

INTRODUCTION
Amyotrophic lateral sclerosis (ALS) causes significant morbidity but despite the fact that it is an incurable disease, symptomatic management has evolved in the past decade, and aggressive symptom management can improve survival and patient quality of life.
CASE 1: PROGRESSIVE NEUROMUSCULAR RESPIRATORY FAILURE
A 48-year-old man diagnosed with limb-onset ALS with right lower extremity weakness and foot drop presents to clinic for 3-month follow-up. During the visit he mentions he is waking up more tired in the morning and getting more fatigued during the day. At his last clinic visit his forced vital capacity (FVC) was 79% upright and his maximal inspiratory pressure (MIP) was −89 mm Hg. Today in clinic his FVC is 61% upright and his MIP is −62 mm Hg.
- Clinical question: What are the criteria required to initiate a patient with ALS on a respiratory assist device (RAD) for noninvasive ventilation support?
- FVC less than 50%
- MIP less than −60
- Overnight oximetry, with oxygen saturation less than 88% for 5 minutes (not continuous)
- Arterial blood gas, with more than 45 mm Hg
- All of the above
Answer: 5.
Discussion
In the United States, under Centers for Medicare & Medicaid Services guidelines, for people living with neuromuscular disease (ALS, muscular dystrophy, or spinal cord injury), noninvasive ventilation can be initiated with only 1 of the above criteria. People with neuromuscular respiratory do not require a polysomnogram or sleep study to qualify for noninvasive ventilation (Fig. 1).
Fig. 1.
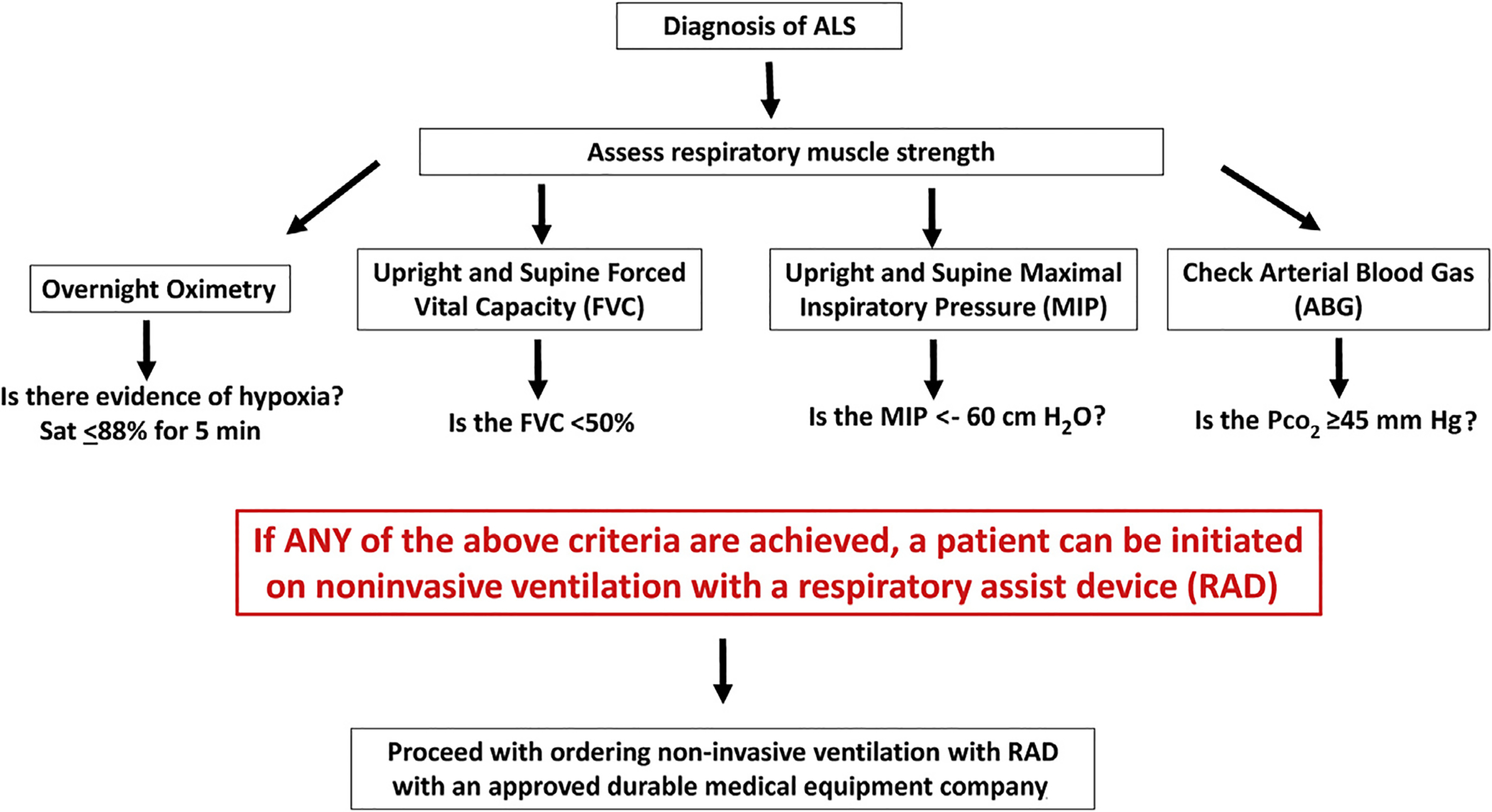
Decision tree, management of respiratory failure in ALS.
