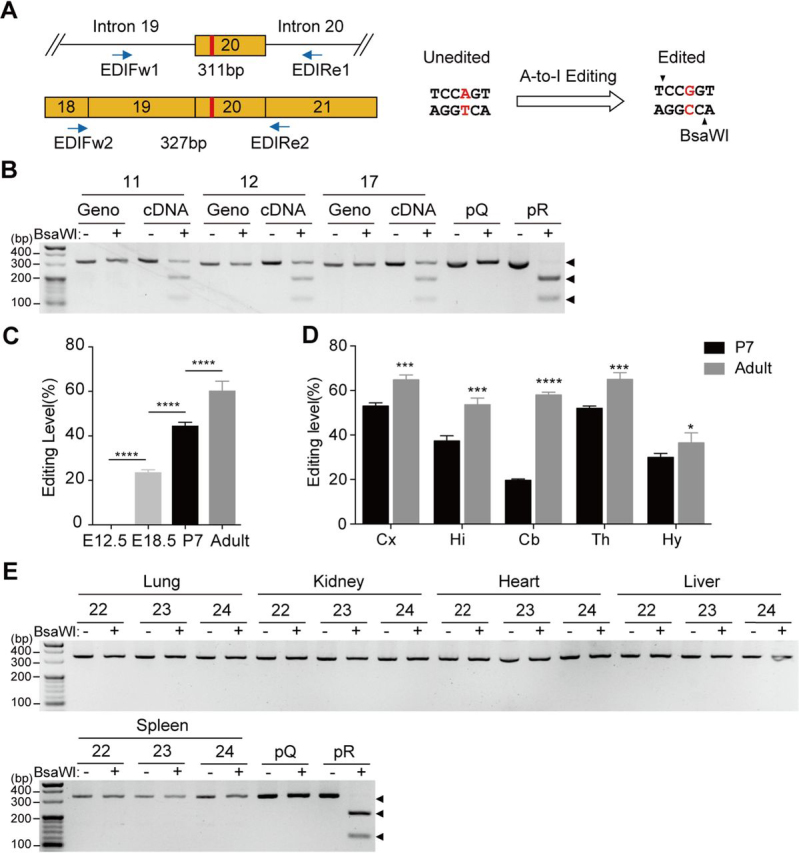
The brain-specific Q/R editing of Tmem63b varies with brain regions and ages.A, schematic demonstration of genomic (upper panel) and cDNA (lower panel) sequence around Tmem63b exon 20 with Q/R editing site marked in red. Primers for amplification are indicated. Q/R editing led to a BsaWI endonuclease recognizing site (right panel). B, Q/R editing of Tmem63b in brains of three adult mice with codes labeled above. The amplified sequences with unedited (Q) and edited (R) forms were indicated by black arrowheads. Plasmids with Q-form (pQ) and R-form (pR) Tmem63b cDNAs served as control to indicate the enzymatic activity of BsaWI. C, quantification of Q/R editing levels in brains of E12.5 (0.0 ± 0.0%, n = 3), E18.5 (23.7 ± 1.2%, n = 3), P7 (44.6 ± 1.5%, n = 5), and adult (60.3 ± 4.2%; n = 6) mice. Data are shown as mean ± S.D. (error bars). ****p < 0.0001; one-way ANOVA, Tukey's post hoc test. D, quantification of Q/R editing levels in cerebral cortex (Cx), hippocampus (Hi), cerebellum (Cb), thalamus (Th), and hypothalamus (Hy) of P7 (black column) and adult (gray column) mice. Data are shown in Table S1 as mean ± S.D. *p < 0.05; ***p < 0.001; ****p < 0.0001; Student's t test, two-tailed. E, Q/R editing of Tmem63b was absent in lungs, kidneys, hearts, livers, and spleens from three adult mice with codes labeled above.
Q/R editing of Tmem63b is catalyzed by Adar2
In mammals, Adar1 (Adar) and Adar2 (Adarb1) catalyze A-to-I editing. To determine whether Adar1 or Adar2 catalyzed the Q/R editing in Tmem63b, we transfected mouse Adar1 or Adar2 cDNA together with a Tmem63b minigene containing the genomic sequence from exon 19 to exon 21 (the editing segment) in HEK293 cells and evaluated the editing efficiency (Fig. 3A). To avoid putative contamination from endogenous Tmem63b in HEK cells, the exogenous RNAs were reverse-transcribed into cDNAs by a primer RTIRES (Internal Ribosome Entry Site) paired to vector sequence (Fig. 3A). Adar2 but not Adar1 transfection resulted in partial digestion of PCR products (403 bp) into two bands (284 bp and 119 bp) by BsaWI (Fig. 3B). To verify that the transfected Adar1 was functional, we coexpressed Adar1 with an Htr2c minigene containing the “A” editing site at exon 5 that is preferentially edited by Adar1 (30) and found that this site was efficiently edited (Fig. S2). To examine whether the Tmem63b Q/R editing was catalyzed by Adar2 in vivo, we generated Adar2 floxed mice (Adar2fl/fl) and bred them with Nestin-cre line (31) to obtain brain-specific Adar2 cKO (Adar2fl/fl;Nestin-Cre) mice (Fig. 3C). The Adar2 cKO mice were postnatally lethal, likely because of inability to edit the Gria2 Q/R site (32). We therefore analyzed the Q/R editing of Tmem63b in P0 mouse brains. The Q/R editing of Tmem63b was completely eliminated in Adar2 cKO brains (Fig. 3, D and E).
Figure 3.
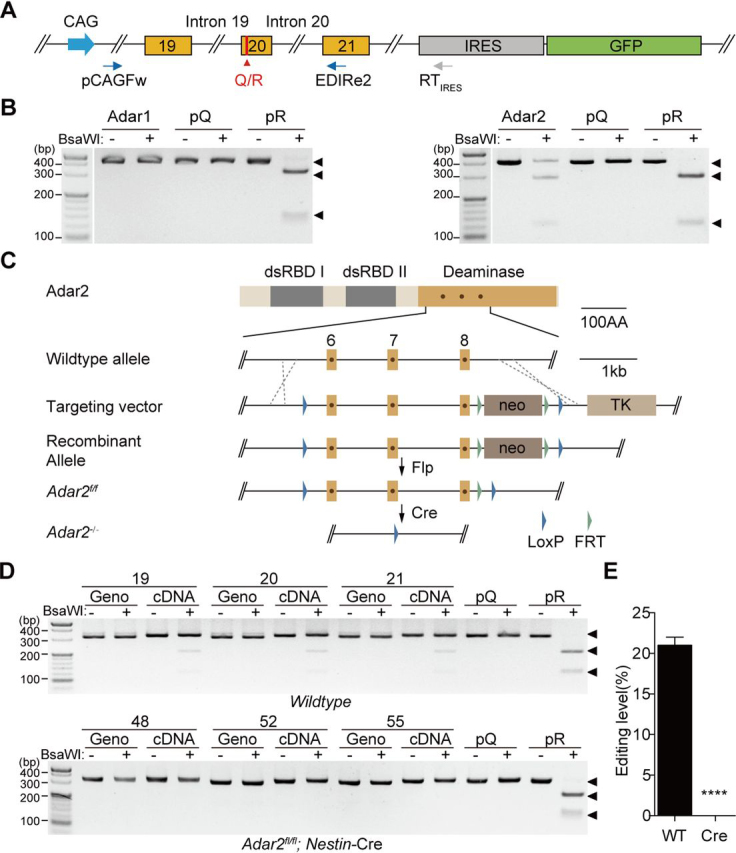
Q/R editing of Tmem63b is catalyzed by Adar2.A, schematic representation of the minigene construct containing the editing segment of Tmem63b. The minigene was driven by CAG promoter and linked with GFP through IRES. pCAGFw and EDIRe2 are primers for amplification. B, Q/R editing of Tmem63b minigene in HEK293 cells expressing Adar1 or Adar2. C, the generation of Adar2 floxed (Adar2fl/fl) mice by homologous recombination techniques. The dots within the targeted exons indicate the three amino acid residues essential for the deaminase activity. dsRBD, dsRNA binding domains. TK, thymidine kinase. neo, neomycin. D, Q/R editing in brains from WT and brain-specific Adar2 knockout P0 mice. The experiments were repeated in three WT mice (labeled as 19, 20, and 21) and three Adar2 cKO mice (labeled as 48, 52, and 55). E, quantification of the Q/R editing levels in brains from WT mice (21.0 ± 1.0%, n = 3) and brain-specific Adar2 knockout mice (0.0 ± 0.0%, n = 3). Data are shown as mean ± S.D. (error bars). ****p < 0.0001; Student's t test, two-tailed. FRT, Flp recognition target.
