

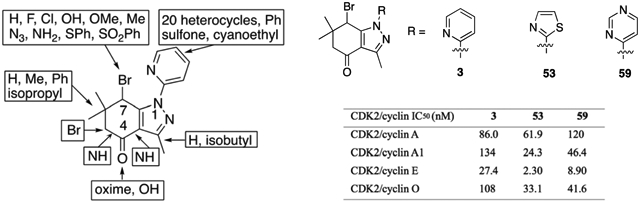
Abstract
Over 50 tetrahydroindazoles were synthesized after 7-bromo-3,6,6-trimethyl-1-(pyridin-2-yl)-5,6,7,7a-tetrahydro-1H-indazol-4(3aH)-one (3) was identified as a hit compound in a high throughput screen for inhibition of CDK2 in complex with cyclin A. The activity of the most promising analogues was evaluated by inhibition of CDK2 enzyme complexes with various cyclins. Analogues 53 and 59 showed 3-fold better binding affinity for CDK2 and 2- to 10-fold improved inhibitory activity against CDK2/cyclin A1, E, and O compared to screening hit 3. The data from the enzyme and binding assays indicate that the binding of the analogues to a CDK2/cyclin complex is favored over binding to free CDK2. Computational analysis was used to predict a potential binding site at the CDK2/cyclin E1 interface.
Keywords: Tetrahydroindazoles, CDK2, Cyclins, Kinase inhibitors
Graphical Abstract
1. Introduction
Cyclin-dependent kinases (CDKs) are a family of serine-threonine protein kinases that regulate progression of cells through the cell cycle [1] and have additional roles such as regulation of transcription, DNA damage repair, and epigenetic functions [2]. There are 21 kinases, named CDK, all of which possess a conserved ATP-binding site, a PSTAIRE-like cyclin-binding domain and an activating T-loop [3]. Activity of CDKs is regulated through multiple mechanisms including association with their partner protein cyclins, and positive and negative phosphorylation events. CDKs have become attractive therapeutic targets for cancer chemotherapy [4]. Several CDK-cyclin complexes are involved in cell cycle control. Among them, the CDK2/cyclin A complex is required for both DNA synthesis, mitosis, meiosis and controls the G1- to S-phase checkpoint [5]. In the area of developing new male non-hormonal contraceptive agents [6–8], targeting cell cycle regulatory processes is a promising approach, which may be achievable via inhibition of the CDK2/cyclin A1 complex or CDK2/Speedy 1 [9]. Spermatogenesis is highly dependent on CDK2 for meiosis [10], and cyclin A1 is a testis-specific isoform of cyclin A [11], whereas Speedy 1 is about 20-fold overexpressed in the testes. Cyclin A1 knockout mice show meiotic arrest associated with reduction of CDK2 activation [12]. In addition, CDK2−/− knockout mice are viable but sterile [10]. Therefore, inhibitors specific for the CDK2/cyclin A1 or CDK2/Speedy 1 complexes may have potential as non-hormonal male contraceptive agents [9, 13].
Inhibitors of kinases can be classified into four major types [14]. Type I and II inhibitors are ATP-competitive and recognize the active (DFG-in) and inactive conformation (DGF-out), respectively. Type III inhibitors are allosteric inhibitors that bind outside of the ATP-binding site, and the fourth type are covalent inhibitors that typically target a residue in the ATP site. Most reported CDK inhibitors possess a broad specificity profile, but new generation CDK inhibitors have been developed with high potency and selectivity. The FDA has approved palbociclib, abemaciclib, and ribociclib, three selective CDK4/CDK6 inhibitors for the treatment of hormone receptor positive, human epidermal growth factor receptor 2 negative, advanced or metastatic breast cancer [4]. The common core structural feature of all three compounds is a N-(5-(piperazin-1-ylmethyl)pyridin-2-yl)pyrimidin-2-amine. In addition, the FDA has given alvocidid (Figure 1) Orphan Drug designation for the treatment of acute myeloid leukemia. Alvocidib, TP-1287 and voruciclib are derivatives of the plant-derived flavone rohitukine [15].
Figure 1.
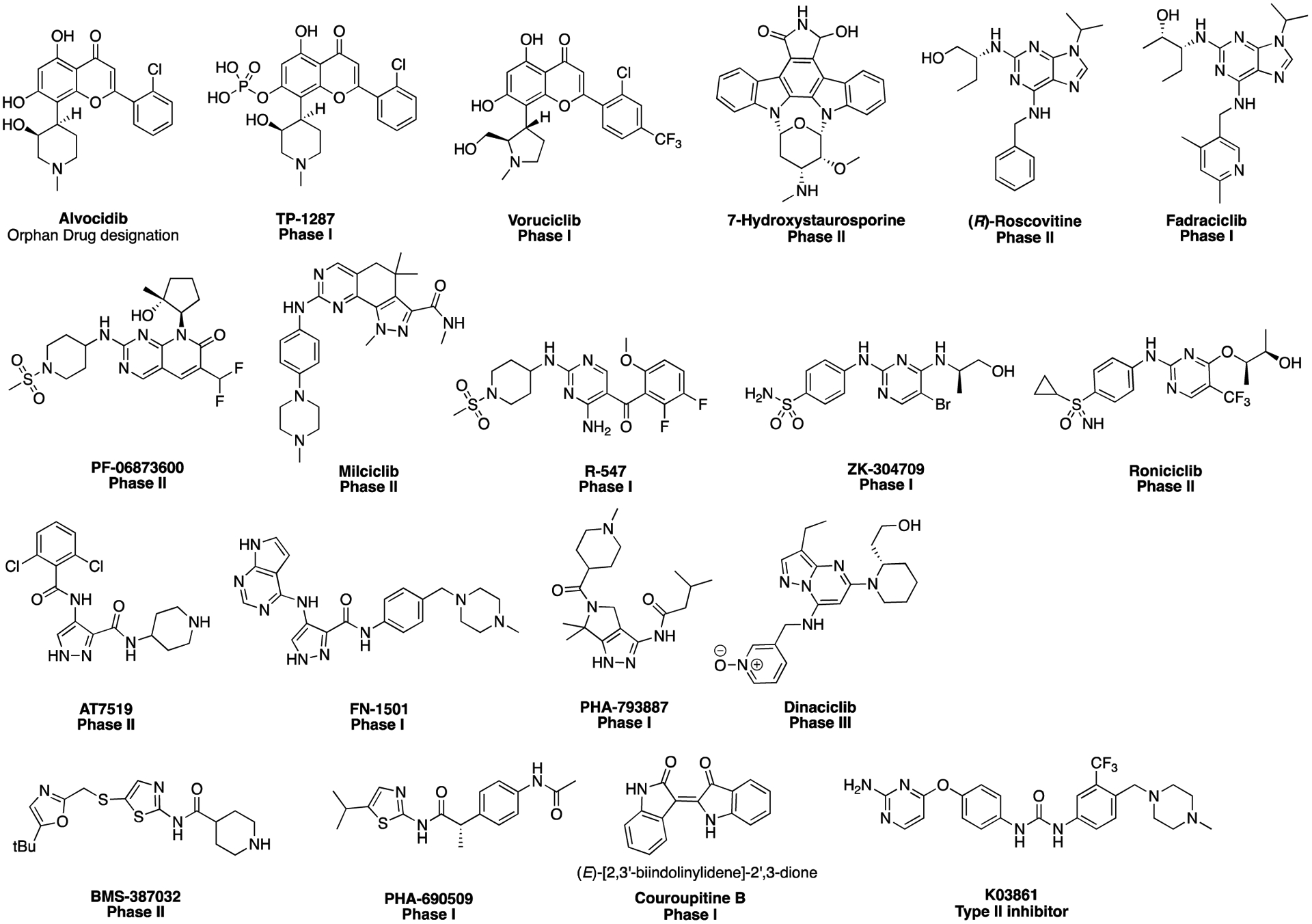
Structures of CDK2 inhibitors in clinical trials and structure of type II inhibitor K03861.
Figure 1 shows the structures of CDK2 inhibitors that have been investigated in clinical trials in the past (highest level shown) or are currently in clinical trials. However, these inhibitors are inhibiting multiple kinases in addition to CDK2. A selective CDK2 inhibitor is not yet available that could be a candidate for investigation as a male contraceptive agent. For a summary of the CDK targets for the compounds shown in Figure 1 see reference [9]. As shown in Figure 1, there a several structural classes of CDK2 inhibitors that target the ATP binding site (type 1 inhibitors). 7-Hydroxystaurosporine is an analog of the natural product staurosporine. (R)-roscovitine and fadraciclib are 9H-purine-2,6-diamines. The hinge-binding aminopyrimidine core structure is common to PF-06873600, milciclib, R-547, ZK-304709 and roniciclib. AT7519, FN-1501 and PHA-793887 are based on a 4-amino-1H-pyrazole-3-carboxamide core structure. AT7519, FN01501 and PHA-793887 are 4-amino-1H-pyrazole-3-carboxamides. Dinaciclib, a 5-(piperidin-1-yl)pyrazolo[1,5-a]pyrimidin-7-amine, is an inhibitor of CDK1/2/5/9, that is currently in phase III clinical trials for several cancers [16]. Compounds BMS-387032 and PHA-690509 are N-acylated thiazol-2-amines. Couroupitine B is a (E)-[2,3’-biindolinylidene]-2’,3-dione. A type II inhibitor of CDK2, K03861, is an aminopyrimidinephenyl urea (Figure 1), but this inhibitor is also promiscuous and not selective for CDK2. Some CDK2 inhibitors with increased selectivity have been identified but none of those have progressed to a clinical trial [9]. In general, they are structurally similar to those shown in Figure 1.
2. Screening
We screened 100,000 compounds for inhibitors of the human CDK2/cyclin A (also named A2) complex (cyclin A is an isoform of cyclin A1 that can be functionally expressed in E. coli) [17]. Staurosporine was used as the positive control in all assays. Among the hit compounds identified, compound 3 (Figure 2) inhibited the enzymatic activity of CDK2/cyclin A with a Ki value of 2.3 μM. The 3,6,6-trimethyl-1-phenyl-6,7-dihydro-1H-indazol-4(5H)-one scaffold of hit 3, appeared promising for a hit to lead campaign since a number of bioactivities have been reported for this privileged scaffold [18–21]. This includes HSP90 inhibitor SNX-5422 [21], which is in phase III clinical trials for cancer chemotherapy [22], and opioid receptor agonists (Figure 2) [23]. Furthermore, indazoles have been identified previously as kinase inhibitors [24–28]. Here, we report structure-activity relationship studies based on hit compound 3 in our efforts to obtain potent and selective CDK2/cyclin inhibitors.
Figure 2.
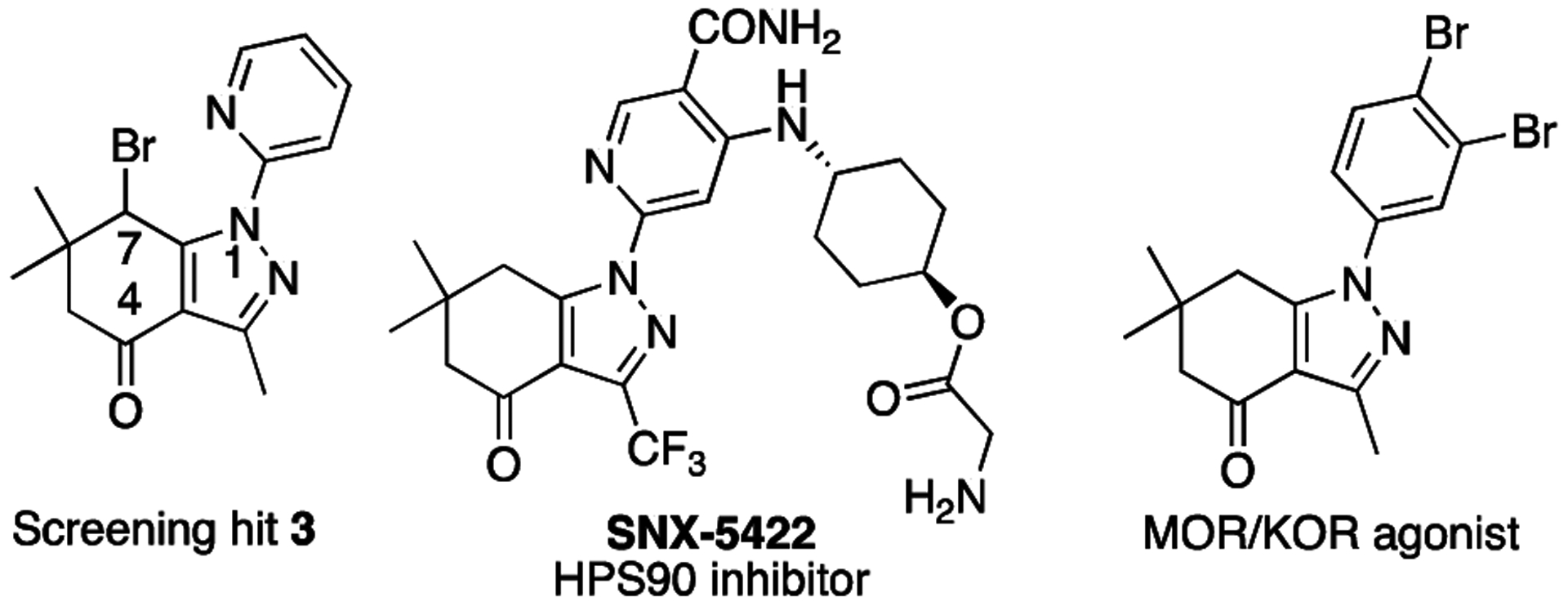
Structures of hit compound 3, HSP90 inhibitor SNX-5422, and an opioid receptor agonist.
3. Chemistry
As shown in Scheme 1, compound 3 was obtained by reaction of commercially available 2-acetyl-5,5-dimethy1-cyclohexane-1,3-dione (1) with 2-hydrazinopyridine and subsequent bromination of intermediate 2 [18]. Modification was initiated at C7 since the C7 allylic bromine offered a facile functional group to generate new analogues to probe steric and electronic influences on activity. A fluoro group was introduced into the C7 position using silver fluoride to furnish compound 4. The 7-methoxy derivative 5 was obtained using silver nitrate and MeOH [18]. The 7-hydroxy compound 6 was prepared from 3 using silver nitrate and water. Subsequent chlorination of compound 6 provided the 7-chloro analogue 7 [29]. The 7-methyl analogue 8 was prepared by the reaction of compound 2 with methyl iodide in the presence of n-butyl lithium [30]. A dimethylamino group was introduced by reaction of 3 with dimethylamine under microwave conditions to afford 7-dimethylamino analogue 9 [18]. The phenylthio group was introduced at C7 to provide compound 10, which was oxidized and furnished sulfone 11. 7-Azido analogue 12 was obtained by substitution reaction of 3 with sodium azide [31]. Hydrogenation of the azido group afforded 7-amino derivative 13.
Scheme 1.
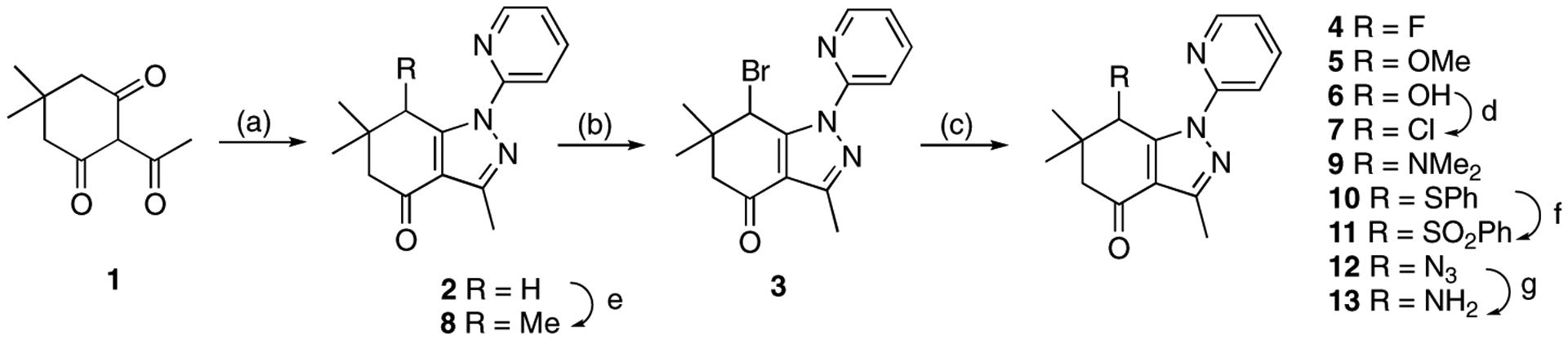
Synthesis of compounds 2-13. Reagents and conditions: 2: (a) 2-hydrazino-pyridine, EtOH, 80 °C, 4 h, 84%. 3: (b) N-bromosuccinimide, CHCl3, 60 °C, 6 h, 61%. 4: c) AgF, CaF2, acetonitrile, 80 °C, 22 h, 12%. 5: (c) AgNO3. MeOH/acetone, rt, 5 days, 21%. 6: (c) AgNO3, MeOH/H2O/acetone, rt, 20 h, 70%; 7: (d) from 6 with SOCl2, pyridine, rt, 2 h, 70%. 8: (e) from 2 with MeI, n-BuLi, −78 °C to rt, 16.5 h, 25%; 9: (c) dimethylamine, THF, microwave at 40 °C, 38 h, 21%. 10: (c) sodium thiophenate, DMF, rt, 7 h, 79%; (f) 11: from 10 with Oxone, H2O/MeOH, rt, 3 h, 68%. 12: (c) NaN3, 80% EtOH, 90–100 °C, 24 h, 43%; 13: (g) from 12 with H2, 10% Pd/C, EtOH, rt, 4 h, 73%.
Modifications at C6 were performed using 5-substituted cyclohexane-1,3-diones 14a-14d to provide compounds 18a-18d (Scheme 2). First, cyclohexenyl acetates 15a-15d were prepared from 14a-14d using acetyl chloride and pyridine. 2-Acetylcyclohexane-1,3-diones 16a −16d were obtained through O-C isomerization of enol esters 15a-15d in the presence of AlCl3 [32]. Tetrahydroindazoles 17a-17d were then prepared by reaction of 16a −16d with 2-hydrazinopyridine in EtOH. Finally, bromination of 17a-17d using NBS provided analogues 18a-18d.
Scheme 2.
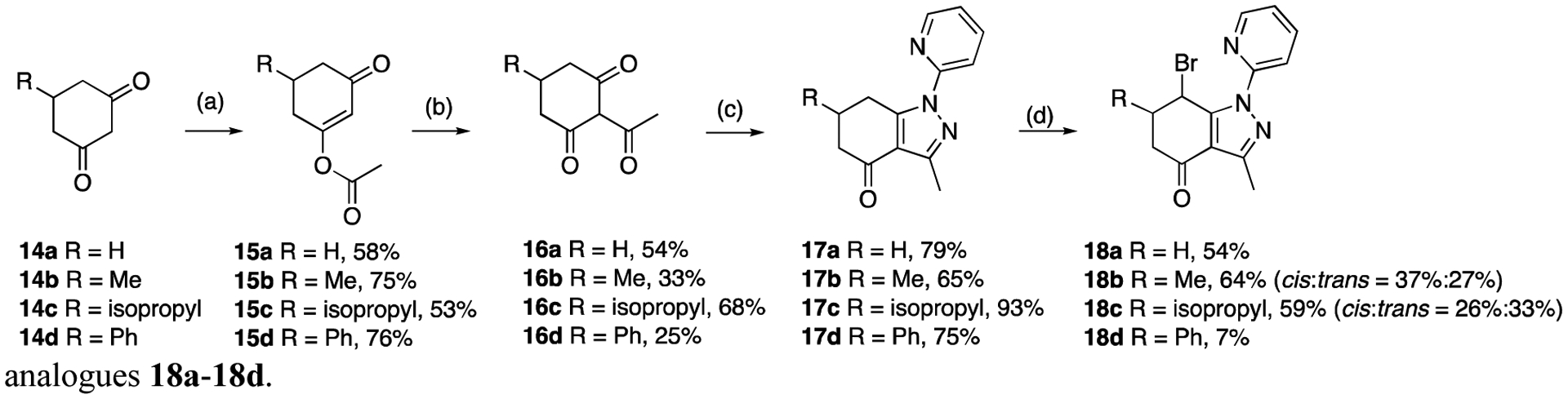
Synthesis of analogues 18a-18d. Reagents and conditions: (a) acetyl chloride, pyridine, CHCl3, rt; (b) AlCl3, CH2Cl2, rt; (c) 2-hydrazinopyridine, EtOH, 80 °C; (d) NBS, CHCl3, 60 °C.
We next prepared the ring-enlarged cyclic amides 19 and 20 by Schmidt reaction of compound 2 using sodium azide and polyphosphoric acid [33]. Bromination of 20 furnished compound 21 in 35% yield (Scheme 3).
Scheme 3.
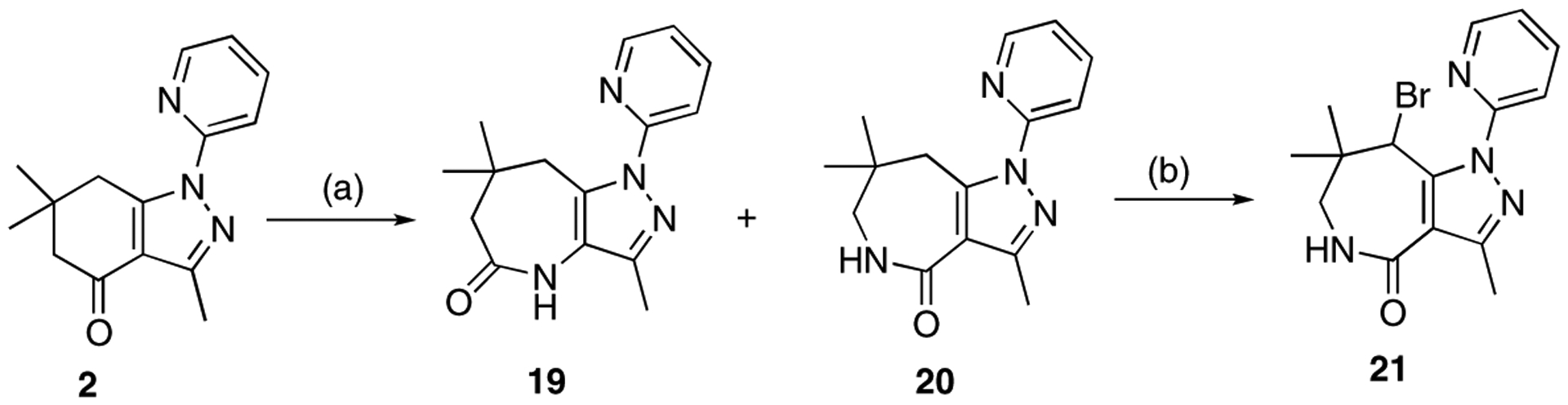
Synthesis of cyclic amides 19-21.Reagents and conditions: (a) sodium azide, polyphosphoric acid, 100 °C, 4.5 h, 17% for 19, 24% for 20; (b) NBS, CHCl3, 60 °C, 6 h, 35% (20 to 21).
Reduction of compound 3 afforded alcohol 22 and oxime 23 was obtained by the reaction of compound 3 with hydroxylamine (Scheme 4) [18].
Scheme 4.
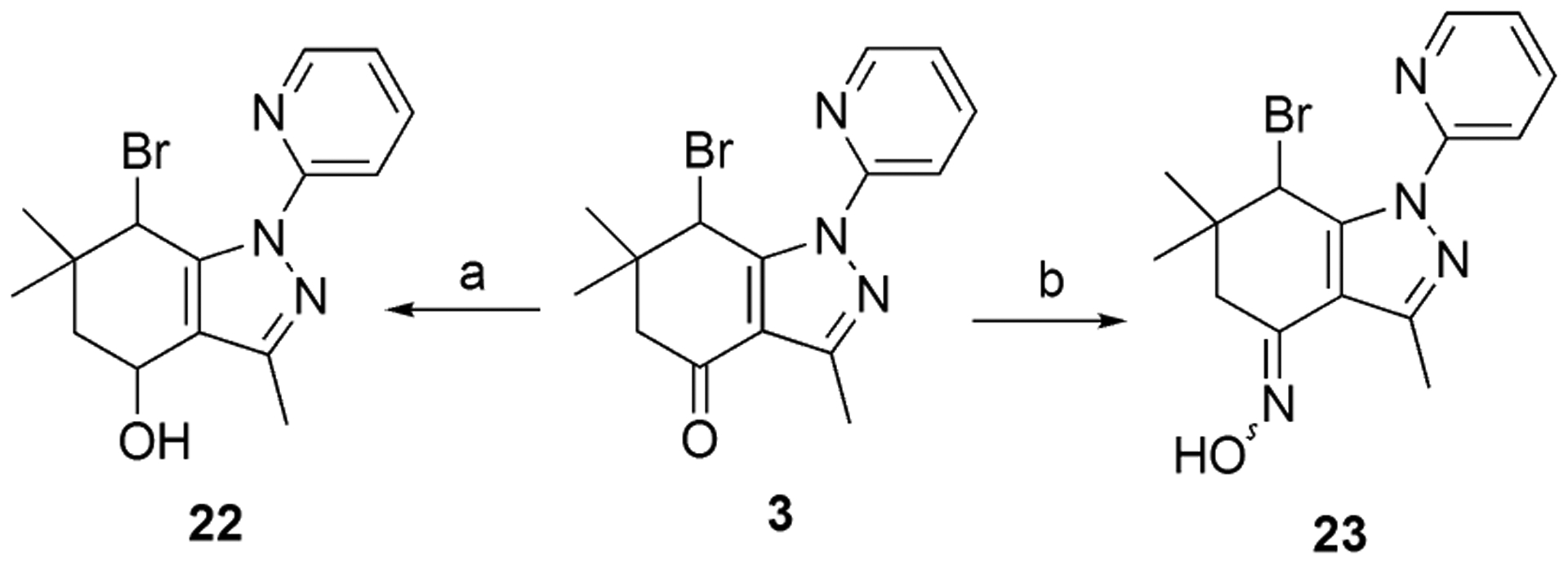
Synthesis of analogues 22 and 23. Reagents and conditions: (a) CeCl3, NaBH4, MeOH/THF, −40 to −10 °C, 30 min, 20%; (b) H2NOH·HCl, NaOAc, 1,4-dioxane, reflux, 6 h, 48%.
To explore modifications at the C3 position, we prepared demethyl analogue 25. In a one-flask reaction under microwave conditions, 5,5-dimethylcyclohexane-1,3-dione, 1,1-dimethoxy-N,N-dimethylmethanamine, and 2-hydrazinopyridine provided compound 24. Subsequent bromination afforded compound 25 (Scheme 5) [34].
Scheme 5.
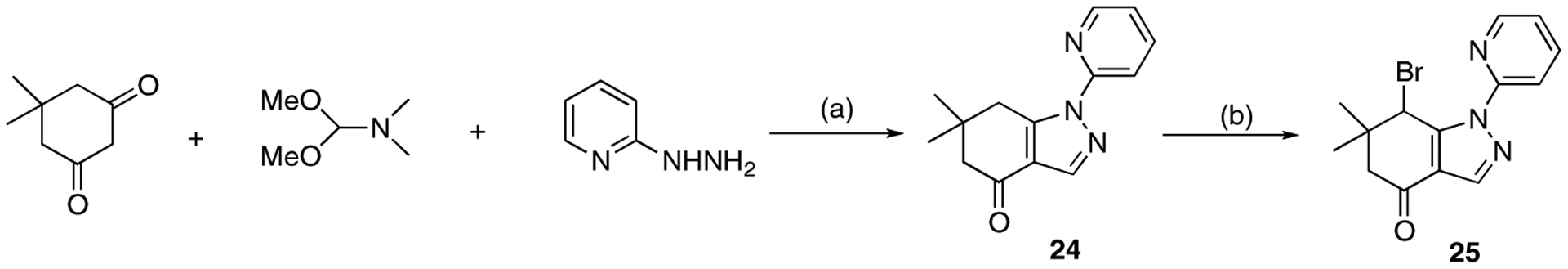
Synthesis of analogue 25. Reagents and conditions: (a) AcOH/H2O, microwave at 200 °C, 5 min, 54%; (b) NBS, CHCl3, 60 °C, 4 h, 29%.
Another modification at C3 was made by introducing an isobutyl group. The procedure includes O-acylation of 5,5-dimethyl cyclohexane-1,3-dione, O-C-isomerization, tetrahydroindazole formation and bromination of 26 to yield analogue 27 (Scheme 6).