Summary
Chronic neuromuscular respiratory failure is the most common cause of morbidity and mortality in ALS patients, due to progressive diaphragm weakness. Diaphragm weakness has an impact on the following respiratory functions:
Inspiratory muscle strength, which contributes most to ventilation
Expiratory muscle strength, required for airway clearance and secretion management
Bulbar muscle function, which protects the airway from recurrent aspiration1
Noninvasive ventilation is proved to extend life in people living with ALS on average 205 days.2
All patients with ALS qualifying and requiring noninvasive ventilation must be initiated on therapy with the proper machine. All patients require a device, either a RAD or home mechanical ventilator, that can provide full (ventilatory) support, meaning the machine provides a back-up respiratory rate. There are various different modes of ventilation available, but every one must include a respiratory rate.
CASE 2: INEFFECTIVE COUGH AND SECRETION MANAGEMENT
A 57-year-old woman with bulbar-onset ALS, with persistent secretions and weak cough, presents to clinic. She currently is on noninvasive ventilation with excellent adherence and compliance. She denies any shortness of breath. She has had progressive difficulty with swallowing and having increased episodes of choking and aspiration. When aspiration occurs, she has difficulty clearing her secretions. Her most recent FVC was 23% with mask, but this value likely reflects difficulty with upper airway weakness.
- With the upcoming cold and flu season, in addition to an annual flu shot, what are other airway clearance options?
- Lung volume recruitment
- Manual assisted cough
- Nebulizer machine
- Suction machine
- High-frequency chest wall oscillation (therapy vest)
- Mechanical insufflation/exsufflation (cough assist)
- All of the above
Answer: 7.
Discussion
Progressive neuromuscular respiratory weakness in ALS leads to impairment with clearing secretions and coughing and can lead to respiratory infections, increasing morbidity and mortality.3 Techniques to assist with secretion clearance are widely recommended and include mucus-mobilizing techniques and assisted cough techniques (Fig. 2).4
Fig. 2.
