

Abstract
Synthesis and characterisation of a dithiadiaza chelator NSNS2A, as well as copper complexes thereof are reported in this paper. Molecular structures of copper (I/II) complexes were calculated using density-functional theory (DFT) further validated by both NMR and EPR spectroscopy. DFT calculations revealed a switch in the orientation of tetragonal distortion upon protonation, which might be responsible for poor stability of Cu(II)NSNS2A complex in aqueous media, whilst the same switch in tetragonal distortion was experimentally observed by changing the solvent. The chelator was radiolabeled with 64Cu and evaluated using PET/MRI in rats. Despite a favorable redox potential to stabilize the cuprous state in vivo, 64Cu(II)NSNS2A complex showed suboptimal stability compared to its tetraazamacrocyclic analogue, 64Cu(TE2A), with a significant 64Cu uptake in the liver.
Introduction
Copper homeostasis in the body remains a subject of an active debate, given its essential role in various biological processes. Many of these processes involve reversible switching between Cu(I) and Cu(II), suggesting an intrinsic capacity of biological systems to stabilize both cupric and cuprous states.1 Endogenous copper complexes may experience similar redox transformations, which affect their in vivo stability but often escape the scrutiny when designing new radiopharmaceuticals, including 64Cu PET tracers. Although very high kinetic inertness and thermodynamic stability was reported for macrocyclic chelators widely used for binding 64Cu in vitro, the residual uptake by the hepatobiliary system was reported in preclinical studies.2 If chemically intact in the body, these hydrophilic chelates would experience an exclusively renal clearance, thereby the residual uptake in the liver suggests a possible transchelation mediated by liver proteins, as was later confirmed by size-exclusion radio-HPLC.3 Substantial progress has been achieved by rigidifying ligand structures to further enhance the stability of their Cu(II) complexes in vitro, giving rise to a cross-bridged CB-TE2A chelator, as well as hexaazamacrobicyclic cage-like sarcophagine family.4 Their use resulted in an increased stability of 64Cu tracers against transchelation in vivo. Although CB-TE2A required harsher radiolabelling conditions, often incompatible with the use of conjugated biological vectors, sarcophagine chelators combined a very high kinetic inertness in vivo with mild radiolabelling conditions, resulting in their rising popularity in preclinical studies.5, 6
Notwithstanding the fact that the lability of Cu(I) species formed by reduction of the Cu(II) complexes was suggested among possible culprits behind the observed in vivo transchelation in complexes with some tetraazamacrocycles,7 only few recent attempts have been made to design ligands that can support both cuprous and cupric states.8–10 For example, the transchelation of copper to intracellular proteins upon reduction to Cu(I) has been suggested as the rationale behind the observed signal enhancement in patients with hypoxic tumours, who were administered with a cell-permeable tracer 64Cu-ATSM.11 Meanwhile, most extracellular chelators have been designed to provide an increased redox stability of the Cu(II) state by decreasing the E1/2 potential to prevent a possible in vivo reduction in the first place.7, 12
Unlike oxophilic cupric ions, larger cuprous ions are known to preferentially form stable tetrahedral complexes with softer donors, such as thiols and thioethers. In biological systems, active sites of cuproenzymes usually share either a tetrahedral N2S2 or five-coordinate N2S2O coordination motifs of copper ions that can reversibly switch between Cu(I) and Cu(II) states.13 For instance, the five-coordinate copper ion in Type 1 copper site in a bacterial blue copper protein azurin is coordinated by two histidine N and one cysteine S atoms in an equatorial plane, as well as by axially bound amide oxygen of glycine and a thioether of methionine (Fig.1). Upon reduction, the coordination polyhedron, best represented as a distorted trigonal bipyramid, experiences only small structural rearrangements to ensure its reversibility and rapid electron transfer.14 Lower reduction potentials were observed for homologous green and red copper proteins with thiolates of cysteine and homocysteine substituted for methionine, respectively, since a stronger axial Cu-S bond with thiolates favours a tetragonal geometry around copper and hence stabilises the cupric state.15
Figure 1.

Coordination environment of the Type 1 copper binding site in azurin.
