

Abstract
Metal–organic frameworks (MOFs) are promising materials for onboard hydrogen storage thanks to the tunable pore size, pore volume, and pore geometry. In consideration of pore structures, the correlation between the pore volume and hydrogen storage capacity is examined and two empirical equations are rationalized to predict the hydrogen storage capacity of MOFs with different pore geometries. The total hydrogen adsorption under 100 bar and 77 K is predicted as ntot = 0.085 × Vp − 0.013 × Vp2 for cage-type MOFs and ntot = 0.076 × Vp − 0.011 × Vp2 for channel-type MOFs, where Vp is the pore volume of corresponding MOFs. The predictions by these empirical equations are validated by several MOFs with an average deviation of 5.4%. Compared with a previous equation for activated carbon materials, the empirical equations demonstrate superior accuracy especially for MOFs with high surface area (i.e., SBET over ≈3000 m2 g−1). Guided by these empirical equations, a highly porous Zr-MOF NPF-200 (NPF: Nebraska Porous Framework) is examined to possess outstanding hydrogen total adsorption capacity (65.7 mmol g−1) at 77 K and record high volumetric working capacity of 37.2 g L−1 between 100 and 5 bar at 77 K.
Keywords: empirical equations, hydrogen storage, metal–organic frameworks (MOFs), pore geometry, pore occupancy
The CO2 emission from combustion of fossil fuel has caused serious environmental issues such as global warming. Hydrogen has been recognized as a promising candidate to replace fossil fuel because of its high energy density (33.3 kWh kg−1),[1] which is three times higher than that of gasoline (11.1 kWh kg−1) and clean combustion that only produce water as by-product. In order to use hydrogen as fuel for automobiles, 5.6 kg of H2 need to be stored safely and efficiently to allow 300 miles driving of fuel cell vehicle with each filling. Storage of hydrogen is very challenging due to its low density and requires compression of hydrogen at very high pressure of 700 bar. Such processes involve significant energy input and expensive carbon fiber tank.[2]
Traditional porous adsorbents such as zeolites[3] and carbon materials[4] and emerging porous materials such as metal–organic frameworks (MOFs),[5] covalent-organic frameworks (COFs),[6] and microporous polymer[7] have been studied to store hydrogen at lower pressure. MOFs constructed by connecting metal ion or cluster with organic linkers are highly tuneable materials in terms of their surface area, pore size, pore geometry, and functional sites[8] and has demonstrated great potential for gas storage and separation.[1,9] The well-defined structures also allow crystallography and computational studies of host–guest interaction to gain fundamental understanding for rational material design.[10]
In 2003, Rosi et al.[11] reported for the first time the H2 adsorption of microporous MOF at high-pressure up to 20 bar. Since then, many MOFs have been examined for H2 storage and great progress has been made. The storage capacities of some highly porous MOFs[12] (>14 wt% under 70 bar, 77 K) are much higher than those of traditional porous materials such as zeolite and activated carbon (<7 wt%).[3,13]
For onboard hydrogen storage for fuel cell vehicles, the minimum usable gas pressure is 5 bar and only the adsorption capacity between pressure of 100 and 5 bar is the usable as working capacity. Therefore, the ideal H2 storage material should exhibit high capacity at 100 bar and low capacity at 5 bar. U.S. Department of Energy (DOE) has set the gravimetric and volumetric working capacity targets of 4.5 wt%; 30 g L−1 (2020), 5.5 wt%; 40 g L−1 (2025), and 6.5 wt%; 50 g L−1 (ultimate).[14] Although several MOFs such as NU-1103 (NU: Northwestern University), UMCM-9 (UMCM: University of Michigan Crystalline Material), and NU-100 possess high gravimetric working capacity reaching the DOE target, simultaneously high volumetric working capacity is still very challenging and the current record by NU-100 is only 35.5 g L−1 between 100 and 5 bar at 77 K.
Working capacity involves two distinct adsorption conditions under which the correlations between adsorption capacity and pore properties are quite different. Understanding the adsorption behavior at high pressure is pivotal to obtain high working capacity and some research endeavours have been performed along this way. For example, the excess H2 adsorption is found to be positively correlated with the MOF surface area.[15] Such correlation is similar to what found previously for carbon materials.[16] Indeed, H2 excess adsorption of MOFs follows this correlation fairly well for those with surface area under ≈ 3000 m2 g−1.[2c] For MOFs with higher surface area, the hydrogen adsorption falls off the trend,[15a,17] which is likely caused by the decreased pore surface occupancy as the surface area and pore volume increase. Such phenomenon has been found in our previous study of high-pressure methane adsorption.[9a,18] Besides, the structural properties of MOFs such as pore size, pore geometry, and aperture size significantly affect the gas adsorption behavior at high pressure.[10a] Full consideration of structural properties can only be realized with extensive computational screening of a great number of MOFs.[10c,15b] Our previous successful discovery of the correlation between pore volume and methane adsorption motivates us to explore such empirical equation for hydrogen adsorption with consideration of pore occupancy and pore geometry.