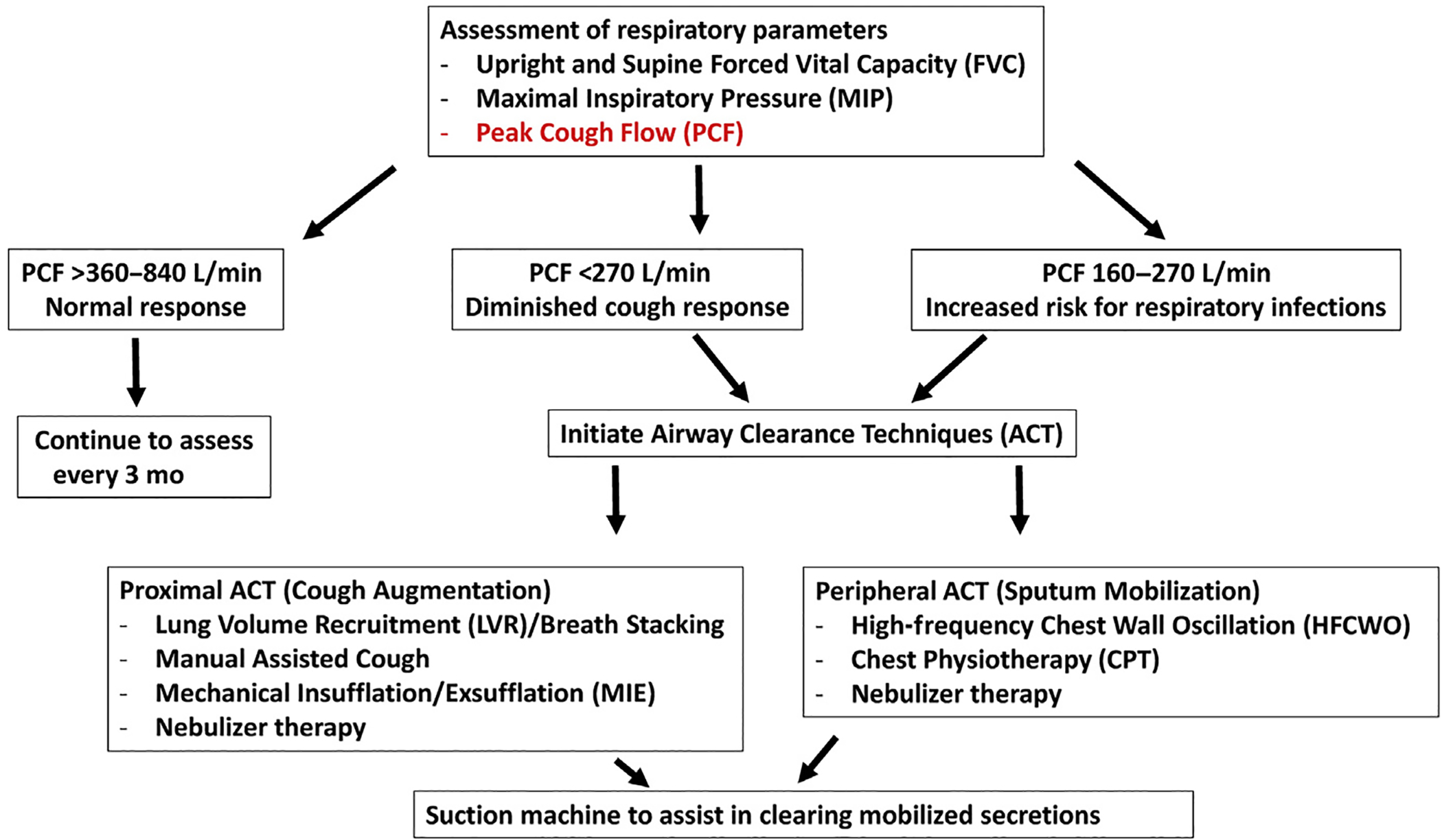
Airway clearance techniques in ALS.
Summary
Assessing effective airway clearance starts with an assessment of peak cough flow. An effective cough is essential to clear airway secretions from the proximal airways. When cough is ineffective secondary to diaphragm muscle weakness, there are a variety of therapies available to help mobilize secretions.5
In addition to respiratory therapies to help mobilize secretions, management of excessive secretions is important. Many people living with ALS struggle with excessive oral secretions, or sialorrhea, and this can come in 2 different types, thick and copious or thin and watery. Management approaches are different in each case.
A classic example of excessive thin watery secretions is a patient with tongue weakness and immobilization, unable to redistribute secretions throughout the mouth. Treatment options include6
Anticholinergic medications (glycopyrrolate, scopolamine, sublingual atropine, and amitriptyline): doses should be escalated as tolerated, and multiple therapies can be used at the same time. With anticholinergic medications, need to monitor for constipation and urinary retention.
Botox injections: performed by otolaryngology surgeons and can be to either or both the submandibular glands and/or parotid glands. This is an effective therapy and typically lasts 2 months to 3 months.7
Ligation: surgical removal of salivary glands
Radiation: evolving therapy, typically requires 5 days of treatment and patient must be able to lay flat8
Excessive thick secretions commonly occur in patients with bulbar disease, who have excessive thick secretions. These can be secondary to medications (anticholinergic), dehydration, or mouth breathing. Treatment options include
Increasing overall hydration status: people living with ALS tend to always not take enough hydration, whether due to difficulty swallowing or difficulty with mobility to get to the washroom.
Increasing oral hydration with nebulizer therapy or humidifiers: with nebulizer therapy, it is recommended to use saline solution to help moisturize thick oral secretions. Increasing humidification in the home if a patient is on noninvasive ventilation is helpful.