Analysis of the downstream ECS
To dissect the critical ECS for Q/R editing of Tmem63b, we analyzed the secondary structure of RNA sequence surrounding editing site using the bioinformatics software RNAstructure (33). A dsRNA duplex containing Q/R editing site was obtained, and the sequence base-paired to the editing region was predicted as ECS (in red frame, Fig. 4A). We then introduced a series of mutations on the predicted ECS and analyzed their effects on Q/R editing in Adar2 coexpressed HEK 293 cells. The editing efficiency in WT sequence was 62%, whereas deletion of 14 consecutive nucleotides from the center of ECS abolished Q/R editing (Mut 1, Fig. 4B). In addition, mutations on single or multiple nucleotides that destabilized the dsRNA duplex also decreased the editing efficiency (Mut 2–Mut 9, Fig. 4B), whereas introduction of the compensatory mutations around the editing site restored the editing efficiency (Mut 10–Mut 12, Fig. 4C), indicating the importance of the hairpin structure formed by the intronic ECS and the exonic editing region. Taken together, the intronic sequence at the proximal 5′ end of intron 20 was demonstrated to be the ECS for Tmem63B Q/R editing.
Figure 4.
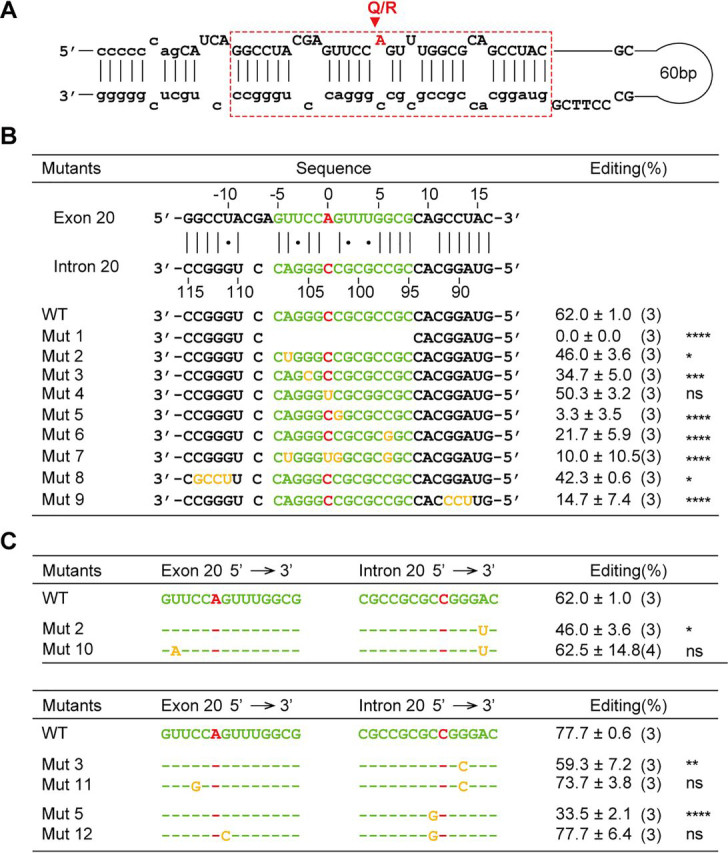
Analysis of downstream ECS essential for Q/R Editing.A, schematic representation of dsRNA hairpin structure formed by the editing region and the ECS, with the ECS marked in red frame and Q/R editing site in red arrowhead. B, Q/R editing of Tmem63b minigenes with mutations on ECS. The extent of Q/R editing was determined as illustrated in Fig. 3A. C, compensatory mutations in the editing region rescue editing efficiency impaired by mutations in ECS. The editing efficiency was quantified by measuring the peak heights ratio at the Q/R site obtained from sequencing chromatograms in the lower table. Data are shown as mean ± S.D. *p < 0.05; **p < 0.01; ***p < 0.001; ****p < 0.0001; ns, not significant; one-way ANOVA, Tukey's post hoc test. The sample numbers of replicates are listed in parentheses.
Alternative splicing of exon 4 is brain-specific
To study alternative splicing of exon 4, a pair of primers, SPFw and SPRe, were designed to amplify the sequence around exon 4 (Fig. 5A). Tmem63b cDNAs with (long-form) or without (short-form) exon 4 were amplified into PCR products of different lengths, 223 bp and 184 bp, respectively. The efficiency of the splicing was calculated as the percentage of short form to total Tmem63b. The short form was detected in brain of adult mouse but not in lung, kidney, liver, and spleen (Fig. 5B). Notably, a weak splicing occurred in about 2% of total heart Tmem63b mRNAs. In the whole brains, ∼80% of the Tmem63b mRNAs were the short form. The splicing efficiency in different brain regions mildly varied from 70 to 80% (Fig. 5D and Fig. S3, A and C). In addition, the splicing efficiency in P7 mouse brains was similar to that in the adult brains, but it was modestly lower in the embryonic brains (E12.5 and E18.5), with the short form accounting for 68% of Tmem63b mRNAs at E12.5. These data demonstrated that the splicing efficiency in Tmem63b pre-mRNA was relatively stable during development (Fig. 5, B and C and Fig. S3, B and D).
Figure 5.

Alternative splicing of Tmem63b exon 4 in mouse brains.A, schematic representation of Tmem63b exons 3–5 in cDNA with exon 4 marked in green. SPFw and SPRe are primers for amplification. B, alternative splicing of Tmem63b exon 4 in brains, lungs, kidneys, hearts, livers, and spleens from three adult mice with codes labeled above. The amplified sequences with long and short forms were indicated by black arrowheads. C, quantification of alternative splicing levels in brains of E12.5 (67.7 ± 0.6%, n = 3), E18.5 (72.0 ± 1.0%, n = 3), P7 (77.0 ± 1.9%, n = 5), and adult (73.0 ± 3.6%, n = 3) mice. Data are shown as mean ± S.D. (error bars) *p < 0.05; ns, not significant; one-way ANOVA, Tukey's post hoc test. D, quantification of alternative splicing levels in cerebral cortex (Cx), hippocampus (Hi), cerebellum (Cb), thalamus (Th), and hypothalamus (Hy) of P7 (black column) and adult (gray column) mice. Data are presented in Table S2 as mean ± S.D. E, left, schematic representation of Tmem63b cDNA sequence from exon 3 to exon 6. Primers for amplification are indicated. Right, quantification of alternative splicing levels in brains of P7 mice by real-time PCR, which were similar to the results of agarose gel electrophoresis. E5, exon 5 (n = 3); E345, exon 3-4-5 without intron interval (18.7 ± 1.6%, n = 3); E35, exon 3-5 without intron interval (77.5 ± 2.6%, n = 3).
Above data suggested that up to ∼80% of Tmem63b mRNAs from mouse brain were short isoforms lacking exon 4. One concern was that the PCR reaction using this pair of primers could prefer one isoform over the other. To perform an independent evaluation on the splicing efficiency, we designed three pairs of primers to amplify the short form, the long form, and the total Tmem63b mRNAs, respectively, and examined the splicing by quantitative real-time PCR (Fig. 5E). The results showed that the long form (exon 3-4-5) and the short form (exon 3-5) were 18.7% and 77.5%, respectively, of the total Tmem63b mRNAs (exon 5) (Fig. 5E). This experiment further verified that around 80% of mRNAs from adult mouse brain were short isoforms. Taken together, the alternative splicing of Tmem63b exon 4 is brain-specific. The splicing efficiency is constant among different brain regions and relatively stable at different development stages.
Distant coupling between alternative splicing and editing in Tmem63b
Previous studies demonstrated that A-to-I editing in exons and splicing of nearby downstream introns could affect each other (22, 23, 24, 25, 26). To test whether the Q/R editing at exon 20 and the splicing of exon 4 in Tmem63b pre-mRNA are also coupled, 40 Tmem63b clones originated from mouse brain were subjected to Sanger sequencing with full length, of which 36 were short cDNA clones lacking exon 4 and the majority of them (33/36) contained the edited R at the Q/R site. In contrast, all four long cDNA clones were not edited at the Q/R site (Fig. 6A). These data suggested that Q/R editing efficiency in long and short isoforms was biased. We further analyzed this phenotype using BsaWI digestion. A pair of primers, with the forward primer base-paired to exon 4 (LFw) and the reverse one downstream of the Q/R site (CRe), was used to amplify the long isoform (2255 bp). The forward primer, with 4 bp of the 3′ end base-paired to exon 5 and the rest of sequence base-paired to exon 3 (SFw), was designed to amplify the short isoform (2246 bp) (Fig. 6B). We found that the fragments amplified from short isoform were largely digested by BsaWI (1593 bp and 653 bp) whereas the digestion bands were not seen in long-form Tmem63b fragments (Fig. 6, C and D). The Q/R editing was also limited to the short-form mRNAs of P7 and E18.5 mouse brains (Fig. S4, B and D), indicating that coupling is also present during development. As expected, the editing efficiency in short form was lower in young brains (Fig. 6E). The editing efficiency in short-form Tmem63b was further analyzed in different brain regions of P7 and adult mice. In adult, the editing was ∼60% in hypothalamus and ∼80% in other brain regions, whereas in P7, the editing in short-form mRNAs was generally lower in all brain regions as compared with those in adult (Fig. 6F and Fig. S4, A and C). To test whether Q/R editing is mutually excluded from the long-form Tmem63b, we subcloned the above fragments amplified from long- or short-form Tmem63b into pMD19-T vectors and subjected them to Sanger sequencing. In 44 clones of the long isoform, 42 contained unedited Q and two clones were edited at the Q/R site. In contrast, 87% (13/15) of the clones amplified from the short isoform were edited to R at the Q/R site (Fig. 6G). Taken together, these results indicated that the alternative splicing of exon 4 and Q/R editing at exon 20 in Tmem63b pre-mRNA were coupled.