Scheme 6.
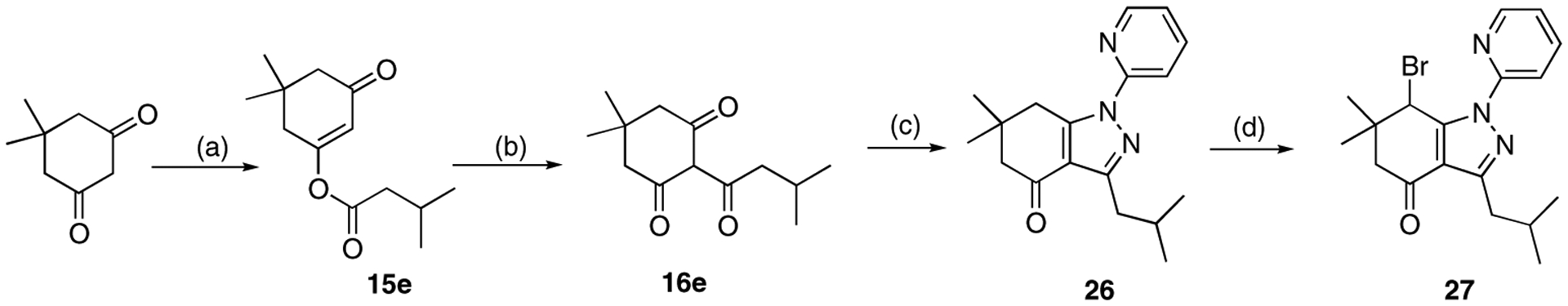
Introducing an isobutyl group at C3. Reagents and conditions: (a) isovaleryl chloride, pyridine, CHCl3, rt, 2 h, 80%; (b) AlCl3, CH2Cl2, rt, 2 h, 51%; (c) 2-hydrazinopyridine, EtOH, 80 °C, 2 h, 95%; (d) NBS, CHCl3, 60 °C, 40 min, 58%.
Modifications at the N1 position with heteroaromatic substituents are shown in Scheme 7. 2-Hydrazinylpyrimidine 28 was prepared by the reaction of 2-chloropyrimidine with hydrazine [35]. Ring formation and bromination of 29 provided imidazole analogue 30. Next, a pyrazine group was introduced at N1. 2-Hydrazinylpyrazine 31 was prepared from 2-chloropyrazine [36]. Tetrahydroindazole formation and subsequent bromination of 32 afforded compound 33. A 4-pyridyl group was also introduced at the N1 position. 4-Hydrazinylpyridine hydrochloride 34 was prepared from 4-chloropyridine hydrochloride by reaction with hydrazine. Ring formation and bromination reaction of 35 furnished compound 36.
Scheme 7.
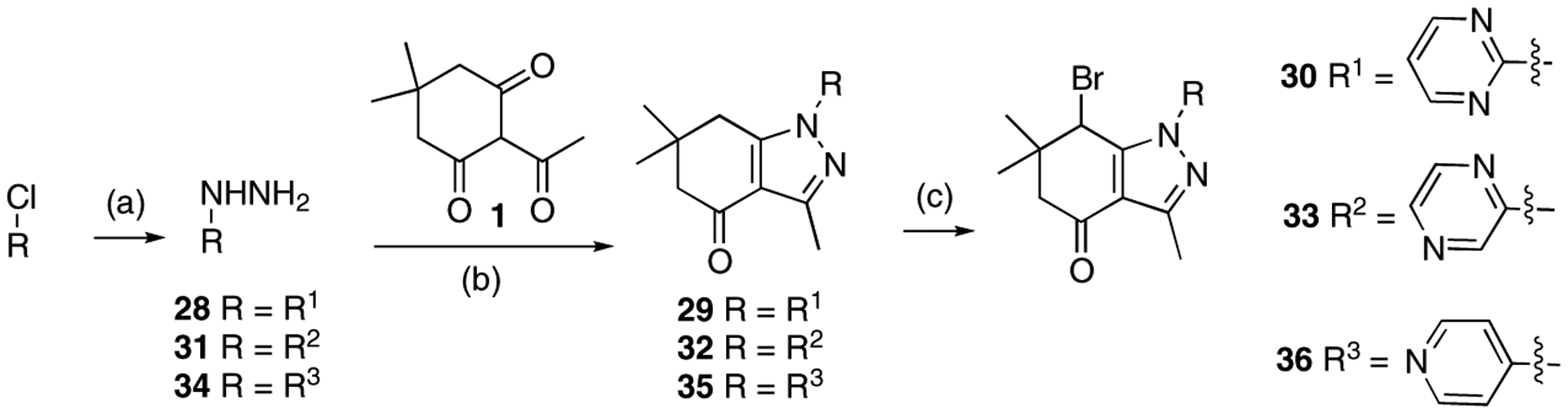
Reagents and conditions: 30: (a) NH2NH2, pyridine, rt, 2.5 h, 41%; (b) EtOH, 80 °C, 2 h, 95%; (c) NBS, CHCl3, 60 °C, 3 h, 50%. 33: (a) 35% NH2NH2·H2O, 65 °C, 12 h, 75%; (b) EtOH, 80 °C, 2 h, 62%; (c) NBS, CHCl3, 60 °C, 2.5 h, 59%. 36: (a) i) NaOH/H2O, extraction with Et2O, ii) NH2NH2·H2O, EtOH, 100 °C, 16 h, 37%; (b) pyridine, EtOH, 80 °C, 2 h, 98%; (c) NBS, CHCl3, 60 °C, 24 h, 93%.
The 3-methylpyridin-2-yl analogue 40 was synthesized in the same way (Scheme 8). Bromination of 38 provided both the 7-bromo analogue 39 and the 5-bromo analogue 40. The 5-methylpyridine-2-yl analogue 43 was synthesized in a similar fashion through hydrazination of 2-bromo-5-methyl pyridine, tetrahydroindazole 42 formation, and bromination. 3-Trifluoromethylpyridin-2-yl analogues 46 and 47 were synthesized similarly, starting from 2-bromo-3-(trifluoromethyl)pyridine. 5-Chloropyridin-2-yl analogue 49 was synthesized using 5-chloro-2-hydrazinopyridine as the starting material. 6-Chloropyridazin-3-yl analogue 51 was synthesized from commercially available 3-chloro-6-hydrazinopyridazine.
Scheme 8.
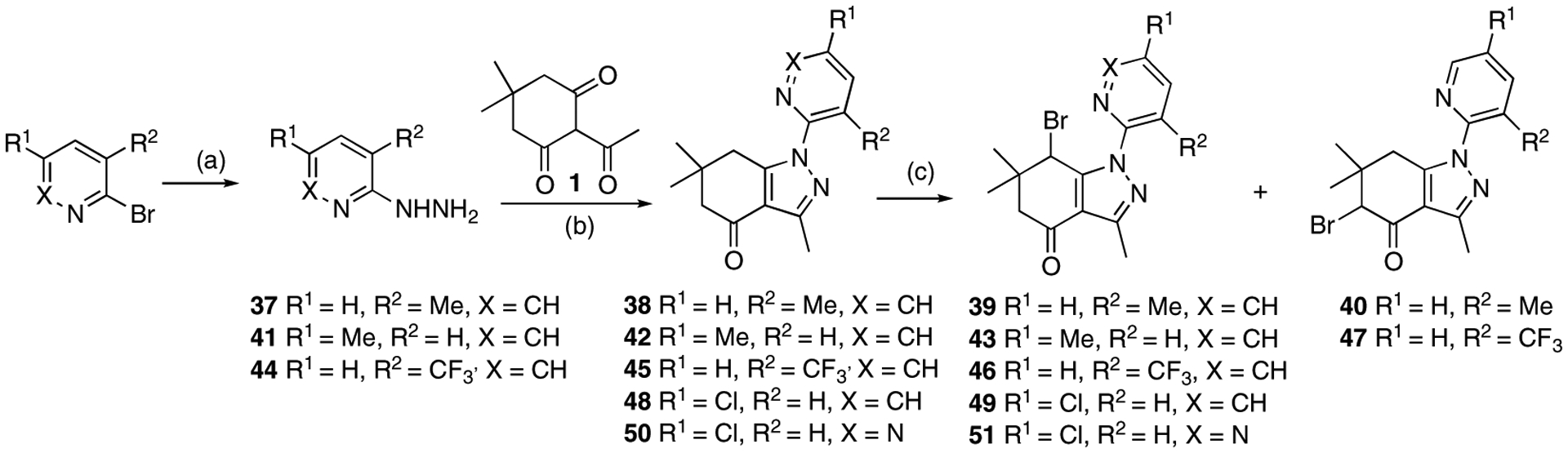
Reagents and conditions: 39 and 40: (a) NH2NH2·H2O, EtOH, 105 °C, 40 h, 36%; (b) EtOH, 80 °C, 2.5 h, 30%; (c) NBS, CHCl3, 60 °C, 6.5 h, 39% for 39, 17% for 40. 43: (a) NH2NH2·H2O, EtOH, 105 °C, 5 days, 79%; (b) EtOH, 80 °C, 2.5 h, 83%; (c) NBS, CHCl3, 60 °C, 2 h, 81%. 46 and 47: (a) NH2NH2·H2O, EtOH, 105 °C, 29 h, 53%; (b) pyridine, EtOH, 80 °C, 3 h, 22%; (c) NBS, CHCl3, 60 °C, 2.5 h, 11% for 46, 10% for 47. 49: (b) EtOH, 80 °C, 3 h, 83%; (c) NBS, CHCl3, 60 °C, 2 h, 60%. 51: b) EtOH, 80 °C, 3 h, 73%; (c) NBS, CHCl3, 60 °C, 2 h, 23%.
A thiazole group was introduced at the N1 position using 2-hydrazinothiazole furnishing analog 53 (Scheme 9). Similarly, a benzothiazole group was also introduced at the N1 position (55). As a nonaromatic substituent, a cyanoethyl group was introduced at N1 furnishing analogue 57. Pyrimidine-4-yl analogue 59 and 5-trifluoromethylpyridine-2-yl compound 61 were synthesized as other analogues modified at N1. The 6-methoxypyridin-2-yl analogue 64 was prepared starting from 2-bromo-6-methoxypyridine, which was converted to 2-hydrazineyl-6-methoxypyridine 62 (NH2NH2·H2O, 120 °C, 3.5 h, 41%) and then converted in two steps to analog 64. 5-Nitropyridin-2-yl compound 66 was synthesized using 2-hydrazinyl-5-nitropyridine as the starting material. Subsequent reduction of the nitro group in 66 yielded pyridinoaniline 67.
Scheme 9.
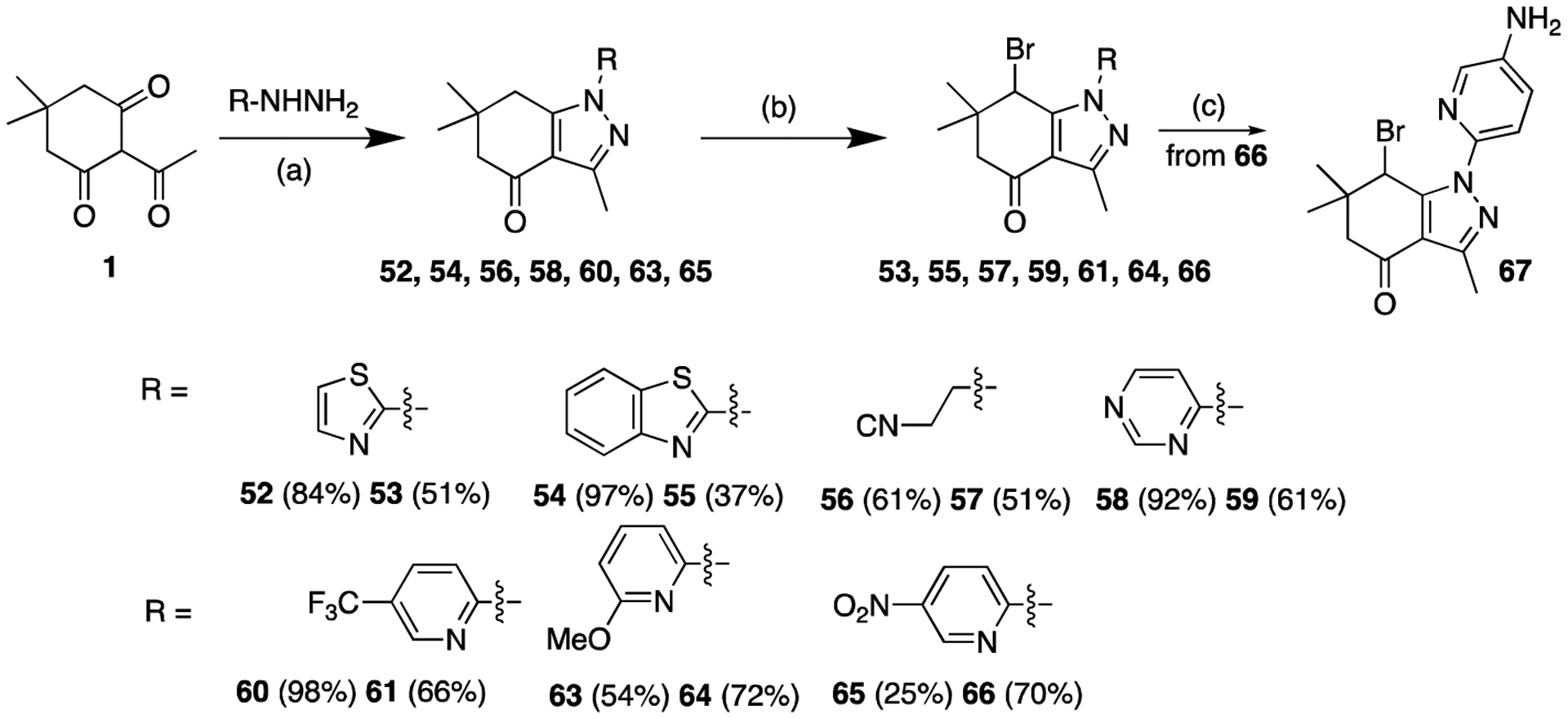
Synthesis of N1-modified analogues. Reaction conditions: (a) EtOH, 80 °C, 2–3 h; (b) NBS, CHCl3, 60 °C, 1.5–5 h; (c) SnCl2-2H2O, EtOH, 70 °C, 15 min, 62%.
A 4-nitrophenylsulfonyl group was introduced at N1 (Scheme 10). Intermediate 68 was synthesized by the reaction of 2-acetyl-5,5-dimethyl-cyclohexane-1,3-dione with hydrazine. Then 4-nitrobenzenesulfonyl chloride was reacted with 68 to furnish compound 69.
Scheme 10.
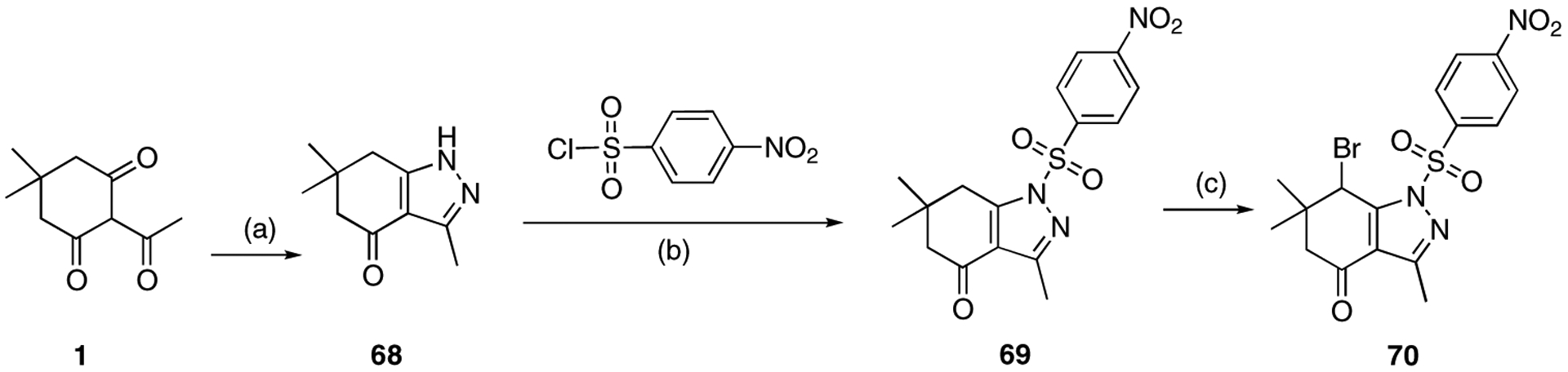
Reagents and conditions: (a) NH2NH2·H2O, EtOH, 80 °C, 4 h, 88%; (b) TEA, CH2Cl2, 0 °C to rt, 5 h, 75%; (c) NBS, CHCl3, 60 °C, 4 h, 56%.
Finally, bromination provided analogue 70.
For further modification at N1, the amino group of compound 67 was coupled with monomethyl malonate and sulfamoyl chloride to afford compounds 71 and 72, respectively (Scheme 11). Methanesulfonyl chloride and acetyl chloride were also coupled to the amino group of compound 67 to provide analogues 73 and 74. The naphthalene-2-yl compound 76 was prepared using naphthalene-2-yl hydrazine hydrochloride (Scheme 12).
Scheme 11.
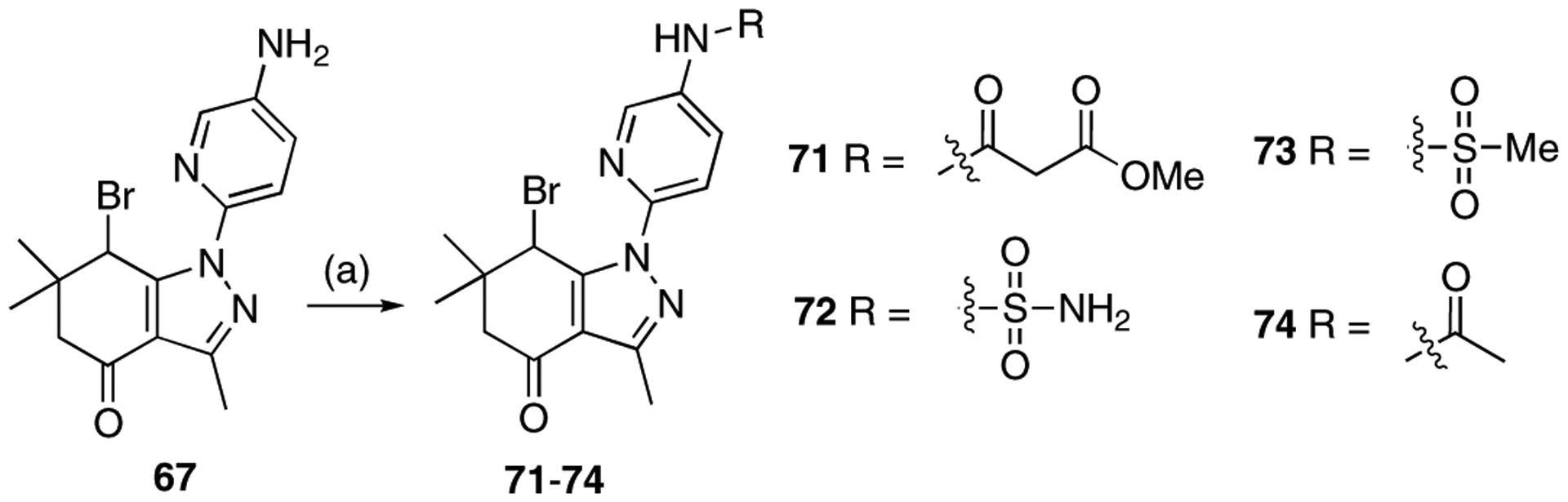
Reagents and conditions: 71: (a) monomethyl malonate, EDC·HCl, HOBT, DIPEA, CH2Cl2, rt, 24 h, 69%; 72: (a) sulfamoyl chloride, dimethylacetamide, rt, 20 h, 89%. 73: (a) methanesulfonyl chloride, pyridine, CH2Cl2, rt, 23 h, 88%; 74: (a) acetyl chloride, TEA, CH2Cl2, rt, 1.5 h, 92%.
Scheme 12.
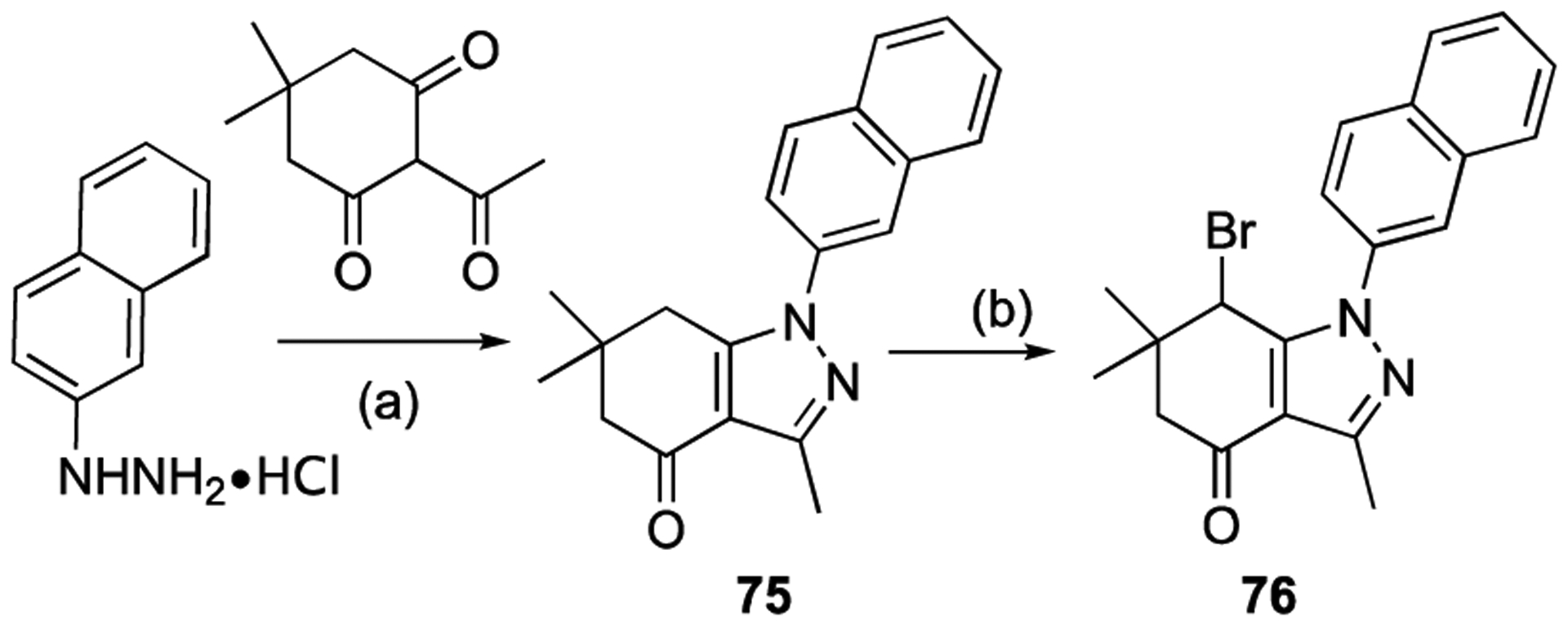
Introducing a naphthalene-2-yl group at N1. Reagents and conditions: (a) pyridine, EtOH, 80 °C, 2 h, 79%; (b) NBS, CHCl3, 60 °C, 3 h, 68%.
4. Inhibition of CDK2 kinase activity-structure activity relationship
The in vitro inhibitory activities of the analogues were determined from dose-response curves using the phosphorylated CDK2/cyclin A complex. Steady-state kinetic analysis of the parent compound indicated ATP-competitive inhibition with a Ki value of 2.3 ± 0.2 μM (Fig. S1).
Modifications at the C7 position:
Analogues carrying chloro- or azido-groups at C7 showed activities similar to 3 (compounds 2, 4-13 in Table 1). In the case of compound 2, which is devoid of a C7 substituent, the activity was reduced by more than 50-fold. Presumably, electron-withdrawing groups in this position provide more favorable binding interactions with the target site.
Table 1.
CDK2/cyclin A inhibitory activities of analogues with modified R groups on C3, C4, C5, C6, C7 and N1
 | |||||||
|---|---|---|---|---|---|---|---|
| Compound | R1 | R2 | R3 | R4 | R5 | R6 | IC50 (μM) |
| 3 | Me | =O | H | Me, Me | Br | H | 2.6 ± 0.1 |
| 2 | Me | =O | H | Me, Me | H | H | 110 ± 5 |
| 4 | Me | =O | H | Me, Me | F | H | 21 ± 0.5 |
| 5 | Me | =O | H | Me, Me | OMe | H | 46 ± 2 |
| 6 | Me | =O | H | Me, Me | OH | H | 110 ± 12 |
| 7 | Me | =O | H | Me, Me | Cl | H | 4.9 ± 0.3 |
| 8 | Me | =O | H | Me, Me | Me | H | 200 ± 16 |
| 9 | Me | =O | H | Me, Me | NMe2 | H | 630 ± 11 |
| 10 | Me | =O | H | Me, Me | SPh | H | 12 ± 1 |
| 11 | Me | =O | H | Me, Me | SO2Ph | H | 107 ± 5 |
| 12 | Me | =O | H | Me, Me | N3 | H | 4.5 ± 0.2 |
| 13 | Me | =O | H | Me, Me | NH2 | H | 350 ± 36 |
| 18a | Me | =O | H | H, H | Br | H | 130 ± 6 |
| 18b-trans | Me | =O | H | Me, H | Br | H | 4.8 ± 0.1 |
| 18b-cis | Me | =O | H | Me, H | Br | H | 31.0 ± 0.9 |
| 18c-trans | Me | =O | H | isopropyl, H | Br | H | 130 ± 7 |
| 18c-cis | Me | =O | H | isopropyl, H | Br | H | 470 ± 80 |
| 18d | Me | =O | H | Ph, H | Br | H | > 1000 |
| 22 | Me | OH | H | Me, Me | Br | H | 560 ± 63 |
| 23 | Me | =NOH | H | Me, Me | Br | H | 59 ± 3 |
| 24 | H | =O | H | Me, Me | H | H | > 1000 |
| 25 | H | =O | H | Me, Me | Br | H | 160 ± 10 |
| 27 | isobutyl | =O | H | Me, Me | Br | H | 820 ± 660 |
| 39 | Me | =O | H | Me, Me | Br | Me | > 1000 |
| 40 | Me | =O | Br | Me, Me | H | Me | 180 ± 20 |
| 46 | Me | =O | H | Me, Me | Br | CF3 | 460 ± 50 |
| 47 | Me | =O | Br | Me, Me | H | CF3 | 230 ± 20 |
| Staurosporine | ~50 | ||||||
Modifications at the C6 position.
Change of the 6-dimethyl moiety and introduction of single methyl, isopropyl or phenyl groups (compounds 18a-18d in Table 1) did not improve inhibitory activity. The trans isomers of methyl and isopropyl substituted compounds 18b and 18c were more potent than their corresponding cis isomers. The phenyl analogue 18d was inactive. Thus, a small alkyl group (Me) and trans configuration seem important for inhibition.
Modifications at C4 (carbonyl):
For carbonyl modification, hydrogen bond donor groups were introduced replacing the carbonyl group with hydroxyl and oxime groups (Table 1). Compounds 22 and 23 displayed only weak inhibitory activity, indicating that the hydrogen bonding acceptor characteristics of the carbonyl group are important for binding.
Modifications at C4/C5:
Cyclic amides were prepared to investigate the effect of a hydrogen bond donor next to the carbonyl group. Both compounds 19 and 20 were inactive, but bromo compound 21 was moderately active (Table 2). This result shows that a hydrogen bond donor at this position does not increase the activity. This result might be related to the structural deformation due to the size of the ring (7-membered lactam ring). 5-Br-substituted compounds 40, 46, and 47 showed low activities (Table 1).
Table 2.