Respiratory failure is the most common cause of death in people living with ALS. Respiratory infections can severely diminish respiratory reserve in patients with already reduced function. Comprehensive airway clearance, from management of excessive secretions to mobilization and removal of excessive secretions, is key to maintain optimal respiratory function.
CASE 3: DYSARTHRIA
A 58-year-old man presents with 1 year of progressive dysarthria and dysphagia. The patient initially was having difficulty pronouncing words. The patient denies word-finding difficulties but instead speech was noted by family to be slurred and much slower than his baseline. The patient also notes that eating is taking longer and occasionally he coughs on dry solids. He denies any weakness of arms and legs but does have generalized fatigue. Additional complaints include bouts of laughter and crying that seem difficult to control. Examination demonstrates weakness of the palate, tongue atrophy, and tongue fasciculation. The speech is very slow and nasal (Video 1).
Clinical Questions
-
1.What currently are the most important interventions to help treat dysarthria in ALS?
- Text-to-speech apps to allow patients to select words and phrases from customized menus
- Exercises to strengthen the muscles of the palate, tongue, and mouth
- Surgical palatal lift and/or prostheses
- Brain-machine interface that can translate thoughts into speech
-
2.When should interventions to treat ALS dysarthria be discussed?
- Before a patient has any symptoms of ALS
- As early as possible within the disease course
- Only once a patient has lost all ability to speak
- When dysarthria progresses despite conservative treatments, such as speech therapy
-
3.Which of the following is correct?
- Speech-to-text apps work the best for patients with severe dysarthria.
- It is important for patients to learn about options, such as voice banking, as soon as possible.
- It is important to avoid talking about interventions for dysarthria until it is absolutely necessary.
- Exercises to strengthen speech muscles can reverse dysarthria in most patients
-
Answers: 1, A; 2, B; 3, B.
Discussion
Dysarthria may be a presenting symptom in ALS with bulbar onset, which includes approximately 20% of patients.9 Nonetheless, eventually at some point in their disease, 80% to 95% of ALS patients cannot meet their daily communication needs with natural speech. This makes it challenging for patients to communicate their needs to caregivers, which is a major factor in quality of life.10 The dysarthria most often is of mixed etiology, with spasticity from corticobulbar dysfunction and flaccidity with atrophy from bulbar dysfunction contributing, although either presentation can predominate early in the disease.11
Summary
The major interventions for dysarthria in ALS focus on training the patient and family to use assistive and augmentative communication.11–13
Low-technology options, such as a writing board, may be used early in the disease in patients with functional limbs.
The most commonly used tools include text-to-speech apps, where patients can type or choose words/phrases on a tablet screen.
If motor function precludes manipulating a tablet screen with the limbs, patients can use eye gaze software that tracks eye movements to control a cursor on the screen.
Proactive, early introduction of these tools is essential for several important reasons.
-
Some of the apps include customizable menus and often have a significant learning curve. This requires patients and their families to practice and tailor the functionality of the program to their needs over time.
Some text-to-speech devices allow patients to bank their own voice, and, for this to be most beneficial, the voice must be recorded when speech is most intelligible. Although patients and families often ask for specific strengthening exercises to improve speech, the role of exercise in the management of bulbar dysfunction in ALS is not well understood.14 Because of the detrimental effect of fatigue, patients usually are taught to save their voice for specific times that are highest priority and to minimize fatigue by using speech amplification devices, such as portable microphones. Additionally, patients and families can reduce fatigue by pursuing nonverbal communication wherever possible. Several centers have reported improved dysarthria with surgical palatal lift or prostheses but the evidence supporting their use is limited.
CASE 4: DYSPHAGIA
A 62-year-old woman, who presented with left foot drop 18 months ago and 6 months ago, developed left hand weakness, dysphagia, and dysarthria. In the last year, the patient’s weight declined from 160 to 125. Initially the patient had decreased appetite but in the last 6 months she has had dysphagia first for solids and then for all oral intake. When she tries to eat, the food gets stuck in her throat and she coughs. She tried to eat more slowly and cut her food up but eventually meals took over an hour. She also complains of excessive drooling and trouble catching her breath during meals.
Clinical Questions
-
1.When should enteral nutrition be discussed with an ALS patient?
- When a patient has reached 50% of the baseline/premorbid weight
- The timing is individualized but should be introduced as early as possible. This is best approached over multiple visits with a goal of having a firm plan well before it is needed urgently.
