
Figure 1. Putative molecular mechanisms contributing to abnormal Ca2+ handling associated with atrial fibrillation (AF).
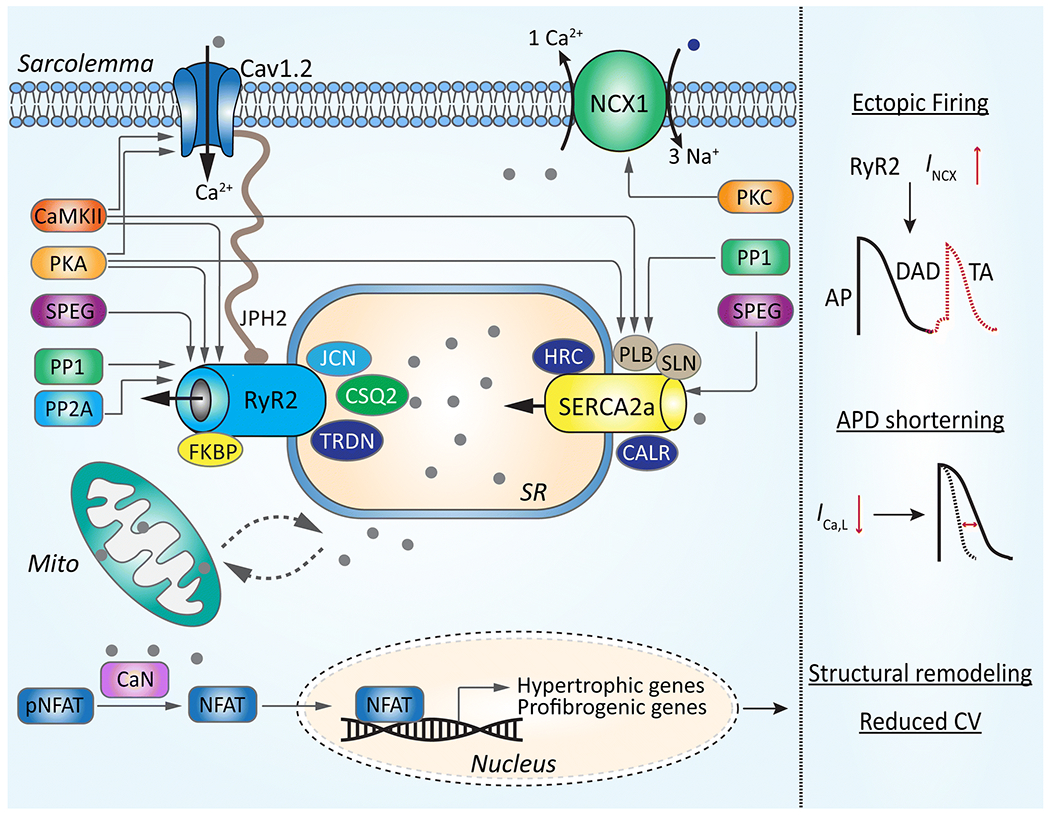
Focal ectopic firing due to the delayed afterdepolarization (DAD)-induced triggered activity (TA), APD shortening, and atrial enlargement as a result of the upregulation of hypertrophic and profibrogenic genes are key events associated with dysregulated Ca2+ handling in atrial cardiomyocytes during AF. DAD-inducing sarcoplasmic reticulum (SR) Ca2+ release (SR Ca2+ leak) is higher due to the enhanced activity of ryanodine receptor type-2 (RyR2). The latter could be a consequence of an impaired interaction with FKBP (FK506-binding protein 12.6) or junctophilin-2 (JPH2), altered phosphorylation by CaMKII, PKA, and SPEG, or a combination of both. The SERCA2a-mediated SR Ca2+ uptake could be increased in pAF because of a reduction of sarcolipin (SLN) or a hyperphosphorylation of phospholamban (PLB). In cAF, the hyperphosphorylation of PLB is associated with the enhanced Inhibitor-I (I-1) mediated inhibition of protein phosphatase type-1 (PP1). These alterations lead to the enhanced SR Ca2+ leak, which can activate the Na+/Ca2+ exchanger 1 (NCX1) and promote DADs. The shortening of APD is partially due to the reduced level of Cav1.2 (α-subunit of L-type Ca2+ channel, LTCC) or increased LTCC dephosphorylation due to enhanced activity of PP1 and PP2A. Together with structural remodeling involving Ca2+-induced calcineurin (CaN)-mediated activation of NFAT (nuclear factor of activated T-cell), a transcription factor associated with the hypertrophy and fibrosis, AP shortening promote AF-maintaining reentry.