Theoretically, the sorbate–sorbent interaction potential in spherical pore is higher than that in cylindrical pore due to more surface atom interaction.[19] Similarly for MOF materials, we speculate that the interaction potential in cages would be higher than that in channels, which would result in higher pore occupancy and adsorption capacity for cage-type MOFs. In this work, we analyzed a series of MOFs from the same measurements[2c] to correlate the pore geometry with hydrogen total adsorption. We found that the cage-type MOFs generally possess higher pore occupancy than that of channel-type MOFs with the same pore volume. Accordingly, taking the pore geometry into accounts, empirical equations have been rationalized for the H2 total adsorption prediction with given pore volume. Guided by these empirical equations, we discovered that a cage-type Zr-MOF NPF-200 (NPF: Nebraska Porous Framework)[20] exhibits record high volumetric working capacity (37.2 g L−1 at 77 K, 100–5 bar) and simultaneously high gravimetric working capacity.
In comparison of computational screening of millions of MOFs by machine learning[15b] strategy, only limited number of MOFs were experimentally measured for H2 storage. Our analysis starts from the MOFs measured under the same conditions[2c] to minimize the inconsistency of measurements. To study the effects of structural properties to H2 adsorption, the crystal structure of MOFs must accurately represent the bulk MOF material, which is not always the case since some MOFs are well known to contain significant amount of crystal defects[21] or become partially collapsed during activation.[22] Therefore, we compared the experimental pore volume and Brunauer–Emmett–Teller (BET) surface area with calculated pore volume and surface area, and large deviations (>10%) have been found for four MOFs as shown in Table S1 in the Supporting Information. We then excluded these four MOFs and used the other six MOFs: HKUST-1 (HKUST: Hong Kong University of Science and Technology), NU-125, NU-1000, UiO-68-Ant (UiO: University of Oslo; Ant: anthracene), Cu-MOF-74, and Zn2(bdc)2(dabco) for the following study.
The pore diameter (D), aperture size (A), and pore diameter to aperture size ratio (D/A) have been measured from their crystal structures. Based on the D/A ratio, these six MOFs can be categorized into two types: cage-type with large D/A ≈1.8 or higher and channel-type with D/A = 1 as shown in Figure 1a,–f and Table 1. We hypothesize that the cage-type MOFs would have stronger sorbate–sorbent interaction potential due to more surface atom interaction. Such hypothesis is based on theoretical calculation of interaction potential in cylindrical and spherical pore geometries. In spherical pore, more surface atoms are in close contact with the guest molecule, which lead to higher interaction potential as demonstrated in Figure S1 in the Supporting Information. Consequently, spherical pore can adsorb more guest molecules under unsaturated conditions, in another word, possess a higher pore occupancy. The closest model of ideal spherical pore is fullerene, while we speculate that the adsorption behavior of cage and channel in MOFs would to some extend follow the principle of spherical and cylindrical pores.
Figure 1.