- Once the FVC has reached less than 30%
- Only once a patient has lost 10% of baseline/premorbid weight despite speech therapist evaluation and nutrition supplementation
-
2.What are the key deciding factors regarding which procedure a patient will have to place a gastrostomy tube?
- FVC
- Patient preference
- Center expertise
- All of the above
-
3.Which of the following is correct?
- To be able to receive gastrostomy via percutaneous endoscopic gastrostomy (PEG), a patient must be able to lay flat.
- Swallowing therapy is able to reverse dysphagia in a majority of patients.
- Once a patient receives gastrostomy, it is essential to no longer eat or drink by mouth to prevent aspiration.
- Consensus guidelines recommend dysphagia screening at least annually in all ALS patients.
-
Answers: 1, B; 2, D; 3, A.
Discussion
Management of weight loss and nutrition in ALS
ALS patients who are malnourished have 30% increase in risk of death for each 5% of weight lost and more than 7-fold increased risk of death overall.15,16 For this reason, ensuring that all ALS patients are treated proactively for dysphagia and weight loss is among the most impactful treatment goals in their overall care plan.
Consensus guidelines support quarterly assessments of dysphagia initially with screening questions.1 Any dysphagia complaints should prompt evaluation by speech therapist, who can perform bedside and videofluoroscopic assessments.
Summary
It is important to stress to patients that the presence of dysphagia does not have to preclude all oral intake.
Especially early in the course of disease, behavioral adaptations, such as modification of food and/or liquid consistency, upright eating, and chin-tuck maneuvers, can minimize the chance of aspiration.17 These techniques also are useful later in the disease because they can enable some patients to experience select foods for pleasure.
Nutritionists can help monitor caloric intake and often recommend nutritional supplements to help maintain body weight
If patients have lost more than 10% of their premorbid bodyweight, enteral nutrition is the appropriate intervention.
Enteral nutrition actually may slow disease progression and improve survival.1 The discussion about enteric nutrition should be started as early in the disease course as is reasonably possible. Patients and their families benefit from time to process all of the options.
The options include
PEG
Radiologically inserted gastrostomy (RIG)18
Importantly, respiratory function is used to help guide the timing of initiation and the specific procedure the patient will receive.19
PEG tube placement requires a patient to receive sedation and is performed in a supine position. This becomes more dangerous in patients with poor respiratory function. For this reason, most centers attempt PEG tube insertion before the FVC declines to less than 50%.1,20
RIG can be performed in an upright position and can be done without sedation.
If FVC is less than 50%, PEG still can be performed with additional support and monitoring by anesthesia. Many centers, however, first stabilize the patient on noninvasive ventilation and then pursue RIG.
Nonetheless, center and patient preference ultimately guide this decision, with the goal of a successful procedure as early as is appropriate.
CASE 5: PSEUDOBULBAR AFFECT
A 58-year-old man presented with bulbar-onset ALS; his symptoms started about 1 year prior to diagnosis with slurred speech that was worse when he was tired. He also noticed episodes of incontrollable laughing with a minimal trigger. His wife said that that certain commercials on TV make him cry easily. He would not describe him-self as an emotional person but these episodes were embarrassing to the patient to the point that he limited his social interactions out of fear of having these sudden and inexplicable outbursts in public (Video 2).
- What is the correct answer?
- These episodes of incontrollable laughing or crying, or pseudobulbar affect (PBA), depend on the patient mood.
- PBA affects only ALS patients.
- The only Food and Drug Administration (FDA)-approved treatments for PBA are selective serotonin reuptake inhibitors (SSRIs) and tricyclic antidepressants (TCAs).
- A combination dextromethorphan and quinidine (Nuedexta) has been shown to reduce the frequency of PBA episodes and improve the quality of life of patients in a large, phase 3, multicenter randomized trial.
Answer: D.
Discussion and Summary
Management of pseudobulbar affect
PBA, also known as emotional lability and pathologic laughing and crying, falls under the umbrella term of involuntary emotional expression disorder.21 It is characterized by sudden outbursts of involuntary and exaggerated laughter and/or crying.22 These episodes are disproportionate or separate from a patient’s undelaying mood or social context.23 PBA affects up to 50% of patients with ALS22 and is more prevalent in patient with the bulbar form of the disease. It has significant impact on quality of life and often leads to social isolation. PBA is not specific to ALS and can be seen in multiple sclerosis, dementia, traumatic brain injury, stroke, and Parkinson disease.23 The anatomic substrate of this syndrome is not well understood but applying advanced neuroimaging and neurophysiologic techniques suggests that, irrespective of the pathology, disturbed circuitry involved in the initiation and modulation of emotional output with cortico-ponto-cerebellar network dysfunction seems central to its pathophysiology.24
SSRIs and TCAs used to be the most frequently used off-label treatment of PBA.