Inspired by unusually intense absorption bands around 600 nm in blue copper proteins, a series of macrocyclic complexes containing sulphur donors was devised in the late 1970s. These complexes featured a reversible redox behaviour with very high reduction potentials, which correlate with the number of sulphur atoms, as well as chelate and macrocyclic ring sizes.16 However, the higher reduction potential comes at a cost of compromised stability of the Cu(II) complex, whilst the stability of Cu(I) species was shown to vary little with the general ligand morphology.17 Therefore, an optimal interplay between the biologically relevant stability constant of the Cu(II) complex and its redox potential should be attained through judicious design of the ligand. For instance, by introducing sulphur donors in thiaaza macrocycles, a fine tuning of the redox potential can easily be achieved.
Although the previous attempt to utilise 12/13/14-membered dithiadiaza macrocycles NEC-SE/P/B (Fig.2) as 64Cu chelators proved unsuccessful, presumably due to a high lability of the Cu(II) complexes,18 alternative ligands that form kinetically inert copper(I/II) complexes can potentially deliver a better in vivo performance. A previously reported dithiadiaza macrocycle [14]aneNSNS19 – a dithiadiaza analogue of cyclam – and its recently reported derivative [14]aneNSNS2POH20 bearing phosphonate-derivatised arms have a reasonably high redox potential to enable their facile reduction in the body (E1/2 = 209 mV vs N.H.E. for [14]aneNSNS), whilst maintaining a sufficient thermodynamic stability (LogKCu(II)L = 15.15). By introducing two N-acetate pendant arms, a potentially six-coordinate ligand NSNS2A is obtained, which is expected to lock a highly flexible NSNS core and form a kinetically inert Cu(II)(NSNS2A) complex suitable for PET imaging. The proposed ligand is reminiscent of a cyclam-derived TE2A,21 which forms highly kinetically inert copper (II) complexes and was successfully tested as 64Cu chelator in preclinical studies.22
Figure 2.

Structures of ligands discussed in the paper.
In the present paper, we report on the synthesis and structural elucidation of Cu(I) and Cu(II) complexes with NSNS2A ligand by means of X-ray crystallography, EPR and NMR spectroscopies augmented by DFT and TDDFT calculations. The chelator was also radiolabelled with 64Cu and injected into Wistar rats to evaluate its biodistribution and pharmacokinetics.
Results and discussion
Dithiadiaza macrocycle [14]aneNSNS was synthesised according to a previously reported procedure.23 It was alkylated to yield an ethyl protected chelator 1, which was hydrolysed to give 2 as an HCl double salt (Scheme 2). Cupric complex Cu(II)(NSNS2A) was synthesized by stirring copper (II) acetate with 2 in water at room temperature for 10 min.
Since radiolabelling of chelators is often carried out in the presence of various buffers, such as acetate and citrate salts, their potential effect on the formation of Cu(II)(NSNS2A) was also investigated. It turned out that the acetate buffer (40 mM, pH = 5.5) did not affect the UV-vis spectrum either at room temperature or after heating at 80°C for 30 min, suggesting no direct interaction between an acetate anion of the buffer and complexed copper ion. At the same time, only minor changes were observed in the UV-vis spectrum of Cu(II)(NSNS2A) in a citrate buffer (0.3 M, pH = 6.0), which did not, however, include d-d transitions, and therefore are unlikely to corroborate any significant changes in the coordination environment of copper ion (Fig.S22 in ESI).
Cuprous complex [Cu(I)(H2NSNS2A)]PF6 was synthesized by reacting Cu(MeCN)4PF6 with 2 under anaerobic conditions. Formation of both complexes was verified by 1H and 13C NMR spectroscopy in deuterated DMSO-d6.