Figure 6.

The coupling between exon 4 alternative splicing and Q/R editing in Tmem63b pre-mRNA.A, summary of the sequencing results of full-length Tmem63b cDNAs from mouse brain. B, schematic representation of Tmem63b exons 3–24 in cDNA. Primers for amplification are indicated. C and D, Q/R editing of long-form and short-form Tmem63b in brains from three adult mice with codes labeled above. The amplified sequences with long and short forms are indicated by black arrowheads. Plasmids with Q-form (pQ) and R-form (pR) Tmem63b cDNAs served as control to indicate the enzymatic activity of BsaWI. E, quantification of Q/R editing levels of short-form Tmem63b in brains of E12.5 (0.0 ± 0%, n = 3), E18.5 (31.3 ± 1.5%, n = 3), P7 (67.0 ± 1.2%, n = 5), and adult (81.7 ± 3.9%, n = 6) mice. Data are shown as mean ± S.D. (error bars). ****p < 0.0001; one-way ANOVA, Tukey's post hoc test. F, quantification of Q/R editing levels of short-form Tmem63b in cerebral cortex (Cx), hippocampus (Hi), cerebellum (Cb), thalamus (Th), and hypothalamus (Hy) of P7 (black column) and adult (gray column) mice. Data are presented in Table S3 as mean ± S.D. **p < 0.01; ****p < 0.0001; Student's t test, two-tailed. G, summary of the sequencing results of long-form and short-form Tmem63b from mouse brains.
Presence of exon 4 suppresses Q/R editing in Tmem63b
We then studied which of the post-transcriptional modifications, the editing or the splicing, was dominant. We reasoned that if the editing determined the splicing efficiency as reported previously (22, 23, 24), then the alternative splicing of exon 4 would be absent in Adar2 cKO mice. However, we found that the efficiency of alternative splicing was not changed (Fig. 7, A and B), suggesting that the editing did not affect the splicing.
Figure 7.

Alternative splicing of exon 4 regulates Q/R editing at exon 20.A, alternative splicing of exon 4 in brains of WT and Adar2 cKO mice. B, quantification of alternative splicing levels in brains of WT (78.0 ± 1.0%, n = 3) and Adar2 cKO (78.7 ± 1.5%, n = 3) mice. Data are shown as mean ± S.D. ns, not significant; Student's t test, two-tailed. C, schematic demonstration of the experiments conducted in CGNs. D, schematic demonstration of the intronic segments spliced out in alternative splicing. E and F, Q/R editing efficiencies of the editing segment when cotransfected with I3 (24.3 ± 7.4%, n = 7), I4 (26.9 ± 5.9%, n = 7), I3E4I4 (22.3 ± 4.6%, n = 7), and vehicle control (CTL) (26.9 ± 7.8%, n = 7) in CGNs. Data are shown as mean ± S.D. (error bars). ns, not significant; one-way ANOVA, Tukey's post hoc test. G, schematic demonstration of WT and mutant minigene constructs fuzing the editing segment and splicing segment. E5Fw and EDIRe2 are primers for amplification. H and I, Q/R editing of fusion minigenes in CGNs. The Q/R editing efficiency was not changed in SQi (17.2 ± 3.3%, n = 5) and SpM (17.0 ± 2.4%, n = 5) but was suppressed in LQi (9.8 ± 3.8%, n = 5) and NvM (12.4 ± 2.6%, n = 5) mutant, compared with the LiQi control (17.4 ± 2.1%, n = 5). Lower panel in (H) shows the splicing levels corresponding to the upper editing event. Data are shown as mean ± S.D. (error bars). *p < 0.05; **p < 0.01; one-way ANOVA, Tukey's post hoc test.
To test the possibility that the splicing regulates the editing, we used primary culture of CGNs. We first examined the coupling of splicing and editing of endogenous Tmem63b in these neurons at DIV5, a time point at which we examined the transfected constructs (Fig. 7C). The splicing efficiency (75.7 ± 2.1%, n = 3, Fig. S5A) was close to that in the whole brain of P7 mice (Fig. 5C). The editing efficiency of endogenous Tmem63b in cultured CGNs was 28.7 ± 2.1% (n = 3, Fig. S5B), close to that in the P7 mouse cerebellum (∼20%, Fig. 2D). Importantly, the editing was only detected in short isoforms using BsaWI digestion, with editing efficiency of 37% (Fig. S5, C and D). We then examined the splicing and editing efficiencies of Tmem63b minigenes transfected in cultured CGNs. The editing segment of Tmem63b (Fig. 3A) was efficiently edited (26.9 ± 7.8%, n = 7) when transfected into CGNs (CTL, Fig. 7, D–F). The alternative splicing might modulate the editing through a trans mechanism, where the introns spliced out from long- or short-form Tmem63b regulate the editing, or a cis mechanism, where the mRNA sequence around the splicing region affects the editing. For the long-form Tmem63b, the introns spliced out are intron 3 (I3) and intron 4 (I4), whereas in short form, the intronic segment spliced out is I3E4I4 (Fig. 7D). We found that all these intronic segments had no effects on the editing efficiency (Fig. 7, E and F), arguing against a trans mechanism. Then we constructed minigenes by fusing the editing segment with DNA segments containing exon 3∼5 varied by alternative splicing of exon 4 (LiQi, LQi and SQi, Fig. 7G). The LiQi (genomic sequence from exon 3 to exon 5 linked with the editing segment) was largely (∼65%) spliced to short form (Fig. 7I, lower panel). The editing efficiency in LiQi (17.4 ± 2.1%, n = 5) was close to SQi (exon 3-5 linked with the editing segment, 17.2 ± 3.3%, n = 5) but higher than LQi (exon 3-4-5 linked with the editing segment, 9.8 ± 3.8%, n = 5), indicating that the splicing regulates the editing in a cis manner. We then wondered whether the editing efficiency can be modified by changing splicing. We first made splicing site mutant (SpM), 5′ splice site (GU) at intron 4 and 3′ splice site (AG) at intron 3 mutated to complimentary nucleotides, which disrupts the inclusion of exon 4 (Fig. 7G). As expected, only the short-form transcript was observed. The editing in SpM (17.0 ± 2.4%, n = 5) was the same as SQi (Fig. 7, H and I). In neurons, multiple splicing factors such as Nova (34), Rbfox (35), nSR100 (36), Ptbp1, and Ptbp2 (37) enhance or silence splicing events through binding to specific RNA motifs. There are Nova binding motifs (YCAY clusters) upstream of exon 4 (Fig. 7G) which could be involved in Nova-mediated exon skipping (38). These sites were then simultaneously mutated to obtain Nova binding motif mutant (NvM) (Fig. 7G). As expected, LiQi harboring these mutations significantly reduced the short isoform (∼40% in NvM versus ∼65% in WT LiQi). The editing efficiency was lower in NvM (12.4 ± 2.6%, n = 5) as compared with WT LiQi (Fig. 7, H and I). Taken together, these data indicate that the splicing of exon 4 regulated Q/R editing through a cis mechanism.
The Q/R editing affects Ca2+ permeability of Tmem63b channel
Tmem63 family are osmosensitive (or mechanosensitive) cation channels (27, 39). Their plant orthologue OSCA channels are also osmosensitive and mechanosensitive (39, 40, 41). The Cryo-EM structures of OSCA1.1 and OSCA1.2 have revealed protein domains that are responsible for differential channel properties, including mechanosensation, gating, ion selectivity, and dimer formation (40, 42, 43). To understand whether the editing and splicing affect the channel functions, we made a homology structural model for Tmem63b based on the cryo-EM structure of OSCA1.2 (Fig. 8, A–C) (42). The Tmem63b Q/R site is located at the intracellular mouth of the channel pore consisting of transmembrane helices M3–M7 (Fig. 8, A and B). A charged arginine residue at this position might interfere with the conductance of polyvalent cations, such as Ca2+. To test this possibility, we examined the Ca2+ conductance relative to Na+ for these four isoforms of Tmem63b in Neuro2a (N2a) cells (27). Tmem63b-mediated currents were recorded using a ramp protocol in whole-cell configuration by switching extracellular solution from 300 mOsm/liter to 170 mOsm/liter (Fig. 8, D and E). We recorded the currents using Na-gluconate in both extracellular and pipette solutions; gluconate was used to avoid the contamination from the endogenous Cl− currents (27). The osmolarity was adjusted by addition of mannitol without changing the ionic concentrations. After Tmem63b currents were induced by 170 mOsm/liter Na+ solution and reached the amplitudes of ∼600 pA (measured at −70 mV), the extracellular solution was switched to 170 mOsm/liter Ca2+ solution. Because most reacting cells burst with continuous exposure to hypotonic solution, we did not try to record the currents at plateau in this experiment (27). The reversal potential in Na+ solution was constant for all four isoforms, −2.1 ± 2.1 mV for QL (the long isoform without editing, n = 15), −2.8 ± 2.8 mV for QS (the short isoform without editing, n = 9), −2.7 ± 2.2 mV for RL (the long isoform with the edited R, n = 7), and −2.9 ± 1.7 mV for RS (the short isoform with the edited R, n = 8). The reversal potential in Ca2+ solution was −10.4 ± 4.9 mV for QL (n = 13), −10.1 ± 7.6 mV for QS (n = 9), −23.4 ± 12.6 mV for RL (n = 7), and −20.3 ± 12.4 mV for RS (n = 8) (Fig. 8, F and G). The calculated Ca2+ permeability relative to Na+ (PCa/PNa) was QL (0.64) ∼ QS (0.72) > RL (0.38) ∼ RS (0.45) (Fig. 8H). These data thus demonstrated that Q/R editing at the intracellular mouth of Tmem63b channel reduces Ca2+ permeability.