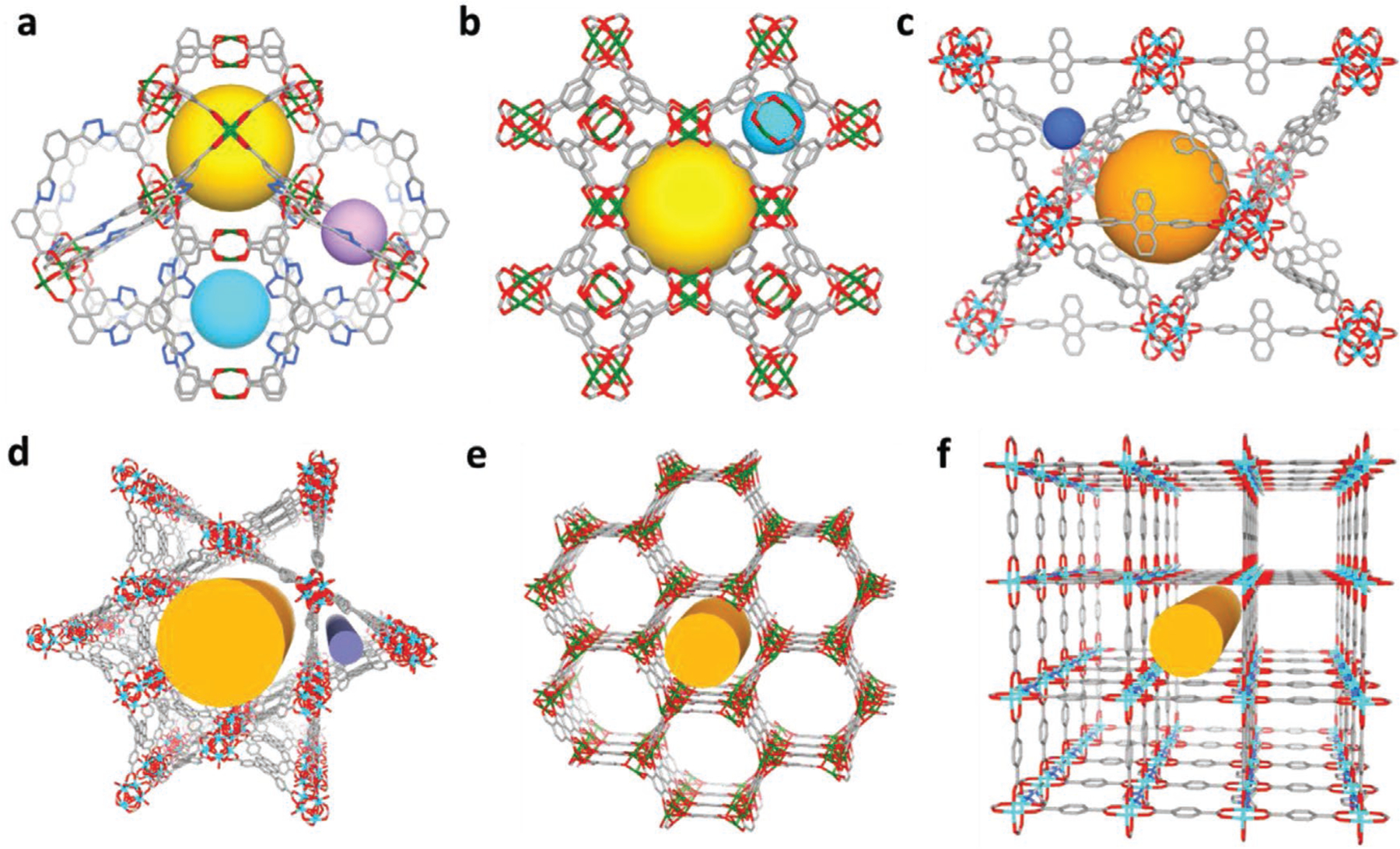
Major channels or cages in the investigated MOFs structures. a) NU-125, b) HKUST-1, c) UiO-68-Ant, d) NU-1000, e) Cu-MOF-74, and f) Zn2(bdc)2(dabco)2.
Table 1.
Comparison of D (pore diameter) (cage or channel size is measured by fitting the largest sphere or cylinder inside the pore considering the Van der Waals radius of atoms), A (aperture), and D/A ratio of cage and channel in MOFs.
| MOF | Cavity-1 |
Cavity-2 |
Cavity-3 |
||||||
|---|---|---|---|---|---|---|---|---|---|
| D [Å] | A [Å] | D/A ratio | D [Å] | A [Å] | D/A ratio | D [Å] | A [Å] | D/A ratio | |
| NU-125 | 20 | 5.3 | 3.8 | 12 | 5.3 | 2.3 | 6.6 | 5.3 | 1.2 |
| HKUST-1 | 12 | 6.5 | 1.8 | 5 | 4.1 | 1.2 | 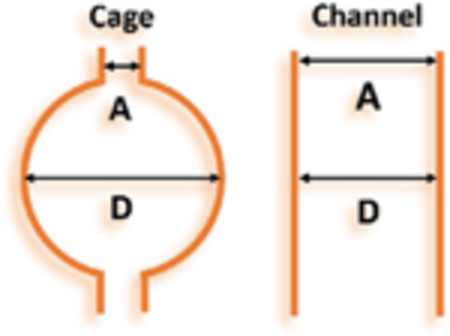 |
||
| UiO-68-Ant | 17 | 4 | 4.25 | 5 | 4 | 1.25 | |||
| NU-1000 | 28.5 | 28.5 | 1 | 7.2 | 7.2 | 1 | |||
| Cu-MOF-74 | 11 | 11 | 1 | ||||||
| Zn2(bdc)2(dabco) | 7.6 | 7.6 | 1 | 3.6 × 7.6 | 3.6 × 7.6 | 1 | |||
At very low temperature such as 30 K, the H2 adsorption can reach saturation at about 4 bar, for MOF-5, and the H2 packing density inside the pore is about 0.087 g cm−3, very close to the density of solid H2 (0.086 g cm−3).[18] Thus the saturated adsorption of hydrogen in MOFs can be estimated by equation: nsat = ρsolid × Vp, where nsat is the saturated total adsorption, ρsolid is the density of solid H2, and Vp is the MOF pore volume. At higher temperature, such as 77 K, the adsorption cannot reach saturation even at high pressure of 100 bar. We can define pore occupancy, O, under 100 bar and 77 K, as total adsorption of H2 under 100 bar and 77 K, ntot, divided by saturated total adsorption nsat (see Table S1, Supporting Information, for calculation details). As shown in Figure 2a and Table S2 in the Supporting Information, for both types of MOFs, the pore occupancy decreases as pore volume increases, which can be attributed to the lower interaction potential of adsorption in large pore center. The interaction potential decreases as the distance between H2 molecule and the pore surface increases. For MOFs with high pore volume, the pore size is larger and the interaction potential of H2 molecule in the pore center is mainly from the weak sorbate–sorbate interaction similar to the bulk gas. As expected, the pore occupancy of cage-type MOFs is higher than that of channel-type MOFs for MOFs in this study, such superiority of cage-type MOFs indicates that adsorption behavior of cage and channel in MOFs indeed follows the previously reported principle of spherical and cylindrical pores. Through data fitting, the pore occupancy is found to be approximately linearly related to the corresponding pore volume, for cage-type MOFs: O = 0.992–0.149 × Vp; for channel-type MOFs: O = 0.881 − 0.128 × Vp. Cross-validation of the fitting revealed the deviation is less than 1.0% which is much smaller than that of fitting all six MOFs in one group as shown in Tables S3 and S4 in the Supporting Information. Thus the total H2 adsorption of a specific MOF can be calculated by the equation: ntot = nsat × O = (0.086 × Vp) × (0.992 – 0.149 × Vp) = 0.085 × Vp – 0.013 × Vp 2 for cage-type MOFs; ntot = nsat × O = (0.086 × Vp) × (0.881 – 0.128 × Vp) =0.076 × Vp – 0.011 × Vp2 for channel-type MOFs with units of g g−1 for ntot and cm3 g−1 for Vp.