Although attempts to obtain single crystals of copper complexes suitable for X-ray analysis proved unsuccessful, slow evaporation of the solvent from the solution containing (1) and Cu(ClO4)2 yielded single crystals of [Cu(1)](ClO4)2 (Fig.3, Table 1). The obtained crystal structure revealed a trigonally distorted octahedral coordination of Cu2+ ion, with an almost perfect [N2S2] plane (Cu-N(1) 2.074(3)Å, Cu-S(1) 2.3186(8)Å) and a straight O-Cu-O bond (Cu-O(1) 2.3256(18)) tilted in [N2O2] plane (angle O(1)-Cu-N(1) 79.22(9)°). In a previously reported crystal structure of [Cu(NSNS)](ClO4)2 with a coordination polyhedron that is best described as an axially elongated octahedron with perpendicular rhombic distortion, slightly shorter bond lengths in equatorial [N2S2] plane were revealed (Cu-N(1) 2.0146(13)Å, Cu-S(1) 2.2954(10)Å), due to much weaker Cu-O bonding (Cu-O 2.576(2)Å). However, a similar, albeit a much smaller tilt of a perfectly straight O-Cu-O bond was still observed, this time in [N2O2] plane (angle O-Cu-S 83.59(6)°).
Figure 3.

The thermal ellipsoid diagram showing the structure of the cationic unit for [Cu(1)](ClO4)2. Hydrogen atoms and counteranions are omitted for clarity. Thermal ellipsoids are set at 35% probability.
Table 1.
Selected bond angles and bond lengths for copper (II) complexes discussed in the paper, as calculated by XRD ([Cu(1)](ClO4)2) or DFT (Cu(II)(NSNS2A) and [Cu(II)(H2NSNS2A)]2+)
| [Cu(1)](ClO4)2 | Cu(II)(NSNS2A) | [Cu(II)(H2NSNS2A)]2+ | |
|---|---|---|---|
| Bond lengths (Å) | |||
| Cu-O | 2.3256(18), 2.3256(18) | 1.997, 1.997 | 2.462, 2.464 |
| Cu-S | 2.3186(8), 2.3186(8) | 2.746, 2.746 | 2.366, 2.366 |
| Cu-N | 2.074(3), 2.074(3) | 2.066, 2.066 | 2.141, 2.142 |
| Bond angles (°) | |||
| N-Cu-N | 180.0(3) | 180.0 | 179.9 |
| N-Cu-S | 88.82, 91.18(8) | 88.1, 91.9, 88.1, 91.9 | 88.4, 91.6, 88.4, 91.6 |
| S-Cu-S | 180.0 | 180.0 | 180.0 |
| O-Cu-O | 180.0 | 180.0 | 180.0 |
| O-Cu-N | 100.78(9), 79.22(9), 100.78(9), 79.22(9) | 95.7, 84.3, 95.7, 84.3 | 103.7, 76.3, 103.6, 76.3 |
| O-Cu-S | 89.85(5), 90.15(5), 89.85(5), 90.15(5) | 91.7, 88.3, 81.7, 88.3 | 90.3, 89.7, 90.3, 89.7 |
| C-S-C | 104.81(16), 104.81(16) | 105.1, 105.1 | 104.4, 104.4 |
| C-N-C | 105.1(3), 105.1(3) | 107.3, 107.3 | 106.6, 106.6 |
In a first approximation, solid state structures of metal complexes are usually assumed to be close to the solutes, as most analytical tools at our disposal can only provide indirect evidences towards a particular structure in solution. Given these circumstances, ab initio quantum chemical calculations can serve as a good starting point to validate and, if necessary, optimize the structure based on spectroscopic experiments. The crystal structure of ([Cu(1)](ClO4)2) was used as an initial input to calculate the molecular structure of Cu(II)(NSNS2A) in aqueous solution at the D3-B3LYP/def2-TZVP level of theory. In the calculated structure, the [14]aneNSNS macrocycle experienced notable conformational changes, with significantly elongated Cu-S bonds (Table 1), partially releasing the strain caused by the rearrangement of the macrocycle upon complexation.
This, in turn, gave rise to a remarkable elongation of Cu-S bonds (2.746Å), while leaving Cu-N bonds almost intact (2.066Å). At the same time, Cu-O bond with anionic pendant arms significantly strengthened, becoming much shorter than the bond in an analogous Cu(TE2A) complex (1.997Å vs 2.263Å).21 The straight O-Cu-O bond is tilted with respect to the N2O2 plane by 84.27°.