Figure 8.
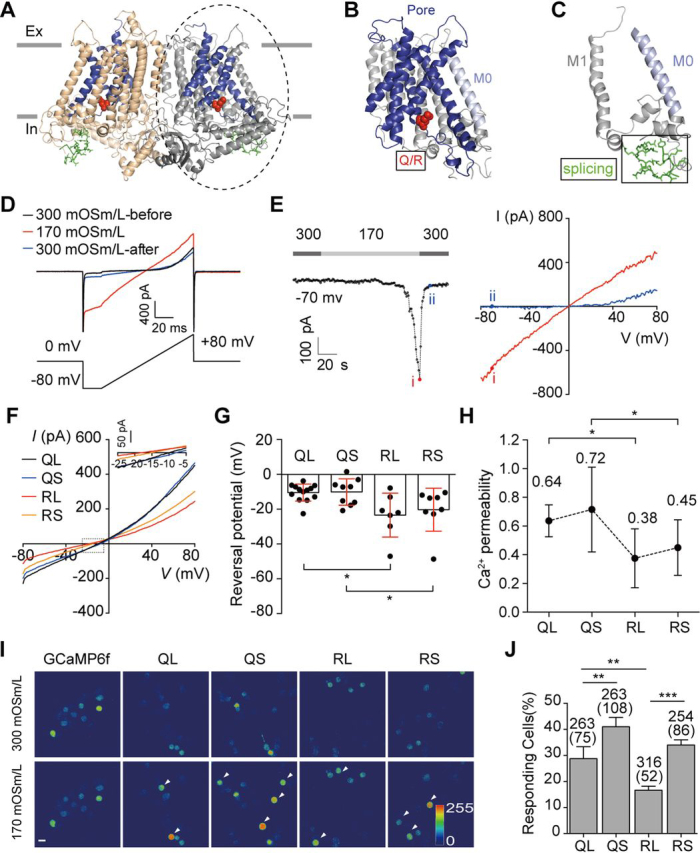
The splicing and the editing in Tmem63b regulate Ca2+ influx in response to hypotonic stimuli.A–C, the 3-dimensional homology model of mouse Tmem63b based on AtOSCA1.2 (PDB ID: 6mjv). The Q/R site (red sphere) is located at the intracellular mouth of channel pore (M3–M7, blue) (B). The exon 4 encoded amino acid in the first intracellular loop (green sticks) (C). D, the raw recording traces of Tmem63b (QL) currents responded to hypotonic stimuli. The cells were holding at 0 mV and recorded in whole-cell configuration by ramp protocol from −80 mV to 80 mV, 100-ms duration, 1 Hz. E, left, hypotonic solution gradually induced Tmem63b (QL) currents (measured at −70 mV). Right, I-V curves at the indicated time during hypotonic (red) to isotonic (blue) solutions. F, the averaged I-V curves after replacing Na+ to Ca2+ in hypotonic perfusion solution. G, the reversal potential of Ca2+ measured by zero-current on I-V plots. QL (−10.4 ± 4.9, n = 13), QS (−10.1 ± 7.6, n = 9), RL (−23.4 ± 12.6, n = 7), RS (−20.3 ± 12.4, n = 8). Data are shown as mean ± S.D. (error bars). *p < 0.05; one-way ANOVA, Tukey's post hoc test. H, the Ca2+ permeability relative to Na+ (PCa/PNa) was QL (0.64) ∼ QS (0.72) > RL (0.38) ∼ RS (0.45). Data are shown as mean ± S.D. (error bars). *p < 0.05; one-way ANOVA, Tukey's post hoc test. I, fluorescent emission images of N2a cells transfected with four Tmem63b isoforms, accompanied with the calcium-sensitive reporter GCaMP6f, in response to 170 mOsm/liter hypotonic stimuli. QL, the long isoform without editing. QS, the short isoform without editing. RL, the long isoform with the edited R. RS, the short isoform with the edited R. White arrowhead marks the responding cells. Scale bar, 10 µm. J, percentage of cells responding to 170 mOsm/liter hypotonic solutions. Numbers of cells tested and responding (in parentheses) are indicated above the bars. QL (28.8 ± 4.6%, n = 4), QS (41.0 ± 3.6%, n = 3), RL (16.7 ± 1.5%, n = 3), RS (34.0 ± 2.0%, n = 3). Data are shown as mean ± S.D. **p < 0.01; ***p < 0.001; ns, not significant; one-way ANOVA, Tukey's post hoc test.
The splicing and editing regulate hypoosmolarity-induced Ca2+ influx
The exon 4 of Tmem63b encoded amino acids in the first intracellular loop 1 between M0 and M1 (Fig. 8, A and C). In AtOSCA1.2, the intracellular segment intracellular loop 2 paralleling plasma membrane is predicted to be involved in mechanosensation (42, 43). We recently demonstrated that Tmem63s are osmosensitive cation channels activated by hypotonic stress and mediate extracellular Ca2+ influx (27). We have established that the most sensitive measurement for the osmosensitivity was the percentage of cells responding to hypotonic stress (27). Interestingly, our measure of osmosensitivity is correlated with the whole-cell currents induced by negative pressure, a direct measure of mechanosensitivity in Tmem63 family proteins (39), indicating that the osmosensitivity reflects the mechanosensitivity. Therefore, we examined the osmosensitivity of the four isoforms of Tmem63b using this established method. In brief, we expressed Tmem63b isoforms accompanied with the calcium reporter GCaMP6f (Tmem63b-P2A-GCaMP6f) in N2a cells (27). GCaMP6f fluorescence was monitored after switching the extracellular osmolarity from 300 mOsm/liter to 170 mOsm/liter. The [Ca2+]i elevation was detected in cells expressing Tmem63b (Fig. 8I), but the ratios of responsive cells varied among four isoforms (Fig. 8J). Control cells that do not express Tmem63b failed to respond to hypotonic stimuli (Fig. 8I; 1/199). Hypoosmolarity-induced Ca2+ influx occurred more frequently in cells expressing the short form of Tmem63b (41% of QS and 34% of RS transfected cells) than in cells expressing the long forms (29% of QL and 17% of RL), indicating that exclusion of exon 4 enhanced the osmosensitivity of Tmem63b channel (Fig. 8J). These data also showed that Q-form Tmem63bs appeared to be more sensitive to osmolarity change than R-forms. Thus, the alternative splicing of exon 4 and the Q/R editing in Tmem63b coordinately regulate Ca2+ influx induced by hypoosmolarity.
Discussion
In this study, we have identified a coupling between the A-to-I editing at the Q/R site of exon 20 and the alternative splicing of exon 4 in Tmem63b pre-mRNAs in the mouse brain. Our study reveals that it displays the following properties. First, the occurrence sites for above post-transcriptional modifications are remote. Second, the alternative RNA splicing plays a dominant role.
In our observation, about 60% of Tmem63b mRNAs are edited at the Q/R site in the adult mouse brain, consistent with a previous study (44). The finding that this RNA editing event is brain-specific is also consistent with those reported recently (1, 45, 46). Furthermore, we demonstrate that the editing efficiencies vary in different brain regions at distinct development stages. Lastly, we show that the editing relies on Adar2, in line with previous observations (1, 46), and that the ECS is localized at the proximal 5′ end of intron 20.
We have identified an alternative splicing of exon 4 in brain-originated Tmem63b mRNAs. The short Tmem63b mRNA isoform lacking exon 4 is the major form in the brain and accounts for about 80% of the total Tmem63b mRNAs. The splicing efficiency is constant in different brain regions and relatively stable at development stages. Like the editing, the alternative splicing also occurs mainly in the brain. Although the short isoform accounts for 2% of the total Tmem63b mRNAs in the heart, it is not observed in other tissues.