Figure 2.

a) Pore occupancy under 100 bar and 77 K versus pore volume (cm3 g−1) of channel-type and cage-type MOFs. The dash lines show the linear fitting results. b) Predicted H2 total adsorption under 100 bar and 77 K versus pore volume using our empirical equations for the two types of MOFs. Vertical line at 3.3 cm3 g−1 indicates the peak total adsorption position. c) Comparison of predicted total adsorption with experimental adsorption of nine MOFs not used in empirical equation fitting. Prediction by our equation is closer to experimental adsorption (diagonal line). d) Comparison of deviations of prediction by our empirical equations and Chahine’s rule. The average deviation of the prediction by our empirical equation and by Chahine’s rule are 5.4% and 21.9%, respectively.
With the obtained empirical equations, the H2 total adsorption at 100 bar and 77 K can be predicted with given pore volume. As shown in Figure 2b, the total adsorption would initially increase as the pore volume increases, until around pore volume of 3.3 cm3 g−1. After that, the total adsorption starts to decrease due to the lower pore occupancy at higher pore volume region. Overall, the adsorption of cage-type MOFs is higher than that of channel-type MOFs with the same pore volume. The superiority of cage-type structure for gas adsorption has also been observed in pore space partition study when large channels are partitioned into cages.[23] In reality, many MOFs would be somewhere between being purely cage-type and being purely channel-type. In this case, the two empirical equations predict a narrow range where the expected H2 uptake might be located. To validate the prediction by these empirical equations, adsorption of another nine MOFs was predicted based on their structural type and compared with their experimental adsorption capacity (Figure 2c and Table S5, Supporting Information). The average deviation of the prediction is 5.4% indicating the reasonable accuracy of this method (Figure 2d). For comparison, the H2 adsorption of these nine MOFs were also predicted using Chahine’s rule which states that the maximum excess adsorption can be predicted from the BET surface area (Scheme S1, Supporting Information). For activated carbon, the H2 excess adsorption Nexcess (wt%) = 1.91 × 10−3 × SA (m2 g−1) determined by fitting experimental data.[16] In comparison with experimental adsorption and prediction by our equations (Figure 2c,d), the predictions of by Cahine’s rule yield large deviation for MOFs with high pore volume such as NU-1101 (30.2%) and NU-1103 (36.9%). The average deviation is 21.9%, much higher than that of our empirical equations (5.4%) as shown in Figure 2d. The superiority of our empirical equations indicates that the pore occupancy and pore geometry merit consideration especially for highly porous MOFs.
In practical application, the ideal material should also exhibit high volumetric adsorption capacity which requires high volumetric surface area for MOFs.[10c] As shown in Table S6 in the Supporting Information, MOFs with ultrahigh pore volume such as NU-1301[24] and DUT-60 (DUT: Dresden University of Technology)[25] exhibits only small volumetric surface area. Considering the prediction results in Figure 2b, MOFs with moderately high pore volume of ≈3.3 cm3 g−1 and high volumetric surface area would likely possess simultaneously high gravimetric and volumetric H2 storage capacity at 100 bar and 77 K.
Base on the above analysis, we projected that a highly porous Zr-MOF NPF-200[20] would be a promising candidate to balance the volumetric capacity and the gravimetric capacity, due to its near-optimal pore volume of 2.17 cm3 g−1 and high volumetric surface area of 2268 m2 cm−3. NPF-200 is considered as a cage-type MOFs since its largest cage with D/A ≈2 contributes the most to the total porosity, as shown in Figure 3a and Table S7 in the Supporting Information. The H2 total capacity of NPF-200 was predicted to be 61.8 mmol g−1 (11.7 wt%) based on the equation for cage-type MOFs. The H2 excess and total adsorption of NPF-200 was predicted by Chahine’s rule as 55.7 and 89.4 mmol g−1, respectively.
Figure 3.