The infrared spectrum in water is in line with the proposed structure, featuring symmetric and asymmetric stretching bands at frequencies typical for coordinated carboxylate groups (νs = 1395 cm−1 and νas = 1592 cm−1, Δ = νas-νs = 197 cm−1).
The absorption spectrum of Cu(II)(NSNS2A) features two absorption bands at λ = 314 nm (ε = 2445 M−1cm−1, pH = 5.0) and λ = 365 nm (ε = 5770 M−1cm−1, pH = 5.0), which were previously assigned to N(δ)→Cu2+(dx2-y2) and S(δ)→Cu2+(dx2-y2) ligand-to-metal charge transfer (LMCT) states, respectively, and can be used to probe the coordination environment of copper.24 These absorption bands are very close to the LMCT transitions observed in [Cu(NSNS)](ClO4)2, suggesting the presence of a square-planar [N2S2] coordination environment in Cu(II)(NSNS2A), although the broad asymmetric band corresponding to d-d transition is bathochromically shifted (λd-d = 597 nm (ε = 98 M−1cm−1, pH = 5.0) in Cu(II)(NSNS2A) compared to Cu(II)(NSNS) (λd-d = 545 nm). The latter can serve as an evidence of the stronger axial interaction, in line with the notion of oxygen atoms of acetate pendant arms being bound to copper.
At the same time, the presence of a relatively intense S(δ)→Cu2+(dx2-y2) band contradicts to the calculated DFT structure of Cu(II)(NSNS2A) (Fig.4), with the dx2-y2 orbital being located in the [N2O2] plane and therefore providing very poor overlap, if any, with δ orbitals of the sulphur atoms. In contrast, the DFT structure of [Cu(II)(H2NSNS2A)]2+ with protonated acetate arms (vide infra) exhibits shorter Cu-S and elongated Cu-O bonds, flipping the dx2-y2 orbital of Cu2+ into [N2S2] plane, consistent with the absorption spectrum in water. A similar orbital switch might be observed if acetate pendant arms of the macrocycle participate in hydrogen bonding with water molecules. Despite our efforts to obtain corresponding DFT structures with water molecules bound via hydrogen bonds, such calculations never converged. The possible role of hydrogen bonding in elongation of Cu-O bonds and rearrangement of the spin density was further elucidated by running absorption spectra in aprotic solvents, which, however, gave inconclusive results. Although the UV-vis spectrum of Cu(II)(NSNS2A) in acetonitrile and 1,4-dioxane showed the expected dramatic decrease of S(δ)→Cu2+(dx2-y2) band (in the case of acetonitrile, down to zero), the spectrum recorded in DMSO was identical to that in water (Fig.S17 in ESI). At the same time, the energy of d-d transitions experienced very little change, suggesting that no direct binding of solvent molecules to the copper ion has occurred. These experimental findings, including the assignment of transition bands, were supported by TDDFT calculations, predicting a disappearance of the S(δ)→Cu2+(dx2-y2) transition in the spectrum of Cu(II)(NSNS2A) when no protonation or other perturbation of acetate arms takes place (Fig.5). A similar phenomenon was previously reported by Archibald for two structural isomers of Cu(TETA) with different tetragonal elongations – one along O-Cu-O direction (crystallised from water) and one along N-Cu-N direction (crystallised from acetonitrile), across the macrocycle.25 It was also hypothesised that hydrogen bonding might be the driving force behind the presence of the conformation with an unusual macrocyclic Jahn-Teller distortion, which possibly reflects the dynamic interchange between different conformations of the complex in solution, eventually leading to poor in vivo stability of Cu(TETA).
Figure 4.

Spin density contour plots (contour value 0.003) of Cu(II)(NSNS2A) on the left and [Cu(II)(H2NSNS2A)]2+ on the right based on optimised geometry from DFT calculations. Atoms are colour-coded as follows: Cu – orange, S – yellow, O – red, N – blue, C – dark grey, H – light grey, hydrogens attached to carbons are omitted for clarity.
Figure 5.

Experimentally observed UV-vis spectra of Cu(II)(NSNS2A) in water and acetonitrile (top) and corresponding calculated spectra for Cu(II)(NSNS2A) and [Cu(II)(H2NSNS2A)]2+ (bottom). The calculated spectra were shifted to match the corresponding experimental spectra.