In 2010, dextromethorphan (20 mg) and quinidine sulfate (10 mg) become the first FDA-approved treatment of PBA. Dextromethorphan is metabolized rapidly by the hepatic first-pass metabolism through cytochrome P450 (CYP)-2D6, and adding a low-dose quinidine as a CYP-2D6 inhibitor increases its bioavailability. A large, phase 3, randomized placebo-controlled trial showed that this combination reduced the frequency and the severity of PBA episodes and improved patient quality of life25; side effects include dizziness, nausea, diarrhea, and somnolence,25 and they can be minimized by starting the treatment at 1 tablet at bedtime for 1 week followed by 1 tablet twice a day. There also is a mild non-arrhythmogenic QT prolongation,25 and caution should be used with this drug in patients with cardiac disease; an electrocardiogram prior and 2 weeks after treatment initiation may be needed to establish safety. A small randomized, blinded, crossover study also showed that dextromethorphan/quinidine improved speech and swallowing in ALS patients, suggesting that it may have a neuroprotective effect26; further study is needed to verify this finding.27
CASE 6: SPASTICITY
A 37-year-old man recently diagnosed with lower limb–onset ALS complains of increasing right leg stiffness, decreased walking endurance, and painful muscle spasms that occur mainly at night. In retrospect, his symptoms began approximately 9 months previously, when he noticed frequent muscle twitching in his legs and felt his right legs stiffen up when he tried to run to catch his train to work in the morning. His symptoms have progressed gradually since then to include difficulty holding a pen or typing with this right hand. On general examination, his blood pressure was 106/68 mm Hg, heart rate was 58 beats per minute, and respiratory rate was 16 breaths per minute. The neurologic examination was notable for diffuse but asymmetric lower extremity weakness without marked muscle atrophy. He also has weakness in the right hand intrinsic muscles, including the first dorsal interosseous and abductor policies brevis but sparing the abductor digiti minimi. He still can overcome mild resistance on manual muscle testing in his right knee extensors, but he walks right knee–flexed during the midstance phase of gait cycle. He abnormally has increased deep tendon reflexes in all limbs and an exaggerated jaw jerk reflex. His sensory examination is normal.
- Regarding the case, what are the clinical features that are most supportive of an ALS diagnosis?
- Progressive motor deficits
- Asymmetric weakness pattern
- Split hand pattern of weakness in right hand (ie, greater involvement of the thenar/lateral hand then of the hypothenar/medial hand intrinsic muscles)1
- Combination of upper and lower motor neuron clinical features
- All of the above
Answer: 5.
Discussion
At different stages in their disease, ALS patients have different functional limitations to be addressed. At this early stage, the patient remains independent for ambulation, but his right leg stiffness and muscle spasms are causing him pain and decreasing his walking endurance. In addition to following American Academy of Neurology guidelines regarding management and care of the patient with ALS, personalized consideration is needed for his upper motor neuron features that include right leg muscle spasticity. Spasticity refers to a velocity-dependent increase in muscle tone. This is demonstrated in Video 3 as there is normal tone with slow passive range of motion of the knee but a clear spastic catch and release near the end range on a more rapid passive motion (see Video 3). This can be graded and monitored most commonly with the Modified Ashworth Scale.28 Symptomatic treatment of spasticity should be goal directed. In this case, the goals are to decrease painful muscle spasms and improve his stiff-legged gait pattern. Conservative treatment with skilled physical therapy that includes stretching and education on proper limb positioning and relaxation techniques should be offered as first-line therapy. Currently there are 4 FDA-approved prescription oral agents for the management of spasticity:
- Baclofen (γ-aminobutyric acid [GABA]B receptor agonist)
- Tizanidine (α2-adrenergic agonist)
- Dantrolene (blocks the release of Ca++ from the sarcoplasmic reticulum in muscle)
- Diazepam (facilitates activation of GABAA receptor subtype)
Answer: 1.