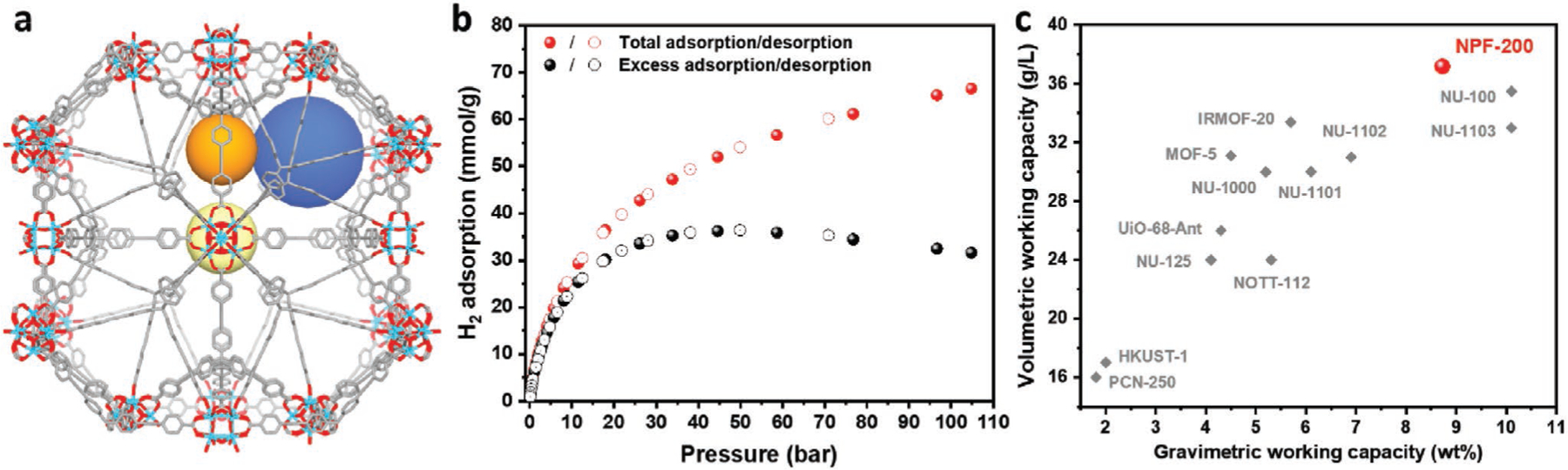
a) Three major cages of NPF-200. b) Hydrogen total and excess adsorption isotherm of NPF-200 up to 100 bar. c) Volumetric and gravimetric working capacity of NPF-200 between 100 and 5 bar at 77 K in comparison with the best MOFs reported to date.
The excess and total H2 adsorption isotherms of NPF-200 were measured experimentally as shown in Figure 3b. The total adsorption at 77 K and 100 bar is 65.7 mmol g−1 which is slightly higher than the predicted value of 61.8 mmol g−1 by our empirical equation (with ≈5.9% deviation). In contrast, the predictions by Chahine’s rule overestimate the H2 excess adsorption and total adsorption by 54% and 40%, respectively.
The total adsorption of NPF-200 is comparable to the best MOF materials such as NU-1102 (54.4 mmol g−1), NU-1103 (73.9 mmol g−1), MOF-210 (88 mmol g−1), and NU-100 (82 mmol g−1). The volumetric adsorption of NPF-200 (51 g L−1) is also very high compared to the best MOFs reported such as NU-1102 (43.8 g L−1), NU-1103 (44 g L−1), MOF-210 (44 g L−1), and NU-100 (49.7 g L−1). The high gravimetric and volumetric adsorption of NPF-200 motivate us to further examine the gravimetric and volumetric working capacities which are 8.7 wt% and 37.2 g L−1 respectively under 100 to 5 bar pressure swing condition at 77 K. To the best of our knowledge, the volumetric working capacity of NPF-200 is the highest among MOF materials under identical conditions as shown in Figure 3c and Table S8 in the Supporting Information. Meanwhile, its gravimetric working capacity is also quite high, only slightly smaller than those of NU-100 and NU-1103. NPF-200 represents a rare example of simultaneously high gravimetric and volumetric working capacities thanks to its near-optimal pore volume indicated by the empirical equation, cage-type pore geometry, and high volumetric surface area.
Lastly, the heat of adsorption (Qst) was calculated from the adsorption isotherms at different temperatures (see Figure S4, Supporting Information). The initial Qst of H2 adsorption in NPF-200 is 4.5 kJ mol−1, which is modest and similar to those of many other classical MOFs. It indicates that H2 adsorption in NPF-200 is based on the typical van der Waals type interaction.[2c,26]
In summary, we analyzed the contribution of pore geometry and pore occupancy to hydrogen total adsorption for a series of MOFs and rationalized two empirical equations to predict the total hydrogen adsorption under 100 bar and 77 K with given MOFs pore volume. Based on the prediction, cage-type MOFs with moderately high pore volume and high volumetric surface area seem to be promising candidates for hydrogen storage, which inspired our discovery of the cage-type Zr-MOF NPF-200 with simultaneously high volumetric and gravimetric working capacities. Especially, the volumetric working capacity (37.2 g L−1) between 100 and 5 bar at 77 K of NPF-200 is the highest among reported MOFs. Considering its excellent structural and water stabilities,[20] NPF-200 is a very promising material for practical H2 storage application. Such encouraging result indicates that prediction based on our empirical equations could provide a quick guidance on MOFs evaluation and consequently accelerate the discovery of MOFs for high-pressure H2 storage. It may also provide valuable guidance for H2 storage using other types of porous materials such as covalent organic frameworks (COFs) and hydrogen bonded organic frameworks (HOFs). It is foreseeable that MOFs with higher storage capacity and working capacity will be discovered in the near future.