CDK2/cyclin A inhibitory activities of C4/C5-modified analogues
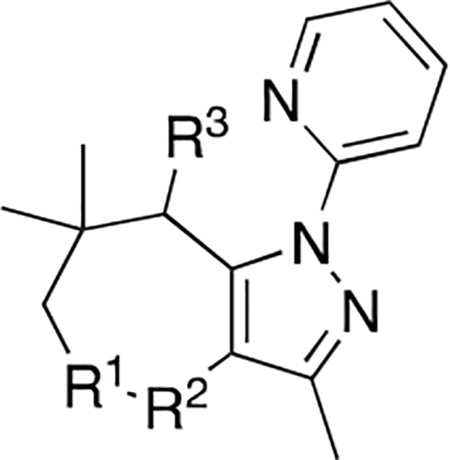 | ||
|---|---|---|
| Compound | R1-R3 | IC50 (μM) |
| 19 | R1= -(CO)-, R2= -NH-, R3=H | > 1000 |
| 20 | R1= -NH-, R2= -(CO)-, R3=H | > 1000 |
| 21 | R1= -NH-, R2= -(CO)-, R3=Br | 12 ± 3 |
Modifications at C3:
Deletion of the C3 methyl group (24) or introduction of a 3-isobutyl group (27) did not provide analogues with better inhibitory activities. The methyl group seemed to be important at this position (Table 1).
Modifications at N1
Many different heterocyclic groups were introduced to modify the N1 position. Among the synthesized analogues, several analogues showed similar or slightly better activities than 3 (IC50: 10.5–1.5 μM) (Table 3).
Table 3.
CDK2/cyclin A inhibitory activities of N1-modified analogues
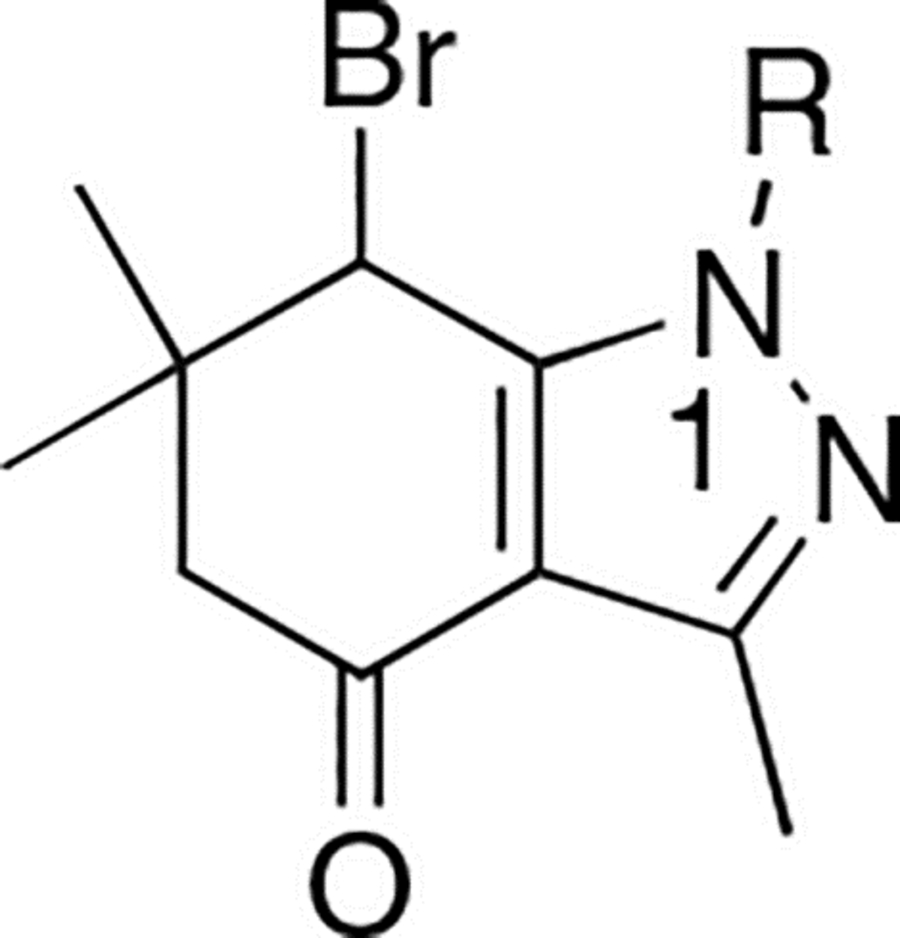 | |||||
|---|---|---|---|---|---|
| Compound | R | IC50 (μM) | Compound | R | IC50 (μM) |
| 3 | 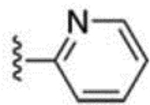 |
2.6 ± 0.1 | 59 | 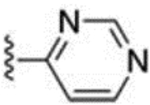 |
1.5 ± 0.03 |
| 30 | 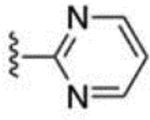 |
9.3 ± 0.2 | 61 | 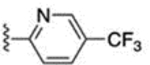 |
6.7 ± 0.1 |
| 33 | 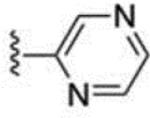 |
97 ± 8 | 64 | 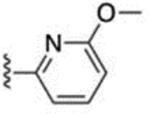 |
2.1 ± 0.3 |
| 36 | 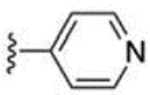 |
10.5 ± 0.2 | 66 | 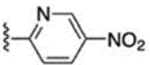 |
3.1 ± 0.2 |
| 39 | 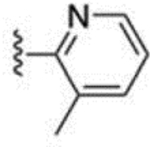 |
> 1000 | 67 | 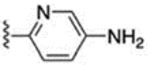 |
4.6 ± 0.2 |
| 43 | 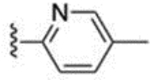 |
7.5 ± 0.8 | 70 | 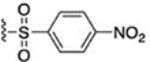 |
> 1000 |
| 46 | 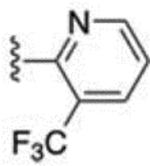 |
460 ± 50 | 71 | 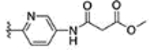 |
2.7 ± 0.06 |
| 49 | 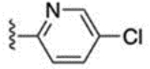 |
3.0 ± 0.2 | 72 | 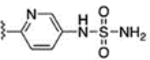 |
2.4 ± 0.05 |
| 51 | 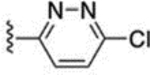 |
26 ± 1 | 73 | 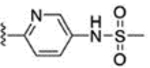 |
19 ± 0.3 |
| 53 | 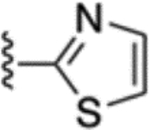 |
2.2 ± 0.3 | 74 | 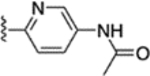 |
3.6 ± 0.03 |
| 55 | 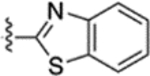 |
> 1000 | 76 | 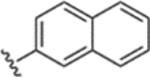 |
37 ± 4 |
| 57 | 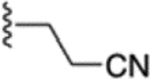 |
> 1000 | Commercial | 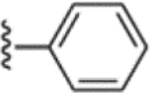 |
56 ± 1.4 |
Compounds with relatively high activity against CDK2/cyclin A were tested against CDK2 complexes with different isoforms of cyclins (Figure 3). Despite the low activity of 18b-cis, it was included in this assay to investigate the role of stereochemistry in the inhibition of the CDK2 complexes. Compounds exerted significant inhibition against CDK2 complexes with specific isoforms of cyclins at 1 μM while 18b-cis did not exhibit significant inhibitory activity against all tested CDK complexes. Cyclins A1, E, and O are highly expressed in testis, implying a potential role in spermatogenesis [37]. The IC50 values against CDK2 with cyclins A, A1, E, and O are shown in Table 8.
Figure 3.
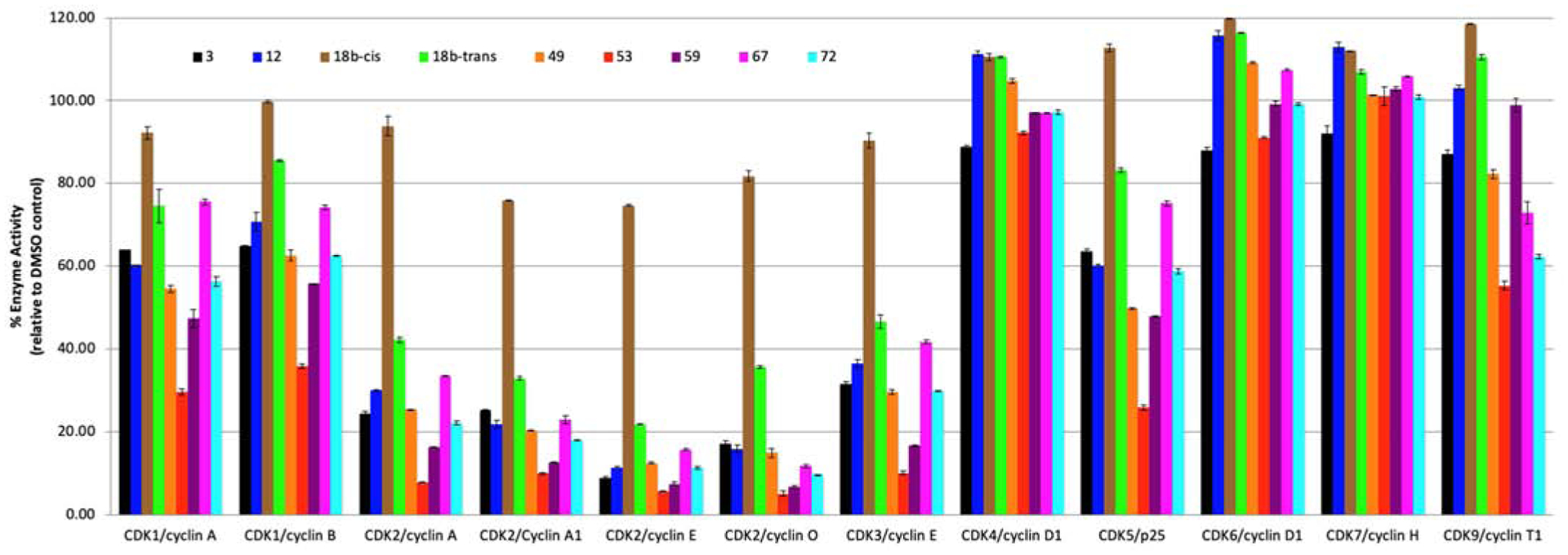
Inhibition of CDK complexes by 3 and its analogues at 1 μM.
When compared with CDK2/cyclins A, A1, and O, the tested compounds exerted enhanced activity against CDK2/cyclin E (Table 4) with IC50 values of 27, 2, and 9 nM against CDK2/cyclin E for 3, 53, 59, respectively. Cyclin E (cyclins E1/E2) is responsible for pairing of chromosomes, the stability of telomere, and the localization of CDK2 in male meiosis [38]. The specific expressions patterns of these cyclins during spermatogenesis in the testis offer opportunities to develop a safe male contraceptive agent selectively aiming at a target in the testis.
Table 4.
IC50 values for 3 and analogues 53 and 59 against CDK2 complexes with cyclin A, cyclin A1, cyclin E, and cyclin O
| CDK2/cyclin | 3 IC50 (μM) | 53 IC50 (μM) | 59 IC50 (μM) | Staurosporine IC50 (μM) |
|---|---|---|---|---|
| CDK2/cyclin A | 0.0860 | 0.0619 | 0.120 | 0.000720 |
| CDK2/cyclin A1 | 0.134 | 0.0243 | 0.0464 | 0.00114 |
| CDK2/cyclin E | 0.0274 | 0.0023 | 0.00890 | 0.00164 |
| CDK2/cyclin O | 0.108 | 0.0331 | 0.0416 | 0.00148 |
However, the binding affinities (Kd) of compounds 3, 53 and 59 for CDK2 in the absence of cyclins are in the low micromolar range (5–15 μM, Table 5). This binding assay is based on the competition of test compounds and an immobilized ligand for the ATP binding site so that the results can represent a compound’s affinity for the ATP binding pocket or an allosteric site that can induce conformational changes to alter the ATP binding pocket. The results suggest that the binding pocket in CDK2 could be altered by cyclins A1, E, and O to enhance the affinity of CDK2 for these compounds. This pocket might be generated by complex formation of CDK2 with these cyclins. The pocket shapes may vary according to cyclin isoform. Based on the data from the enzyme assays and the binding assay (Tables 4 and 5), it was concluded that the binding of the analogues to a complex of CDK2 with a cyclin is favored compared to binding to free CDK2.
Table 5.
Binding affinity of 3 and analogues 53 and 59 for CDK2
| 3 Kd (μM) | 53 Kd (μM) | 59 Kd (μM) | |
|---|---|---|---|
| CDK2 | 15 | 4.8 | 4.5 |
To obtain insights about their activity against cell growth, antiproliferation assay of 3, 53, and 59 against MCF-7 cell line were performed. None of these compounds showed antiproliferative activities up to 200 μM. In addition, since 3 is an analogue of SNX-5422, an HSP90 inhibitor, compound 3 was tested against HSP90 but did not exhibit any activity (data not shown).
MDCK Cell Permeability Assay
To investigate whether the inactivity of these compounds in the antiproliferation assay was due to poor membrane permeability, lead compound 3 was tested to determine its cell permeability using the MDCK cell line (Table 6). A high-permeability compound (metoprolol) and two moderate-permeability compounds (digoxin and imatinib) were used as references. Compound 3 exerted high permeability (Papp (A-B)), better than digoxin and imatinib. However, 3 also exhibited a higher rate in the assay for Papp (B-A) than digoxin and imatinib, but not as high as metoprolol. Compound 3 showed a lower efflux ratio compared to the reference compounds. The recovery rates of 3 for both assays (AP-BL and BL-AP) were approximately 50% that might result from compound 3 being trapped in the lysosome [39]. The data suggests that compound 3 should be cellularly active and does not explain why this compound does not inhibit MCF-7 cell proliferation.
Table 6.
Permeability results of test compounds in MDCK cell line
| Compound | Papp (A-B) (10−6, cm/s) | Papp (B-A) (10−6, cm/s) | Efflux ratio | Recovery (%) AP-BL | Recovery (%) BL-AP |
|---|---|---|---|---|---|
| Metoprolol | 26.9 | 21.2 | 0.79 | 90.7 | 90.4 |
| Digoxin | 1.08 | 5.41 | 5.01 | 77.1 | 83.7 |
| Imatinib | 7.69 | 6.50 | 0.84 | 56.6 | 69.9 |
| 3 | 20.1 | 9.52 | 0.47 | 52.4 | 49.8 |
Computational Simulations
In order to obtain insights into possible binding modes for compounds 3, 53, and 59, we performed computational simulations using the crystal structures of CDK2 and of the CDK2/cyclin E1 complex (Figure 4). We first searched for potential binding sites using FTMap [40] and then selected the protein-protein interface between CDK2 and cyclin E for the docking simulations because the experimental result had shown that the compounds exerted much higher affinity for CDK2 in the presence of cyclins compared to CDK2. In addition, the ATP binding site and other potential binding sites did not show significant differences between CDK2 and CDK2/cyclin E1. We then performed the docking simulations of compounds 3, 53, and 59 with the selected binding sites (Figure 5). The predicted binding poses of these compounds at the CDK2/cyclin E1 interface are shown in Figure 5. In CDK2, they occupy the binding site formed by Arg126, Ala149, Arg150, Gly153, Val154, Tyr179, and Tyr180 (Figure 5 a, b, and c) and form a H-bond interaction with Arg150 through the C4 carbonyl oxygen. The binding site in CDK2 is formed by a conformational change of the activation loop of CDK2 from an active conformation to an inactive conformation (Figure 5a). This site is occupied by the activation loop of CDK2 and disappears when CDK2 forms a complex with cyclins. The MM-GBSA relative binding free energies of 3, 53, and 59 for CDK2 are −37.3 kcal/mol, −41.7 kcal/mol, and −39.3 kcal/mol. In the CDK2/cyclin E1 complex, they occupy a binding site in the interface of CDK2 and cyclin E1, formed by Val156, Glu172, Cys177, Lys178, Tyr179, Ser181, Ala183, Trp227, Pro228, Met233, Asp270, Pro271, and Asn272 of CDK2 and Leu251 and Leu252 of cyclin E1 (Figure 5 d, e, and f). The MM-GBSA relative binding free energies of 3, 53, and 59 for CDK2/cyclin E1 are −44.0 kcal/mol, −45.0 kcal/mol, −45.5 kcal/mol, respectively. In CDK2/cyclin E1, the C4 carbonyl oxygen did not form any H-bonding interaction in the binding site. In the predicted binding modes in CDK2/cyclin E1, the nitrogen in the heteroaromatic ring at the N1 position of compound 59 forms a H-bonding interaction with Ser181 of CDK2 while compound 3 cannot form such a H-bond (pyrimidine of 59 vs pyridine of 3). This could explain why compound 59 exerts higher potency against CDK2/cyclin E1 than compound 3. The predicted binding modes of compounds 3, 53, and 59 in CDK2 and CDK2/cyclin E1 are consistent with their affinities for CDK2 and CDK2/cyclin E1, suggesting that these two pockets in the interface domain of CDK2/cyclins might be the potential pockets for these compounds and that the CDK2/cyclin E1 is predicted to be the preferred one based on the calculations of the relative free binding energies. We also performed the same docking simulations with the ATP bind sites but did not find any differences between them that would explain the observed disparities in binding affinities for CDK2 and the CDK2/cyclin E1 complex.
Figure 4.
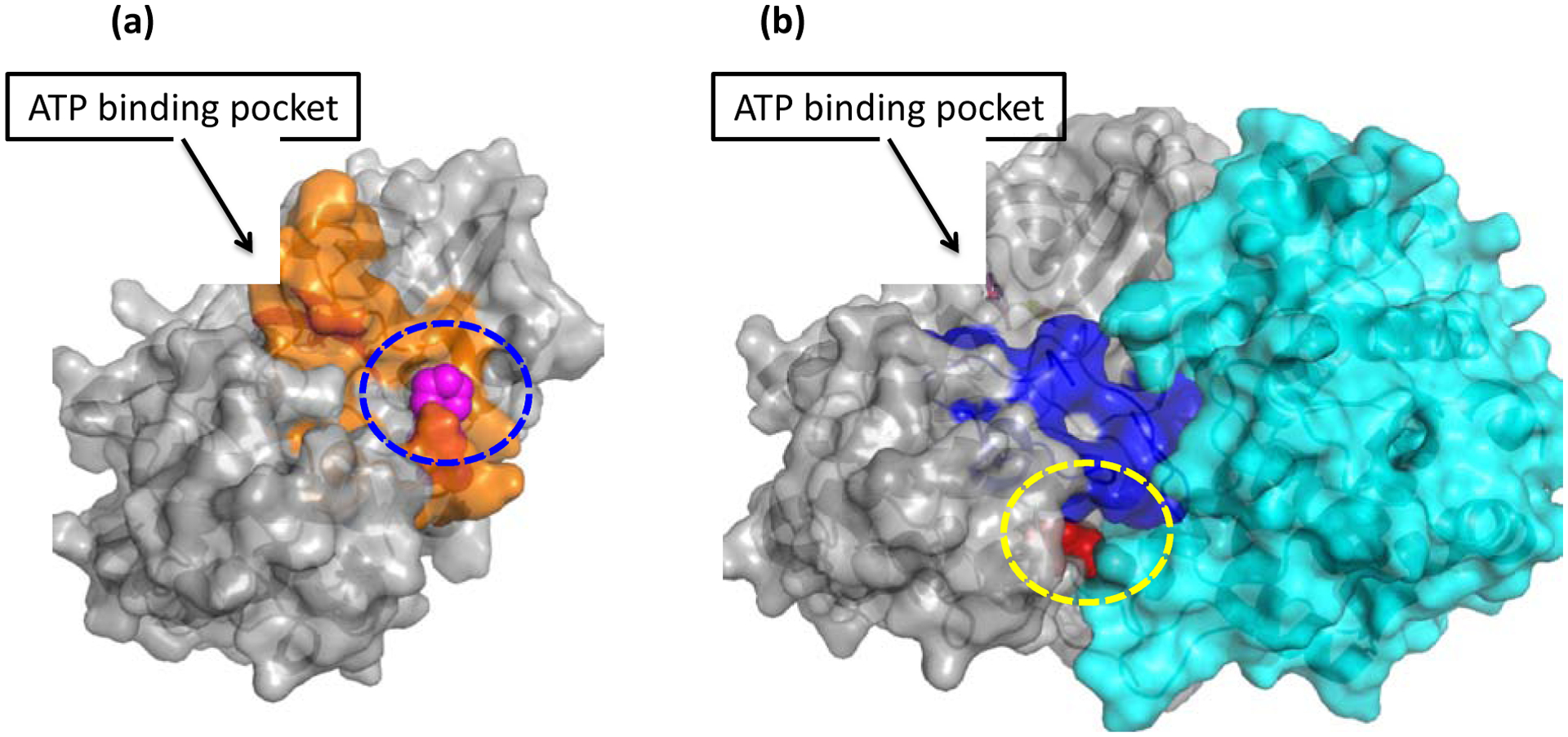
FT-mapping of the protein-protein interface region of CDK2 interacting with cyclins: (a) CDK2 with ATP (PDB ID: 1B38) with the activation loop in the inactive conformation (orange, mapping solvents (magenta) in the blue circle) and (b) a complex of CDK2 with cyclin E1 (cyan) and dinaciclib (PDB ID: 5L2W) with the activation loop in the active conformation (blue, mapping solvents (red) in the yellow circle).
Figure 5.
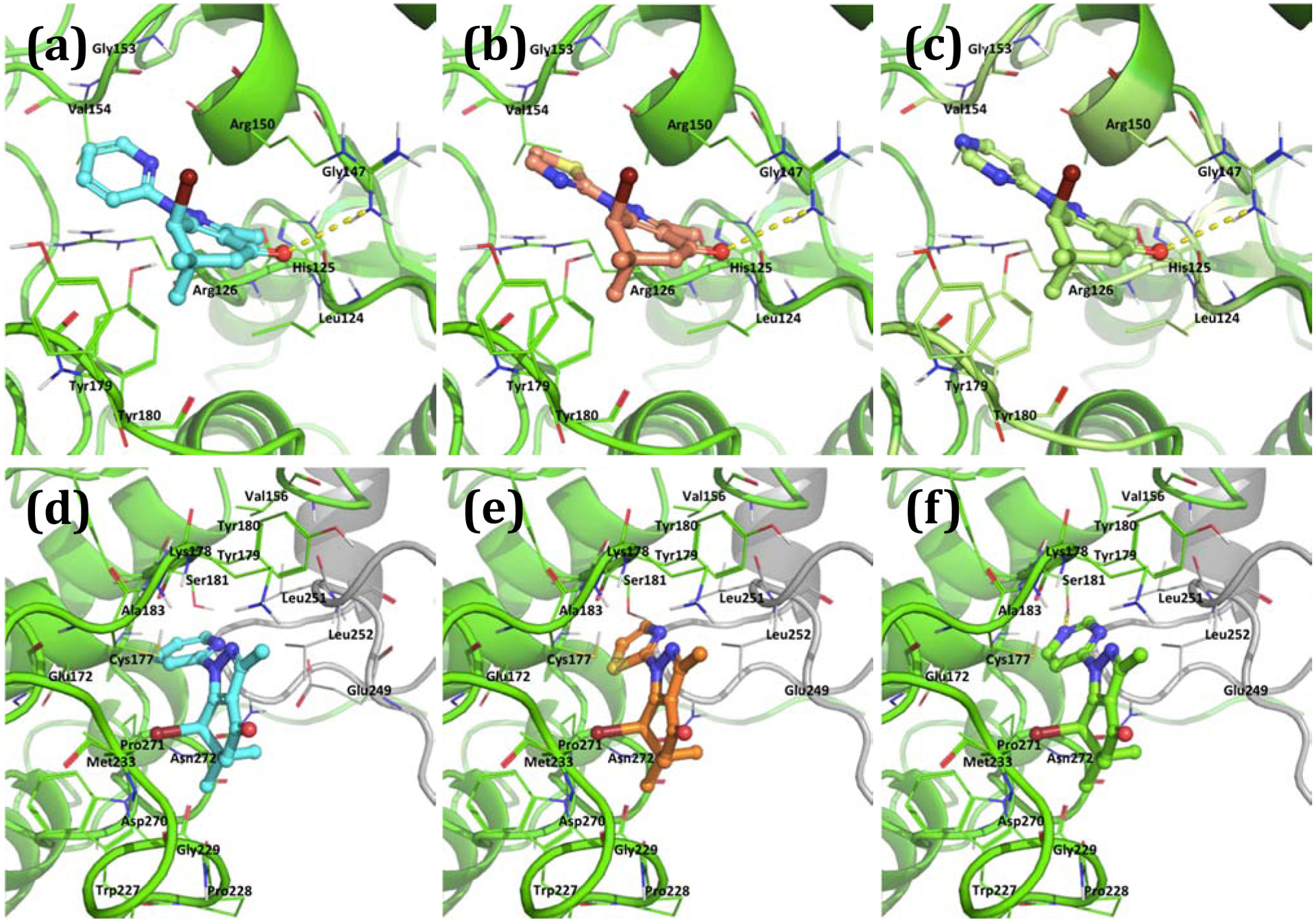
Predicted binding modes of 3 (cyan), 53 (brown), and 59 (green) in CDK2 (a, b and c) and the complex of CDK2 (green) with cyclin E1 (gray) (d, e and f).
5. Conclusion
Compound 3, a 4,5,6,7-tetrahydroindazole, was identified as a CDK2/cyclin A inhibitor from a high throughput screening campaign of a library of 100,000 diverse compounds. SAR studies with over 50 analogues based on hit compound 3 were performed by modifying eight different positions. The in vitro inhibitory activities of the analogues were determined from dose-response curves using the phosphorylated CDK2/cyclin A complex. Replacing the C7 bromine with electron-withdrawing substituents Cl and N3 provided compounds with activity similar to 3, whereas the introduction of C7 electron donating groups reduced potency. Removal of the C6 gem-dimethyl group and attachment of larger C6 groups (isopropyl and phenyl) reduced activity. The 6,7-trans monomethyl analogue of 18 retained activity and was 10-fold more active than the corresponding cis analogues. Reduction of the C4 carbonyl to the alcohol reduced activity >200-fold and oxime formation reduced activity 30-fold. Schmidt reaction-generated seven-membered amide analogue 21 had 30-fold reduced activity. Deletion of the C3 methyl group or introduction of a 3-isobutyl group did not provide analogues with better inhibitory activities. 5-Br-substituted compounds showed low activities. Different heterocyclic groups were introduced to modify the N1 position. Several analogues showed similar or in the case of 53 and 59 slightly better activities than 3. In summary, none of the analogues had significantly superior potency over the parent compound against CDK2/cyclin A. Nine compounds were tested for inhibition of 12 CDK/cyclin complexes. Compounds 3, 53, 59 displayed significant inhibitory activities against CDK2/cyclin E with IC50 values of 27, 2, and 9 nM respectively. However, the binding affinities (Kd) of compounds 3, 53 and 59 for CDK2 in the absence of cyclins were in the low micromolar range. The three compounds did not inhibit MCF-7 cancer cell proliferation. Lead compound 3 was tested to determine its cell permeability using the MDCK cell line. The data suggested that compound 3 could be cellularly active, but the low recovery rates of 3 for both assays (AP-BL and BL-AP) of approximately 50% suggested that compound 3 might being trapped in the lysosome. As compounds 3, 53 and 59 inhibit the enzymatic activity of the activated CDK2/cyclin A complex, phosphorylated in vitro by CDK-activating kinase 1 [17]), but shows less binding potential towards free CDK2, these compounds may interact with the intact CDK2/cyclin complexes at a site different from the ATP site. CDK2 undergoes large conformational changes upon interaction with cyclins, and it is conceivable that these compounds exert inhibitory activity through interactions with a conformer of CDK2 induced by its interaction with cyclins. Co-crystal structures of CDK2-inhibitor complexes have been determined for numerous ATP site directed inhibitors including compounds that bind to a recently identified allosteric pocket [41, 42]. While co-crystal structures of CDK2 were readily obtained for other hit compounds [17], which all bind to the ATP site of CDK2, hit 3 and its analogues failed to show binding potential in co-crystallization and in-diffusion experiments. In order to gain insights into possible binding modes for compounds 3, 53, and 59, we performed computational simulations using the crystal structures of CDK2 and the CDK2/cyclin E1 complex. The docking studies indicated that the compounds could bind at the interface between CDK2 and cyclin E1 and that the predicted binding mode based on the CDK2/cyclin E1 complex is favored over the one generated from CDK2.
6. Experimental Section
General procedures.
Commercially available chemicals were used as purchased without further purification. All solvents were dried before use. All reactions with air- or moisture-sensitive reagents were carried out under nitrogen atmosphere. The 1H-NMR spectra were obtained at 400 MHz. For 1H NMR spectra, chemical shifts were given in parts per million (ppm) and were referenced to tetramethyl silane (TMS) peak as an internal standard or the residual solvent peak. 13C NMR spectra were recorded at 100 MHz. Chemical shifts were reported in ppm and were referenced to the appropriate residual solvent peak. High-resolution mass spectrometry (HRMS) was performed by the University of Minnesota mass spectroscopy facility. Column chromatography was performed on silica gel (SiliaFlash P60 230–400 mesh).