The Q/R editing and alternative splicing events in Tmem63b are coupled, in which the alternative splicing of exon 4 plays a dominant role. It has been shown that the exonic A-to-I editing and the splicing of nearby downstream introns often affect each other (22, 23, 24, 25, 26). This could well be explained by interference between the editing machinery and the spliceosomes that may simultaneously act on the adjacent loci on the pre-mRNA. To our knowledge, the interplay between an A-to-I editing and a distant alternative splicing has not been reported before. Our mechanistic analysis reveals that the Q/R editing of Tmem63b requires Adar2 binding to the hairpin structure formed by ECS and the editing region. Genetic deletion of Adar2 abolishes the Q/R editing, and mutations on the ECS impair the editing activity (Fig. 3, C–E and Fig. 4). We suspect that the splicing of exon 4 may regulate the stability of hairpin structure around the editing region or affect Adar2 capability in binding to the structure. Recent evidence has shown that Pin1 promotes the activity of Adar2 and increases the editing efficiencies (47). WWP2 and AIPM2 regulate the degradation of Adar2 to affect RNA editing (1, 47). The splicing factor SRSF9 downregulates Adar2-mediated RNA editing (46, 48). Interestingly, SRSF9 inhibits the Tmem63b Q/R editing in the mouse brain (46). Thus, it is possible that the splicing of exon 4 regulates the Q/R editing through some of the above factors.
We have demonstrated that the exon 4 alternative splicing and the Q/R editing regulate the osmosensitivity of Tmem63b. The Q/R site is located at the inner mouth of the Tmem63b channel pore (Fig. 8, A and B). A positively charged residue at this site reduces the permeability of divalent cation Ca2+ (Fig. 8H). A similar situation happens in α-amino-3-hydroxy-5-methyl-4-isoxazolepropionic acid receptor GluA2 and kainate receptors GluK1 and GluK2, where the Q/R editing occurs at the center of the channel pore and dictates the Ca2+ permeability (4, 49). The splicing site is located in intracellular loop 1 (Fig. 8, A and C). The structure studies on OSCA channels suggest that the α-helices in intracellular loop 2 paralleling membrane may involve mechanodetection and channel gating (42, 43). Our data demonstrated that the short-form Tmem63s are more sensitive to osmotic changes than long forms. The inclusion of exon 4 may interfere with the α-helices in intracellular loop 2 to change the mechanosensitivity of the channel or regulate osmosensitivity through undetermined mechanisms. It is very likely that the overall Ca2+ influx is determined by the Ca2+ permeability of the Q/R site combined with the mechanosensitivity of splicing sequences; thus, the apparent osmosensitivity reading by Ca2+ influx is QS > QL ∼ RS > RL. Future studies are required to dissect physiological functions of Tmem63b isoforms in the brain.
Experimental procedures
Mice
All animal studies were approved by the Institutional Animal Care and Use Committee of Model Animal Research Center of Nanjing University and performed in accordance with guidelines for humane treatment of animals. All animals were housed in 12 h of light and 12 h of dark cycles at 25 ± 1 °C, with water and food obtained ad libitum. The Adar2fl/+ mouse line with loxP site flanked exons 6–8 was generated through the bacterial artificial chromosome homologous recombination in embryonic stem cells at GemPharmatech (Nanjing). The founder mice were backcrossed with C57BL/6J mice for over five generations followed by breeding with Nestin-Cre mice (31) to obtain the brain-specific Adar2 knockout mice (Adar2fl/fl; Nestin-Cre). Genotypes were determined by PCR with the following primers: 5′-AGTCATTCCTCCTAGCCTTTT-3′ (forward) and 5′-TTATCACCTTGGCATCTTTG-3′ (reverse) to confirm the presence of the loxP site, 5′-ATTTGCCTGCATTACCGGTC-3′ (forward) and 5′-ATCAACGTTTTCTTTTCGG-3′ (reverse) for the presence of Cre.
Cloning of Tmem63b, Adar1, and Adar2 from mouse brains
The full-length coding sequences of Tmem63b (NM_198167.3), Adar1 (Adar, NM_019655.3), and Adar2 (Adarb1, NM_001024837.2) were amplified from mouse brain cDNAs, using specific primers (Table S4), by PrimeSTAR® HS DNA Polymerase (Takara Bio, R01A) and subcloned into pCAGGS vectors by Ligation-Free Cloning Kit (abm, E001) (50). For measuring the cytoplasmic calcium concentration, the free calcium indicator GCaMP6f was fused to the C-terminal of Tmem63b through a P2A linker (Tmem63b-P2A-GCaMP6f), leading to the separate expression of Tmem63b and GCaMP6f (27).
Generation of minigene constructs
In the study of A-to-I editing enzyme, the editing segment of Tmem63b and the Htr2c minigene with 211 bp spanning exon 5 and intron 5 were obtained from mouse genomic DNAs by specific primers (Table S4). The nucleotide in the “B” editing site of Htr2c was mutated from the unedited A to the edited G. To explore the ECS, a series of mutations on the editing segment and ECS were obtained through overlapping PCR and corresponding primers (Table S4). In the study of the coupling between alternative splicing of exon 4 and A-to-I editing at the Q/R site, the splicing segments (exon 3∼5) amplified from mouse genomic DNAs and brain cDNAs were fused to the editing segment (LiQi, LQi and SQi) by overlapping PCR and corresponding primers (Table S4). The mutations on SpMs and NvMs were made on the WT LiQi minigene. The intronic segments spliced out from long- or short-form Tmem63b, including I3, I4, and I3E4I4, were obtained from mouse genomic DNAs by specific primers (Table S4). These minigenes were subcloned into pCAGGS vectors by Ligation-Free Cloning Kit (abm, E001).
Cell culture and transfection
HEK 293 cells were cultured in DMEM with 10% FBS (Gibco). The minigenes were transfected into cells using lipofectamine 2000 according to the manufacturer's instruction. The cells were collected for analysis 24–48 h after transfection. CGNs were acutely dissected and cultured following the protocols reported in previous studies (51). In brief, CGNs were prepared from cerebellums of WT mice at P6-8 and plated at a density of 5 × 105 cells/cm2 in 10-cm dishes. The cells were cultured in Basal Medium Eagle–based medium containing 10% FBS, 22 mm KCl, 2% B27, 2 mm l-glutamine, and 1% penicillin-streptomycin (all from Gibco) for 2 days. At DIV 2, primary cultured neurons were transfected with Tmem63b minigenes using lipofectamine 2000, and the medium was replaced with MEM-based medium containing 5 mg/ml d-glucose (Sigma-Aldrich), 1% ITS (insulin-transferrin-sodium selenite) (Sigma-Aldrich), 2 mm l-glutamine, and 1% penicillin-streptomycin. CGNs were collected for analysis 48–72 h after transfection.
RNA isolation and quantitative real-time PCR
For cloning and quantification of mRNA isoforms of Tmem63b in tissues, total RNA samples from mouse tissues were extracted by TRIzol (Invitrogen) and reverse-transcribed into cDNAs using HiScript® 1st Strand cDNA Synthesis Kit (Vazyme Biotech, R111) and oligo(dT)18 primer. For cultured cells transfected with Tmem63b or Htr2c minigenes, the extracted total RNAs were digested by RNase-free DNaseI (Life Technologies, AM2238) at 37 °C for 1 h to remove the endogenous genomic and transfected DNAs. Then the RNAs were reverse-transcribed into cDNAs by specific reverse transcription primer, 5′-GAATGCTCGTCAAGAAGAC-3′ (RTIRES), using HiScript® 1st Strand cDNA Synthesis Kit (Vazyme, R111). Only the RNAs from exogenous constructs could be reverse-transcribed. The RT (minus) controls of each experiment were carried out to assess the amount of DNA contaminations in RNA preparations.
For quantitative real-time PCR, the RNA samples from mouse brains were reverse-transcribed into cDNAs using HiScript Q RT SuperMix for qPCR (+gDNA wiper) Kit (Vazyme Biotech, R123). cDNAs of different Tmem63b isoforms were quantified by Applied Biosystems StepOnePlus Real-Time PCR system (Life Technologies) using AceQ® qPCR SYBR® Green Master (High ROX Premixed) (Vazyme Biotech, Q141) reagents with specific primer pairs: exon 3-4-5, 5′-ACAGATGCAGACAGGCTTC-3′ and 5′-TTGGTCAAAGTCGACAGAGC-3′; exon 3-5, 5′-TGACAGATGCAGACAGTGTG-3′ and 5′-TTGGTCAAAGTCGACAGAGC-3′; exon 5, 5′-ATCAGCTATGCATGGGGAC-3′ and 5′-CCAGGAACAGAAGCCATTG-3′; and Gapdh, 5′-TGAACGGGAAGCTCACTGG-3′ and 5′-TCCACCACCCTGTTGCTGTA-3′.
