
Fig. 3 |. NAD+ metabolism in ageing.
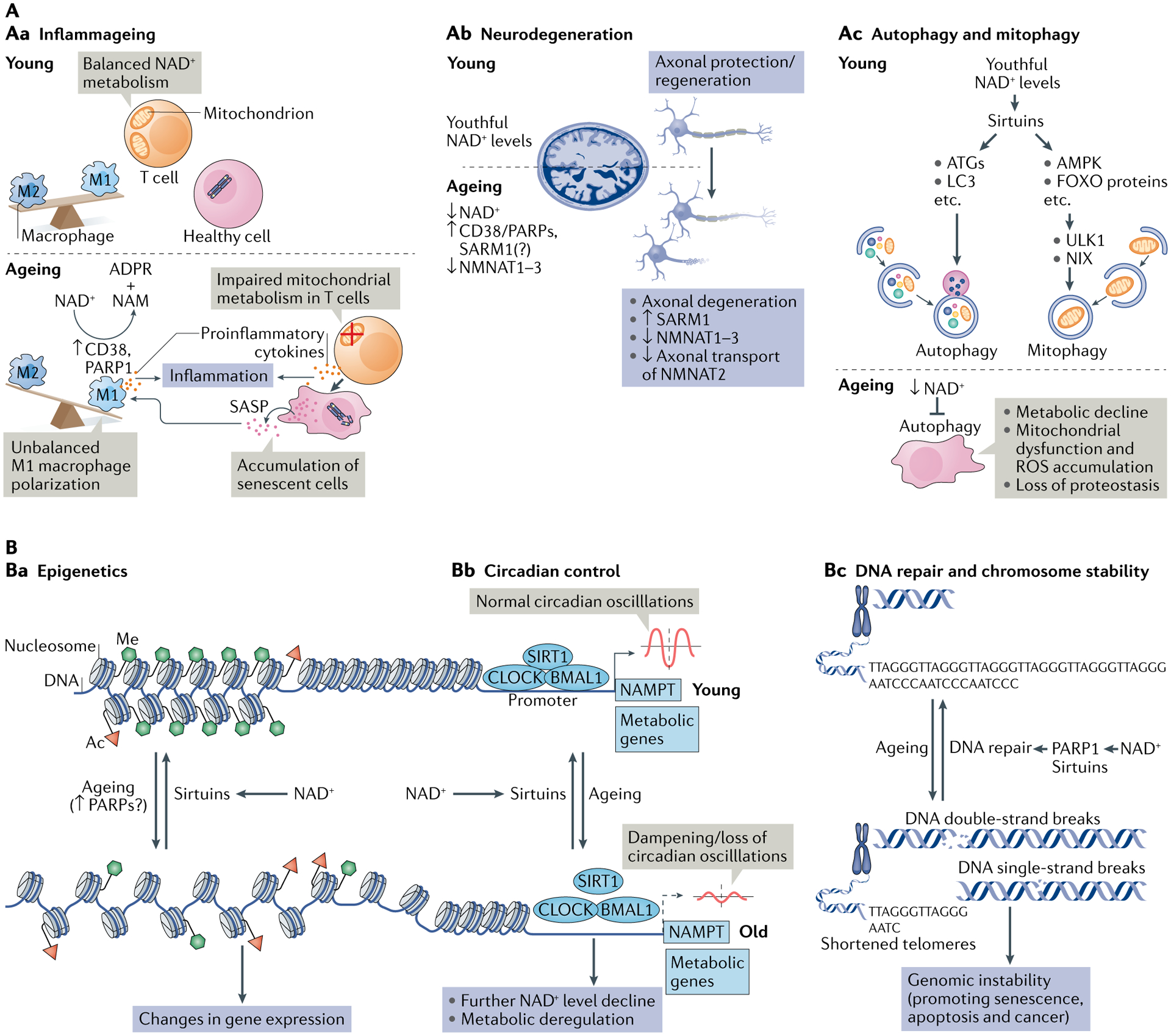
A | Decreased nicotinamide adenine dinucleotide (NAD+) levels have been implicated in various processes associated with ageing (see also Supplementary Box 2). Aa | Ageing is associated with aberrant proinflammatory immune cell activation or ‘inflammageing’, leading to sustained low-grade inflammation. This is caused in part by the accumulation of senescent cells, which via a senescence-associated secretory phenotype (SASP) promote the phenotypic polarization of macrophages towards a proinflammatory M1 state, thereby driving inflammation. There is evidence that in response to the SASP in these macrophages expression of the NAD+-consuming enzymes CD38 and poly(ADP-ribose) polymerases (PARPs) increases, leading to NAD+ level decline, and that these mechanisms importantly contribute to the decrease of NAD+ levels in ageing. In addition, it has been shown that in aged T cells mitochondrial function declines, and this leads to increased secretion of proinflammatory cytokines that promote the state of inflammation and also induce senescence. Ab | Axonal degeneration, which is a precursor to many age-related neuronal disorders, is characterized by rapid NAD+ depletion. During normal physiological conditions, NAD+ biosynthetic enzymes, nicotinamide mononucleotide adenylyltransferases (NMNATs), are protective against axonal degeneration, and their expression supports maintenance of axons and prevents neurodegeneration. In particular, NMNAT2 is an important survival factor in axons, to which it needs to be constantly delivered from the soma — where it is synthesized — to account for its rapid turnover, and these transport processes are disturbed during axonal degeneration. Moreover, the NAD+-consuming enzyme SARM1 is activated by axonal injury and mediates axonal degeneration by promoting NAD+ degradation. Ac | Autophagy is a key cellular catabolic process that allows cells to adapt to variable nutrient availability and serves in cellular quality control, allowing removal of defective organelles and proteins. Autophagy is regulated downstream of NAD+ levels via sirtuins (mostly SIRT1). Decline of NAD+ levels reduces overall autophagic flux as well as selective removal of mitochondria via mitophagy, suggesting that defective autophagy can be a consequence of NAD+ depletion during ageing, contributing to cell dysfunction. B | Because NAD+ is a cofactor for various enzymes, loss of NAD+ impacts a plethora of cellular processes. For example, NAD+ is required for the activity of epigenetic regulators such as SIRT1, and decline in its level causes changes in histone modifications, thereby affecting chromatin organization and function in gene expression. There is evidence that the ageing-associated loss of NAD+ is related to increased expression of PARPs, which can be caused by increased levels of DNA damage and the need for DNA repair during ageing (panel Ba). NAD+ also affects transcriptional activity of the core clock components CLOCK and BMAL, thereby regulating circadian expression of important metabolic genes as well as nicotinamide phosphoribosyltransferase (NAMPT), which in turn is required for circadian oscillation in NAD+ levels (panel Bb). Decreased NAD+ levels also interfere with the activity of PARPs and sirtuins in DNA repair, leading to genomic instability: a hallmark of ageing and cancer (panel Bc). ADPR, ADP-ribose; ATG, autophagy-related protein; FOXO, forkhead box protein O; ROS, reactive oxygen species.