Biochemical methods.
Reagents and compounds for biochemical experiments were purchased from Sigma–Aldrich unless otherwise indicated. Expression, purification and activation of the CDK2/cyclin A complex were performed as described [41]. Protein concentration was determined using the Coomassie reagent from BioRad with bovine serum albumin as a standard.
Enzyme assay.
The synthetic peptide PKTPKKAKKL [43] served as a substrate for the activated CDK2/cyclin A complex, and the formation of ADP from ATP was coupled to the oxidation of NADH using pyruvate kinase (PK) and lactate dehydrogenase (LDH), monitored at 340 nm [17]. HTS and dose-response assays were carried out in 384-well plates at room temperature in 50 mM Tris/HCl buffer (pH 7.5) containing 10 mM MgCl2, 0.24 mM NADH, 5 mM DTT, 6 U mL−1 LDH, 10 U mL−1 PK, 1 mM phosphoenolpyruvate, 5 % (v/v) DMSO, and 0.01 mg mL−1 activated CDK-cyclin A complex (specific activity 6 U/mg). Inhibitor was added to the mixture, and the reaction was started by the addition of 0.15 mM ATP and 0.5 mM peptide substrate. IC50 values were obtained by fitting the data to equation 1:
| (equation 1) |
where A/A0 is the activity remaining relative to the control without inhibitor (A0), [I] is the concentration of the inhibitor, and n is the Hill slope coefficient. Non-linear regression analysis was performed using SigmaPlot (Systat Software). The IC50 data were generated by conduction three independent assays.
Enzyme assays with CDK2 cyclin complexes was performed by Reaction Biology.
Reaction Biology protocol: The kinase assay method is briefly described below. The substrates histone H1 (20 μM) for most CDK/cyclins, RB protein (3 μM,) for CDK4/cyclin D1 and CDK6/cyclin D1, myelin basic protein (20 μM) for CDK7/cyclin H, peptide substrate, (KTFCGTPEYLAPEVRREPRILSEEEQEMFRDFDYIADWC, 20 μM) for CDK9/cyclin T1 and ATP (10 μM) were added to the base reaction buffer containing 20 mM HEPES (pH 7.5), 10 mM MgCl2, 1 mM EGTA, 0.02% Brij35, 0.02 mg/mL BSA, 0.1 mM Na3VO4, 2 mM DTT, and 1% DMSO. Cofactors were added to the substrate solution in the base reaction buffer followed by CDK complexes. Compound solutions in DMSO were added to the kinase reaction mixture, and then the reaction mixture was incubated at rt for 20 min. After that, 33P-ATP was added to the reaction mixture. Following incubating the reaction mixture at rt for 2 h, reactions were spotted on P81 ion exchange paper and the kinase activity was detected by filter-binding method. The kinase profiling was carried out at 1 mM single dose in duplicate. The IC50 data were determined at a 10-dose single assay.
Kinase binding assay for Kd determining was performed by DiscoverX.
DiscoverX protocol: The kinase binding assay was performed according to the reported method [44]. For the assay, an E. coli host from the BL21 strain was used to prepare kinase-tagged T7 phage strains. Following growing to log-phase, E. coli were infected with T7 phage, and then incubated with shaking at 32 °C until lysis. Cell debris was removed from the lysates by centrifugation and filtration. Affinity resins for kinase binding assay were prepared by treating streptavidin-coated magnetic beads with biotinylated small molecule ligands for 30 min at room temperature. To remove unbound ligands and to reduce non-specific binding, the liganded beads were treated with excess biotin and washed with blocking buffer (SeaBlock (Pierce), 1% BSA, 0.05% Tween 20, 1 mM DTT). Binding reactions were carried out by combining a kinase, liganded affinity beads, and test compounds in 1x binding buffer (20% SeaBlock, 0.17x PBS, 0.05% Tween 20, 6 mM TDD).
Kd values were measured with an 11-point, 3-fold compound dilution series with three DMSO control points. Following acoustic transfer of compounds in 100% DMSO, the compounds were diluted directly into the assays maintaining the final concentration of DMSO was 0.9%. Binding reactions were performed in polypropylene 384-well plate with each well with a final volume of 0.02 mL. Following 1 h incubation of the assay plate at rt with shaking, the affinity beads were washed with wash buffer (1x PBS, 0.05% Tween 20). After that, the beads were resuspended in elution buffer (1x PBS, 0.05% Tween 20, 0.5 mM non-biotinylated affinity ligand) and incubated at rt with shaking for 30 min. The kinase concentrations in the eluates were then measured by qPCR. The binding constants (Kd values) were calculated with a standard dose-response curve using the Hill equation shown below. The possible effects of the aggregation of the molecules of hemoglobin on its dissociation curves [45].
| (equation 2) |
The Hill Slope was set to −1.
Curves were fitted using a non-linear least square fit with the Levenberg-Marquardt algorithm [46]. The assay was performed in duplicate.
Antiproliferation assay.
Upon harvesting MCF-7 cells, 5000 cells were plated out in each well of 96-well plates. Following overnight incubation at 37 °C in an incubator with 5% CO2, cells were inoculated with various concentrations of compounds. Compounds 3, 53, and 59 were tested at 8 different concentrations ranges from 0.00256 μM to 200 μM. Staurosporine (IC50 = 282 nM) was used as a positive control at 8 different concentrations ranges from 0.000256 μM to 20 μM. After the cells were incubated in the incubator (37 °C, 5% CO2) for 72 h, 10 μL of alamarBlue was added in each well of the plates. Following 90 min incubation at 37 °C, optical density of fluorescence (excitation at 530 nm, emission at 590 nm) from each well was recorded to determine the population of the proliferating cells.
MDCK assay (performed by Pharmaron).
Pharmaron protocol: The MDCK plate from the incubator was washed twice with pre-warmed HBSS (10 mM HEPES, pH 7.4), and then incubated at 37 °C for 30 minutes. The stock solutions of a test compound and controls were diluted in DMSO to prepare 0.2 mM solutions, and further diluted with HBSS (10 mM HEPES, pH 7.4) to prepare 1 μM working solutions. The final concentration of DMSO in the incubation system was 0.5%. The rate of drug transport in the apical to basolateral direction was measured by adding 75 μL of 1 μM working solutions of test compound and controls to the Transwell inserts (apical compartment) and adding 235 μL of HBSS (10 mM HEPES, pH 7.4) to the wells in the receiver plate (basolateral compartment). The assay was performed in duplicate. The rate of drug transport in the basolateral to apical direction was measured by adding 235 μL of 1 μM working solutions of a test compound and controls to the receiver plate wells (basolateral compartment) and adding 75 μL of HBSS (10 mM HEPES, pH 7.4) to the Transwell inserts (apical compartment). Time 0 samples were prepared by transferring 50 μL of 1 μM working solution to wells of the 96-deepwell plate, followed by the addition of 200 μL cold methanol containing appropriate internal standards (100 nM alprazolam, 200 nM labetalol, 200nM caffeine and 200 nM diclofenac). Following incubating the plates at 37 °C for 2 h, 50 μL samples from donor sides (apical compartment for Ap→Bl flux, and basolateral compartment for Bl→Ap) and receiver sides (basolateral compartment for Ap→Bl flux, and apical compartment for Bl→Ap) were transferred to wells of a new 96-well plate. Then, 200 μL cold methanol containing appropriate internal standards (100 nM alprazolam, 200 nM labetalol, 200nM caffeine and 200 nM diclofenac) to the wells of the new 96-well plate. Samples were mixed by vortexing for 5 minutes, and then centrifuged at 3,220 g for 40 min. 100 μL of the supernatants were mixed with an appropriate volume of ultra-pure water before LC-MS/MS analysis. For the measurement of Lucifer Yellow leakage after 2 h transport, the stock solution of Lucifer yellow in water was prepared and then diluted with HBSS (10 mM HEPES, pH 7.4) to prepare the final concentration of 100 μM. 100 μL of the Lucifer yellow solution was added to each Transwell insert (apical compartment), and 300 μL of HBSS (10 mM HEPES, pH 7.4) to the wells in the receiver plate (basolateral compartment). The plates were Incubated at 37 °C for 30 min. 80 μL of samples from the apical and basolateral wells (using the basolateral access holes) were transferred to wells of new 96 wells plates. The Lucifer Yellow fluorescence (to monitor monolayer integrity) signal was measured in a fluorescence plate reader (excitation at 485 nm excitation and emission at 530 nm). The MDCK cell monolayers were checked for integrity and were found to be intact. The assay was performed in duplicate.
Experimental protocol for Molecular Modeling.
CDK2 (PDB ID: 1B38) and CDK2/cyclin E1 (PDB ID: 5L2W) crystal structures were imported from the PDB data bank (www.rcsb.org). FTMap solvent mapping was performed at ftmap.bu.edu [40]. Imported PDB structures were prepared for the computational simulations by assigning bond orders and H atoms, generating zero-order bonds to metals and metal binding states, removing waters, and reassigning H-bond networks followed by performing restrained minimization of H atoms. Grids were generated in the protein-protein interface domain using Glide: van der Waals radius scaling factor 1.0, partial charge cutoff 0.25, and the grid box dimension of 15 Å × 15 Å × 15 Å. Ligands were prepared for the docking simulations using LigPrep [47]. Docking simulations were performed in the SP mode followed by the XP mode using Glide [48]. MM-GBSA relative binding free energy calculations of the compounds in the target proteins were performed with Prime [49].
Chemistry.
3,6,6-Trimethyl-1-(pyridin-2-yl)-6,7-dihydro-1H-indazol-4(5H)-one (2) [18].
Commercially available 2-acetyl-5,5-dimethy1-cyclohexane-1,3-dione (1, 500 mg, 2.74 mmo1), and 2-hydrazino-pyridine (300 mg, 2.75 mmo1) were dissolved in ethanol (15 m1). The solution was stirred at 80 °C for 4 h. After evaporation of the solvent, the residue was purified by silica column chromatography (EtOAc:hexanes 1:1) to afford compound 2 (586 mg, 84 %) as a white solid. Mp = 125–127 °C; 1H NMR (400 MHz, CDCl3) δ 8.45 (ddd, J = 4.9, 1.9, 0.8 Hz, 1H), 7.94 (dt, J = 8.3, 0.9 Hz, 1H), 7.83 (ddd, J = 8.3, 7.4, 1.8 Hz, 1H), 7.23 (ddd, J = 7.3, 4.9, 1.0 Hz, 1H), 3.30 (s, 2H), 2.55 (s, 3H), 2.40 (s, 2H), 1.14 (s, 6H); 13C NMR (100 MHz, CDCl3) δ 194.1, 152.7, 150.6, 150.2, 147.7, 138.6, 121.7, 118.0, 115.6, 52.5, 39.0, 35.4, 28.6, 13.5; HRMS (ESI) calcd for [C15H17N3O + H]+ 256.1450, found 256.1453.
7-Bromo-3,6,6-trimethyl-1-(pyridin-2-yl)-6,7-dihydro-1H-indazol-4(5H)-one (3) [18].
To a solution of compound 2 (300 mg, 1.18 mmol) in CHCl3 (8 mL) was added N-bromosuccinimide (209 mg, 1.18 mmol) and the mixture was stirred at 60 °C for 6 h. After cooling the reaction mixture, CH2Cl2 (10 mL) was added. The solution was washed with water (2 × 10 mL), dried over anhydrous Na2SO4, and evaporated under reduced pressure. The residue was purified by silica column chromatography (EtOAc:hexanes 1:5) to afford compound 3 as a white solid (238 mg, 61%). Mp = 149–154 °C; 1H NMR (400 MHz, CDCl3) δ 8.50 (ddd, J = 4.9, 1.8, 0.8 Hz, 1H), 8.00 (d, J = 8.3 Hz, 1H), 7.85 (ddd, J = 8.3, 7.4, 1.9 Hz, 1H), 7.27 (ddd, J = 7.4, 4.8, 1.0 Hz, 1H), 6.39 (d, J = 1.3 Hz, 1H), 2.87 (dd, J = 16.9, 0.8 Hz, 1H), 2.55 (s, 3H), 2.27 (dd, J = 16.8, 1.4 Hz, 1H), 1.40 (s, 3H), 1.26 (s, 3H); 13C NMR (100 MHz, CDCl3) δ 192.9, 152.6, 150.1, 149.6, 147.9, 138.7, 122.0, 116.6, 115.4, 52.9, 48.7, 39.7, 29.6, 25.4, 13.5; HRMS (ESI) calcd for [C15H16BrN3O + H]+ 334.0555, found 334.0546.
7-Fluoro-3,6,6-trimethyl-1-(pyridin-2-yl)-6,7-dihydro-1H-indazol-4(5H)-one (4) [18].
To a solution of compound 3 (150 mg, 0.449 mmol) in acetonitrile (7 mL) were added silver fluoride (114 mg, 0.898 mmol) and calcium fluoride (140 mg, 1.80 mmol). The mixture was stirred at 80 °C for 22 h. After cooling the reaction mixture, the precipitate was filtered and the filtrate was evaporated under reduced pressure. The crude product was purified by silica column chromatography (EtOAc:hexanes 1:2) to afford compound 4 as a white solid (13 mg, 12%). Mp = 91–99 °C; 1H NMR (400 MHz, CDCl3) δ 8.52 (ddd, J = 4.8, 1.8, 0.7 Hz, 1H), 8.00 (d, J = 8.3 Hz, 1H), 7.86 (ddd, J = 8.2, 7.4, 1.8 Hz, 1H), 7.28 (ddd, J = 7.4, 4.9, 1.0 Hz, 1H), 6.28 (d, J = 48.2 Hz, 1H), 2.86 (d, J = 16.6 Hz, 1H), 2.57 (s, 3H), 2.27 (dd, J = 16.5, 1.0 Hz, 1H), 1.32 (d, J = 2.0 Hz, 3H), 1.05 (s, 3H); 13C NMR (100 MHz, CDCl3) δ 193.5, 152.2, 150.0, 148.2, 144.7, 138.8, 122.4, 118.4, 115.4, 88.2, 86.4, 47.7, 39.2, 39.0, 25.4, 25.0, 13.4; HRMS (ESI) calcd for [C15H16FN3O + Na]+ 296.1170, found 296.1173.
7-Methoxy-3,6,6-trimethyl-1-(pyridin-2-yl)-6,7-dihydro-1H-indazol-4(5H)-one (5) [18].
A solution of 0.1 N silver nitrate in methanol (1.45 mL, 0.145 mmol) was added to a solution of compound 3 (24.2 mg, 0.0724 mmol) in acetone (1.45 mL). After stirring for 24 h, 1.75 ml of 0.1 N silver nitrate solution in methanol was added. The reaction mixture was stirred for 4 days and then filtered. The solvent was evaporated and the residue was purified by preparative TLC (silica gel, EtOAc:hexanes 1:2) to afford compound 5 as a white solid (4.3 mg, 21%). Mp = 99–104 °C; 1H NMR (400 MHz, CDCl3) δ 8.48 (ddd, J = 4.9, 1.8, 0.7 Hz, 1H), 7.98 (d, J = 8.3 Hz, 1H), 7.86 (ddd, J = 8.2, 7.4, 1.8 Hz, 1H), 7.27 (ddd, J = 7.4, 4.8, 1.0 Hz, 1H), 5.32 (s, 1H), 3.37 (s, 3H), 2.87 (d, J = 16.8 Hz, 1H), 2.55 (s, 3H), 2.14 (dd, J = 16.8, 0.7 Hz, 1H), 1.26 (s, 3H), 1.02 (s, 3H); 13C NMR (100 MHz, CDCl3) δ 194.4, 153.0, 150.2, 149.8, 147.5, 138.8, 122.3, 117.6, 116.1, 77.8, 59.1, 47.9, 40.2, 26.2, 25.8, 13.5; HRMS (ESI) calcd for [C16H19N3O2 + Na]+ 308.1369, found 308.1364.
7-Hydroxy-3,6,6-trimethyl-1-(pyridin-2-yl)-6,7-dihydro-1H-indazol-4(5H)-one (6) [18].
A solution of 0.1 N silver nitrate in H2O (6.0 mL, 0.60 mmol) was added to a solution of compound 3 (100 mg, 0.299 mmol) in acetone (6.0 mL). After stirring for 15 h, 3.0 mL of 0.1 N silver nitrate solution in H2O was added. The reaction mixture was then stirred for 5 h. The reaction mixture was filtered through Celite and evaporated to remove acetone. The residual water solution was extracted with EtOAc (3 × 10 mL). The organic layer was dried over anhydrous Na2SO4 and evaporated. The residue was purified by preparative TLC (silica gel, EtOAc:hexanes 1:2) to afford compound 6 as a white solid (57 mg, 70%). Mp = 130–133 °C; 1H NMR (400 MHz, CDCl3) δ 8.46 (d, J = 5.0 Hz, 1H), 8.04 (d, J = 8.3 Hz, 1H), 7.94 (td, J = 7.8, 1.6 Hz, 1H), 7.32 (t, J = 6.2 Hz, 1H), 5.70 (s, 1H), 4.67 (s, 1H), 2.86 (d, J = 16.0 Hz, 1H), 2.55 (s, 3H), 2.23 (d, J = 16.0 Hz, 1H), 1.27 (s, 3H), 1.07 (s, 3H); 13C NMR (100 MHz, CDCl3) δ 194.2, 151.8, 150.9, 150.3, 147.4, 139.8, 122.2, 117.2, 116.1, 68.7, 48.8, 38.5, 26.7, 25.1, 13.4; HRMS (ESI) calcd for [C15H17N3O2 + Na]+ 294.1213, found 294.1202.
7-Chloro-3,6,6-trimethyl-1-(pyridin-2-yl)-6,7-dihydro-1H-indazol-4(5H)-one (7).
To a solution of compound 6 (30 mg, 0.11 mmol) and pyridine (36 μL, 0.44 mmol) in Et2O (0.5 mL) was added thionyl chloride (16 μL, 0.22 mmol) at 0 °C. The reaction mixture was stirred at room temperature for 2 h. The mixture was then poured to ice-cold H2O (5 mL) and extracted with EtOAc (10 mL). The organic layer was washed with H2O (5 mL), brine (2 × 5 mL), NH4Cl (2 × 5 mL) and dried over anhydrous Na2SO4. After evaporation under reduced pressure, the residue was purified by silica column chromatography (EtOAc:hexanes 1:2) to furnish compound 7 as a white solid (22 mg, 70%). Mp = 141–146 °C; 1H NMR (400 MHz, CDCl3) δ 8.50 (dd, J = 4.9, 1.2 Hz, 1H), 8.00 (d, J = 8.3 Hz, 1H), 7.86 (ddd, J = 8.3, 7.5, 1.9 Hz, 1H), 7.27 (ddd, J = 7.3, 5.0, 0.9 Hz, 1H), 6.22 (d, J = 1.0 Hz, 1H), 2.93 (d, J = 16.8 Hz, 1H), 2.55 (s, 3H), 2.26 (dd, J = 16.8, 1.2 Hz, 1H), 1.37 (s, 3H), 1.20 (s, 3H); 13C NMR (100 MHz, CDCl3) δ 193.1, 152.5, 150.1, 148.7, 147.9, 138.8, 122.1, 116.9, 115.3, 59.6, 47.8, 39.9, 27.7, 26.3, 13.5; HRMS (ESI) calcd for [C15H16ClN3O + Na]+ 312.0874, found 312.0843.
3,6,6,7-Tetramethyl-1-(pyridin-2-yl)-6,7-dihydro-1H-indazol-4(5H)-one (8).
To a solution of compound 2 (70 mg, 0.27 mmol) in THF (1 mL) was slowly added n-BuLi (175 μL, 0.28 mmol, 1.6 M solution in hexanes) at −78 °C. The reaction mixture was stirred at −78 °C for 30 min and further stirred at 0 °C for 1 h. After the mixture was cooled down to −78 °C, methyl iodide (21 μL, 0.33 mmol) was added. The mixture was stirred at −78 °C for 30 min and further stirred at room temperature for 16 h. The mixture was then poured into cold brine (0.5 mL) and extracted with EtOAc (5 mL). The organic layer was washed with brine (2 × 5 mL) and dried over anhydrous Na2SO4. After evaporation under reduced pressure, the residue was purified by preparative TLC (silica gel, EtOAc:hexanes 1:2) to afford compound 8 as a pale yellow solid (19 mg, 25%). Mp = 80–83 °C; 1H NMR (400 MHz, CDCl3) δ 8.50 (ddd, J = 4.9, 1.8, 0.8 Hz, 1H), 7.94 (dt, J = 8.3, 0.8 Hz, 1H), 7.83 (ddd, J = 8.3, 7.4, 1.9 Hz, 1H), 7.22 (ddd, J = 7.3, 4.9, 1.0 Hz, 1H), 3.36 (d, J = 18.2 Hz, 1H), 3.24 (d, J = 18.2 Hz, 1H), 2.55 (s, 3H), 2.35 (q, J = 7.1 Hz, 1H), 1.17 (d, J = 7.1 Hz, 3H), 1.15 (s, 3H), 1.00 (s, 3H); 13C NMR (100 MHz, CDCl3) δ 197.4, 152.7, 150.5, 149.5, 147.7, 138.6, 121.6, 117.2, 115.5, 52.8, 38.3, 37.8, 29.0, 23.5, 13.5, 10.2; HRMS (ESI) calcd for [C16H19N3O + Na]+ 292.1420, found 292.1413.
7-(Dimethylamino)-3,6,6-trimethyl-1-(pyridin-2-yl)-6,7-dihydro-1H-indazol-4(5H)-one (9) [18].
To a solution of compound 3 (60 mg, 0.18 mmol) in THF (1.8 mL) was added 30% dimethylamine in ethanol (3.6 mL). The mixture was stirred at 40 °C with 300 W power in a microwave apparatus for 38 h and then evaporated. The residue was diluted with CH2Cl2 (10 mL), washed with water (5 mL) and brine (2 × 5 mL), dried over anhydrous Na2SO4, and evaporated. The residue was purified by preparative TLC (silica gel, EtOAc:hexanes 1:1) to afford compound 9 as a pale yellow solid (11 mg, 21%). Mp = 59–67 °C; 1H NMR (400 MHz, CDCl3) δ 8.46 (ddd, J = 4.8, 1.8, 0.8 Hz, 1H), 7.94 (d, J = 8.3 Hz, 1H), 7.85 (ddd, J = 8.2, 7.4, 1.8 Hz, 1H), 7.26 (m, 1H), 4.67 (s, 1H), 2.82 (d, J = 17.3 Hz, 1H), 2.57 (s, 3H), 2.25 (s, 6H), 2.11 (d, J = 17.3 Hz, 1H), 1.23 (s, 3H), 1.02 (s, 3H); 13C NMR (100 MHz, CDCl3) δ 194.9, 152.8, 150.1, 150.0, 147.5, 138.6, 122.1, 118.9, 116.2, 64.6, 48.7, 44.3, 40.7, 27.8, 27.0, 13.6; HRMS (ESI) calcd for [C17H22N4O + H]+ 299.1866, found 299.1868.
3,6,6-Trimethyl-7-(phenylthio)-1-(pyridin-2-yl)-6,7-dihydro-1H-indazol-4(5H)-one (10) [18].
A solution of compound 3 (160 mg, 0.48 mmol), and sodium thiophenate (78 mg, 0.60 mmol) in dry DMF (10 ml) was stirred at room temperature for 7 h. The reaction mixture was then poured into brine (10 mL) and extracted with CH2Cl2 (2 × 5 mL). The combined organic layer was washed with brine (2 × 10 mL), dried over anhydrous Na2SO4 and evaporated. The residue was purified by silica column chromatography (EtOAc:hexanes 1:5) to furnish compound 10 as a solid (138 mg, 79%). Mp = 60–88 °C; 1H NMR (400 MHz, CDCl3) δ 8.25 (ddd, J = 4.8, 1.6, 0.8 Hz, 1H), 7.77 (d, J = 8.2 Hz, 1H), 7.74 (dd, J = 7.0, 1.8 Hz, 1H), 7.26–7.24 (m, 2H), 7.15–7.11 (m, 4H), 5.82 (d, J = 1.1 Hz, 1H), 2.99 (dd, J = 17.1, 0.6 Hz, 1H), 2.53 (s, 3H), 2.18 (dd, J = 17.1, 1.2 Hz, 1H), 1.30 (s, 3H), 1.22 (s, 3H); 13C NMR (100 MHz, CDCl3) δ 193.4, 152.7, 150.3, 150.0, 147.4, 138.5, 134.3, 132.5, 128.6, 127.5, 121.8, 117.5, 115.7, 52.1, 48.6, 40.3, 28.1, 27.7, 13.6; HRMS (ESI) calcd for [C21H21N3OS + Na]+ 386.1298, found 386.1301.
3,6,6-Trimethyl-7-(phenylsulfonyl)-1-(pyridin-2-yl)-6,7-dihydro-1H-indazol-4(5H)-one (11) [18].
To a solution of compound 10 (98 mg, 0.27 mmol) in MeOH (2.5 mL) was slowly added a solution of oxone (826 mg, 1.34 mmol) in H2O (2.5 mL) at 0 °C. The reaction mixture was stirred at room temperature for 3 h and then diluted with H2O (10 mL). The mixture was extracted with CH2Cl2 (3 × 10 mL) and the combined organic layer was washed with brine (2 × 10 mL), dried over anhydrous Na2SO4 and evaporated. The residue was purified by preparative TLC (silica gel, EtOAc:hexanes 1:1) to afford compound 11 as a white solid (72 mg, 68%). Mp = 160–164 °C; 1H NMR (400 MHz, CDCl3) δ 8.33 (ddd, J = 4.9, 1.8, 0.8 Hz, 1H), 7.59 (ddd, J = 8.3, 7.4, 1.9 Hz, 1H), 7.38 (d, J = 8.3 Hz, 1H), 7.33 (m, 3H), 7.18–7.10 (m, 3H), 6.29 (s, 1H), 3.65 (d, J = 18.0 Hz, 1H), 2.58 (s, 3H), 2.27 (d, J = 18.0 Hz, 1H), 1.66 (s, 3H), 1.19 (s, 3H); 13C NMR (100 MHz, CDCl3) δ 193.2, 152.4, 150.5, 147.0, 142.4, 139.0, 138.5, 133.7, 128.6, 128.3, 122.1, 119.2, 115.6, 67.9, 48.7, 40.1, 30.8, 27.9, 13.6; HRMS (ESI) calcd for [C21H21N3O3S + Na]+ 418.1196, found 418.1214.
7-Azido-3,6,6-trimethyl-1-(pyridin-2-yl)-6,7-dihydro-1H-indazol-4(5H)-one (12).
A mixture of compound 3 (250 mg, 0.748 mmol) and sodium azide (194 mg, 2.98 mmol) in 80% EtOH (5mL) was stirred at 90–100 °C for 24 h, and then evaporated. The residue was purified by preparative TLC (silica gel, EtOAc:hexanes 1:2) to afford compound 12 as a yellowish solid, which was further purified by recrystallization from 60% EtOH (4mL) to give pure compound 12 as a white solid (95 mg, 43%). Mp = 125–131 °C; 1H NMR (400 MHz, CDCl3) δ 8.49 (ddd, J = 4.9, 1.8, 0.8 Hz, 1H), 8.04 (dt, J = 8.3, 0.8 Hz, 1H), 7.87 (ddd, J = 8.3, 7.4, 1.9 Hz, 1H), 7.28 (ddd, J = 7.3, 4.8, 1.0 Hz, 1H), 5.53 (d, J = 0.8 Hz, 1H), 2.77 (dd, J = 16.6, 0.6 Hz, 1H), 2.55 (s, 3H), 2.18 (dd, J = 16.6, 1.1 Hz, 1H), 1.32 (s, 3H), 1.07 (s, 3H); 13C NMR (100 MHz, CDCl3) δ 193.2, 152.3, 150.1, 147.7, 147.0, 139.0, 122.1, 117.7, 114.8, 63.4, 47.9, 39.7, 26.7, 26.0, 13.5; HRMS (ESI) calcd for [C15H16N6O + Na]+ 319.1278, found 319.1286.
7-Amino-3,6,6-trimethyl-1-(pyridin-2-yl)-6,7-dihydro-1H-indazol-4(5H)-one (13).