Experimental Section
Powder X-ray diffraction (PXRD) data were taken with a PANalytical Empyrean diffractometer with a PIXcel 3D detector. The copper target X-ray tube was set to 45 kV and 40 mA. N2 adsorption isotherms at 77 K was measured under liquid nitrogen bath with Micromeritics ASAP (Accelerated Surface Area and Porosimetry System) 2020 surface area analyzer. High-pressure hydrogen sorption measurements were performed using a computer-controlled Sieverts apparatus, details of which can be found in a previous publication.[27] Research grade hydrogen with purity of 99.999% was used for high-pressure measurements. PLATON[28] was used to calculate the pore volume using a probe of 1.8 Å in radius, which corresponds to the kinetic diameter of N2, to enable accurate comparison with experimental pore volume measured by N2 adsorption at 77 K. Simulated BET surface area values are taken from ref. [2c]. Cage or channel size is measured by fitting largest sphere or cylinder inside the pore considering the Van der Waals radius of atoms.
For high-pressure studies, excess adsorption and total adsorption are frequently used to evaluate the gas storage capacity of porous material. Excess adsorption is approximately the amount of gas molecules interacting with pore surface. Total adsorption is the total amount of gas molecules inside the pore. Excess adsorption is determined experimentally and total adsorption is calculated with the following equation: ntot = nex + ρbulk (P,T) × Vp, where ρbulk is obtained from the National Institute of Standardsand Technology (NIST) Refprop database[29] and Vp is usually determined from the N2 adsorption isotherm at 77 K. The H2 density ρbulk = 15.528 mol L−1 under 100 bar and 77 K. Weight percent (wt%) in this work is calculated by mass of H2/(mass of H2 + mass of material).
The structures and synthesis details of six MOFs (NU-125,[30] HKUST-1,[31] UiO-68-Ant,[32] NU-1000,[33] Cu-MOF-74,[2c] and Zn2(bdc)2(dabco)[34]) investigated for empirical formula derivation can be found in literatures.
Highly crystalline NPF-200 sample was synthesized and activated by supercritical CO2 following the previously reported procedures.[20] The PXRD and nitrogen adsorption match well with the previous measurements, indicating the phase purity and thorough activation of the sample. The measured pore volume is 2.17 cm3 g−1, very close to the calculated value of 2.10 cm3 g−1 by Platon. More details can be found in Figures S2 and S3 in the Supporting Information.
Supplementary Material
Acknowledgements
This work was supported by an Award AX-1730 from Welch Foundation.
Footnotes
The ORCID identification number(s) for the author(s) of this article can be found under https://doi.org/10.1002/adma.201907995.
Supporting Information
Supporting Information is available from the Wiley Online Library or from the author.
Conflict of Interest
The authors declare no conflict of interest.
Contributor Information
Xin Zhang, Department of Chemistry, University of Texas at San Antonio, One UTSA Circle, San Antonio, TX 78249-0698, USA.
Rui-Biao Lin, Department of Chemistry, University of Texas at San Antonio, One UTSA Circle, San Antonio, TX 78249-0698, USA.
Jing Wang, Department of Chemistry, University of Texas at San Antonio, One UTSA Circle, San Antonio, TX 78249-0698, USA; College of Chemical Engineering, Zhejiang University of Technology, Hangzhou 310014, China.
Bin Wang, Department of Chemistry, University of Texas at San Antonio, One UTSA Circle, San Antonio, TX 78249-0698, USA.
Bin Liang, Department of Chemistry, University of Texas at San Antonio, One UTSA Circle, San Antonio, TX 78249-0698, USA.
Taner Yildirim, NIST Center for Neutron Research, Gaithersburg, Maryland 20899-6102, USA.
Jian Zhang, The Molecular Foundry, Lawrence Berkeley National Laboratory Berkeley, Berkeley, CA 94720, USA.
Wei Zhou, NIST Center for Neutron Research, Gaithersburg, Maryland 20899-6102, USA.
Banglin Chen, Department of Chemistry, University of Texas at San Antonio, One UTSA Circle, San Antonio, TX 78249-0698, USA.