Summary
There is no high-quality evidence to choose 1 class of antispasticity agent over another in ALS. In ALS multidisciplinary clinics, however, the most common antispasticity medication is baclofen29,30 The initial dose is 5 mg to 10 mg, 2 to 3 times a day, and can be titrated up to 20 mg, 4 times a day, but the highest doses rarely are needed for ALS. In addition, for this case, the relatively low normal blood pressure value is an argument against selecting tizanidine. Tizanidine is in the same drug class as common antihypertensive medications, such as clonidine, and therefore could increase risk for orthostatic hypotension. In theory, dantrolene has the lowest risk of central nervous system side effects because its mechanism of action is peripheral, but in practice it is rarely used in ALS patients over concerns that it exacerbates weakness and its association with hepatic toxicity.31 Diazepam tends to be avoided due to concerns for respiratory depression.30
Supplementary Material
KEY POINTS.
Multidisciplinary care of patients with amyotrophic lateral sclerosis (ALS) in specialized clinics improves survival.
Early initiation of noninvasive ventilation improves quality of life and prolongs survival.
Early implementation of airway clearance techniques in patients with bulbar ALS may improve noninvasive ventilation compliance and prevent respiratory infections.
Addressing dysarthria and pseudobulbar affect may improve social isolation of ALS patients.
Spasticity may be difficult to treat in ALS patients and may require the trial of different agents.
Footnotes
Video content accompanies this article at http://www.neurologic.theclinics.com.
SUPPLEMENTARY DATA
Supplementary data to this article can be found online at https://doi.org/10.1016/j.ncl.2020.03.013.
REFERENCES
- 1.Miller RG, Jackson CE, Kasarskis EJ, et al. Practice parameter update: the care of the patient with amyotrophic lateral sclerosis: drug, nutritional, and respiratory therapies (an evidence-based review): report of the Quality Standards Subcommittee of the American Academy of Neurology. Neurology 2009;73(15):1218–26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Bourke SC, Tomlinson M, Williams TL, et al. Effects of non-invasive ventilation on survival and quality of life in patients with amyotrophic lateral sclerosis: a randomised controlled trial. Lancet Neurol 2006;5(2):140–7. [DOI] [PubMed] [Google Scholar]
- 3.Hanayama K, Ishikawa Y, Bach JR. Amyotrophic lateral sclerosis. Successful treatment of mucous plugging by mechanical insufflation-exsufflation. Am J Phys Med Rehabil 1997;76(4):338–9. [DOI] [PubMed] [Google Scholar]
- 4.Lechtzin N, Wolfe LF, Frick KD. The impact of high-frequency chest wall oscillation on healthcare use in patients with neuromuscular diseases. Ann Am Thorac Soc 2016;13(6):904–9. [DOI] [PubMed] [Google Scholar]
- 5.Boitano LJ. Management of airway clearance in neuromuscular disease. Respir Care 2006;51(8):913–22 [discussion: 22–4]. [PubMed] [Google Scholar]
- 6.Jackson CE, McVey AL, Rudnicki S, et al. Symptom management and end-of-life care in amyotrophic lateral sclerosis. Neurol Clin 2015;33(4):889–908. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Squires N, Humberstone M, Wills A, et al. The use of botulinum toxin injections to manage drooling in amyotrophic lateral sclerosis/motor neurone disease: a systematic review. Dysphagia 2014;29(4):500–8. [DOI] [PubMed] [Google Scholar]
- 8.Assouline A, Levy A, Abdelnour-Mallet M, et al. Radiation therapy for hypersalivation: a prospective study in 50 amyotrophic lateral sclerosis patients. Int J Radiat Oncol Biol Phys 2014;88(3):589–95. [DOI] [PubMed] [Google Scholar]
- 9.Swinnen B, Robberecht W. The phenotypic variability of amyotrophic lateral sclerosis. Nat Rev Neurol 2014;10(11):661–70. [DOI] [PubMed] [Google Scholar]
- 10.Korner S, Sieniawski M, Kollewe K, et al. Speech therapy and communication device: impact on quality of life and mood in patients with amyotrophic lateral sclerosis. Amyotroph Lateral Scler Frontotemporal Degener 2013;14(1):20–5. [DOI] [PubMed] [Google Scholar]