To a solution of compound 12 (20 mg, 0.068 mmol) in EtOH (0.7 mL) was added 10% Pd/C (2 mg). The reaction mixture was stirred under a H2 atmosphere at room temperature for 4 h. The mixture was filtered through Celite and concentrated under reduced pressure. The residue was purified by preparative TLC (silica gel, MeOH: CH2Cl2 1:9) to afford compound 13 as a pale yellow solid (13 mg, 73%). 1H NMR (400 MHz, CDCl3) δ 8.48 (ddd, J = 4.9, 1.8, 0.8 Hz, 1H), 7.99 (dt, J = 8.3, 0.8 Hz, 1H), 7.88 (ddd, J = 8.3, 7.4, 1.9 Hz, 1H), 7.27 (ddd, J = 7.3, 5.0, 1.0 Hz, 1H), 4.27 (s, 1H), 2.83 (d, J = 16.7 Hz, 1H), 2.55 (s, 3H), 2.19 (dd, J = 16.7 Hz, 1H), 2.19 (br s, 2H), 1.26 (s, 3H), 1.02 (s, 3H); 13C NMR (100 MHz, CDCl3) δ 193.9, 153.8, 152.3, 150.3, 147.8, 139.0, 121.9, 116.3, 115.7, 53.7, 47.7, 38.3, 27.2, 26.5, 13.5; HRMS (ESI) calcd for [C15H18N4O + H]+ 271.1553, found 271.1538.
3-Oxocyclohex-1-en-1-yl Acetate (15a). General procedure for the formation of 3-acyloxy-2-cyclohexenones.
(Compounds 15a-15e are also commercially available.) Acetyl chloride (1.02 mL, 14.3 mmol) was added to a solution of cyclohexane-1,3-dione (14a, 1.00 g, 8.92 mmol) and pyridine (1.08 ml, 13.4 mmol) in CHCl3 (31 mL). The reaction mixture was stirred at room temperature for 16 h. The mixture was then washed with H2O (30 mL), 0.1 N HCl (30 mL), saturated NaHCO3 (30 mL) and H2O (30 mL), dried over anhydrous Na2SO4, and evaporated. The residue was purified by silica column chromatography (EtOAc:hexanes 1:2) to afford compound 15a as a yellowish oil (790 mg, 58%). 1H NMR (400 MHz, CDCl3) δ 5.91 (t, J = 1.2 Hz, 1H), 2.53 (td, J = 3.1, 1.2 Hz, 2H), 2.41 (t, J = 6.7 Hz, 2H), 2.22 (s, 3H), 2.06 (quin, J = 6.5 Hz, 2H); 13C NMR (100 MHz, CDCl3) δ 199.6, 169.7, 167.4, 117.6, 36.7, 28.3, 21.3, 21.2.
5-Methyl-3-oxocyclohex-1-en-1-yl Acetate (15b).
Prepared from 5-methylcyclohexane-1,3-dione (14b, 1.50 g, 11.9 mmol) according to the general procedure. Yield: 75% (1.50 g); colorless oil. 1H NMR (400 MHz, CDCl3) δ 5.90 (d, J = 1.7 Hz, 1H), 2.49 (m, 2H), 2.33 (m, 2H), 2.22 (s, 3H), 2.12 (m, 1H), 1.11 (d, J = 6.3 Hz, 3H); 13C NMR (100 MHz, CDCl3) δ 199.6, 169.1, 167.4, 117.2, 45.0, 36.4, 29.1, 21.3, 20.8.
5-Isopropyl-3-oxocyclohex-1-en-1-yl Acetate (15c).
Prepared from 5-isopropylcyclohexane-1,3-dione (14c, 1.00 g, 6.49 mmol) according to the general procedure. Yield: 53% (674 mg); colorless oil. 1H NMR (400 MHz, CDCl3) δ 5.90 (s, 1H), 2.50 (dd, J = 16.0, 3.8 Hz, 1H), 2.43 (m, 2H), 2.23 (s, 3H), 2.14 (dd, J = 16.0, 13.3 Hz, 1H), 1.97 (m, 1H), 1.63 (m, 1H), 0.95 (s, 3H), 0.93 (s, 3H); 13C NMR (100 MHz, CDCl3) δ 200.0, 169.7, 167.4, 117.2, 40.8, 40.3, 32.3, 31.8, 21.3, 19.5.
5-Oxo-1,2,5,6-tetrahydro-[1,1’-biphenyl]-3-yl Acetate (15d).
Prepared from 5-phenylcyclohexane-1,3-dione (14d, 2.00 g, 10.6 mmol) according to the general procedure. Yield: 76% (1.85 g); white solid. Mp = 181–188 °C; 1H NMR (400 MHz, CDCl3) δ 7.35 (m, 2H), 7.27 (m, 3H), 6.01 (d, J = 2.2 Hz, 1H), 3.44 (m, 1H), 2.88 (ddd, J = 17.9, 11.1, 2.3 Hz, 1H), 2.67 (m, 3H), 2.23 (s, 3H); 13C NMR (100 MHz, CDCl3) δ 198.6, 168.7, 167.4, 142.1, 128.9, 127.3, 126.7, 117.4, 43.8, 39.6, 36.1, 21.3.
5,5-Dimethyl-3-oxocyclohex-1-en-1-yl 3-Methylbutanoate (15e).
Isovaleryl chloride (1.14 mL, 9.27 mmol) was added to a solution of 5,5-dimethylcyclohexane-1,3-dione (1.00 g, 7.13 mmol) and pyridine (692 μl, 8.56 mmol) in CHCl3 (25 mL). The reaction mixture was stirred at room temperature for 2 h. The mixture was then washed with H2O (30 mL), 0.1 N HCl (30 mL), and saturated NaHCO3 (30 mL), dried over anhydrous Na2SO4, and evaporated. The residue was purified by silica column chromatography (EtOAc:hexanes 1:1) to afford compound 15e as a colorless oil (1.28 g, 80%). 1H NMR (400 MHz, CDCl3) δ 5.89 (s, 1H), 2.41 (s, 2H), 2.34 (d, J = 7.1 Hz, 2H), 2.27 (s, 2H), 2.16 (m, 1H), 1.11(s, 6H), 1.01 (d, J = 6.6 Hz, 6H); 13C NMR (100 MHz, CDCl3) δ 199.4, 169.7, 168.2, 116.6, 50.8, 43.3, 42.3, 33.2, 28.2, 25.7, 22.3.
2-Acetylcyclohexane-1,3-dione (16a); general procedure for the formation of 2-acylcyclohexane-1,3-diones.
To a suspension of anhydrous aluminum chloride (1.35 g, 10.1 mmol) in 1,2-dichloroethane (15 mL) was added a solution of 3-oxocyclohex-1-en-1-yl acetate (15a, 780 mg, 5.06 mmol) in 1,2-dichloroethane (2 mL). The reaction mixture was stirred at room temperature for 2 h. The mixture was then poured into a mixture of ice (8 g) and concentrated HCl (8 mL) and extracted with CHCl3 (4 × 17 mL). The combined organic layer was washed with H2O (30 mL), dried over anhydrous Na2SO4, and evaporated. The residue was purified by silica column chromatography (EtOAc:hexanes 1:2) to afford compound 16a as a colorless oil (687 mg, 54%) [50]. 1H NMR (400 MHz, CDCl3) δ 2.67 (t, J = 6.4, 2H), 2.61 (s, 3H), 2.49 (t, J = 6.6, 2H), 1.98 (quin, J = 6.5, 2H); 13C NMR (100 MHz, CDCl3) δ 203.1, 198.6, 195.4, 113.4, 38.6, 33.3, 28.8, 19.0.
2-Acetyl-5-methylcyclohexane-1,3-dione (16b).
Prepared from 5-methyl-3-oxocyclohex-1-en-1-yl acetate (15b, 1.40 g, 8.33 mmol) according to the general procedure. Yield: 33% (456 mg); pale yellow solid. Mp = 44–46 °C; 1H NMR (400 MHz, CDCl3) δ 2.70 (ddd, J = 17.8, 4.0, 2.1 Hz, 1H), 2.61 (s, 3H), 2.57 (dd, J = 12.5, 2.0 Hz, 1H), 2.38 (dd, J = 17.9, 11.0 Hz, 1H), 2.21 (m, 2H), 1.09 (d, J = 6.3 Hz, 3H); 13C NMR (100 MHz, CDCl3) δ 202.9, 198.1, 195.3, 112.9, 46.8, 41.1, 26.4, 20.8, 14.2; HRMS (ESI) calcd for [C9H12O3 + H]+ 169.0865, found 169.0863.
2-Acetyl-5-isopropylcyclohexane-1,3-dione (16c).
Prepared from 5-isopropyl-3-oxocyclohex-1-en-1-yl acetate (15c, 600 mg, 3.06 mmol) according to the general procedure. Yield: 68% (406 mg); yellowish oil. 1H NMR (400 MHz, CDCl3) δ 2.68 (ddd, J = 17.8, 4.0, 2.4 Hz, 1H), 2.60 (s, 3H), 2.58 (dt, J = 16.5, 3.0 Hz, 1H), 2.44 (dd, J = 17.8, 11.9 Hz, 1H), 2.20 (dd, J = 16.3, 12.7 Hz, 1H), 1.87 (m, 1H), 1.60 (m, 1H), 0.95 (s, 3H), 0.94 (s, 3H); 13C NMR (100 MHz, CDCl3) δ 202.9, 198.6, 195.7, 112.9, 42.7, 37.5, 37.1, 31.6, 28.7, 19.3; HRMS (ESI) calcd for [C11H16O3 + H]+ 197.1178, found 197.1185.
2-Acetyl-5-phenylcyclohexane-1,3-dione (16d).
Prepared from 5-phenyl-3-oxocyclohex-1-en-1-yl acetate (16d, 500 mg, 2.17 mmol) according to the general procedure. Yield: 25% (127 mg); white solid. Mp = 100–104 °C; 1H NMR (400 MHz, CDCl3) δ 7.38–7.35 (m, 2H), 7.34–7.21 (m, 3H), 3.37 (m, 1H), 2.91 (m, 2H), 2.80 (m, 1H), 2.69 (dd, J = 16.5, 12.4 Hz, 1H), 2.65 (s, 3H); 13C NMR (100 MHz, CDCl3) δ 203.0, 197.8, 194.5, 141.7, 129.0, 127.3, 126.5, 113.0, 45.8, 40.5, 36.8, 28.8; HRMS (ESI) calcd for HRMS (ESI) calcd for [C14H14O3 + H]+ 231.1021, found 231.1028.
3-Methyl-1-(pyridin-2-yl)-6,7-dihydro-1H-indazol-4(5H)-one (17a).
General procedure for the formation of 4,5,6,7-tetrahydroindazoles. A mixture of 2-acetylcyclohexane-1,3-dione (16a, 350 mg, 2.27 mmo1) and 2-hydrazinopyridine (263 mg, 2.41 mmo1) in ethanol (13 mL) was stirred at 80° C for 2 h. The solvent was then removed by evaporation. The residue was purified by silica column chromatography (EtOAc:hexanes 3:1) to afford compound 17a as a pale yellow solid (405 mg, 79%). Mp = 189–195 °C; 1H NMR (400 MHz, CDCl3) δ 8.44 (dd, J = 4.9, 1.0 Hz, 1H), 7.93 (d, J = 8.3 Hz, 1H), 7.83 (ddd, J = 8.2, 7.4, 1.8 Hz, 1H), 7.22 (ddd, J = 7.3, 4.9, 0.9 Hz, 1H), 3.43 (t, J = 6.2 Hz, 2H), 2.55 (s, 3H), 2.52 (t, J = 6.5 Hz, 2H), 2.17 (quin, J = 6.4 Hz, 2H); 13C NMR (100 MHz, CDCl3) δ 194.7, 152.6, 151.6, 150.5, 147.7, 138.6, 121.7, 118.9, 115.6, 38.4, 25.2, 23.6, 13.6; HRMS (ESI) calcd for [C13H13N3O + Na]+ 250.0951, found 250.0960.
3,6-Dimethyl-1-(pyridin-2-yl)-6,7-dihydro-1H-indazol-4(5H)-one (17b).
Prepared from 2-acetyl-5-methylcyclohexane-1,3-dione (16b, 358 mg, 2.13 mmol) according to the general procedure. Yield: 65% (331 mg); pale yellow solid. Mp = 111–113 °C; 1H NMR (400 MHz, CDCl3) δ 8.46 (ddd, J = 4.9, 1.8, 0.8 Hz, 1H), 7.93 (dt, J = 8.3, 0,9 Hz, 1H), 7.83 (ddd, J = 8.3, 7.4, 1.9 Hz, 1H), 7.23 (ddd, J = 7.3, 4.8, 1.0 Hz, 1H), 3.67 (dd, J = 17.9, 4.5 Hz, 1H), 2.93 (dd, J = 17.9, 10.2 Hz, 1H), 2.54 (s, 3H), 2.54 (dd, J = 15.5, 3.1 Hz, 2H), 2.41 (m, 1H), 2.29 (dd, J = 15.6, 11.7 Hz, 1H), 1.20 (d, J = 6.5 Hz, 3H); 13C NMR (100 MHz, CDCl3) δ 194.4, 152.6, 151.3, 150.3, 147.7, 138.6, 121.7, 118.7, 115.6, 46.8, 33.2, 31.4, 21.2, 13.6; HRMS (ESI) calcd for [C14H15N3O + Na]+ 264.1107, found 264.1115.
6-Isopropyl-3-methyl-1-(pyridin-2-yl)-6,7-dihydro-1H-indazol-4(5H)-one (17c).
Prepared from 2-acetyl-5-isopropylcyclohexane-1,3-dione (16c, 330 mg, 1.68 mmol) according to the general procedure. Yield: 93% (421 mg); pale yellow solid. Mp = 93–96 °C; 1H NMR (400 MHz, CDCl3) δ 8.46 (dd, J = 4.8, 0.7 Hz, 1H), 7.92 (d, J = 8.3 Hz, 1H), 7.84 (t, J = 7.8 Hz, 1H), 7.23 (dd, J = 7.3, 4.9 Hz, 1H), 3.62 (dd, J = 17.9, 4.4 Hz, 2H), 2.98 (dd, J = 17.9, 11.4 Hz, 1H), 2.57 (dd, J = 13.2, 9.8 Hz, 1H), 2.54 (s, 3H), 2.33 (dd, J = 16.1, 12.9 Hz, 1H), 2.09 (m, 1H), 1.77 (m, 1H), 1.02 (d, J = 2.4 Hz, 3H), 1.00 (d, J = 2.4 Hz, 3H); 13C NMR (100 MHz, CDCl3) δ 194.8, 152.6, 151.8, 150.2, 147.8, 138.6, 121.7, 118.9, 115.7, 42.7, 42.2, 31.9, 28.8, 19.8, 19.6, 13.6; HRMS (ESI) calcd for [C16H19N3O + Na]+ 292.1420, found 292.1431.
3-Methyl-6-phenyl-1-(pyridin-2-yl)-6,7-dihydro-1H-indazol-4(5H)-one (17d).
Prepared from 2-acetyl-5-phenylcyclohexane-1,3-dione (16d, 290 mg, 1.26 mmol) according to the general procedure. Yield: 75% (285 mg); white solid. Mp = 163–179 °C; 1H NMR (400 MHz, CDCl3) δ 8.38 (d, J = 4.8 Hz, 1H), 7.94 (d, J = 8.3 Hz, 1H), 7.82 (td, J = 4.0, 1.6 Hz, 1H), 7.40–7.28 (m, 5H), 7.20 (dd, J = 7.3, 4.9 Hz, 1H), 3.93 (dd, J = 17.3, 4.0 Hz, 1H), 3.52 (m, 1H), 3.41 (dd, J = 17.4, 11.4 Hz, 1H), 2.86 (dd, J = 16.3, 12.5 Hz, 1H), 2.75 (dd, J = 16.3, 3.8 Hz, 1H), 2.58 (s, 3H); 13C NMR (100 MHz, CDCl3) δ 193.3, 152.5, 150.8, 150.4, 147.8, 143.1, 138.6, 128.8, 127.1, 127.0, 121.8, 118.8, 115.5, 45.6, 42.2, 33.1, 13.6; HRMS (ESI) calcd for [C19H17N3O + Na]+ 326.1264, found 326.1266.
7-Bromo-3-methyl-1-(pyridin-2-yl)-6,7-dihydro-1H-indazol-4(5H)-one (18a). General bromination procedure.
A mixture of 3-methyl-1-(pyridin-2-yl)-6,7-dihydro-1H-indazol-4(5H)-one (17a, 100 mg, 440 mmo1) and N-bromosuccinimide (102 mg, 0.572 mmo1) in CHCl3 (4 mL) was stirred at 60 °C for 20 min. After cooling the reaction mixture to room temperature, the solvent was removed by evaporation. The residue was purified by silica column chromatography (EtOAc:hexanes 1:2) to afford compound 18a as a white solid (73 mg, 54%). Mp = decompose at 292 °C; 1H NMR (400 MHz, CDCl3) δ 8.50 (ddd, J = 4.9, 1.9, 0.8 Hz, 1H), 7.99 (dt, J = 8.3, 0.9 Hz, 1H), 7.86 (ddd, J = 8.3, 7.4, 1.9 Hz, 1H), 7.27 (ddd, J = 7.4, 4.8, 1.0 Hz, 1H), 6.73 (t, J = 3.2 Hz, 1H), 2.99 (m, 1H), 2.67 (m, 1H), 2.59 (m, 1H), 2.56 (m, 1H), 2.55 (s, 3H); 13C NMR (100 MHz, CDCl3) δ 193.2, 152.5, 150.3, 148.9, 147.9, 138.8, 122.1, 118.0, 115.3, 40.8, 35.3, 33.6, 13.6; HRMS (ESI) calcd for [C13H12BrN3O + H]+ 306.0242, found 306.0244.
7-Bromo-3,6-dimethyl-1-(pyridin-2-yl)-6,7-dihydro-1H-indazol-4(5H)-one (18b).
6,7-Cis and trans isomers were prepared from 3,6-dimethyl-1-(pyridin-2-yl)-6,7-dihydro-1H-indazol-4(5H)-one (17b, 100 mg, 0.414 mmol) according to the general procedure. Yield: cis-isomer; 37% (49 mg), trans-isomer; 27% (36 mg); pale yellow solids. Cis-isomer: mp = 100–105 °C; 1H NMR (400 MHz, CDCl3) δ 8.50 (ddd, J = 4.9, 1.9, 0.8 Hz, 1H), 7.99 (dt, J = 8.3, 0.9 Hz, 1H), 7.86 (ddd, J = 8.3, 7.4, 1.9 Hz, 1H), 7.27 (ddd, J = 7.4, 4.8, 1.0 Hz, 1H), 6.55 (d, J = 2.4 Hz, 1H), 2.63 (dd, J = 16.5, 11.8 Hz, 1H), 2.54 (s, 3H), 2.48 (m, 1H), 2.40 (ddd, J = 16.5, 3.3, 1.0 Hz, 1H), 1.30 (d, J = 6.4 Hz, 3H); 13C NMR (100 MHz, CDCl3) δ 193.1, 152.5, 150.2, 150.1, 147.9, 138.8, 122.1, 117.8, 115.4, 49.6, 43.2, 36.3, 19.7, 13.5; HRMS (ESI) calcd for [C14H14BrN3O + H]+ 320.0398, found 320.0390. Trans-isomer: mp = 118–122 °C; 1H NMR (400 MHz, CDCl3) δ 8.50 (ddd, J = 4.9, 1.9, 0.8 Hz, 1H), 7.99 (dt, J = 8.3, 0.9 Hz, 1H), 7.85 (ddd, J = 8.3, 7.5, 1.9 Hz, 1H), 7.27 (ddd, J = 7.4, 4.9, 1.0 Hz, 1H), 6.50 (dd, J = 2.3, 0.9 Hz, 1H), 3.18 (dd, J = 16.8, 4.6 Hz, 1H), 2.93 (m, 1H), 2.55 (s, 3H), 2.42 (ddd, J = 16.9, 2.5, 1.1 Hz, 1H), 1.21 (d, J = 7.2 Hz, 3H); 13C NMR (100 MHz, CDCl3) δ 192.8, 152.5, 150.2, 147.9, 147.4, 138.7, 122.1, 117.0, 115.3, 45.3, 42.1, 39.8, 18.9, 13.6; HRMS (ESI) calcd for [C14H14BrN3O + H]+ 320.0398, found 320.0393.
7-Bromo-6-isopropyl-3-methyl-1-(pyridin-2-yl)-6,7-dihydro-1H-indazol-4(5H)-one (18c).
6,7-Cis and trans isomers were prepared from 6-isopropyl-3-methyl-1-(pyridin-2-yl)-6,7-dihydro-1H-indazol-4(5H)-one (17c, 200 mg, 0.743 mmol) according to the general procedure. Yield: cis-isomer; 26% (34 mg), trans-isomer; 33% (42 mg); pale yellow solid. Cis-isomer: mp = 127–131 °C; 1H NMR (400 MHz, CDCl3) δ 8.51 (ddd, J = 4.9, 1.8, 0.7 Hz, 1H), 8.00 (dt, J = 8.4, 0.8 Hz, 1H), 7.86 (ddd, J = 8.3, 7.5, 1.9 Hz, 1H), 7.27 (ddd, J = 7.4, 4.9, 1.0 Hz, 1H), 6.72 (d, J = 1.9 Hz, 1H), 2.72 (ddd, J = 17.2, 3.4, 1.0 Hz, 1H), 2.56 (m, 1H), 2.54 (s, 3H), 1.85 (m, 1H), 1.12 (d, J = 6.2 Hz, 3H), 1.07 (d, J = 6.4 Hz, 3H); 13C NMR (100 MHz, CDCl3) δ 193.3, 152.6, 150.2, 150.1, 147.9, 138.8, 122.0, 118.0, 115.4, 48.4, 46.6, 40.1, 30.5, 20.5, 20.0, 13.5; HRMS (ESI) calcd for [C16H18BrN3O + Na]+ 370.0525, found 370.0516. Trans-isomer: mp = 76–84 °C; 1H NMR (400 MHz, CDCl3) δ 8.51 (ddd, J = 4.9, 1.8, 0.7 Hz, 1H), 7.99 (d, J = 8.3 Hz, 1H), 7.86 (ddd, J = 8.3, 7.4, 1.9 Hz, 1H), 7.27 (ddd, J = 7.4, 4.8, 1.0 Hz, 1H), 6.91 (dd, J = 2.2, 1.2 Hz, 1H), 3.17 (dd, J = 17.0, 4.6 Hz, 1H), 2.72 (ddd, J = 17.0, 2.5, 1.3 Hz, 1H), 2.54 (s, 3H), 2.38 (m, 1H), 1.68 (m, 1H), 1.02 (d, J = 6.0 Hz, 3H), 1.01 (d, J = 6.4 Hz, 3H); 13C NMR (100 MHz, CDCl3) δ 193.0, 152.6, 150.0, 148.0, 147.8, 138.8, 122.1, 117.9, 115.3, 52.1, 43.8, 39.1, 30.1, 21.6, 21.5, 13.5; HRMS (ESI) calcd for [C16H18BrN3O + Na]+ 370.0525, found 370.0524.
7-Bromo-3-methyl-6-phenyl-1-(pyridin-2-yl)-6,7-dihydro-1H-indazol-4(5H)-one (18d).
Prepared from 3-methyl-6-phenyl-1-(pyridin-2-yl)-6,7-dihydro-1H-indazol-4(5H)-one (17d, 200 mg, 0.743 mmol) according to the general procedure. Yield: 7% (8 mg); yellow solid. 1H NMR (400 MHz, CDCl3) δ 8.47 (ddd, J = 4.9, 1.9, 0.8 Hz, 1H), 7.97 (dt, J = 8.3, 0.9 Hz, 1H), 7.82 (ddd, J = 8.3, 7.4, 1.9 Hz, 1H), 7.28–7.18 (m, 5H), 6.95 (dd, J = 2.4, 1.0 Hz, 1H), 4.09 (m, 1H), 3.47 (dd, J = 17.3, 5.4 Hz, 1H), 2.95 (ddd, J = 17.2, 2.4, 1.1 Hz, 1H), 2.56 (s, 3H); 13C NMR (100 MHz, CDCl3) δ 192.4, 152.4, 150.2, 147.9, 147.3, 140.0, 138.7, 128.9, 127.6, 127.4, 122.0, 118.1, 115.1, 49.6, 44.2, 40.4, 13.6; HRMS (ESI) calcd for [C19H16BrN3O + H]+ 382.0555, found 382.0569.
3,7,7-Trimethyl-1-(pyridin-2-yl)-4,6,7,8-tetrahydropyrazolo[4,3-b]azepin-5(1H)-one (19) and 3,7,7-Trimethyl-1-(pyridin-2-yl)-5,6,7,8-tetrahydropyrazolo[4,3-c]azepin-4(1H)-one (20).
A mixture of compound 2 (400 mg, 1.57 mmol) and polyphosphoric acid (6.7 g) was vigorously stirred at 100 °C for 1.5 h. Sodium azide (102 mg, 1.57 mmol) was then added portionwise with stirring to the homogenized solution at 100 °C for 2 h. The solution was vigorously stirred at 100 °C for 1 h. After cooling, the solution was poured into crushed ice (67 g) and the mixture was diluted with NH4OH solution (H2O: conc. NH4OH=1:1, 12 mL). The solution having some precipitation was left at room temperature for 19 h. The slurry was filtered, washed with H2O and dried. The crude compound was purified by silica column chromatography (EtOAc:hexanes 3:1) to afford compound 19 (73 mg, 17%) and 20 (103 mg, 24%) as white solids. Compound 19: mp = 217–219 °C; 1H NMR (400 MHz, CDCl3) δ 8.40 (ddd, J = 4.9, 1.8, 0.9 Hz, 1H), 7.84–7.76 (m, 1H), 7.42 (br s, 1H), 7.17 (ddd, J = 6.9, 4.9, 1.5 Hz, 1H), 3.29 (s, 2H), 2.50 (s, 2H), 2.29 (s, 3H), 1.19 (s, 6H); 13C NMR (100 MHz, CDCl3) δ 172.8, 153.6, 147.3, 141.0, 138.3, 132.4, 121.0, 120.0, 115.9, 48.5, 40.5, 34.2, 29.3, 11.0; HRMS (ESI) calcd for [C15H18N4O + H]+ 271.1559, found 271.1562. Compound 20: mp = 149–154 °C; 1H NMR (400 MHz, CDCl3) δ 8.45 (dt, J = 4.8, 1.3 Hz, 1H), 7.83 (m, 2H), 7.24 (dd, J = 8.8, 4.5 Hz, 1H), 6.06 (br s, 1H), 3.27 (s, 2H), 3.00 (d, J = 5.8 Hz, 2H), 2.54 (s, 3H), 1.10 (s, 6H); 13C NMR (100 MHz, CDCl3) δ 168.8, 153.0, 151.9, 147.5, 144.4, 138.5, 122.0, 117.6, 115.2, 53.0, 38.8, 37.2, 26.9, 13.7; HRMS (ESI) calcd for [C15H18N4O + H]+ 271.1559, found 271.1569.
8-Bromo-3,7,7-trimethyl-1-(pyridin-2-yl)-5,6,7,8-tetrahydropyrazolo[4,3-c]azepin-4(1H)-one (21).
To a solution of compound 20 (70 mg, 0.26 mmol) in CHCl3 (1.8 mL) was added N-bromosuccinimide (60 mg, 0.34 mmol) and the mixture was stirred at 60 °C for 6 h. After cooling the reaction mixture, CH2Cl2 (10 mL) was added. The solution was washed with water (10 mL) and brine (10 mL), dried over anhydrous Na2SO4, and evaporated under reduced pressure. The residue was purified by preparative TLC (silica gel, EtOAc:hexanes 4:1) to afford compound 21 as a pale yellow solid (42 mg, 35%). Mp = 193–195 °C; 1H NMR (400 MHz, CDCl3) δ 8.49 (ddd, J = 4.9, 1.7, 0.9 Hz, 1H), 7.90–7.82 (m, 2H), 7.30 (ddd, J = 6.8, 5.0, 1.7 Hz, 1H), 6.76 (d, J = 1.48 Hz, 1H), 6.01 (br d, J = 5.24 Hz, 1H), 3.95 (dd, J = 14.7, 2.8 Hz, 1H), 2.86 (ddd, J = 14.7, 7.3, 1.5 Hz, 1H), 2.58 (s, 3H), 1.36 (s, 3H), 1.30 (s, 3H); 13C NMR (100 MHz, CDCl3) δ 166.0, 153.7, 153.3, 147.6, 142.4, 138.9, 122.4, 118.3, 113.8, 56.6, 48.6, 36.4, 29.4, 22.9, 15.1; HRMS (ESI) calcd for [C15H17BrN4O + H]+ 349.0664, found 349.0646.