Quantification of RNA editing
For quantification of Q/R editing in mouse tissues, the genomic sequences spanning the editing site (311 bp) were amplified by primers EDIFw1 (forward) and EDIRe1 (reverse). The cDNA sequences spanning the Q/R site (327 bp) were amplified by primers EDIFw2 (forward) and EDIRe2 (reverse). In the study of A-to-I editing enzyme and ECS in HEK293 cells, the cDNA sequences of transfected Tmem63b minigene (403 bp) were amplified by primers pCAGFw (forward) and EDIRe2 (reverse). The cDNA sequence of transfected Htr2c minigene (340 bp) was amplified by primers pCAGFw (forward) and pCAGRe (reverse). In the experiment to study the coupling between alternative splicing of exon 4 and Q/R editing at exon 20 in mouse brain cDNAs, the primers LFw (forward) and CRe (reverse) were designed to amplify the long-form (2255 bp) Tmem63b, whereas the short-form (2246 bp) Tmem63b fragments were amplified by SFw (forward) and CRe (reverse). In the experiment to explore the regulation of splicing to editing in CGNs, the cDNA sequences of transfected Tmem63b minigene were amplified by following primers: pCAGFw and EDIRe2 for the study of trans regulation, resulting in the PCR a product of 403 bp; and E5Fw (forward) and EDIRe2 (reverse) for the study of cis regulation, resulting in the PCR a product of 334 bp. The primers used in above amplification were shown in Table S4.
For Tmem63b, the amplified fragments were incubated with BsaWI endonuclease at 60 °C for 1 h and resolved by agarose gel electrophoresis. For Htr2c, the amplified fragments were incubated with BtsI endonuclease at 55 °C for 1 h and resolved by agarose gel electrophoresis. The gels were scanned by Gel Imaging System (Tanon). The abundance of each band was analyzed by ImageJ software (National Institutes of Health). The efficiency of editing was displayed by the ratio of integrated densities of BsaWI enzymed bands to control bands. In analysis of ECS, because the BsaWI recognition site is disrupted when the mutations are nearby Q/R site, the editing efficiencies of these mutants were quantified by measuring the ratio of peak heights obtained from sequencing chromatograms.
Quantification of mRNA splicing isoforms
A pair of primers with forward (SPFw) and reverse (SPRe) were designed to amplify the cDNA sequence near the splicing region (Table S4). Tmem63b cDNAs with (long-form) or without (short-from) exon 4 were amplified into different lengths (223 bp and 184 bp), followed by agarose gel electrophoresis. The long (309 bp) and short (270 bp) isoforms from LiQi minigenes were amplified by pCAGFw (forward) and E5Re (reverse), followed by agarose gel electrophoresis (Table S4). The gels were scanned by Gel Imaging System (Tanon). The abundance of each band was analyzed by ImageJ software (National Institutes of Health). The values were normalized according to bands size difference. The efficiency of exon 4 alternative splicing was displayed by the ratio of integrated densities of short form bands to total.
Cytoplasmic Ca2+ measurements
The cytoplasmic calcium concentration was monitored by free calcium indicator GCaMP6f (27). Tmem63b-P2A-GCaMP6f vectors were transfected into N2a cells mounted on the coverslip. GCaMP6f vector was used as a control. 40 h after transfection, the cells were perfused with isotonic extracellular solution (in mm): 65 NaCl, 5 KCl, 1 CaCl2, 1 MgCl2, and 10 HEPES (pH 7.4 adjusted with NaOH; 300 mOsm/liter adjusted with mannitol). The isotonic solution was exchanged to 170 mOsm/liter hypotonic solution without changing the ionic concentrations by a peristaltic pump (BT100-2J, Longer Precision Pump Co., Ltd., Hebei, China) at a constant speed. The osmolarity was measured by a vapor pressure osmometer (Wescor Vapro Model 5600). The cytoplasmic calcium fluorescence was recorded at 1 Hz for 10 min by laser scanning confocal microscope (Leica Microsystems, TCS SP2) at room temperature (24 ± 2 °C) using 488-nm illumination. The change of fluorescence was normalized by the ratio of real-time intensity (Ft) relative to the initial value (F0). The cells with Ft/F0 > 1.5 were considered as positive cells responding to osmolarity changes.
N2a cell electrophysiology
Whole-cell path clamp recordings were performed on transfected N2a cells under ramp protocol. The recording electrodes had the resistance of 8∼10 MΩ when filled with the pipette solution composed of (in mm) 80 Na-gluconate, 130 mannitol, and 10 HEPES (pH 7.4 with NaOH, 300 mOsm/liter). The cells were perfused with isotonic extracellular solution containing (in mm) 80 Na-gluconate, 1 Ca-gluconate, 10 HEPES, and 130 mannitol (pH 7.4 with NaOH, 300 mOsm/liter). The hypotonic extracellular solution without changing the ionic concentration contains (in mm) 80 Na-gluconate, 1 Ca-gluconate, and 10 HEPES (pH 7.4 with NaOH, 170 mOsm/liter). For measuring the Ca2+ permeability, the Na+-rich hypotonic extracellular solution was changed to Ca2+-rich solution containing (in mm) 40 Ca-gluconate, 40 mannitol, and 10 HEPES (pH 7.4 with Ca(OH)2, 170 mOsm/liter). The currents were collected using an Axopatch 200B amplifier and Digidata 1550 digitizer (Molecular Devices) at the sampling rate of 10 kHz and were low-pass filtered at 1 kHz. The current data were analyzed using pClamp 10 software. The cells were holding at 0 mV before application of 100-ms ramp from −80 mV to 80 mV every 1 s. The cells with a membrane resistance below 800 MΩ or series resistance above 10 MΩ were discarded. The Ca2+ permeability relative to Na+ (PCa/PNa) was calculated as PCa/PNa = [Na+]iexp(ΔVrevF/RT) (1+exp(ΔVrevF/RT)) / 4[Ca2+]o, where the ΔVrev is measured the shift in reversal potential, [Na+]i is the intracellular Na+ concentration, and [Ca2+]o is the concentration of the extracellular substituting Ca2+. The reversal potential was measured in 7–13 cells for each isoform.
RNA secondary structure prediction
The secondary structures of RNA around the editing site and the ECS were predicated by RNAstructure software (Version 6.0.1). The sequence 200 bases downstream and upstream of the editing site was imported for analysis. The lowest free energy structure and a set of low free energy structures were predicted according to following restrictions on parameters: Maximum % Energy Difference, Maximum Number of Structures, and Window Size. The parameters were set as previously reported (33). The Maximum % Energy Difference was 10%. The Maximum Number of Structures was 20. The Window Size was 3.
Homology modeling
Protein sequence of mouse Tmem63b was aligned to AtOSCA1.2, the plant homolog of Tmem63b with Cryo-EM structure solved (42, 43). The homology modeling of mouse Tmem63b was obtained using SWISS-MODEL server (53) with the template of AtOSCA1.2 (PDB ID: 6mjv). The model was depicted by PyMol program.
Statistical analysis
All data are presented as mean ± standard deviation (S.D.) in at least three independent experiments. Statistical analyses were performed using GraphPad Prism version 6.0c software and analyzed using one-way analysis of variance (ANOVA) or unpaired t test if not otherwise stated. p values less than 0.05 were considered statistically significant. *p < 0.05; **p < 0.01; ***p < 0.001; ****p < 0.0001. p ≥ 0.05 was denoted as “ns.”
Data availability
All data supporting our conclusions are contained within this article and in the supporting information.
Acknowledgments
We thank all members in Shi lab for helpful discussion.
National Key R&D Program of China (2019YFA0801603) to Yun Stone Shi
National Natural Science Foundation of China (NSFC) (91849112) to Yun Stone Shi
National Natural Science Foundation of China (NSFC) (31571060) to Yun Stone Shi
Natural Science Foundation of Jiangsu Province (Jiangsu Natural Science Foundation) (BK20140018) to Yun Stone Shi
Natural Science Foundation of Jiangsu Province (Jiangsu Natural Science Foundation) (BE2019707) to Yun Stone Shi
MOE | Fundamental Research Funds for the Central Universities (Fundamental Research Fund for the Central Universities) (0903-14380029) to Yun Stone Shi
Edited by Ronald C. Wek
Footnotes
This article contains supporting information.
Author contributions—D. W., Y.-Y. Z., Y.-Y. S., C. Y., W.-M. C., and X.-H. T. investigation; D. W. writing-original draft; L. Z., Y. L., Z. G., and G.-q, C. resources; Y. X., J.-J. Y., and Y. S. S. supervision; J.-J. Y. and Y. S. S. conceptualization; Y. S. S. funding acquisition; Y. S. S. writing-review and editing.
Funding and additional information—This work is supported by the National Key R&D Program of China Grant 2019YFA0801603 (to Y. S. S), the National Natural Science Foundation of China Grants 91849112 and 31571060 (to Y. S. S.), the Natural Science Foundation of Jiangsu Province Grants BK20140018 and BE2019707 (to Y. S. S.), and the Fundamental Research Funds for the Central Universities Grant 0903-14380029 (to Y. S. S.).
- Adar
- adenosine deaminases acting on RNA
- ECS
- editing site complementary sequence
- CGN
- cerebellar granule neurons
- I3
- intron 3
- I4
- intron 4
- E4
- extron 4
- SpM
- splicing site motif
- NvM
- Nova binding motif
- N2a
- Neuro2a
- ANOVA
- analysis of variance.
Contributor Information
Jian-Jun Yang, Email: yjyangjj@126.com.
Yun Stone Shi, Email: yunshi@nju.edu.cn.