The EPR spectra are characterized by slightly to moderately rhombic g- and A-tensors, the latter one being the tensor of the hyperfine interaction (hfi) of the copper nucleus (see Fig. 6 and Table 2). The principal axes X, Y, and Z of the tensors correspond to the similar axes defining the real d-orbital set of the Cu(II) ion, with the orbital carrying the unpaired electron. At least three out of four EPR turning points corresponding to (gz, Az) are readily observable at the low-field side of the spectra for Cu(II)(NSNS2A).
Figure 6.

CW X-band EPR spectrum at 77K (solid line) with a fitted curve (dashed lines) for Cu(II)(NSNS2A) (left). 45% glycerol/55% 0.1 HEPES solution (pH = 7.0). CW X-band EPR spectrum at 298K for Cu(II)(NSNS2A) in water (right).
Table 2.
Principal g-values and copper hyperfine couplings, A, obtained from EPR spectral fitting and calculated with DFT (calc.).
| Complex | gz | gy | gx | Az (MHz) | Ay (MHz) | Ax (MHz) |
|---|---|---|---|---|---|---|
| Cu(II)(NSNS2A) (pH = 7.0) | 2.182 | 2.063 | 2.036 | −504 | −76 | −13 |
| Cu(II)(NSNS2A) calc. | 2.162 | 2.058 | 2.039 | −557 | 30 | −17 |
| Cu(II)(H2NSNS2A) calc. | 2.120 | 2.041 | 2.035 | −508 | −42 | −39 |
The square-planar geometry observed in a solid state can often experience distortions in solution. It has been shown that the gz/Az ratio can serve as a qualitative measure of the tetrahedral distortion in square-planar copper complexes. If Az is expressed in cm−1 (1 cm−1 ≈ 30 GHz), then gz/Az < 130 cm correspond to the planar geometry, the values between 130 – 150 cm indicate a small to moderate departure from planarity, and still larger values (even as high as 250) result from a considerable distortion.24, 26–28 As an example, gz/Az = 122 cm observed for Cu(II)(NSNS)) corresponds to a planar geometry.19 For the complex Cu(II)(NSNS2A) studied in this work, the gz/Az ratio of 130 cm suggests that the tetrahedral distortion of the [N2S2] plane is relatively insignificant. Indeed, the DFT calculations show that both protonated and deprotonated complexes do not have any tetrahedral distortion.
A large body of evidence suggests that the coordination geometry of copper complexes strongly correlates with their redox behaviour. Indeed, it has been shown that a substitution of sulphur atoms for nitrogen atoms resulted in a significant increase of redox potentials.29 Since Cu(I) species predominantly form tetrahedral complexes, a tetrahedral distortion of Cu(II) complexes would likely facilitate redox processes. For Cu(II)(NSNS2A), a quasi-reversible redox behaviour was observed (E1/2=146 mV vs Ag/AgCl electrode, respectively, Fig.7, pH = 7.0). This value is much higher than the one previously reported for Cu(II)(TE2A) (E1/2= −1.10 V vs Ag/AgCl electrode).22
Figure 7.

Cyclic voltammograms of copper complexes Cu(NSNS2A) upon variation of scanning rate (A, 0.1M HEPES, 0.5M KNO3, pH = 7.0) and pH (B, 0.1M HEPES, 0.5M KNO3). The inset in Fig.7A shows a linear dependence between the anodic peak current and the square root of scan rates.