7-Bromo-3,6,6-trimethyl-1-(pyridin-2-yl)-4,5,6,7-tetrahydro-1H-indazol-4-ol (22) [18].
To a solution of compound 3 (50 mg, 0.15 mmol) in MeOH/THF (3 mL/0.3 mL) was added CeCl3·7H2O (84 mg, 0.22 mmol). After the mixture was cooled to −40 °C, NaBH4 (7 mg, 0.19 mmol) was added. The reaction mixture was stirred at −10 °C for 30 min and then quenched with saturated NH4Cl (2 mL). After MeOH and THF were evaporated, the aqueous layer was extracted with EtOAc (3 × 5 mL). The combined organic layer was washed with water (2 × 10 mL) and brine (2 × 10 mL), dried over anhydrous Na2SO4, and evaporated under reduced pressure. The residue was purified by preparative TLC (silica gel, EtOAc:hexanes 1:1) to afford compound 22 as a pale yellow solid (10 mg, 20%). Mp = 65–70 °C; 1H NMR (400 MHz, CDCl3) δ 8.43 (ddd, J = 4.9, 1.8, 0.8 Hz, 1H), 7.87 (d, J = 8.3 Hz, 1H), 7.77 (ddd, J = 8.3, 7.4, 1.8 Hz, 1H), 7.16 (ddd, J = 7.3, 4.9, 1.0 Hz, 1H), 6.18 (s, 1H) 4.92 (t, J = 8.4 Hz, 1H), 2.42 (s, 3H), 1.98 (d, J = 8.7 Hz, 2H), 1.30 (s, 3H), 1.16 (s, 3H); 13C NMR (100 MHz, CDCl3) δ 153.1, 148.8, 147.6, 141.3, 138.4, 120.9, 119.1, 114.8, 63.4, 55.4, 42.4, 38.2, 30.6, 24.0, 13.0; HRMS (ESI) calcd for [C15H18BrN3O + Na]+ 358.0525, found 358.0529.
7-Bromo-3,6,6-trimethyl-1-(pyridin-2-yl)-6,7-dihydro-1H-indazol-4(5H)-one Oxime (23) [18].
A mixture of compound 3 (150 mg, 0.449 mmol), hydroxylamine hydrochloride (156 mg, 2.24 mmol) and sodium acetate (221 mg, 2.69 mmol) in 1,4-dioxane (25 mL) was refluxed for 4 h. Additional hydroxylamine hydrochloride (75 mg, 1.08 mmol) and sodium acetate (111 mg, 1.35 mmol) were added and then the mixture was refluxed for 2 h. After cooling, the mixture was poured into ice water (80 mL) and extracted with CH2Cl2 (2 × 50 mL). The combined organic layer was washed with brine (2 × 50 mL), dried over anhydrous Na2SO4, and evaporated under reduced pressure. The residue was purified by preparative TLC (silica gel, EtOAc:hexanes 1:4) to afford compound 23 as a white solid (76 mg, 48%). Mp = 177–181 °C; 1H NMR (400 MHz, CDCl3) δ 8.69 (br s, 1H), 8.47 (dd, J = 4.9, 1.1 Hz, 1H), 7.95 (d, J = 8.4 Hz, 1H), 7.81 (ddd, J = 8.4, 7.3, 1.8 Hz, 1H), 7.20 (ddd, J = 7.3, 4.9, 0.9 Hz, 1H), 6.36 (d, J = 1.0 Hz, 1H), 3.11 (d, J = 17.1 Hz, 1H), 2.56 (d, J = 17.1 Hz, 1H), 2.50 (s, 3H), 1.41 (s, 3H), 1.18 (s, 3H); 13C NMR (100 MHz, CDCl3) δ 152.9, 151.1, 147.7, 147.4, 143.6, 138.5, 121.2, 114.7, 113.3, 54.1, 37.0, 32.7, 29.7, 25.6, 15.0; HRMS (ESI) calcd for [C15H17BrN4O + H]+ 349.0664, found 349.0658.
6,6-Dimethyl-1-(pyridin-2-yl)-6,7-dihydro-1H-indazol-4(5H)-one (24).
To a solution of 5,5-dimethylcyclohexane-1,3-dione (700 mg, 4.99 mmol) in dimethylformamide dimethyl acetal (DMFDMA, 796 μL, 5.99 mmol) were added 2-hydrazino pyridine (561 mg, 5.14 mmol), H2O (12 mL) and AcOH (743 μL, 13.0 mmol). The mixture, contained in a sealed vial, was submitted to microwave irradiation for 5 min at 200 °C. After cooling the reaction mixture, the precipitate was filtered and washed with H2O. The crude compound was purified by silica column chromatography (EtOAc:hexanes 1:2) to afford compound 24 (648 mg, 54%) as a yellow solid. Mp = 111–116 °C; 1H NMR (400 MHz, CDCl3) δ 8.48 (dd, J = 4.9, 1.7 Hz, 1H), 8.07 (s, 1H), 7.99 (d, J = 8.2 Hz, 1H), 7.86 (td, J = 4.1, 1.8 Hz, 1H), 7.27 (ddd, J = 7.3, 4.9, 0.8 Hz, 1H), 3.35 (s, 2H), 2.43 (s, 2H), 1.16 (s, 3H); 13C NMR (100 MHz, CDCl3) δ 193.4, 152.7, 150.0, 147.7, 138.7, 138.6, 122.1, 120.5, 115.7, 52.1, 38.8, 35.5, 28.6; HRMS (ESI) calcd for [C14H15N3O + Na]+ 264.1107, found 264.1120.
7-Bromo-6,6-dimethyl-1-(pyridin-2-yl)-6,7-dihydro-1H-indazol-4(5H)-one (25).
Prepared from compound 24 (100 mg, 0.414 mmol) according to the general procedure for bromination. Yield: 29% (39 mg); white solid. Mp = 158–161 °C; 1H NMR (400 MHz, CDCl3) δ 8.53 (ddd, J = 4.8, 1.7, 0.7 Hz, 1H), 8.04 (s, 1H), 8.03 (d, J = 9.2 Hz, 1H), 7.89 (ddd, J = 8.2, 7.5, 1.8 Hz, 1H), 7.31 (ddd, J = 7.4, 4.9, 0.9 Hz, 1H), 6.40 (s, 1H), 2.89 (dd, J = 16.9, 0.7 Hz, 2H), 2.31 (dd, J = 16.9, 1.3 Hz, 2H), 1.42 (s, 3H), 1.26 (s, 3H); 13C NMR (100 MHz, CDCl3) δ 192.1, 152.6, 149.0, 147.9, 138.9, 138.4, 122.4, 119.4, 115.5, 52.3, 48.4, 39.7, 29.6, 25.4; HRMS (ESI) calcd for [C14H14BrN3O + Na]+ 342.0212, found 342.0185.
5,5-Dimethyl-2-(3-methylbutanoyl)cyclohexane-1,3-dione (16e).
Prepared from compound 15e (1.10 g, 4.90 mmol) according to the general procedure for the formation of 2-acylcyclohexane-1,3-diones. Yield: 51% (558 mg); yellow oil. 1H NMR (400 MHz, CDCl3) δ 2.92 (d, J = 6.9 Hz, 2H), 2.54 (s, 2H), 2.36 (s, 2H), 2.14 (m, 1H), 1.08(s, 6H), 0.97 (d, J = 6.7 Hz, 6H); 13C NMR (100 MHz, CDCl3) δ 205.0, 198.2, 195.1, 112.2, 52.7, 48.6, 47.1, 30.6, 28.2, 25.6, 22.7; HRMS (ESI) calcd for [C13H20O3 + H]+ 225.1491, found 225.1487.
3-Isobutyl-6,6-dimethyl-1-(pyridin-2-yl)-6,7-dihydro-1H-indazol-4(5H)-one (26).
Prepared from compound 16e (450 mg, 2.01 mmol) according to the general procedure for the formation of 4,5,6,7-tetrahydroindazoles. Yield: 95% (567 mg); pale orange solid. Mp = 89–93 °C; 1H NMR (400 MHz, CDCl3) δ 8.45 (ddd, J = 4.9, 1.8, 0.8 Hz, 1H), 7.96 (dt, J = 8.3, 0.8 Hz, 1H), 7.83 (ddd, J = 8.3, 7.4, 1.9 Hz, 1H), 7.23 (ddd, J = 7.3, 4.9, 1.0 Hz, 1H), 3.30 (s, 2H), 2.83 (d, J = 7.2 Hz, 2H), 2.39 (s, 2H), 2.15 (m, 1H), 1.14 (s, 6H), 0.97 (d, J = 6.7 Hz, 6H); 13C NMR (100 MHz, CDCl3) δ 193.8, 153.6, 152.8, 150.6, 147.7, 138.6, 121.6, 117.8, 115.9, 52.7, 39.0, 36.4, 35.3, 28.6, 28.0, 22.5; HRMS (ESI) calcd for [C18H23N3O + Na]+ 320.1733, found 320.1732.
7-Bromo-3-isobutyl-6,6-dimethyl-1-(pyridin-2-yl)-6,7-dihydro-1H-indazol-4(5H)-one (27).
Prepared from compound 26 (150 mg, 0.504 mmol) according to the general procedure for bromination. Yield: 58% (110 mg); yellowish oil. 1H NMR (400 MHz, CDCl3) δ 8.51 (ddd, J = 4.8, 1.9, 0.8 Hz, 1H), 8.01 (dt, J = 8.3, 0.9 Hz, 1H), 7.85 (ddd, J = 8.3, 7.4, 1.9 Hz, 1H), 7.27 (ddd, J = 7.4, 4.9, 1.0 Hz, 1H), 6.41 (d, J = 1.4 Hz, 1H), 2.87 (dd, J = 16.8, 0.9 Hz, 2H), 2.83 (dd, J = 7.2, 2.2 Hz, 2H), 2.26 (dd, J = 16.9, 1.4 Hz, 2H), 2.15 (m, 1H), 1.39 (s, 3H), 1.26 (s, 3H), 0.98 (d, J = 1.4 Hz, 3H), 0.97 (d, J = 1.4 Hz, 3H); 13C NMR (100 MHz, CDCl3) δ 192.6, 153.5, 152.7, 149.5, 147.8, 138.7, 122.0, 116.5, 115.6, 53.0, 48.9, 39.6, 36.3, 29.6, 27.9, 25.4, 22.5; HRMS (ESI) calcd for [C18H22BrN3O + H]+ 376.1024, found 376.1010.
2-Hydrazinylpyrimidine (28).
To a solution of 2-chloropyrimidine (1.0 g, 8.7 mmol) in pyridine (22 mL) was added dropwise anhydrous hydrazine (3.56 mL, 113 mmol) at 0 °C. After adding additional pyridine (11 mL), the mixture was stirred at room temperature for 2.5 h. Pyridine and hydrazine were removed under reduced pressure and then H2O (6 mL) was added to the residue. The resulting white precipitate was filtered and dried to give compound 28 as a white solid (392 mg, 41%). Mp = 93–107 °C; 1H NMR (400 MHz, DMSO-d6) δ 8.28 (d, J = 4.6 Hz, 2H), 8.10 (br s, 1H), 6.57 (t, J = 4.7 Hz, 1H), 4.12 (br s, 2H); 13C NMR (100 MHz, DMSO-d6) δ 164.3, 157.8, 110.4.
3,6,6-Trimethyl-1-(pyrimidin-2-yl)-6,7-dihydro-1H-indazol-4(5H)-one (29).
Prepared from compound 28 (123 mg, 1.12 mmol) and 2-acetyl-5,5-dimethy1-cyclohexane-1,3-dione (1, 200 mg, 1.10 mmo1) according to the general procedure for the formation of 4,5,6,7-tetrahydroindazoles. Yield: 95% (268 mg); pale yellow solid. Mp = 130–139 °C; 1H NMR (400 MHz, CDCl3) δ 8.81 (d, J = 4.8 Hz, 2H), 7.27 (t, J = 4.8 Hz, 1H), 3.30 (s, 2H), 2.60 (s, 3H), 2.42 (s, 2H), 1.16 (s, 6H); 13C NMR (100 MHz, CDCl3) δ 194.0, 158.7, 156.9, 152.2, 151.5, 118.9, 118.8, 52.3, 39.4, 35.4, 28.6, 13.7; HRMS (ESI) calcd for [C14H16N4O + H]+ 257.1402, found 257.1397.
7-Bromo-3,6,6-trimethyl-1-(pyrimidin-2-yl)-6,7-dihydro-1H-indazol-4(5H)-one (30).
Prepared from compound 29 (100 mg, 0.390 mmol) according to the general procedure for bromination. Yield: 50% (65 mg); white solid. Mp = 147–158 °C; 1H NMR (400 MHz, CDCl3) δ 8.86 (d, J = 4.8 Hz, 2H), 7.32 (t, J = 4.8 Hz, 1H), 6.26 (d, J = 1.3 Hz, 1H), 2.89 (dd, J = 16.9, 0.8 Hz, 1H), 2.61 (s, 3H), 2.30 (dd, J = 16.9, 1.4 Hz, 1H), 1.41 (s, 3H), 1.26 (s, 3H); 13C NMR (100 MHz, CDCl3) δ 192.8, 158.8, 156.6, 151.4, 151.1, 119.2, 117.5, 52.8, 48.6, 39.7, 29.7, 25.4, 13.7; HRMS (ESI) calcd for [C14H15BrN4O + H]+ 335.0507, found 335.0522.
2-Hydrazinylpyrazine (31).
To 35% hydrazine hydrate (4.48 g, 48.9 mmol) was added dropwise 2-chloropyrazine (779 μL, 8.73 mmol) at 65 °C. The reaction mixture was stirred at 65 °C for 12 h. After cooling, the reaction mixture was extracted with 10% isopropyl alcohol/CH2Cl2 (5 ×10 mL). The combined organic layer was dried over anhydrous Na2SO4 and evaporated to give compound 31 as a brown solid (718 mg, 75%). 1H NMR (400 MHz, DMSO-d6) δ 8.08 (d, J = 1.4 Hz, 1H), 7.91 (dd, J = 2.7, 1.5 Hz, 1H), 7.89 (br s, 1H), 7.68 (d, J = 2.8 Hz, 1H), 4.20 (br s, 2H); 13C NMR (100 MHz, DMSO-d6) δ 157.4, 141.3, 131.8, 131.1.
3,6,6-Trimethyl-1-(pyrazin-2-yl)-6,7-dihydro-1H-indazol-4(5H)-one (32).
Prepared from compound 31 (185 mg, 1.68 mmol) and 2-acetyl-5,5-dimethy1-cyclohexane-1,3-dione (1, 300 mg, 1.65 mmo1) according to the general procedure for the formation of 4,5,6,7-tetrahydroindazoles. Yield: 62% (261 mg); yellow solid. Mp = 179–186 °C; 1H NMR (400 MHz, CDCl3) δ 9.32 (d, J = 1.4 Hz, 1H), 8.51 (d, J = 2.6 Hz, 1H), 8.40 (dd, J = 2.6, 1.4 Hz, 1H), 3.27 (s, 2H), 2.56 (s, 3H), 2.41 (s, 2H), 1.16 (s, 6H); 13C NMR (100 MHz, CDCl3) δ 193.8, 151.4, 151.2, 148.9, 141.8, 141.3, 138.2, 118.5, 52.4, 38.8, 35.4, 28.6, 13.5; HRMS (ESI) calcd for [C14H16N4O + H]+ 257.1402, found 257.1411.
7-Bromo-3,6,6-trimethyl-1-(pyrazin-2-yl)-6,7-dihydro-1H-indazol-4(5H)-one (33).
Prepared from compound 32 (100 mg, 0.390 mmol) according to the general procedure for bromination. Yield: 59% (77 mg); white solid. Mp = 140–150 °C; 1H NMR (400 MHz, CDCl3) δ 9.36 (d, J = 1.3 Hz, 1H), 8.56 (d, J = 2.5 Hz, 1H), 8.46 (dd, J = 2.5, 1.4 Hz, 1H), 6.18 (d, J = 1.2 Hz, 1H), 2.88 (d, J = 16.8 Hz, 1H), 2.56 (s, 3H), 2.29 (dd, J = 16.9, 1.3 Hz, 1H), 1.40 (s, 3H), 1.26 (s, 3H); 13C NMR (100 MHz, CDCl3) δ 192.5, 151.1, 150.3, 148.7, 142.2, 141.4, 137.9, 117.2, 52.1, 48.7, 39.7, 29.6, 25.4, 13.5; HRMS (ESI) calcd for [C14H15BrN4O + H]+ 335.0507, found 335.0498.
4-Hydrazinylpyridine Hydrochloride (34).
4-Chloropyridine hydrochloride (3g, 20 mmol) was dissolved in 2N NaOH (12 mL). The neutralized 4-chloropyridine was extracted with Et2O (4 × 12 mL) and the combined organic layer was dried over anhydrous Na2SO4 and evaporated. The residue was dissolved in EtOH (7 mL) with 80 % hydrazine hydrate (4.26 g, 68 mmol). The mixture was stirred for 16 h at 100 °C. After cooling, the EtOH was evaporated. To the residue was added MeOH (10 mL) and the resulting slurry was filtered to afford compound 34 as a light brown solid (1.08 g, 37%). Mp = 205–217 °C; 1H NMR (400 MHz, DMSO-d6) δ 8.83 (br s, 1H), 8.03 (d, J = 5.2 Hz, 2H), 6.79 (d, J = 5.6 Hz, 2H), 4.59 (br s, 2H); 13C NMR (100 MHz, DMSO-d6) δ 158.1, 143.4, 105.5.
3,6,6-Trimethyl-1-(pyridin-4-yl)-6,7-dihydro-1H-indazol-4(5H)-one (35).
A mixture of 2-acetyl-5,5-dimethy1-cyclohexane-1,3-dione (1, 350 mg, 1.92 mmo1), 4-hydrazino-pyridine hydrochloride (285 mg, 1.96 mmo1) and pyridine (233 μL, 2.88 mmol) in ethanol (15 m1) was stirred at 80 °C for 2 h. The solvent was then removed by evaporation. The residue was purified by silica column chromatography (MeOH:CH2Cl2 1:9) to afford compound 35 as a pale yellow solid (478 mg, 98%). Mp = 105–111 °C; 1H NMR (400 MHz, CDCl3) δ 8.74 (dd, J = 4.8, 1.5 Hz, 2H), 7.51 (dd, J = 4.7, 1.6 Hz, 2H), 2.92 (s, 2H), 2.55 (s, 3H), 2.43 (s, 2H), 1.14 (s, 6H); 13C NMR (100 MHz, CDCl3) δ 193.2, 151.2, 151.1, 149.3, 145.4, 118.2, 116.5, 52.2, 37.9, 35.9, 28.4, 13.4; HRMS (ESI) calcd for [C15H17N3O + H]+ 256.1450, found 256.1454.
7-Bromo-3,6,6-trimethyl-1-(pyridin-4-yl)-6,7-dihydro-1H-indazol-4(5H)-one (36).
Prepared from compound 35 (222 mg, 0.870 mmol) according to the general procedure for bromination. Yield: 93% (270 mg); white solid. Mp = 168–173 °C; 1H NMR (400 MHz, CDCl3) δ 8.80 (dd, J = 4.7, 1.5 Hz, 1H), 7.77 (dd, J = 4.6, 1.6 Hz, 1H), 4.97 (d, J = 1.1 Hz, 1H), 2.89 (dd, J = 17.1, 0.8 Hz, 1H), 2.55 (s, 3H), 2.31 (dd, J = 17.1, 1.2 Hz, 1H), 1.39 (s, 3H), 1.15 (s, 3H); 13C NMR (100 MHz, CDCl3) δ 192.0, 151.5, 151.2, 149.0, 145.2, 117.1, 116.7, 50.8, 48.3, 40.4, 29.5, 25.2, 13.4; HRMS (ESI) calcd for [C15H16BrN3O + H]+ 334.0555, found 334.0562.
2-Hydrazinyl-3-methylpyridine (37).
A solution of 2-bromo-3-methyl pyridine (1.54 g, 8.98 mmol) and 80% hydrazine hydrate (74.5 mmol, 4.72 g) in EtOH (3 mL) was stirred at 105 °C for 40 h. After evaporating EtOH, the resulting slurry was filtered and washed with diisopropyl ether (9 mL). The solid was collected, dissolved in CH2Cl2, dried over anhydrous Na2SO4 and evaporated to give compound 37 as a white solid (394 mg, 36%). Mp = 122–130 °C; 1H NMR (400 MHz, CDCl3) δ 8.08 (dd, J = 5.0, 0.9 Hz, 1H), 7.25 (m, 1H), 6.65 (dd, J = 7.1, 5.1 Hz, 1H), 5.51 (br s, 1H), 4.03 (br s, 2H), 2.08 (s, 3H),; 13C NMR (100 MHz, CDCl3) δ 158.4, 144.9, 137.1, 116.9, 114.4, 16.2.
3,6,6-Trimethyl-1-(3-methylpyridin-2-yl)-6,7-dihydro-1H-indazol-4(5H)-one (38).
Prepared from compound 37 (213 mg, 1.73 mmol) and 2-acetyl-5,5-dimethy1-cyclohexane-1,3-dione (1, 300 mg, 1.65 mmo1) according to the general procedure for the formation of 4,5,6,7-tetrahydroindazoles. Yield: 30% (133 mg); pale yellow solid. Mp = 100–103 °C; 1H NMR (400 MHz, CDCl3) δ 8.40 (d, J = 4.7 Hz, 1H), 7.73 (d, J = 7.7 Hz, 1H), 7.31 (dd, J = 7.6, 4.7 Hz, 1H), 2.71 (s, 2H), 2.54 (s, 3H), 2.39 (s, 2H), 2.33 (s, 3H), 1.11 (s, 6H); 13C NMR (100 MHz, CDCl3) δ 193.6, 151.0, 149.9, 149.4, 146.3, 140.7, 130.1, 124.1, 116.2, 52.6, 36.4, 35.7, 28.4, 17.9, 13.4; HRMS (ESI) calcd for [C16H19N3O + H]+ 270.1606, found 270.1601.
7-Bromo-3,6,6-trimethyl-1-(3-methylpyridin-2-yl)-6,7-dihydro-1H-indazol-4(5H)-one (39) and 5-Bromo-3,6,6-trimethyl-1-(3-methylpyridin-2-yl)-6,7-dihydro-1H-indazol-4(5H)-one (40).
Two compounds (39 and 40) were obtained from compound 38 (50 mg, 0.18 mmol) according to the general procedure for bromination. Yield for 39: 39% (25 mg), yield for 40: 17% (11 mg); white solids. Compound 39: Mp = 150–160 °C; 1H NMR (400 MHz, CDCl3) δ 8.41 (dd, J = 4.6, 1.0 Hz, 1H), 7.76 (dd, J = 7.6, 0.7 Hz, 1H), 7.36 (dd, J = 7.6, 4.8 Hz, 1H), 5.36 (d, J = 0.8 Hz, 1H), 2.80 (d, J = 16.6 Hz, 1H), 2.54 (s, 3H), 2.35 (s, 3H), 2.27 (dd, J = 16.7, 1.0 Hz, 1H), 1.32 (s, 3H), 1.31 (s, 3H); 13C NMR (100 MHz, CDCl3) δ 192.4, 150.0, 149.6, 149.0, 146.3, 141.1, 130.6, 124.5, 114.9, 49.8, 48.6, 39.6, 29.5, 25.5, 18.1, 13.3; HRMS (ESI) calcd for [C16H18BrN3O + H]+ 348.0711, found 348.0703. Compound 40: Mp = 135–149 °C; 1H NMR (400 MHz, CDCl3) δ 8.41 (dd, J = 4.7, 1.0 Hz, 1H), 7.73 (dd, J = 7.6, 0.8 Hz, 1H), 7.33 (dd, J = 7.6, 4.8 Hz, 1H), 4.14 (s, 1H), 3.02 (d, J = 17.3 Hz, 1H), 2.60 (d, J = 17.5 Hz, 1H), 2.56 (s, 3H), 2.33 (s, 3H), 1.28 (s, 3H), 1.21 (s, 3H); 13C NMR (100 MHz, CDCl3) δ 186.8, 150.7, 149.7, 149.4, 146.4, 140.9, 130.0, 124.2, 113.2, 62.5, 38.9, 33.0, 27.9, 24.7, 18.0, 13.4; HRMS (ESI) calcd for [C16H18BrN3O + H]+ 348.0711, found 348.0709.
2-Hydrazinyl-5-methylpyridine (41).
A solution of 2-bromo-5-methyl pyridine (1.00 g, 5.81 mmol) and 80% hydrazine hydrate (3.64 g, 58.1 mmol) in EtOH (2 mL) was stirred at 105 °C for 5 days. After evaporating EtOH, the residue was extracted with 10% isopropyl alcohol/CH2Cl2 (4 × 5 mL). The combined organic layer was washed with H2O (2.5 mL) and the water layer was extracted with CH2Cl2 (5 mL). The combined organic layer was dried over anhydrous Na2SO4 and evaporated to give compound 41 as a red oil (563 mg, 79%). 1H NMR (400 MHz, CDCl3) δ 7.96 (dd, J = 1.3, 0.7 Hz, 1H), 7.32 (dd, J = 8.3, 2.1 Hz, 1H), 6.63 (d, J = 8.4 Hz, 1H), 5.68 (s, 1H), 3.73 (s, 2H), 2.21 (s, 3H); 13C NMR (100 MHz, CDCl3) δ 159.0, 145.8, 139.2, 123.4, 107.3, 17.4.
3,6,6-Trimethyl-1-(5-methylpyridin-2-yl)-6,7-dihydro-1H-indazol-4(5H)-one (42).
Prepared from compound 41 (355 mg, 2.88 mmol) and 2-acetyl-5,5-dimethy1-cyclohexane-1,3-dione (1, 500 mg, 2.74 mmo1) according to the general procedure for the formation of 4,5,6,7-tetrahydroindazoles. Yield: 83% (616 mg); white solid. Mp = 122–129 °C; 1H NMR (400 MHz, CDCl3) δ 8.27 (dd, J = 1.6, 0.7 Hz, 1H), 7.80 (d, J = 8.4 Hz, 1H), 7.64 (ddd, J = 8.4, 2.3, 0.6 Hz, 1H), 3.25 (s, 2H), 2.54 (s, 3H), 2.39 (s, 2H), 2.38 (s, 3H), 1.13 (s, 6H); 13C NMR (100 MHz, CDCl3) δ 194.1, 150.6, 150.3, 149.9, 147.7, 139.1, 131.4, 117.7, 115.2, 52.5, 38.8, 35.4, 28.6, 17.9, 13.5; HRMS (ESI) calcd for [C16H19N3O + H]+ 270.1606, found 270.1607.
7-Bromo-3,6,6-trimethyl-1-(5-methylpyridin-2-yl)-6,7-dihydro-1H-indazol-4(5H)-one (43).
Prepared from compound 42 (50 mg, 0.18 mmol) according to the general procedure for bromination. Yield: 81% (105 mg); yellowish oil. 1H NMR (400 MHz, CDCl3) δ 8.32 (dd, J = 1.6, 0.7 Hz, 1H), 7.87 (d, J = 8.4 Hz, 1H), 7.65 (dd, J = 8.4, 2.3 Hz, 1H), 6.36 (d, J = 1.3 Hz, 1H), 2.86 (dd, J = 16.8, 0.8 Hz, 1H), 2.54 (s, 3H), 2.39 (s, 3H), 2.26 (dd, J = 16.8, 1.4 Hz, 1H), 1.39 (s, 3H), 1.26 (s, 3H); 13C NMR (100 MHz, CDCl3) δ 192.9, 150.5, 149.8, 149.2, 147.9, 139.3, 131.8, 116.4, 114.9, 52.9, 48.7, 39.7, 29.7, 25.4, 18.0, 13.5; HRMS (ESI) calcd for [C16H18BrN3O + H]+ 348.0711, found 348.0707.
2-Hydrazinyl-3-(trifluoromethyl)pyridine (44).