- 11.Beukelman D, Fager S, Nordness A. Communication support for people with ALS. Neurol Res Int 2011;2011:714693. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Hanson EK, Beukelman DR, Yorkston KM. Communication support through multimodal supplementation: a scoping review. Augment Altern Commun 2013;29(4): 310–21. [DOI] [PubMed] [Google Scholar]
- 13.Brownlee A, Palovcak M. The role of augmentative communication devices in the medical management of ALS. NeuroRehabilitation 2007;22(6):445–50. [PubMed] [Google Scholar]
- 14.Plowman EK. Is there a role for exercise in the management of bulbar dysfunction in amyotrophic lateral sclerosis? J Speech Lang Hear Res 2015;58(4):1151–66. [DOI] [PubMed] [Google Scholar]
- 15.Desport JC, Preux PM, Truong TC, et al. Nutritional status is a prognostic factor for survival in ALS patients. Neurology 1999;53(5):1059–63. [DOI] [PubMed] [Google Scholar]
- 16.Marin B, Desport JC, Kajeu P, et al. Alteration of nutritional status at diagnosis is a prognostic factor for survival of amyotrophic lateral sclerosis patients. J Neurol Neurosurg Psychiatry 2011;82(6):628–34. [DOI] [PubMed] [Google Scholar]
- 17.Dorst J, Ludolph AC, Huebers A. Disease-modifying and symptomatic treatment of amyotrophic lateral sclerosis. Ther Adv Neurol Disord 2018;11. 1756285617734734. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.ProGas Study Group. Gastrostomy in patients with amyotrophic lateral sclerosis (ProGas): a prospective cohort study. Lancet Neurol 2015;14(7):702–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Yang B, Shi X. Percutaneous endoscopic gastrostomy versus fluoroscopic gastrostomy in amyotrophic lateral sclerosis (ALS) sufferers with nutritional impairment: a meta-analysis of current studies. Oncotarget 2017;8(60):102244–53. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.EFNS Task Force on Diagnosis and Management of Amyotrophic Lateral Sclerosis, Andersen PM, Abrahams S, Borasio GD, et al. EFNS guidelines on the clinical management of amyotrophic lateral sclerosis (MALS)–revised report of an EFNS task force. Eur J Neurol 2012;19(3):360–75. [DOI] [PubMed] [Google Scholar]
- 21.Cummings JL, Arciniegas DB, Brooks BR, et al. Defining and diagnosing involuntary emotional expression disorder. CNS Spectr 2006;11(S6):1–7. [DOI] [PubMed] [Google Scholar]
- 22.Gallagher JP. Pathologic laughter and crying in ALS: a search for their origin. Acta Neurol Scand 1989;80(2):114–7. [DOI] [PubMed] [Google Scholar]
- 23.Schiffer R, Pope LE. Review of pseudobulbar affect including a novel and potential therapy. J Neuropsychiatry Clin Neurosci 2005;17(4):447–54. [DOI] [PubMed] [Google Scholar]
- 24.Finegan E, Chipika RH, Li Hi Shing S, et al. Pathological crying and laughing in motor neuron disease: pathobiology, screening, intervention. Front Neurol 2019; 10:260. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Pioro EP, Brooks BR, Cummings J, et al. Dextromethorphan plus ultra low-dose quinidine reduces pseudobulbar affect. Ann Neurol 2010;68(5):693–702. [DOI] [PubMed] [Google Scholar]
- 26.Smith R, Pioro E, Myers K, et al. Enhanced bulbar function in amyotrophic lateral sclerosis: the nuedexta treatment trial. Neurotherapeutics 2017;14(3):762–72. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Green JR, Allison KM, Cordella C, et al. Additional evidence for a therapeutic effect of dextromethorphan/quinidine on bulbar motor function in patients with amyotrophic lateral sclerosis: A quantitative speech analysis. Br J Clin Pharmacol 2018;84(12):2849–56. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Bohannon RW, Smith MB. Interrater reliability of a modified Ashworth scale of muscle spasticity. Phys Ther 1987;67(2):206–7. [DOI] [PubMed] [Google Scholar]
- 29.Mayadev AS, Weiss MD, Distad BJ, et al. The amyotrophic lateral sclerosis center: a model of multidisciplinary management. Phys Med Rehabil Clin N Am 2008; 19(3):619–31, xi. [DOI] [PubMed] [Google Scholar]
- 30.Rocha JA, Reis C, Simoes F, et al. Diagnostic investigation and multidisciplinary management in motor neuron disease. J Neurol 2005;252(12):1435–47. [DOI] [PubMed] [Google Scholar]
- 31.Utili R, Boitnott JK, Zimmerman HJ. Dantrolene-associated hepatic injury. Incidence and character. Gastroenterology 1977;72(4 Pt 1):610–6. [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.