A quasi-reversible redox behaviour was previously reported for [Cu(NSNS)]2+ complex in an aqueous solution with a significantly lower half wave potential (E1/2 =−230mV or E1/2 =−260mV vs Ag/AgCl) and a larger peak-to-peak separation (ΔEp) that might be indicative of larger structural rearrangements upon reduction/oxidation in the absence of pendant arms.19 At the same time, alkylation of secondary amines was shown to increase the redox potential of copper complexes, supposedly by destabilising the Cu(II) species as a result of lower donor capacity of tertiary amines.26 On the other hand, the stability of the Cu(II) species can be potentially increased, for instance, by introducing pendant arms, which is expected to lead to a significant decrease of a redox potential. In the case of Cu(NSNS2POH) complex bearing two phosphonate arms, a decreased half wave potential (E1/2 =−310 mV vs Ag/AgCl) was reported.20 Therefore, the much higher E1/2 values observed for Cu(II)(NSNS2A) might be indicative of either lower thermodynamic stability of the Cu(II) complex, or higher stability of the Cu(I) species, although the latter was previously shown to have a much smaller impact.17 However, a high stability constant of the complex does not necessarily translate into a higher stability in vivo, as, for example, has been shown for 64Cu(TETA).30 Instead, transchelation challenge experiments are often performed that can remotely mimic conditions experienced by the tracer in vivo, in the presence of various endogenous chelators. In the present case, a 1-hour incubation of Cu(II)(NSNS2A) with 1 eq. of EDTA (0.1M MES, pH = 5.5, Fig.9) serving as a metal scavenger resulted in almost complete transchelation with the loss of the LMCT band in the UV spectrum. On the other hand, analogous tetraaza macrocycles show no metal loss under the same conditions. By following the Benesi-Hildebrand approach,31 a conditional dissociation constant for Cu(II)(NSNS2A) was estimated (KD = 2.1(±0.34)x10−6 M, pH = 5.0, T = 298K, Fig.8), which is higher than the values reported for Cu(II)(NSNS) and related unsubstituted thiaaza macrocycles.19
Figure 9.

Optimised structures of Cu(I/II)(NSNS2A)-/0 and its protonated version [Cu(I/II)(H2NSNS2A)]+/2+. Geometry was optimized using D3-B3LYP/def2-TZVP in a continuum model of water (SMD parametrization).
Figure 8.

A least-square fit of the binding curve for Cu(II)(NSNS2A) (42 μM ClO4−, 0.2 mM Cu2+, pH = 5.0, left) and dissociation curves for Cu(II)(NSNS2A) in the presence of 1.0 and 12.5 equivalents of EDTA (0.1M MES, pH = 5.5, right).
To determine the thermodynamic stability constant, protonation constants of the ligand were estimated by 1H NMR pH titration experiments (Fig.S23 and Fig.S24). Chemical shifts of macrocyclic protons were plotted as a function of pH, revealing an average logKH1 = 7.7 for both tertiary amines, whilst logKH2 = 2.5 was calculated for pendant acetate arms by monitoring a change in a chemical shift of methylene protons. Unfortunately, the resolution of our titration experiment did not allow to distinguish between the first and the second protonation constants, and therefore the obtained values were treated as the average protonation constants for both tertiary amines and both acetate arms. The obtained value KCu(II)L = 1.2 ×1011 M−1 for Cu(II)(NSNS2A) was used to calculate the stability constant KCu(I)L for Cu(I)(NSNS2A), using the following equation:
where Ef is a redox potential of the complex vs SHE and is the concentration potential of the aquated Cu(II/I) redox couple, for which a value of 0.13 V was utilised.17 The resultant stability constant logKCu(I)L = 14.7 is very similar to the values previously reported by Rorabacher for closely related unsubstituted thiaaza macrocyclic systems, where the average value logKCu(I)L = 13.8 was reported.17 This value was shown to vary little with the ligand morphology, regardless of the ring size, as well as the type and number of donor atoms. This correlation now seems to hold true also for complexes with N-substituted ligands, however additional examples should be studied in order to support this conclusion.
Cu(II)(NSNS2A) complex can be reduced in aqueous solutions by sodium dithionate, although a complete reduction was not observed even in the presence of a large excess of the reductant (Fig.S15 in ESI).
The diamagnetic complex [Cu(I)(H2NSNS2A)]+ was characterized by 1H and 13C NMR spectroscopy. The presence of five broadened multiplets corresponding to the [14]aneNSNS macrocycle backbone indicates the retention of its time-averaged symmetry (C2) upon complexation. NMR spectra of [Cu(I)(H2NSNS2A)]+ showed no changes when heated up to 65°C, as shown in Fig.S2 (ESI).