A solution of 2-bromo-3-(trifluoromethyl)pyridine (500 mg, 2.21 mmol) and 80% hydrazine hydrate (22.1 mmol, 1.38 g) in EtOH (0.8 mL) was stirred at 105 °C for 29 h. After EtOH was removed by evaporation, H2O (2.5 mL) was added to the residue. The resulting slurry was filtered, washed with H2O (0.4 mL) and diisopropyl ether (0.5 mL). The filtered solid was dried under vacuum to afford compound 44 as a pale yellow solid (419 mg, 53%). 1H NMR (400 MHz, DMSO-d6) δ 11.90 (br s, 1H), 8.32 (dd, J = 4.5, 1.6 Hz, 1H), 8.10 (dd, J = 7.9, 1.6 Hz, 1H), 6.94 (dd, J = 7.9, 4.6 Hz, 1H), 5.53 (br s, 2H); 13C NMR (100 MHz, DMSO-d6) δ 152.3, 148.4, 148.1, 129.5, 113.7, 106.1; HRMS (ESI) calcd for [C6H6F3N3 + H]+ 178.0592, found 178.0599.
3,6,6-Trimethyl-1-(3-(trifluoromethyl)pyridin-2-yl)-6,7-dihydro-1H-indazol-4(5H)-one (45).
Prepared from compound 44 (380 mg, 1.78 mmol) and 2-acetyl-5,5-dimethy1-cyclohexane-1,3-dione (1, 324 mg, 1.78 mmo1) according to the general procedure for the formation of 4,5,6,7-tetrahydroindazoles. Yield: 22% (128 mg); white solid. Mp = 82–90 °C; 1H NMR (400 MHz, CDCl3) δ 8.76 (dd, J = 4.8, 1.4 Hz, 1H), 8.23 (dd, J = 8.0, 1.6 Hz, 1H), 7.59 (ddd, J = 7.9, 4.8, 0.7 Hz, 1H), 2.68 (s, 2H), 2.54 (s, 3H), 2.40 (s, 2H), 1.10 (s, 3H); 13C NMR (100 MHz, CDCl3) δ 193.5, 151.8, 151.7, 150.1, 137.5, 124.1, 123.5, 120.8, 116.8, 116.1, 52.6, 36.1, 35.8, 28.3, 13.3; HRMS (ESI) calcd for [C16H16F3N3O + H]+ 324.1324, found 324.1318.
7-Bromo-3,6,6-trimethyl-1-(3-(trifluoromethyl)pyridin-2-yl)-6,7-dihydro-1H-indazol-4(5H)-one (46) and 5-Bromo-3,6,6-trimethyl-1-(3-(trifluoromethyl)pyridin-2-yl)-6,7-dihydro-1H-indazol-4(5H)-one (47).
Two compounds (46 and 47) were obtained from compound 45 (50 mg, 0.15 mmol) according to the general procedure for bromination. Yield for 46: 11% (6.9 mg), Yield for 47: 10% (6.4 mg); pale yellow solids. Compound 46: mp = 140–152 °C; 1H NMR (400 MHz, CDCl3) δ 8.77 (dd, J = 4.8, 1.5 Hz, 1H), 8.26 (dd, J = 7.9, 1.6 Hz, 1H), 7.60 (ddd, J = 7.9, 4.8, 0.5 Hz, 1H), 5.39 (d, J = 1.1 Hz, 1H), 2.82 (dd, J = 16.7, 0.7 Hz, 1H), 2.54 (s, 3H), 2.29 (dd, J = 16.7, 1.2 Hz, 1H), 1.32 (s, 3H), 1.30 (s, 3H); 13C NMR (100 MHz, CDCl3) δ 192.4, 151.6, 150.2, 149.8, 138.1, 124.1, 123.4, 122.6, 120.7, 115.9, 49.1, 48.6, 39.7, 29.4, 25.5, 13.3; HRMS (ESI) calcd for [C16H15BrF3N3O + H]+ 402.0429, found 402.0422. Compound 47: mp = 148–161 °C; 1H NMR (400 MHz, CDCl3) δ 8.78 (dd, J = 4.8, 1.5 Hz, 1H), 8.24 (dd, J = 8.0, 1.7 Hz, 1H), 7.60 (ddd, J = 7.9, 4.8, 0.7 Hz, 1H), 4.14 (s, 1H), 3.03 (d, J = 17.2 Hz, 1H), 2.55 (s, 3H), 2.49 (d, J = 17.2 Hz, 1H), 1.28 (s, 3H), 1.19 (s, 3H); 13C NMR (100 MHz, CDCl3) δ 186.7, 151.9, 151.4, 150.1, 137.6, 124.3, 123.5, 120.8, 113.9, 62.1, 39.0, 35.5, 32.8, 27.9, 24.4, 13.3; HRMS (ESI) calcd for [C16H15BrF3N3O + H]+ 402.0429, found 402.0448.
1-(5-Chloropyridin-2-yl)-3,6,6-trimethyl-6,7-dihydro-1H-indazol-4(5H)-one (48).
Prepared from 5-chloro-2-hydrazinopyridine (300 mg, 2.09 mmol) and 2-acetyl-5,5-dimethy1-cyclohexane-1,3-dione (1, 363 mg, 1.99 mmo1) according to the general procedure for the formation of 4,5,6,7-tetrahydroindazoles. Yield: 83% (478 mg); pale orange solid. Mp = 144–151 °C; 1H NMR (400 MHz, CDCl3) δ 8.39 (dd, J = 2.5, 0.5 Hz, 1H), 7.93 (dd, J = 8.8, 0.5 Hz, 1H), 7.79 (dd, J = 8.8, 2.5 Hz, 1H), 3.26 (s, 2H), 2.54 (s, 3H), 2.39 (s, 2H), 1.14 (s, 6H); 13C NMR (100 MHz, CDCl3) δ 194.0, 150.9, 150.7, 150.5, 146.3, 138.3, 129.4, 118.2, 116.4, 52.4, 39.0, 35.4, 28.6, 13.5; HRMS (ESI) calcd for [C15H16ClN3O + H]+ 290.1060, found 290.1055.
7-Bromo-1-(5-chloropyridin-2-yl)-3,6,6-trimethyl-6,7-dihydro-1H-indazol-4(5H)-one (49).
Prepared from compound 48 (150 mg, 0.52 mmol) according to the general procedure for bromination. Yield: 60% (77 mg); white solid. Mp = 117–123 °C; 1H NMR (400 MHz, CDCl3) δ 8.46 (d, J = 2.5 Hz, 1H), 7.98 (d, J = 8.8 Hz, 1H), 7.82 (dd, J = 8.8, 2.6 Hz, 1H), 6.26 (d, J = 1.1 Hz, 1H), 2.86 (d, J = 16.9 Hz, 1H), 2.53 (s, 3H), 2.27 (dd, J = 16.8, 1.2 Hz, 1H), 1.39 (s, 3H), 1.25 (s, 3H); 13C NMR (100 MHz, CDCl3) δ 192.8, 150.7, 150.4, 149.6, 146.5, 138.5, 129.9, 116.8, 116.2, 52.7, 48.7, 39.7, 29.6, 25.4, 13.5; HRMS (ESI) calcd for [C15H15BrClN3O + H]+ 368.0165, found 368.0174.
1-(6-Chloropyridazin-3-yl)-3,6,6-trimethyl-6,7-dihydro-1H-indazol-4(5H)-one (50).
Prepared from 3-chloro-6-hydrazinopyridazine (300 mg, 2.08 mmol) and 2-acetyl-5,5-dimethy1-cyclohexane-1,3-dione (1, 360 mg, 1.98 mmo1) according to the general procedure for the formation of 4,5,6,7-tetrahydroindazoles. Yield: 73% (420 mg); pale yellow solid. Mp = 140–153 °C; 1H NMR (400 MHz, CDCl3) δ 8.22 (d, J = 9.2 Hz, 1H), 7.65 (d, J = 9.2 Hz, 1H), 3.39 (s, 2H), 2.54 (s, 3H), 2.42 (s, 2H), 1.15 (s, 6H); 13C NMR (100 MHz, CDCl3) δ 194.0, 155.4, 154.6, 151.7, 130.5, 122.4, 119.0, 52.4, 39.2, 35.4, 28.5, 13.5; HRMS (ESI) calcd for [C14H15ClN4O + H]+ 291.1013, found 291.1007.
7-Bromo-1-(6-chloropyridazin-3-yl)-3,6,6-trimethyl-6,7-dihydro-1H-indazol-4(5H)-one (51).
Prepared from compound 50 (150 mg, 0.52 mmol) according to the general procedure for bromination. Yield: 23% (30 mg); white solid. Mp = 179–192 °C; 1H NMR (400 MHz, CDCl3) δ 8.24 (d, J = 9.2 Hz, 1H), 7.68 (d, J = 9.2 Hz, 1H), 6.30 (d, J = 1.2 Hz, 1H), 2.88 (dd, J = 16.8, 0.7 Hz, 1H), 2.54 (s, 3H), 2.31 (dd, J = 16.9, 1.3 Hz, 1H), 1.39 (s, 3H), 1.26 (s, 3H); 13C NMR (100 MHz, CDCl3) δ 192.7, 155.3, 155.0, 151.6, 150.6, 130.7, 122.3, 117.7, 52.1, 48.6, 39.7, 29.5, 25.3, 13.5; HRMS (ESI) calcd for [C14H14BrClN4O + H]+ 369.0118, found 369.0108.
3,6,6-Trimethyl-1-(thiazol-2-yl)-6,7-dihydro-1H-indazol-4(5H)-one (52).
Prepared from 2-hydrazinothiazole (440 mg, 2.41 mmol) and 2-acetyl-5,5-dimethy1-cyclohexane-1,3-dione (1, 300 mg, 2.61 mmo1) according to the general procedure for the formation of 4,5,6,7-tetrahydroindazoles. Yield: 84% (529 mg); pale yellow solid. Mp = 126–131 °C; 1H NMR (400 MHz, CDCl3) δ 7.59 (d, J = 3.5 Hz, 1H), 7.13 (d, J = 3.5 Hz, 1H), 3.28 (s, 2H), 2.53 (s, 3H), 2.41 (s, 2H), 1.17 (s, 6H); 13C NMR (100 MHz, CDCl3) δ 193.6, 161.5, 151.4, 150.2, 140.4, 118.2, 116.2, 52.6, 37.6, 35.4, 28.6, 13.4; HRMS (ESI) calcd for [C13H15N3OS + H]+ 262.1014, found 262.1016.
7-Bromo-3,6,6-trimethyl-1-(thiazol-2-yl)-6,7-dihydro-1H-indazol-4(5H)-one (53).
Prepared from compound 52 (150 mg, 0.52 mmol) according to the general procedure for bromination. Yield: 51% (66 mg); white solid. Mp = 167–174 °C; 1H NMR (400 MHz, CDCl3) δ 7.66 (d, J = 3.5 Hz, 1H), 7.18 (d, J = 3.5 Hz, 1H), 6.03 (d, J = 1.2 Hz, 1H), 2.89 (dd, J = 16.9, 0.8 Hz, 1H), 2.53 (s, 3H), 2.28 (dd, J = 16.9, 1.4 Hz, 1H), 1.40 (s, 3H), 1.23 (s, 3H); 13C NMR (100 MHz, CDCl3) δ 192.3, 160.5, 151.3, 149.0, 140.8, 116.6, 50.8, 48.8, 39.7, 29.5, 25.3, 13.3; HRMS (ESI) calcd for [C13H14BrN3OS + H]+ 340.0119, found 340.0109.
1-(Benzo[d]thiazol-2-yl)-3,6,6-trimethyl-6,7-dihydro-1H-indazol-4(5H)-one (54).
Prepared from 2-hydrazinobenzothiazole (350 mg, 2.12 mmol) and 2-acetyl-5,5-dimethy1-cyclohexane-1,3-dione (1, 357 mg, 1.96 mmo1) according to the general procedure for the formation of 4,5,6,7-tetrahydroindazoles. Yield: 97% (594 mg); white solid. Mp = 234–240 °C; 1H NMR (400 MHz, CDCl3) δ 7.93 (d, J = 8.1 Hz, 1H), 7.85 (d, J = 8.0 Hz, 1H), 7.49 (ddd, J = 8.0, 7.4, 1.3 Hz, 1H), 7.38 (m, 1H), 3.41 (s, 2H), 2.55 (s, 3H), 2.44 (s, 2H), 1.21 (s, 6H); 13C NMR (100 MHz, CDCl3) δ 193.7, 160.3, 151.8, 151.2, 132.8, 126.5, 125.1, 122.7, 121.5, 118.9, 52.7, 37.9, 35.5, 28.6, 13.5; HRMS (ESI) calcd for [C17H17N3OS + H]+ 312.1171, found 312.1172.
1-(Benzo[d]thiazol-2-yl)-7-bromo-3,6,6-trimethyl-6,7-dihydro-1H-indazol-4(5H)-one (55).
Prepared from compound 54 (150 mg, 0.48 mmol) according to the general procedure for bromination. Yield: 37% (47 mg); white solid. Mp = 206–218 °C; 1H NMR (400 MHz, CDCl3) δ 8.01 (d, J = 8.2 Hz, 1H), 7.87 (d, J = 8.0 Hz, 1H), 7.52 (ddd, J = 8.0, 7.4, 1.2 Hz, 1H), 7.41 (m, 1H), 6.16 (d, J = 1.2 Hz, 1H), 2.92 (dd, J = 16.8, 0.7 Hz, 1H), 2.56 (s, 3H), 2.31 (dd, J = 16.9, 1.3 Hz, 1H), 1.45 (s, 3H), 1.27 (s, 3H); 13C NMR (100 MHz, CDCl3) δ 192.4, 159.3, 151.8, 151.3, 149.9, 132.8, 126.7, 125.4, 123.1, 121.5, 117.3, 50.7, 48.9, 39.8, 29.5, 25.3, 13.4; HRMS (ESI) calcd for [C17H16BrN3OS + H]+ 390.0276, found 390.0290.
3-(3,6,6-Trimethyl-4-oxo-4,5,6,7-tetrahydro-1H-indazol-1-yl)propanenitrile (56).
Prepared from cyanoethylhydrazine (426 mg, 5.00 mmol) and 2-acetyl-5,5-dimethy1-cyclohexane-1,3-dione (1, 844 mg, 4.63 mmo1) according to the general procedure for the formation of 4,5,6,7-tetrahydroindazoles. Yield: 61% (657 mg); pale yellow solid. Mp = 57–63 °C; 1H NMR (400 MHz, CDCl3) δ 4.25 (t, J = 6.3 Hz, 2H), 2.95 (t, J = 6.3 Hz, 2H), 2.72 (s, 2H), 2.46 (s, 3H), 2.35 (s, 2H), 1.14 (s, 6H); 13C NMR (100 MHz, CDCl3) δ 193.0, 150.0, 149.9, 116.9, 115.8, 52.4, 44.2, 35.7, 35.2, 28.5, 28.2, 19.0, 13.3; HRMS (ESI) calcd for [C13H17N3O + H]+ 232.1450, found 232.1460.
3-(7-Bromo-3,6,6-trimethyl-4-oxo-4,5,6,7-tetrahydro-1H-indazol-1-yl)propanenitrile (57).
Prepared from compound 56 (300 mg, 1.30 mmol) according to the general procedure for bromination. Yield: 51% (205 mg); white solid. Mp = 94–106 °C; 1H NMR (400 MHz, CDCl3) δ 5.01 (d, J = 1.1 Hz, 1H), 4.36 (m, 2H), 3.11 (m, 1H), 2.94 (m, 1H), 2.76 (dd, J = 16.7, 0.9 Hz, 1H), 2.46 (s, 3H), 2.23 (dd, J = 16.7, 1.2 Hz, 1H), 1.36 (s, 3H), 1.22 (s, 3H); 13C NMR (100 MHz, CDCl3) δ 191.8, 149.9, 148.6, 116.8, 114.4, 48.8, 48.1, 44.5, 39.6, 29.7, 25.5, 18.7, 13.3; HRMS (ESI) calcd for [C13H16BrN3O + H]+ 310.0555, found 310.0557.
3,6,6-Trimethyl-1-(pyrimidin-4-yl)-6,7-dihydro-1H-indazol-4(5H)-one (58).
Prepared from 4-hydrazinopyrimidine (300 mg, 2.72 mmol) and 2-acetyl-5,5-dimethy1-cyclohexane-1,3-dione (1, 460 mg, 2.52 mmo1) according to the general procedure for the formation of 4,5,6,7-tetrahydroindazoles. Yield: 92% (592 mg); white solid. Mp = 145–148 °C; 1H NMR (400 MHz, CDCl3) δ 9.05 (d, J = 1.0 Hz, 1H), 8.78 (d, J = 5.6 Hz, 1H), 7.97 (dd, J = 5.6, 1.2 Hz, 1H), 3.40 (s, 2H), 2.54 (s, 3H), 2.41 (s, 2H), 1.17 (s, 6H); 13C NMR (100 MHz, CDCl3) δ 193.9, 158.7, 158.6, 158.0, 152.1, 151.7, 119.2, 111.2, 52.4, 39.4, 35.4, 28.6, 13.6; HRMS (ESI) calcd for [C14H16N4O + H]+ 257.1402, found 257.1391.
7-Bromo-3,6,6-trimethyl-1-(pyrimidin-4-yl)-6,7-dihydro-1H-indazol-4(5H)-one (59).
Prepared from compound 58 (200 mg, 0.780 mmol) according to the general procedure for bromination. Yield: 61% (160 mg); white solid. Mp = 156–162 °C; 1H NMR (400 MHz, CDCl3) δ 9.13 (d, J = 1.1 Hz, 1H), 8.82 (d, J = 5.6 Hz, 1H), 8.00 (dd, J = 5.7, 1.2 Hz, 1H), 6.34 (d, J = 1.2 Hz, 1H), 2.88 (d, J = 16.9 Hz, 1H), 2.54 (s, 3H), 2.30 (dd, J = 16.7, 1.4 Hz, 1H), 1.42 (s, 3H), 1.25 (s, 3H); 13C NMR (100 MHz, CDCl3) δ 192.6, 159.0, 158.3, 158.1, 151.6, 151.0, 117.8, 111.2, 52.4, 48.7, 39.7, 29.6, 25.3, 13.6; HRMS (ESI) calcd for [C14H15BrN4O + H]+ 335.0507, found 335.0515.
3,6,6-Trimethyl-1-(5-(trifluoromethyl)pyridin-2-yl)-6,7-dihydro-1H-indazol-4(5H)-one (60).
Prepared from 2-hydrazinyl-5-(trifluoromethyl)pyridine (300 mg, 1.69 mmol) and 2-acetyl-5,5-dimethy1-cyclohexane-1,3-dione (1, 285 mg, 1.57 mmo1) according to the general procedure for the formation of 4,5,6,7-tetrahydroindazoles. Yield: 98% (495 mg); white solid. Mp = 117–121 °C; 1H NMR (400 MHz, CDCl3) δ 8.72 (br s, 1H), 8.13 (d, J = 8.7 Hz, 1H), 8.05 (dd, J = 8.8, 2.3 Hz, 1H), 3.34 (s, 2H), 2.55 (s, 3H), 2.41 (s, 2H), 1.16 (s, 6H); 13C NMR (100 MHz, CDCl3) δ 194.0, 151.5, 151.1, 145.1, 135.8, 124.7, 124.4, 124.0, 118.7, 114.9, 52.4, 39.3, 35.4, 28.6, 13.6; HRMS (ESI) calcd for [C16H16F3N3O + H]+ 324.1324, found 324.1317.
7-Bromo-3,6,6-trimethyl-1-(5-(trifluoromethyl)pyridin-2-yl)-6,7-dihydro-1H-indazol-4(5H)-one (61).
Prepared from compound 60 (120 mg, 0.371 mmol) according to the general procedure for bromination. Yield: 66% (99 mg); semi solid. 1H NMR (400 MHz, CDCl3) δ 8.80 (br s, 1H), 8.18 (d, J = 8.7 Hz, 1H), 8.07 (dd, J = 8.7, 2.3 Hz, 1H), 6.31 (d, J = 1.2 Hz, 1H), 2.88 (d, J = 16.9 Hz, 1H), 2.55 (s, 3H), 2.29 (dd, J = 16.9, 1.3 Hz, 1H), 1.41 (s, 3H), 1.25 (s, 3H); 13C NMR (100 MHz, CDCl3) δ 192.7, 151.0, 150.3, 145.3, 136.0, 124.8, 124.6, 124.4, 117.3, 114.9, 52.7, 48.7, 39.7, 29.6, 25.4, 13.5; HRMS (ESI) calcd for [C16H15BrF3N3O + H]+ 402.0429, found 402.0423.
2-Hydrazinyl-6-methoxypyridine (62).
A mixture of 2-bromo-6-methoxypyridine (3.00 g, 16.0 mmol) and 80% hydrazine hydrate (160 mmol, 9.98 g) was stirred at 120 °C for 3.5 h. Excess hydrazine was removed under reduced pressure and then H2O (3 mL) was added to the residue. The aqueous mixture was extracted with EtOAc (4 × 8 mL). The combined organic layer was washed with saturated NaHCO3 (5 mL), dried over anhydrous Na2SO4 and evaporated. The residue was distilled under vacuum (12 mbar) at 100 °C to afford compound 62 as a dark brown oil (909 mg, 41%). 1H NMR (400 MHz, CDCl3) δ 7.42 (t, J = 7.9 Hz, 1H), 6.23 (d, J = 7.9 Hz, 1H), 6.12 (dd, J = 8.0, 0.4 Hz, 1H), 5.69 (br s, 1H), 3.87 (s, 3H), 3.79 (br s, 2H); 13C NMR (100 MHz, CDCl3) δ 163.6, 160.4, 140.2, 98.9, 97.7, 53.3.
1-(6-Methoxypyridin-2-yl)-3,6,6-trimethyl-6,7-dihydro-1H-indazol-4(5H)-one (63).
Prepared from compound 62 (300 mg, 2.16 mmol) and 2-acetyl-5,5-dimethy1-cyclohexane-1,3-dione (1, 364 mg, 2.00 mmo1) according to the general procedure for the formation of 4,5,6,7-tetrahydroindazoles. Yield: 54% (310 mg); light orange solid. Mp = 108–115 °C; 1H NMR (400 MHz, CDCl3) δ 7.71 (t, J = 7.9 Hz, 1H), 7.50 (d, J = 7.8 Hz, 1H), 6.68 (d, J = 8.1 Hz, 1H), 3.96 (s, 3H), 3.33 (s, 2H), 2.54 (s, 3H), 2.40 (s, 2H), 1.14 (s, 6H); 13C NMR (100 MHz, CDCl3) δ 193.9, 162.8, 150.4, 150.2, 150.1, 140.9, 117.8, 108.3, 107.2, 53.9, 52.4, 39.3, 35.3, 28.7, 13.5; HRMS (ESI) calcd for [C16H19N3O3 + H]+ 286.1556, found 286.1548.
7-Bromo-1-(6-methoxypyridin-2-yl)-3,6,6-trimethyl-6,7-dihydro-1H-indazol-4(5H)-one (64).
Prepared from compound 63 (150 mg, 0.526 mmol) according to the general procedure for bromination. Yield: 72% (137 mg); white solid. Mp = 138–144 °C; 1H NMR (400 MHz, CDCl3) δ 7.73 (t, J = 8.0 Hz, 1H), 7.60 (d, J = 7.8 Hz, 1H), 6.72 (d, J = 8.1 Hz, 1H), 6.31 (s, 1H), 4.04 (s, 3H), 2.88 (d, J = 16.9 Hz, 1H), 2.54 (s, 3H), 2.28 (d, J = 16.9 Hz, 1H), 1.38 (s, 3H), 1.24 (s, 3H); 13C NMR (100 MHz, CDCl3) δ 192.7, 163.0, 150.3, 150.2, 149.1, 140.9, 116.5, 108.9, 107.1, 53.9, 53.1, 48.5, 39.7, 29.9, 25.5, 13.5; HRMS (ESI) calcd for [C16H18BrN3O2 + H]+ 364.0661, found 364.0652.
3,6,6-Trimethyl-1-(5-nitropyridin-2-yl)-6,7-dihydro-1H-indazol-4(5H)-one (65).
Prepared from 2-hydrazinyl-5-nitropyridine (1.00 g, 6.49 mmol) and 2-acetyl-5,5-dimethy1-cyclohexanes-1,3-dione (1, 1.10 g, 6.01 mmo1) according to the general procedure for the formation of 4,5,6,7-tetrahydroindazoles. Yield: 25% (448 mg); pale yellow solid. Mp = 174–181 °C; 1H NMR (400 MHz, CDCl3) δ 9.30 (dd, J = 2.7, 0.5 Hz, 1H), 8.60 (dd, J = 9.1, 2.7 Hz, 1H), 8.20 (dd, J = 9.1, 0.5 Hz, 1H), 3.37 (s, 2H), 2.55 (s, 3H), 2.42 (s, 2H), 1.17 (s, 6H); 13C NMR (100 MHz, CDCl3) δ 193.9, 156.0, 152.1, 151.9, 144.3, 141.8, 133.9, 119.3, 115.0, 52.3, 39.5, 35.4, 28.6, 13.6; HRMS (ESI) calcd for [C15H16N4O3 + H]+ 301.1301, found 301.1295.
7-Bromo-3,6,6-trimethyl-1-(5-nitropyridin-2-yl)-6,7-dihydro-1H-indazol-4(5H)-one (66).
Prepared from compound 65 (180 mg, 0.599 mmol) according to the general procedure for bromination. Yield: 70% (158 mg); white solid. Mp = 119–122 °C; 1H NMR (400 MHz, CDCl3) δ 9.38 (d, J = 2.5 Hz, 1H), 8.62 (dd, J = 9.1, 2.6 Hz, 1H), 8.25 (d, J = 9.1 Hz, 1H), 6.27 (s, 1H), 2.88 (d, J = 16.9 Hz, 1H), 2.55 (s, 3H), 2.31 (d, J = 16.9 Hz, 1H), 1.42 (s, 3H), 1.26 (s, 3H); 13C NMR (100 MHz, CDCl3) δ 192.6, 155.6, 151.8, 150.9, 144.4, 142.1, 134.1, 117.9, 115.1, 52.6, 48.6, 39.7, 29.6, 25.4, 13.6; HRMS (ESI) calcd for [C15H15BrN4O3 + H]+ 379.0406, found 379.0420.
1-(5-Aminopyridin-2-yl)-7-bromo-3,6,6-trimethyl-6,7-dihydro-1H-indazol-4(5H)-one (67).
To a solution of compound 66 (60 mg, 0.158 mmol) in EtOH (1 mL) was added SnCl2·2H2O (179 mg, 0.791 mmol). The reaction mixture was stirred at 70 °C for 15 min. After cooling, the reaction mixture was poured into icy water (13 mL) and neutralized with 5% NaHCO3 (2.8 mL). The aqueous solution was extracted with CH2Cl2 (3 × 15 mL). The combined organic layer was washed with brine (20 mL) and dried over anhydrous Na2SO4. The residue was purified by preparative TLC (silica gel, EtOAc:hexanes 2:1) to afford compound 67 as a pale yellow solid (34 mg, 62%). Mp = 163–173 °C; 1H NMR (400 MHz, CDCl3) δ 7.94 (d, J = 2.7 Hz, 1H), 7.73 (d, J = 8.8 Hz, 1H), 7.14 (dd, J = 8.7, 2.9 Hz, 1H), 6.22 (s, 1H), 3.83 (br s, 2H), 2.85 (d, J = 16.9 Hz, 1H), 2.53 (s, 3H), 2.25 (dd, J = 16.8, 1.1 Hz, 1H), 1.37 (s, 3H), 1.25 (s, 3H); 13C NMR (100 MHz, CDCl3) δ 192.8, 149.4, 148.3, 144.5, 141.6, 134.2, 124.2, 116.4, 115.8, 52.7, 48.7, 39.7, 29.7, 25.4, 13.4; HRMS (ESI) calcd for [C15H17BrN4O + H]+ 349.0664, found 349.0649.
3,6,6-Trimethyl-6,7-dihydro-1H-indazol-4(5H)-one (68).
A solution of 80% hydrazine hydrate (32.4 mmol, 2.06 g) and 2-acetyl-5,5-dimethyl-cyclohexanes-1,3-dione (1, 2.00 g, 11.0 mmo1) in EtOH (25 mL) was stirred at room temperature for 4 h. EtOH was then removed by evaporation. To the residue was added CH2Cl2 (30 mL), and then the organic solution was washed with H2O (20 mL) and brine (20 mL) and dried over anhydrous Na2SO4. After evaporation, the residue was purified by silica column chromatography (MeOH:CH2Cl2 5:95) to afford compound 68 as a pale yellow solid (1.71 g, 88%). Mp = 93–107 °C; 1H NMR (400 MHz, CDCl3) δ 2.71 (s, 2H), 2.56 (s, 3H), 2.37 (s, 2H), 1.11 (s, 6H); 13C NMR (100 MHz, CDCl3) δ 194.6, 154.2, 145.0, 114.6, 53.0, 36.4, 35.6, 28.4, 12.0; HRMS (ESI) calcd for [C10H14N2O + H]+ 179.1184, found 179.1185.