Supplementary Material
References
- 1.Tan M.H., Li Q., Shanmugam R., Piskol R., Kohler J., Young A.N., Liu K.I., Zhang R., Ramaswami G., Ariyoshi K., Gupte A., Keegan L.P., George C.X., Ramu A., Huang N. Dynamic landscape and regulation of RNA editing in mammals. Nature. 2017;550:249–254. doi: 10.1038/nature24041. 29022589. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Basilio C., Wahba A.J., Lengyel P., Speyer J.F., Ochoa S. Synthetic polynucleotides and the amino acid code. V. Proc. Natl. Acad. Sci. U. S. A. 1962;48:613–616. doi: 10.1073/pnas.48.4.613. 13865603. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Sommer B., Köhler M., Sprengel R., Seeburg P.H. RNA editing in brain controls a determinant of ion flow in glutamate-gated channels. Cell. 1991;67:11–19. doi: 10.1016/0092-8674(91)90568-J. 1717158. [DOI] [PubMed] [Google Scholar]
- 4.Köhler M., Burnashev N., Sakmann B., Seeburg P.H. Determinants of Ca2+ permeability in both TM1 and TM2 of high affinity kainate receptor channels: diversity by RNA editing. Neuron. 1993;10:491–500. doi: 10.1016/0896-6273(93)90336-P. 7681676. [DOI] [PubMed] [Google Scholar]
- 5.Hoopengardner B., Bhalla T., Staber C., Reenan R. Nervous system targets of RNA editing identified by comparative genomics. Science. 2003;301:832–836. doi: 10.1126/science.1086763. 12907802. [DOI] [PubMed] [Google Scholar]
- 6.Burns C.M., Chu H., Rueter S.M., Hutchinson L.K., Canton H., Sanders-Bush E., Emeson R.B. Regulation of serotonin-2C receptor G-protein coupling by RNA editing. Nature. 1997;387:303–308. doi: 10.1038/387303a0. 9153397. [DOI] [PubMed] [Google Scholar]
- 7.Behm M., Öhman M. RNA editing: A contributor to neuronal dynamics in the mammalian brain. Trends Genet. 2016;32:165–175. doi: 10.1016/j.tig.2015.12.005. 26803450. [DOI] [PubMed] [Google Scholar]
- 8.Liu S.J., Zukin R.S. Ca2+-permeable AMPA receptors in synaptic plasticity and neuronal death. Trends Neurosci. 2007;30:126–134. doi: 10.1016/j.tins.2007.01.006. 17275103. [DOI] [PubMed] [Google Scholar]
- 9.Whitney N.P., Peng H., Erdmann N.B., Tian C., Monaghan D.T., Zheng J.C. Calcium-permeable AMPA receptors containing Q/R-unedited GluR2 direct human neural progenitor cell differentiation to neurons. FASEB J. 2008;22:2888–2900. doi: 10.1096/fj.07-104661. 18403631. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Peng P.L., Zhong X., Tu W., Soundarapandian M.M., Molner P., Zhu D., Lau L., Liu S., Liu F., Lu Y. ADAR2-dependent RNA editing of AMPA receptor subunit GluR2 determines vulnerability of neurons in forebrain ischemia. Neuron. 2006;49:719–733. doi: 10.1016/j.neuron.2006.01.025. 16504947. [DOI] [PubMed] [Google Scholar]
- 11.Higuchi M., Single F.N., Köhler M., Sommer B., Sprengel R., Seeburg P.H. RNA editing of AMPA receptor subunit GluR-B: a base-paired intron-exon structure determines position and efficiency. Cell. 1993;75:1361–1370. doi: 10.1016/0092-8674(93)90622-W. 8269514. [DOI] [PubMed] [Google Scholar]
- 12.Nilsen T.W., Graveley B.R. Expansion of the eukaryotic proteome by alternative splicing. Nature. 2010;463:457–463. doi: 10.1038/nature08909. 20110989. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Pan Q., Shai O., Lee L.J., Frey B.J., Blencowe B.J. Deep surveying of alternative splicing complexity in the human transcriptome by high-throughput sequencing. Nat. Genet. 2008;40:1413–1415. doi: 10.1038/ng.259. 18978789. [DOI] [PubMed] [Google Scholar]
- 14.Wang E.T., Sandberg R., Luo S., Khrebtukova I., Zhang L., Mayr C., Kingsmore S.F., Schroth G.P., Burge C.B. Alternative isoform regulation in human tissue transcriptomes. Nature. 2008;456:470–476. doi: 10.1038/nature07509. 18978772. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Treisman R., Orkin S.H., Maniatis T. Specific transcription and RNA splicing defects in five cloned β-thalassaemia genes. Nature. 1983;302:591–596. doi: 10.1038/302591a0. 6188062. [DOI] [PubMed] [Google Scholar]
- 16.Rockenstein E.M., McConlogue L., Tan H., Power M., Masliah E., Mucke L. Levels and alternative splicing of amyloid beta protein precursor (APP) transcripts in brains of APP transgenic mice and humans with Alzheimer's disease. J. Biol. Chem. 1995;270:28257–28267. doi: 10.1074/jbc.270.47.28257. 7499323. [DOI] [PubMed] [Google Scholar]
- 17.Raj T., Li Y.I., Wong G., Humphrey J., Wang M., Ramdhani S., Wang Y.C., Ng B., Gupta I., Haroutunian V., Schadt E.E., Young-Pearse T., Mostafavi S., Zhang B., Sklar P. Integrative transcriptome analyses of the aging brain implicate altered splicing in Alzheimer's disease susceptibility. Nat. Genet. 2018;50:1584–1592. doi: 10.1038/s41588-018-0238-1. 30297968. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Kashima T., Manley J.L. A negative element in SMN2 exon 7 inhibits splicing in spinal muscular atrophy. Nat. Genet. 2003;34:460–463. doi: 10.1038/ng1207. 12833158. [DOI] [PubMed] [Google Scholar]
- 19.Kim H.J., Kim N.C., Wang Y.D., Scarborough E.A., Moore J., Diaz Z., MacLea K.S., Freibaum B., Li S., Molliex A., Kanagaraj A.P., Carter R., Boylan K.B., Wojtas A.M., Rademakers R. Mutations in prion-like domains in hnRNPA2B1 and hnRNPA1 cause multisystem proteinopathy and ALS. Nature. 2013;495:467–473. doi: 10.1038/nature11922. 23455423. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Perrone B., La Cognata V., Sprovieri T., Ungaro C., Conforti F.L., Andò S., Cavallaro S. Alternative splicing of ALS genes: Misregulation and potential therapies. Cell. Mol. Neurobiol. 2020;40:1–14. doi: 10.1007/s10571-019-00717-0. 31385134. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Song X., Zeng Z., Wei H., Wang Z. Alternative splicing in cancers: From aberrant regulation to new therapeutics. Semin. Cell Dev. Biol. 2018;75:13–22. doi: 10.1016/j.semcdb.2017.09.018. 28919308. [DOI] [PubMed] [Google Scholar]
- 22.Penn A.C., Balik A., Greger I.H. Steric antisense inhibition of AMPA receptor Q/R editing reveals tight coupling to intronic editing sites and splicing. Nucleic Acids Res. 2013;41:1113–1123. doi: 10.1093/nar/gks1044. 23172291. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Schoft V.K., Schopoff S., Jantsch M.F. Regulation of glutamate receptor B pre-mRNA splicing by RNA editing. Nucleic Acids Res. 2007;35:3723–3732. doi: 10.1093/nar/gkm314. 17517775. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Bratt E., Öhman M. Coordination of editing and splicing of glutamate receptor pre-mRNA. RNA. 2003;9:309–318. doi: 10.1261/rna.2750803. 12592005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Licht K., Kapoor U., Mayrhofer E., Jantsch M.F. Adenosine to inosine editing frequency controlled by splicing efficiency. Nucleic Acids Res. 2016;44:6398–6408. doi: 10.1093/nar/gkw325. 27112566. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Licht K., Kapoor U., Amman F., Picardi E., Martin D., Bajad P., Jantsch M.F. A high resolution A-to-I editing map in the mouse identifies editing events controlled by pre-mRNA splicing. Genome Res. 2019;29:1453–1463. doi: 10.1101/gr.242636.118. 31427386. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Du H., Ye C., Wu D., Zang Y.Y., Zhang L., Chen C., He X.Y., Yang J.J., Hu P., Xu Z., Wan G., Shi Y.S. The cation channel TMEM63B is an osmosensor required for hearing. Cell Rep. 2020;31 doi: 10.1016/j.celrep.2020.107596. 32375046. [DOI] [PubMed] [Google Scholar]