The observed C2 symmetry for DMSO-d6 solutions of the complex can either suggest the presence of two tetrahedral species which are in fast exchange resulting in an averaged octahedral conformation or the presence of genuine square-planar [NSNS] coordination around Cu(I) ion. Although the presence of broadened peaks in 1H NMR spectra, whose width is insensitive to temperature variation within studied range, makes the former proposition unlikely, the latter seems also hardly plausible due to the paucity of such precedents in the literature.32–35 Indeed, the examples of square-planar Cu(I) complexes are extremely scarce, as diamagnetic Cu(I) favours tetrahedral coordination. However a distorted square-planar coordination was previously reported for another 14-membered macrocycle in a solid state.32, 36 Surprisingly, DFT calculation performed for [Cu(I)(H2NSNS2A)]+ revealed a perfect square-planar coordination of Cu(I) (Fig.9), featuring long Cu-N bonds (2.403 Å). On the other hand, the deprotonated species Cu(I)(NSNS2A), which might be observed at higher pH exhibits a typical tetrahedral coordination with Cu(I) bound to two nitrogen atoms and one sulphur atoms of the macrocycle, as well as to the oxygen atom of the pendant acetate arm (Fig.9).
Although the retention of square-planar coordination for both Cu(I) and Cu(II) species in solution might have far-reaching implications, including unusual photophysical performance involving strongly red-shifted metal-to-ligand charge transfer (MLCT) state and elevated redox potential, one should treat these initial findings very cautiously. However, even if square-planar coordination of [Cu(I)(H2NSNS2A)]+ in DMSO solution in the absence of competing anions can be proved by other techniques, such as EXAFS, this is unlikely to be the case for biologically relevant aqueous systems with multiple competing anions that can bind to the Cu(I) ion, especially at elevated pH values, when deprotonated Cu(I)(NSNS2A) is predicted to adopt a tetrahedral coordination environment. Our attempts to detect any unusually red-shifted absorption bands for cuprous species were futile and require closer inspection in future studies.
Variation of pH within the range of 3–7 revealed a sharpening of reduction/oxidation peaks for Cu(II)(NSNS2A) as the pH decreased that was initially attributed to the protonation of the acetate arms. The presence of only subtle changes in the EPR and UV spectra at different pH (Table 2; Fig.S16 and Fig.S21 in ESI) rules out any significant structural transformations around the copper ion upon anticipated protonation of acetate arms, in accord with the previously reported copper (II) complex with [14]-ane-[NSNS2Bz2A].37 In the present case, in the calculated structure of [Cu(II)(H2NSNS2A)]2+ with protonated acetate arms, the copper ion retains a square-planar [N2S2] coordination (Fig.9), whereas protonation weakens Cu-O bonds, leading to an elongation of the octahedron. This results in a significant shortening of Cu-S bonds and elongation of Cu-N bonds. Such pronounced changes should have clearly resulted in a more noticeable changes in the EPR spectra, as well as in UV spectra and cyclic voltammetry, suggesting that the protonation of acetate arms at pH above 3 is unlikely. No LMCT bands were detected in the UV-vis spectrum at pH below 2.5, suggesting full dissociation of the complex upon protonation of the acetate arms or tertiary nitrogen atoms of the macrocycle. On a closer inspection, a formation of a precipitate was observed at pH just slightly above 7.0, suggesting that the observed broadening of reduction/oxidation peaks might be a result of partial aggregation at higher pH.