3,6,6-Trimethyl-1-((4-nitrophenyl)sulfonyl)-6,7-dihydro-1H-indazol-4(5H)-one (69).
To a solution of compound 68 (500 mg, 2.81 mmol) and triethylamine (1.17 mL, 8.42 mmol) in CH2Cl2 (12 mL) was added a solution of 4-nitrobenzenesulfonyl chloride (933 mg, 4.21 mmol) in CH2Cl2 (12 mL). The mixture was stirred at room temperature for 5 h. CH2Cl2 (100 mL) was then added and the mixture was washed with saturated NaHCO3 (2 × 60 mL). The organic layer was dried over anhydrous Na2SO4 and evaporated under reduced pressure. The residue was purified by silica column chromatography (EtOAc:hexanes 1:3) to afford compound 69 as a pale yellow solid (766 mg, 75%). Mp = 150–164 °C; 1H NMR (400 MHz, CDCl3) δ 8.41 (d, J = 8.9 Hz, 2H), 8.22 (d, J = 8.9 Hz, 2H), 3.11 (s, 2H), 2.42 (s, 3H), 2.36 (s, 2H), 1.14 (s, 6H); 13C NMR (100 MHz, CDCl3) δ 192.9, 153.9, 153.8, 151.2, 142.6, 129.5, 124.7, 118.8, 52.1, 37.1, 35.5, 28.4, 13.7; HRMS (ESI) calcd for [C16H17N3O5S + H]+ 364.0967, found 364.0976.
7-Bromo-3,6,6-trimethyl-1-((4-nitrophenyl)sulfonyl)-6,7-dihydro-1H-indazol-4(5H)-one (70).
Prepared from compound 69 (180 mg, 0.599 mmol) according to the general procedure for bromination. Yield: 56% (103 mg); white solid. Mp = 146–150 °C; 1H NMR (400 MHz, CDCl3) δ 8.43 (d, J = 1.3 Hz, 1H), 5.63 (d, J = 1.1 Hz, 1H), 2.77 (dd, J = 17.1, 0.6 Hz, 1H), 2.44 (s, 3H), 2.28 (dd, J = 17.1, 1.2 Hz, 1H), 1.39 (s, 3H), 1.20 (s, 3H); 13C NMR (100 MHz, CDCl3) δ 191.8, 153.6, 152.7, 151.4, 141.7, 130.7, 124.4, 116.8, 49.4, 48.2, 40.0, 29.4, 25.1, 13.7; HRMS (ESI) calcd for [C15H18BrN5O3S + H]+ 442.0072, found 442.0067.
Methyl 3-((6-(7-Bromo-3,6,6-trimethyl-4-oxo-4,5,6,7-tetrahydro-1H-indazol-1-yl)pyridin-3-yl)amino)-3-oxopropanoate (71).
To a solution of compound 67 (70 mg, 0.20 mmol) and monomethyl malonate (23 μL, 0.22 mmol) in CH2Cl2 (2 mL) were added EDC·HCl (46 mg, 0.24 mmol), HOBT (14% H2O, 38 mg, 0.24 mmol) and N,N-diisopropylethylamine (42 μL, 0.24 mmol). The solution was stirred at room temperature for 24 h. After adding CH2Cl2 (8 mL), the mixture was washed with saturated NH4Cl (2 × 8 mL), saturated NaHCO3 (8 mL) and brine (8 mL). The organic layer was dried over anhydrous Na2SO4 and evaporated under reduced pressure. The residue was purified by preparative TLC (silica gel, MeOH:CH2Cl2 5:95) to afford compound 71 as a yellowish semisolid (62 mg, 69%). 1H NMR (400 MHz, CDCl3) δ 9.58 (s, 1H), 8.69 (d, J = 2.5 Hz, 1H), 8.19 (dd, J = 8.9, 2.6 Hz, 1H), 7.97 (d, J = 8.9 Hz, 1H), 6.31 (d, J = 1.1 Hz, 1H), 3.84 (s, 3H), 3.55 (s, 2H), 2.86 (d, J = 16.8 Hz, 1H), 2.54 (s, 3H), 2.26 (dd, J = 16.8, 1.3 Hz, 1H), 1.39 (s, 3H), 1.26 (s, 3H); 13C NMR (100 MHz, CDCl3) δ 192.9, 170.4, 163.2, 150.0, 149.2, 148.6, 139.1, 133.0, 130.0, 116.5, 115.5, 52.9, 52.7, 48.7, 40.8, 39.7, 29.6, 25.4, 13.5; HRMS (ESI) calcd for [C19H21BrN4O4 + H]+ 449.0824, found 449.0839.
N-(6-(7-Bromo-3,6,6-trimethyl-4-oxo-4,5,6,7-tetrahydro-1H-indazol-1-yl)pyridin-3-yl)sulfamoylamide (72).
A solution of compound 67 (70 mg, 0.20 mmol) and sulfamoyl chloride (26 mg, 0.22 mmol) in dimethylacetamide (0.5mL) was stirred at room temperature for 20 h. After adding EtOAc (5 mL), the solution was washed with brine (2 × 5 mL), dried over anhydrous Na2SO4 and evaporated under reduced pressure. The residue was purified by preparative TLC (silica gel, MeOH:CH2Cl2 5:95) to afford compound 72 as a white solid (76 mg, 89%). Mp = 148–160 °C; 1H NMR (400 MHz, DMSO-d6) δ 10.07 (s, 1H), 8.35 (d, J = 2.6 Hz, 1H), 7.87 (d, J = 8.9 Hz, 1H), 7.76 (dd, J = 8.9, 2.6 Hz, 1H), 7.40 (s, 2H), 6.30 (s, 1H), 2.67 (d, J = 16.9 Hz, 1H), 2.42 (s, 3H), 2.25 (d, J = 16.9 Hz, 1H), 1.31 (s, 3H), 1.17 (s, 3H); 13C NMR (100 MHz, DMSO-d6) δ 191.7, 148.6, 148.3, 146.0, 137.0, 135.3, 127.9, 115.6, 115.5, 59.7, 53.2, 48.1, 28.8, 24.4, 13.1; HRMS (ESI) calcd for [C15H18BrN5O3S + H]+ 428.0392, found 428.0409.
N-(6-(7-Bromo-3,6,6-trimethyl-4-oxo-4,5,6,7-tetrahydro-1H-indazol-1-yl)pyridin-3-yl)methanesulfonamide (73).
To a solution of compound 67 (70 mg, 0.20 mmol) and pyridine (23 μL, 0.28 mmol) in CH2Cl2 (2 mL) was added methanesulfonyl chloride (22 μL, 0.28 mmol). The mixture was stirred at room temperature for 23 h. CH2Cl2 (7 mL) was then added and the solution was washed with H2O (5 mL) and brine (5 mL). The organic layer was dried over anhydrous Na2SO4 and evaporated under reduced pressure. The residue was purified by preparative TLC (silica gel, EtOAc:hexanes 3:1) to afford compound 73 as a white solid (76 mg, 88%). Mp = 173–178 °C; 1H NMR (400 MHz, CDCl3) δ 8.40 (d, J = 2.6 Hz, 1H), 8.04 (d, J = 8.9 Hz, 1H), 7.82 (dd, J = 8.9, 2.8 Hz, 1H), 6.91 (s, 1H), 6.26 (d, J = 1.0 Hz, 1H), 3.11 (s, 3H), 2.87 (d, J = 16.9 Hz, 1H), 2.54 (s, 3H), 2.29 (dd, J = 16.9, 1.3 Hz, 1H), 1.40 (s, 3H), 1.25 (s, 3H); 13C NMR (100 MHz, CDCl3) δ 192.9, 150.4, 149.9, 149.5, 140.3, 131.8, 131.4, 116.7, 116.1, 52.7, 48.7, 40.2, 39.7, 29.6, 25.4, 13.5; HRMS (ESI) calcd for [C16H19BrN4O3S + H]+ 427.0473, found 427.0457.
N-(6-(7-Bromo-3,6,6-trimethyl-4-oxo-4,5,6,7-tetrahydro-1H-indazol-1-yl)pyridin-3-yl)acetamide (74).
To a solution of compound 67 (70 mg, 0.20 mmol) and triethylamine (42 μL, 0.30 mmol) in CH2Cl2 (1.5 mL) was added acetyl chloride (21 μL, 0.30 mmol). The mixture was stirred at room temperature for 1.5 h. CH2Cl2 (6 mL) was then added and the solution was washed with saturated NaHCO3 (6 mL) and saturated NH4Cl (6 mL). The organic layer was dried over anhydrous Na2SO4 and evaporated under reduced pressure. The residue was purified by preparative TLC (silica gel, EtOAc:hexanes 3:1) to afford compound 74 as a white solid (72 mg, 92%). Mp = 96–107 °C; 1H NMR (400 MHz, CDCl3) δ 8.61 (d, J = 2.3 Hz, 1H), 8.16 (dd, J = 8.9, 2.3 Hz, 1H), 7.95 (d, J = 8.9 Hz, 1H), 7.56 (br s, 1H), 6.29 (s, 1H), 2.87 (d, J = 16.9 Hz, 1H), 2.54 (s, 3H), 2.26 (d, J = 16.9 Hz, 1H), 2.25 (s, 3H), 1.39 (s, 3H), 1.25 (s, 3H); 13C NMR (100 MHz, CDCl3) δ 193.0, 168.6, 150.0, 149.2, 148.4, 138.8, 133.4, 129.8, 116.4, 115.6, 52.7, 48.7, 39.7, 29.6, 25.4, 24.4, 13.5; HRMS (ESI) calcd for [C17H19BrN4O2 + H]+ 391.0770, found 391.0785.
3,6,6-Trimethyl-1-(naphthalen-2-yl)-6,7-dihydro-1H-indazol-4(5H)-one (75).
A mixture of 2-acetyl-5,5-dimethy1-cyclohexanes-1,3-dione (1, 200 mg, 1.10 mmo1), naphthalen-2-ylhydrazine hydrochloride (224 mg, 1.15 mmo1) and pyridine (133 μL, 1.65 mmol) in ethanol (7 m1) was stirred at 80° C for 2 h. The solvent was then removed by evaporation. CH2Cl2 (18 mL) was then added and the solution was washed with saturated NaHCO3 (6 mL) and saturated NH4Cl (9 mL). The organic layer was dried over anhydrous Na2SO4 and evaporated under reduced pressure. The residue was purified by silica column chromatography (EtOAc:hexanes 1:3) to afford compound 75 as a light red solid (264 mg, 79%). Mp = 127–131 °C; 1H NMR (400 MHz, CDCl3) δ 7.97 (d, J = 8.8 Hz, 1H), 7.91 (m, J = 4.2 Hz, 3H), 7.65 (dd, J = 8.7, 2.1 Hz, 1H), 7.56 (m, 2H), 2.86 (s, 2H), 2.59 (s, 3H), 2.43 (s, 2H), 1.11 (s, 6H); 13C NMR (100 MHz, CDCl3) δ 193.4, 150.0, 149.2, 136.1, 133.1, 132.4, 129.5, 128.1, 127.9, 127.2, 126.8, 122.1, 121.8, 117.0, 52.5, 37.3, 35.9, 28.4, 13.4; HRMS (ESI) calcd for [C20H20N2O + H]+ 305.1654, found 305.1665.
7-Bromo-3,6,6-trimethyl-1-(naphthalen-2-yl)-6,7-dihydro-1H-indazol-4(5H)-one (76).
Prepared from compound 75 (100 mg, 0.329 mmol) according to the general procedure for bromination. Yield: 68% (86 mg); pale yellow solid. Mp = 65–75 °C; 1H NMR (400 MHz, CDCl3) δ 8.18 (d, J = 2.0 Hz, 1H), 8.01 (d, J = 8.8 Hz, 1H), 7.94 (m, 2H), 7.74 (dd, J = 8.7, 2.2 Hz, 1H), 7.60 (m, 2H), 4.93 (d, J = 1.2 Hz, 1H), 2.90 (dd, J = 17.1, 0.7 Hz, 1H), 2.59 (s, 3H), 2.29 (dd, J = 17.1, 1.3 Hz, 1H), 1.33 (s, 3H), 1.17 (s, 3H); 13C NMR (100 MHz, CDCl3) δ 192.2, 150.1, 149.1, 135.5, 133.1, 132.9, 129.8, 128.4, 127.9, 127.3, 127.2, 123.0, 122.6, 115.4, 50.8, 48.5, 40.3, 29.5, 25.2, 13.4; HRMS (ESI) calcd for [C20H19BrN2O + H]+ 383.0759, found 383.0777.
Supplementary Material
Highlights.
Tetrahydroindazoles were identified as cyclin dependent kinase inhibitors
Compounds 3, 53 and 59 were submicromolar inhibitors of activated CDK2/cyclin complexes
Binding affinities of compounds 3, 53 and 59 for free CDK2 were found to be in the micromolar range
Based on assay date and computational analysis, the compounds may interact with intact CDK2/cyclin complexes at a site different from the ATP site
The three compounds did not inhibit MCF-7 cancer cell line proliferation
Acknowledgments
We gratefully acknowledge financial support from the Contraception Research Branch of the Eunice Kennedy Shriver National Institute of Child Health and Human Development (NICHD) through contract HHSN275200503400C. We thank Drs. H. K. Kim, Diana Blithe, and Min Lee from NICHD for their support of this project.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References and Notes
- [1].Harper JW, Adams PD, Cyclin-dependent kinases, Chem. Rev, 101 (2001) 2511–2526. [DOI] [PubMed] [Google Scholar]
- [2].Lim S, Kaldis P, Cdks, cyclins and CDKIs: Roles beyond cell cycle regulation, Development, 140 (2013) 3079–3093. [DOI] [PubMed] [Google Scholar]
- [3].Malumbres M, Cyclin-dependent kinases, Genome Biol, 15 (2014) 122. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [4].Mariaule G, Belmont P, Cyclin-dependent kinase inhibitors as marketed anticancer drugs: Where are we now? A short survey, Molecules, 19 (2014) 14366–14382. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [5].Weinberg RA, The retinoblastoma protein and cell cycle control, Cell, 81 (1995) 323–330. [DOI] [PubMed] [Google Scholar]
- [6].Murdoch FE, Gooldberg E, Male contraception: Another holy grail, Bioorg. Med. Chem. Letters, 24 (2014) 419–424. [DOI] [PubMed] [Google Scholar]
- [7].Page ST, Amory JK, Bremner WJ, Advances in male contraception, Endocr. Rev, 29 (2008) 465–493. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [8].Tash JS, Attardi B, Hild SA, Chakrasali R, Jakkaraj SR, Georg GI, A novel potent indazole carboxylic acid derivative blocks spermatogenesis and is contraceptive in rats after a single oral dose, Biol. Reprod, 78 (2008) 1127–1138. [DOI] [PubMed] [Google Scholar]
- [9].Faber EB, Wang N, Georg GI, Review of rationale and progress towards targeting cyclin-dependent kinase 2 (CDK2) for male contraception, Biol. Reprod, 103 (2020) 357–367. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [10].Ortega S, Prieto I, Odajima J, Martin A, Dubus P, Sotillo R, Barbero JL, Malumbres M, Barbacid M, Cyclin-dependent kinase 2 is essential for meiosis but not for mitotic cell division in mice, Nat. Genet, 35 (2003) 25–31. [DOI] [PubMed] [Google Scholar]
- [11].Yang R, Muller C, Huynh V, Fung YK, Yee AS, Koeffler HP, Functions of cyclin A1 in the cell cycle and its interactions with transcription factor E2F-1 and the Rb family of proteins, Molecular and Cellular Biology, 19 (1999) 2400–2407. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [12].Liu D, Matzuk MM, Sung WK, Guo Q, Wang P, Wolgemuth DJ, Cyclin A1 is required for meiosis in the male mouse, Nat. Genet, 20 (1998) 377–380. [DOI] [PubMed] [Google Scholar]
- [13].Johnston DS, Goldberg E, Preclinical contraceptive development for men and women, Biol. Reprod, 103 (2020) 147–156. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [14].Zhang J, Yang PL, Gray NS, Targeting cancer with small molecule kinase inhibitors, Nat. Rev. Cancer, 9 (2009) 28–39. [DOI] [PubMed] [Google Scholar]
- [15].Khadem S, Maries RJ, Chromone and flavonoid alkaloids: Occurrence and bioactivity, Molecules, 17 (2012) 191–206. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [16].Parry D, Guzi T, Shanahan F, Davis N, Prabhavalkar D, Wiswel l.D., Seghezzi W, Paruch K, Dwyer M, Doll, Nomeir, Windsor, Fischmann, Wang Y, Oft M, Chen T, Kirschmeier P, Lees E, Dinaciclib (SCH 727965), a novel and potent cyclin-dependent kinase inhibitor, Mol. Cancer Ther, 8 (2012) 2344–2353. [DOI] [PubMed] [Google Scholar]
- [17].Schonbrunn E, Betzi S, Alam R, Martin MP, Becker A, Han H, Francis R, Chakrasali R, Jakkaraj S, Kazi A, Sebti SM, Cubitt CL, Gebhard AW, Hazlehurst LA, Tash JS, Georg GI, Development of highly potent and selective diaminothiazole inhibitors of cyclin-dependent kinases, J. Med. Chem, 56 (2013) 3768–3782. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [18].Pevarello P, Villa M, Varasi M, Isacchi A, 4,5,6,7-Tetrahydroindazole derivatives and their preparation method and use in treating cell proliferative disorders or Alzheimer’s disease, in, (Pharmacia & Upjohn S.p.A., Italy). WO 2000069846, 2000. [Google Scholar]
- [19].Ju H-Q, Wang S-X, Xiang Y-F, Liu Z, Liu J-Y, Chen Z-P, Zeng F-L, Xia M, Liu Z-H, Xing G-W, Wang S-Y, Wang Y-F, BJ-B11, a novel Hsp90 inhibitor, induces apoptosis in human chronic myeloid leukemia K562 cells through the mitochondria-dependent pathway, Eur. J. Pharmacol, 666 (2011) 26–34. [DOI] [PubMed] [Google Scholar]
- [20].Ju H-Q, Xiang Y-F, Xin B-J, Pei Y, Lu J-X, Wang Q-L, Xia M, Qian C-W, Ren Z, Wang S-Y, Wang Y-F, Xing G-W, Synthesis and in vitro anti-HSV-1 activity of a novel Hsp90 inhibitor BJ-B11, Bioorg. Med. Chem. Lett, 21 (2011) 1675–1677. [DOI] [PubMed] [Google Scholar]
- [21].Huang KH, Veal JM, Fadden RP, Rice JW, Eaves J, Strachan J-P, Barabasz AF, Foley BE, Barta TE, Ma W, Silinski MA, Hu M, Partridge JM, Scott A, DuBois LG, Freed T, Steed PM, Ommen AJ, Smith ED, Hughes PF, Woodward AR, Hanson GJ, McCall WS, Markworth CJ, Hinkley L, Jenks M, Geng L, Lewis M, Otto J, Pronk B, Verleysen K, Hall SE, Discovery of novel 2-aminobenzamide inhibitors of heat shock protein 90 as potent, selective and orally active antitumor agents, J. Med. Chem, 52 (2009) 4288–4305. [DOI] [PubMed] [Google Scholar]
- [22].Reddy N, Voorhees PM, Houk BE, Brega N, Hinson JM, Jillela A, Phase I trial of the HSP90 inhibitor PF-04929113 (SNX5422) in adult patients with recurrent, refractory hematologic malignancies, Clin. Lymphoma Myeloma Leuk, 13 (2013) 385–391. [DOI] [PubMed] [Google Scholar]
- [23].Cheng M-F, Ou L-C, Chen S-C, Chang W-T, Law P-Y, Loh HH, Chao Y-S, Shih C, Yeh S-H, Ueng S-H, Discovery, structure-activity relationship studies, and anti-nociceptive effects of 1-phenyl-3,6,6-trimethyl-1,5,6,7-tetrahydro-4H-indazol-4-one as novel opioid receptor agonists, Biorg. Med. Chem, 22 (2014) 4694–4703. [DOI] [PubMed] [Google Scholar]
- [24].Goodman KB, Cui H, Dowdell SE, Gaitanopoulos DE, Ivy RL, Sehon CA, Stavenger RA, Wang GZ, Viet AQ, Xu W, Ye G, Semus SF, Evans C, Fries HE, Jolivette LJ, Kirkpatrick RB, Dul E, Khandekar SS, Yi T, Jung DK, Wright LL, Smith GK, Behm DJ, Bentley R, Doe CP, Hu E, Lee D, Development of dihydropyridone indazole amides as selective Rho-kinase inhibitors, J. Med. Chem, 50 (2007) 6–9. [DOI] [PubMed] [Google Scholar]
- [25].Fraley ME, Steen JT, Brnardic EJ, Arrington KL, Spencer KL, Hanney BA, Kim Y, Hartman GD, Stirdivant SM, Drakas BA, Rickert K, Walsh ES, Hamilton K, Buser CA, Hardwick J, Tao W, Beck SC, Mao X, Lobell RB, Sepp-Lorenzino L, Yan Y, Ikuta M, Munshi SK, Kuo LC, Kreatsoulas C, 3-(Indol-2-yl)indazoles as Chek1 kinase inhibitors: Optimization of potency and selectivity via substitution at C6, Bioorg. Med. Chem. Lett, 16 (2006) 6049–6053. [DOI] [PubMed] [Google Scholar]
- [26].Kusakabe K.-i. Ide N, Daigo Y, Tachibana Y, Itoh T, Yamamoto T, Hashizume H, Hato Y, Higashino K, Okano Y, Sato Y, Inoue M, Iguchi M, Kanazawa T, Ishioka Y, Dohi K, Kido Y, Sakamoto S, Yasuo K, Maeda M, Higaki M, Ueda K, Yoshizawa H, Baba Y, Shiota T, Murai H, Nakamura Y, Indazole-based potent and cell-active Mps1 kinase inhibitors: Rational design from pan-kinase inhibitor anthrapyrazolone (SP600125), J. Med. Chem, 56 (2013) 4343–4356. [DOI] [PubMed] [Google Scholar]
- [27].Akritopoulou-Zanze I, Wakefield BD, Gasiecki A, Kalvin D, Johnson EF, Kovar P, Djuric SW, Scaffold oriented synthesis. Part 4: Design, synthesis and biological evaluation of novel 5-substituted indazoles as potent and selective kinase inhibitors employing heterocycle forming and multicomponent reactions, Bioorg. Med. Chem. Lett, 21 (2011) 1480–1483. [DOI] [PubMed] [Google Scholar]
- [28].Gavara L, Suchaud V, Nauton L, Théry V, Anizon F, Moreau P, Identification of pyrrolo[2,3-g]indazoles as new Pim kinase inhibitors, Bioorg. Med. Chem. Lett, 23 (2013) 2298–2301. [DOI] [PubMed] [Google Scholar]
- [29].Paryzek Z, Wydra R, Tetracyclic triterpenes. VIII. The skeletal rearrangement of 3-beta-acetoxy-9-alpha,11-alpha-epoxy-5-alpha-lanostan-7-one: 13- and 10-methyl group migration, Can. J. Chem, 63 (1985) 1280–1286. [Google Scholar]
- [30].Yoshimoto M, Ishida N, Hiraoka T, A new general method for carbon-carbon condensation of conjugated enaminoketones at the γ-position, Tetrahedron Lett, 14 (1973) 39–42. [Google Scholar]
- [31].Strakovs A, Strakova I, Krasnova A, Tonkiha N, Petrova M, 1-Phenyl-6,6-dimethyl-4-oxo-5-(acylhydrazono)-4,5,6,7-tetrahydroindazoles, Latvijas Kimijas Zurnals, (1994) 738–741. [Google Scholar]
- [32].Akhrem AA, Lakhvich FA, Budai SI, Khlebnicova TS, Petrusevich II, A new simple synthesis of 2-acylcyclohexane-1,3-diones, Synthesis, (1978) 925–927. [Google Scholar]
- [33].Delyatitskaya LG, Petrova MV, Grinberga S, Tonkikh NN, Strakov AY, 3,6,6-Trimethyl-4-oxo-1-(2-pyridyl)-4,5,6,7-tetrahydroindazole in Schmidt reaction and Beckmann rearrangement conditions for the 4-hydroxyimino derivative, Chem. Heterocycl. Compd, 36 (2000) 728–732. [Google Scholar]
- [34].Molteni V, Hamilton MM, Mao L, Crane CM, Termin AP, Wilson DM, A new simple synthesis of 2-acylcyclohexane-1,3-diones, Synthesis, 12 (2002) 1669–1674. [Google Scholar]
- [35].Srinivasan V, Jebaratnam DJ, Budil DE, Toward enediyne mimics: Methanolysis of azoesters and a bisazoester, J. Org. Chem, 64 (1999) 5644–5649. [DOI] [PubMed] [Google Scholar]
- [36].Hansen KB, Balsells J, Dreher S, Hsiao Y, Kubryk M, Palucki M, Rivera N, Steinhuebel D, Armstrong JD III, Askin D, Grabowski EJJ, First generation process for the preparation of the DPP-IV inhibitor sitagliptin, Org. Process Res. Dev, 9 (2005) 634–639. [Google Scholar]
- [37].Diederichs S, Bäumer N, Schultz N, Hamra FK, Schrader MG, Sandstede M-L, Berdel WE, Serve H, Müller-Tidow C, Expression patterns of mitotic and meiotic cell cycle regulators in testicular cancer and development, Int. J. Cancer, 116 (2005) 207–217. [DOI] [PubMed] [Google Scholar]
- [38].Martinerie L, Manterola M, Chung SS, Panigrahi SK, Weisbach M, Vasileva A, Geng Y, Sicinski P, Wolgemuth DJ, Mammalian E-type cyclins control chromosome pairing, telomere stability and CDK2 localization in male meiosis, PLoS Genet., 10 (2014) e1004165. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [39].Bednarczyk D, Sanghvi MV, The impact of assay recovery on the apparent permeability, a function of lysosomal trapping, Xenobiotica, 50 (2020) 753–760. [DOI] [PubMed] [Google Scholar]
- [40].Kozakov D, Grove LE, Hall DR, Bohnuud T, Mottarella SE, Luo L, Xia B, Beglov D, Vajda S, The FTMap family of web servers for determining and characterizing ligand-binding hot spots of proteins, Nat. Protoc, 10 (2015) 733–755. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [41].Betzi S, Alam R, Martin M, Lubbers DJ, Han H, Jakkaraj SR, Georg GI, Schonbrunn E, Discovery of a potential allosteric ligand binding site in CDK2, ACS Chem. Biol, 6 (2011) 492–501. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [42].Martin MP, Alam R, Betzi S, Ingles DJ, Zhu J-Y, Schonbrunn E, A novel approach to the discovery of small-molecule ligands of CDK2, ChemBioChem, 13 (2012) 2128–2136. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [43].Stevenson-Lindert LM, Fowler P, Lew J, Substrate specificity of CDK2-cyclin A: what is optimal?, J. Biol. Chem, 278 (2003) 50956–50960. [DOI] [PubMed] [Google Scholar]
- [44].Fabian MA, Biggs WH, Treiber DK, Atteridge CE, Azimioara MD, Benedetti MG, Carter TA, Ciceri P, Edeen PT, Floyd M, Ford JM, Galvin M, Gerlach JL, Grotzfeld RM, Herrgard S, Insko DE, Insko MA, Lai AG, Lélias J-M, Mehta SA, Milanov ZV, Velasco AM, Wodicka LM, Patel HK, Zarrinkar PP, Lockhart DJ, A small molecule–kinase interaction map for clinical kinase inhibitors, Nat. Biotechnol, 23 (2005) 329–336. [DOI] [PubMed] [Google Scholar]
- [45].Hill AV, A new mathematical treatment of changes of ionic concentration in muscle and nerve under the action of electric currents, with a theory as to their mode of excitation, J. Physiol, 40 (1910) 190–224. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [46].Levenberg K, A method for the solution of certain non-linear problems in least squares, Quart. Appl. Math, 2 (1944) 164–168. [Google Scholar]
- [47].LigPrep, in, Schrödinger LLC, 2018.
- [48].Glide, Version 7.8, in, Schrödingee LLC, 2018.
- [49].Prime, in, Schrödinger LLC, 2018.
- [50].Matoba K, Tachi M, Itooka T, Yamazaki T, Reaction of aliphatic dicarboxylic acids with acyl chlorides in the presence of aluminum chloride, Chem. Pharm. Bull, 34 (1986) 2007–2012. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.