References
- [1].Suh MP, Park HJ, Prasad TK, Lim DW, Chem. Rev 2012, 112, 782. [DOI] [PubMed] [Google Scholar]
- [2].a) O’Malley K, Ordaz G, Adams J, Randolph K, Ahn CC, Stetson NT, J. Alloys Compd 2015, 645, S419; [Google Scholar]; b) Allendorf MD, Hulvey Z, Gennett T, Ahmed A, Autrey T, Camp J, Cho ES, Furukawa H, Haranczyk M, Head-Gordon M, Jeong S, Karkamkar A, Liu DJ, Long JR, Meihaus KR, Nayyar IH, Nazarov R, Siegel DJ, Stavila V, Urban JJ, Veccham SP, Wood BC, Energy Environ. Sci 2018, 11, 2784; [Google Scholar]; c) García-Holley P, Schweitzer B, Islamoglu T, Liu Y, Lin L, Rodriguez S, Weston MH, Hupp JT, Gómez-Gualdrón DA, Yildirim T, Farha OK, ACS Energy Lett 2018, 3, 748. [Google Scholar]
- [3].Langmi HW, Book D, Walton A, Johnson SR, Al-Mamouri MM, Speight JD, Edwards PP, Harris IR, Anderson PA, J. Alloys Compd 2005, 637, 404. [Google Scholar]
- [4].Yang Z, Xia Y, Mokaya R, J. Am. Chem. Soc 2007, 129, 1673. [DOI] [PubMed] [Google Scholar]
- [5].Kapelewski MT, Runčevski T, Tarver JD, Jiang HZH, Hurst KE, Parilla PA, Ayala A, Gennett T, FitzGerald SA, Brown CM, Long JR, Chem. Mater 2018, 30, 8179. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [6].Han SS, Furukawa H, Yaghi OM, Goddard WA 3rd, J. Am. Chem. Soc 2008, 130, 11580. [DOI] [PubMed] [Google Scholar]
- [7].Ghanem BS, Hashem M, Harris KDM, Msayib KJ, Xu M, Budd PM, Chaukura N, Book D, Tedds S, Walton A, McKeown NB, Macromolecules 2010, 43, 5287. [Google Scholar]
- [8].a) Feng L, Wang KY, Day GS, Zhou HC, Chem. Soc. Rev 2019, 48, 4823; [DOI] [PubMed] [Google Scholar]; b) Kalmutzki MJ, Hanikel N, Yaghi OM, Sci. Adv 2018, 4, eaat9180; [DOI] [PMC free article] [PubMed] [Google Scholar]; c) Chen Z, Hanna SL, Redfern LR, Alezi D, Islamoglu T, Farha OK, Coord. Chem. Rev 2019, 386, 32; [Google Scholar]; d) Li B, Wen HM, Cui Y, Zhou W, Qian G, Chen B, Adv. Mater 2016, 28, 8819. [DOI] [PubMed] [Google Scholar]
- [9].a) Li B, Wen H-M, Zhou W, Xu JQ, Chen B, Chem 2016, 1, 557; [Google Scholar]; b) Li H, Li L, Lin R-B, Zhou W, Zhang Z, Xiang S, Chen B, Energy-Chem 2019, 1, 100006; [Google Scholar]; c) Barnett BR, Gonzalez MI, Long JR, Trends Chem 2019, 1, 159; [Google Scholar]; d) Adil K, Belmabkhout Y, Pillai RS, Cadiau PM Bhatt, Assen AH, Maurin G, Eddaoudi M, Chem. Soc. Rev 2017, 46, 3402; [DOI] [PubMed] [Google Scholar]; e) Wen HM, Li B, Li L, Lin RB, Zhou W, Qian G, Chen B, Adv. Mater 2018, 30, 1704792. [DOI] [PubMed] [Google Scholar]
- [10].a) Cho HS, Yang J, Gong X, Zhang YB, Momma K, Weckhuysen M, Deng H, Kang JK, Yaghi OM, Terasaki O, Nat. Chem 2019, 11, 562; [DOI] [PubMed] [Google Scholar]; b) Yildirim T, Hartman MR, Phys. Rev. Lett 2005, 95, 215504; [DOI] [PubMed] [Google Scholar]; c) Gómez-Gualdrón DA, Colón YJ, Zhang X, Wang TC, Chen Y-S, Hupp JT, Yildirim T, Farha OK, Zhang J, Snurr RQ, Energy Environ. Sci 2016, 9, 3279; [Google Scholar]; d) Sung Cho H, Deng H, Miyasaka K, Dong Z, Cho M, Neimark AV, Ku Kang J, Yaghi OM, Terasaki O, Nature 2015, 527, 503; [DOI] [PubMed] [Google Scholar]; e) Wang B, Wang P, Xie LH, Lin RB, Lv J, Li JR, Chen B, Nat. Commun 2019, 10, 3861. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [11].Rosi NL, Eckert J, Eddaoudi M, Vodak DT, Kim J, O’Keeffe M, Yaghi OM, Science 2003, 300, 1127. [DOI] [PubMed] [Google Scholar]
- [12].a) Furukawa H, Ko N, Go YB, Aratani N, Choi SB, Choi E, Yazaydin AO, Snurr RQ, O’Keeffe M, Kim J, Yaghi OM, Science 2010, 329, 424; [DOI] [PubMed] [Google Scholar]; b) Farha OK, Yazaydin AO, Eryazici I, Malliakas CD, Hauser BG, Kanatzidis MG, Nguyen ST, Snurr RQ, Hupp JT, Nat. Chem 2010, 2, 944. [DOI] [PubMed] [Google Scholar]