- 28.Schulz A., Müller N.V., van de Lest N.A., Eisenreich A., Schmidbauer M., Barysenka A., Purfürst B., Sporbert A., Lorenzen T., Meyer A.M., Herlan L., Witten A., Ruhle F., Zhou W., de Heer E. Analysis of the genomic architecture of a complex trait locus in hypertensive rat models links Tmem63c to kidney damage. eLife. 2019;8 doi: 10.7554/eLife.42068. 30900988. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Yan H., Helman G., Murthy S.E., Ji H., Crawford J., Kubisiak T., Bent S.J., Xiao J., Taft R.J., Coombs A., Wu Y., Pop A., Li D., de Vries L.S., Jiang Y. Heterozygous variants in the mechanosensitive ion channel TMEM63A result in transient hypomyelination during infancy. Am. J. Hum. Genet. 2019;105:996–1004. doi: 10.1016/j.ajhg.2019.09.011. 31587869. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Fukuda M., Oyama Y., Nishitarumizu A., Omura M., Nose K., Deshimaru M. Identification of an RNA element for specific coordination of A-to-I RNA editing on HTR2C pre-mRNA. Genes Cells. 2015;20:834–846. doi: 10.1111/gtc.12272. 26259820. [DOI] [PubMed] [Google Scholar]
- 31.Tronche F., Kellendonk C., Kretz O., Gass P., Anlag K., Orban P.C., Bock R., Klein R., Schütz G. Disruption of the glucocorticoid receptor gene in the nervous system results in reduced anxiety. Nat. Genet. 1999;23:99–103. doi: 10.1038/12703. 10471508. [DOI] [PubMed] [Google Scholar]
- 32.Higuchi M., Maas S., Single F.N., Hartner J., Rozov A., Burnashev N., Feldmeyer D., Sprengel R., Seeburg P.H. Point mutation in an AMPA receptor gene rescues lethality in mice deficient in the RNA-editing enzyme ADAR2. Nature. 2000;406:78–81. doi: 10.1038/35017558. 10894545. [DOI] [PubMed] [Google Scholar]
- 33.Reuter J.S., Mathews D.H. RNAstructure: software for RNA secondary structure prediction and analysis. BMC Bioinformatics. 2010;11:129. doi: 10.1186/1471-2105-11-129. 20230624. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Ule J., Ule A., Spencer J., Williams A., Hu J.S., Cline M., Wang H., Clark T., Fraser C., Ruggiu M., Zeeberg B.R., Kane D., Weinstein J.N., Blume J., Darnell R.B. Nova regulates brain-specific splicing to shape the synapse. Nat. Genet. 2005;37:844–852. doi: 10.1038/ng1610. 16041372. [DOI] [PubMed] [Google Scholar]
- 35.Lovci M.T., Ghanem D., Marr H., Arnold J., Gee S., Parra M., Liang T.Y., Stark T.J., Gehman L.T., Hoon S., Massirer K.B., Pratt G.A., Black D.L., Gray J.W., Conboy J.G. Rbfox proteins regulate alternative mRNA splicing through evolutionarily conserved RNA bridges. Nat. Struct. Mol. Biol. 2013;20:1434–1442. doi: 10.1038/nsmb.2699. 24213538. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Calarco J.A., Superina S., O'Hanlon D., Gabut M., Raj B., Pan Q., Skalska U., Clarke L., Gelinas D., van der Kooy D., Zhen M., Ciruna B., Blencowe B.J. Regulation of vertebrate nervous system alternative splicing and development by an SR-related protein. Cell. 2009;138:898–910. doi: 10.1016/j.cell.2009.06.012. 19737518. [DOI] [PubMed] [Google Scholar]
- 37.Boutz P.L., Stoilov P., Li Q., Lin C.H., Chawla G., Ostrow K., Shiue L., Ares M., Black D.L. A post-transcriptional regulatory switch in polypyrimidine tract-binding proteins reprograms alternative splicing in developing neurons. Genes Dev. 2007;21:1636–1652. doi: 10.1101/gad.1558107. 17606642. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Ule J., Stefani G., Mele A., Ruggiu M., Wang X., Taneri B., Gaasterland T., Blencowe B.J., Darnell R.B. An RNA map predicting Nova-dependent splicing regulation. Nature. 2006;444:580–586. doi: 10.1038/nature05304. 17065982. [DOI] [PubMed] [Google Scholar]
- 39.Murthy S.E., Dubin A.E., Whitwam T., Jojoa-Cruz S., Cahalan S.M., Mousavi S.A.R., Ward A.B., Patapoutian A. OSCA/TMEM63 are an evolutionarily conserved family of mechanically activated ion channels. eLife. 2018;7 doi: 10.7554/eLife.41844. 30382938. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Zhang M., Wang D., Kang Y., Wu J.X., Yao F., Pan C., Yan Z., Song C., Chen L. Structure of the mechanosensitive OSCA channels. Nat. Struct. Mol. Biol. 2018;25:850–858. doi: 10.1038/s41594-018-0117-6. 30190597. [DOI] [PubMed] [Google Scholar]
- 41.Yuan F., Yang H., Xue Y., Kong D., Ye R., Li C., Zhang J., Theprungsirikul L., Shrift T., Krichilsky B., Johnson D.M., Swift G.B., He Y., Siedow J.N., Pei Z.M. OSCA1 mediates osmotic-stress-evoked Ca2+ increases vital for osmosensing in Arabidopsis. Nature. 2014;514:367–371. doi: 10.1038/nature13593. 25162526. [DOI] [PubMed] [Google Scholar]
- 42.Jojoa-Cruz S., Saotome K., Murthy S.E., Tsui C.C.A., Sansom M.S., Patapoutian A., Ward A.B. Cryo-EM structure of the mechanically activated ion channel OSCA1.2. eLife. 2018;7 doi: 10.7554/eLife.41845. 30382939. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Liu X., Wang J., Sun L. Structure of the hyperosmolality-gated calcium-permeable channel OSCA1.2. Nat. Commun. 2018;9 doi: 10.1038/s41467-018-07564-5. 30498218. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Danecek P., Nellåker C., McIntyre R.E., Buendia-Buendia J.E., Bumpstead S., Ponting C.P., Flint J., Durbin R., Keane T.M., Adams D.J. High levels of RNA-editing site conservation amongst 15 laboratory mouse strains. Genome Biol. 2012;13:26. doi: 10.1186/gb-2012-13-4-r26. 22524474. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Gal-Mark N., Shallev L., Sweetat S., Barak M., Billy Li J., Levanon E.Y., Eisenberg E., Behar O. Abnormalities in A-to-I RNA editing patterns in CNS injuries correlate with dynamic changes in cell type composition. Sci. Rep. 2017;7 doi: 10.1038/srep43421. 28266523. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Shanmugam R., Zhang F., Srinivasan H., Charles Richard J.L., Liu K.I., Zhang X., Woo C.W.A., Chua Z.H.M., Buschdorf J.P., Meaney M.J., Tan M.H. SRSF9 selectively represses ADAR2-mediated editing of brain-specific sites in primates. Nucleic Acids Res. 2018;46:7379–7395. doi: 10.1093/nar/gky615. 29992293. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Marcucci R., Brindle J., Paro S., Casadio A., Hempel S., Morrice N., Bisso A., Keegan L.P., Del Sal G., O'Connell M.A. Pin1 and WWP2 regulate GluR2 Q/R site RNA editing by ADAR2 with opposing effects. EMBO J. 2011;30:4211–4222. doi: 10.1038/emboj.2011.303. 21847096. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Huang H., Kapeli K., Jin W., Wong Y.P., Arumugam T.V., Koh J.H., Srimasorn S., Mallilankaraman K., Chua J.J.E., Yeo G.W., Soong T.W. Tissue-selective restriction of RNA editing of CaV1.3 by splicing factor SRSF9. Nucleic Acids Res. 2018;46:7323–7338. doi: 10.1093/nar/gky348. 29733375. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Burnashev N., Monyer H., Seeburg P.H., Sakmann B. Divalent ion permeability of ampa receptor channels is dominated by the edited form of a single subunit. Neuron. 1992;8:189–198. doi: 10.1016/0896-6273(92)90120-3. 1370372. [DOI] [PubMed] [Google Scholar]
- 50.Duan G.F., Ye Y., Xu S., Tao W., Zhao S., Jin T., Nicoll R.A., Shi Y.S., Sheng N. Signal peptide represses GluK1 surface and synaptic trafficking through binding to amino-terminal domain. Nat. Commun. 2018;9 doi: 10.1038/s41467-018-07403-7. 30451858. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Shi Y., Suh Y.H., Milstein A.D., Isozaki K., Schmid S.M., Roche K.W., Nicoll R.A. Functional comparison of the effects of TARPs and cornichons on AMPA receptor trafficking and gating. Proc. Natl. Acad. Sci. U. S. A. 2010;107:16315–16319. doi: 10.1073/pnas.1011706107. 20805473. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Bordoli L., Kiefer F., Arnold K., Benkert P., Battey J., Schwede T. Protein structure homology modeling using SWISS-MODEL workspace. Nat. Protoc. 2009;4:1–13. doi: 10.1038/nprot.2008.197. 19131951. [DOI] [PubMed] [Google Scholar]
Uncited reference
- 52.Eggington J.M., Greene T., Bass B.L. Predicting sites of ADAR editing in double-stranded RNA. Nat. Commun. 2011;2:319. doi: 10.1038/ncomms1324. 21587236. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
All data supporting our conclusions are contained within this article and in the supporting information.