- [13].a) Dundar-Tekkaya E, Yurum Y, Int. J. Hydrogen Energy 2016, 41, 9789; [Google Scholar]; b) Hirscher M, Panella B, J. Alloys Compd 2005, 399, 404. [Google Scholar]
- [14].Zhang W, Huang H, Liu D, Yang Q, Xiao Y, Ma Q, Zhong C, Microporous Mesoporous Mater 2013, 171, 118. [Google Scholar]
- [15].a) Ahmed A, Liu YY, Purewal J, Tran LD, Wong-Foy AG, Veenstra M, Matzger AJ, Siegel DJ, Energy Environ. Sci 2017, 10, 2459; [Google Scholar]; b) Ahmed A, Seth S, Purewal J, Wong-Foy AG, Veenstra M, Matzger AJ, Siegel DJ, Nat. Commun 2019, 10, 1568. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [16].Panella B, Hirscher M, Roth S, Carbon 2005, 43, 2209. [Google Scholar]
- [17].Gomez-Gualdron DA, Wang TC, Garcia-Holley P, Sawelewa RM, Argueta E, Snurr RQ, Hupp JT, Yildirim T, Farha OK, ACS Appl. Mater. Interfaces 2017, 9, 33419. [DOI] [PubMed] [Google Scholar]
- [18].He Y, Zhou W, Yildirim T, Chen B, Energy Environ. Sci 2013, 6, 2735. [Google Scholar]
- [19].a) Rege SU, Yang RT, AlChE J 2000, 46, 734. [Google Scholar]; b) Yang RT, Adsorbents: Fundamentals and Applications, John Wiley and Sons, Inc., Hoboken, NJ, USA: 2003. [Google Scholar]
- [20].Zhang X, Zhang X, Johnson JA, Chen Y-S, Zhang J, J. Am. Chem. Soc 2016, 138, 8380. [DOI] [PubMed] [Google Scholar]
- [21].a) Liu L, Chen Z, Wang J, Zhang D, Zhu Y, Ling S, Huang KW, Belmabkhout Y, Adil K, Zhang Y, Slater B, Eddaoudi M, Han Y, Nat. Chem 2019, 11, 622; [DOI] [PubMed] [Google Scholar]; b) Howarth AJ, Peters AW, Vermeulen NA, Wang TC, Hupp JT, Farha OK, Chem. Mater 2017, 29, 26. [Google Scholar]
- [22].Kaye SS, Dailly A, Yaghi OM, Long JR, J. Am. Chem. Soc 2007, 129, 14176. [DOI] [PubMed] [Google Scholar]
- [23].a) Chen S, Zhang J, Wu T, Feng P, Bu X, J. Am. Chem. Soc 2009, 131, 16027; [DOI] [PubMed] [Google Scholar]; b) Zhai QG, Bu X, Zhao X, Li DS, Feng P, Acc. Chem. Res 2017, 50, 407; [DOI] [PubMed] [Google Scholar]; c) Ye Y, Ma Z, Lin RB, Krishna R, Zhou W, Lin Q, Zhang Z, Xiang S, Chen B, J. Am. Chem. Soc 2019, 141, 4130. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [24].Li P, Vermeulen NA, Malliakas CD, Gomez-Gualdron DA, Howarth AJ, Mehdi BL, Dohnalkova A, Browning ND, O’Keeffe M, Farha OK, Science 2017, 356, 624. [DOI] [PubMed] [Google Scholar]
- [25].Honicke IM, Senkovska I, Bon V, Baburin IA, Bonisch N, Raschke S, Evans JD, Kaskel S, Angew. Chem., Int. Ed 2018, 57, 13780. [DOI] [PubMed] [Google Scholar]
- [26].Dinca M, Long JR, Angew. Chem., Int. Ed 2008, 47, 6766. [DOI] [PubMed] [Google Scholar]
- [27].Zhou W, Wu H, Hartman MR, Yildirim T, J. Phys. Chem. C 2007, 111, 16131. [Google Scholar]
- [28].Spek AL, J. Appl. Crystallogr 2003, 36, 7. [Google Scholar]
- [29].He Y, Chen F, Li B, Qian G, Zhou W, Chen B, Coord. Chem. Rev 2018, 373, 167. [Google Scholar]
- [30].Wilmer CE, Farha OK, Yildirim T, Eryazici I, Krungleviciute V, Sarjeant AA, Snurr RQ, Hupp JT, Energy Environ. Sci 2013, 6, 1158. [Google Scholar]
- [31].Chui SS-Y, Lo SM-F, Charmant JPH, Orpen AG, Williams ID, Science 1999, 283, 1148. [DOI] [PubMed] [Google Scholar]
- [32].Wang C, Volotskova O, Lu K, Ahmad M, Sun C, Xing L, Lin W, J. Am. Chem. Soc 2014, 136, 6171. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [33].Mondloch JE, Bury W, Fairen-Jimenez D, Kwon S, DeMarco EJ, Weston MH, Sarjeant AA, Nguyen ST, Stair PC, Snurr RQ, Farha OK, Hupp JT, J. Am. Chem. Soc 2013, 135, 10294. [DOI] [PubMed] [Google Scholar]
- [34].Dybtsev DN, Chun H, Kim K, Angew. Chem., Int. Ed 2004, 43, 5033. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.