Radiolabelled Cu-64 complex, 64Cu(II)(NSNS2A), was synthesized by mixing 64CuCl2 with the ligand dissolved in 0.3M ammonium citrate buffer (pH = 6.0) and heating the reaction mixture at 80°C for 10 min. The progress of the radiolabelling was monitored by radio-HPLC using a HILIC column (Fig.S18 in ESI). As a control probe, a tetraaza analogue, 64Cu(TE2A), was selected, which was previously evaluated in preclinical studies.22, 38
Each radiotracer was injected in three Wistar rats, who were dynamically scanned for 60 min using a 4.7T PET/MRI scanner and then euthanized 90 min post injection (Fig.10). Eleven different tissue specimens were harvested post-mortem and their radioactivity was counted. The half-life in blood was fitted with a biexponential decay curve (t1/2 = 1.2 min/9.4 min for 64Cu(II)(NSNS2A) and 0.7 min/11.8 min for 64Cu(TE2A)). Rapid clearance from blood was followed by a renal filtration as well as an uptake into the liver for 64Cu(II)(NSNS2A), whereas 64Cu(TE2A) was predominantly filtered by the kidneys. The latter finding is in line with a previously reported study and is typical for other renally excreted tetraaza macrocyclic copper-64 complexes.22 As well as renal filtration, there also appeared to be some uptake into the renal cortex in the case of 64Cu(II)(NSNS2A), as the kidney signal did not decline at later time points.
Figure 10.

Coronal PET/MR (A) and PET (B) images of a rat injected with 64Cu(II)(NSNS2A) 30 min post injection and a corresponding time-activity curve (E). Coronal PET/MR (C) and PET (D) images of a rat injected with 64Cu(TE2A) 30 min post injection and a corresponding time-activity curves for blood, liver, and kidneys (F).
For 64Cu(II)(NSNS2A), the PET signal from the liver plateaued at ca. 8 min post injection and remained unchanged for the rest of the scan. The absence of clearance from the liver suggests a transchelation/dissociation of the 64Cu(II) and binding to liver proteins in hepatocytes and a long-term retention. Some evidence of hepatobiliary elimination is also observed by the appearance of the signal in the intestines with time. By comparing the residual radioactivity (in %ID/g) in various organs at 90 minutes post injection (Fig.11), a much lower tracer uptake in all tissues was observed for 64Cu(TE2A) than for 64Cu(II)(NSNS2A). In fact, for 64Cu(TE2A), a very low uptake in the liver detected 90 min post injection (0.12 %ID/g) is in agreement with the previously reported value (0.073 %ID/g 24h post injection), consistent with a very high in vitro stability of the complex.22 On the other hand, a remarkably higher liver uptake for 64Cu(II)(NSNS2A) (2.15 %ID/g) is comparable with the reported the values for similar dithiadiaza macrocyclic complexes 64Cu(NEC-SE/SP/SB) reported by Lewis and Anderson (ca. 3 %ID/g).18 Similar numbers were reported for copper-67 chloride in rats,39 reflecting a low in vivo stability of these tracers with an apparent metal loss occurring shortly after injection.
Figure 11.

Biodistribution data (%ID/g) for 64Cu(II)(NSNS2A) (N=3) and 64Cu(TE2A) (N=3) at 90 min post injection.
Conclusions
Copper (I)/(II) complexes with a new dithiadiaza macrocyclic ligand NSNS2A were reported and their structures in solution were thoroughly investigated by a combination of spectroscopic techniques and DFT calculations. The latter predicted the change in the orientation of tetragonal distortion in Cu(II)(NSNS2A) upon protonation and possibly hydrogen bonding of pendant acetate arms, supposedly leading to a relatively high lability of the complex in aqueous media. A very unusual square-planar coordination of copper (I) ion was proposed for the cuprous complex [Cu(I)(H2NSNS2A)]+ which, however, requires further validation in future studies. In vivo study performed with a radiolabelled compound 64Cu(II)(NSNS2A) revealed a suboptimal stability that is much lower than exhibited by its tetraaza analogue, in line with in vitro experiments. Given these results along with the findings previously reported for a similar [N2S2] system,18 we can conclude that the stability of copper complexes with sulphur-containing macrocycles appears to be insufficient for their successful use in PET imaging.
Experimental section
1H and 13C NMR spectra were recorded on Varian 400 MHz and JEOL 500 MHz systems. FTIR spectra were collected on a Jasco 4600 spectrophotometer running Spectra Manager CFR®, where samples were prepared as a neat oil or solid on a ZnSe window using attenuated total reflectance (ATR). Melting points were obtained on a MeltTemp melting point apparatus and are uncorrected. High-resolution mass spectra (HRMS) were recorded on a time-of-flight JEOL JMST1000LC spectrometer with a DART ion source. Liquid chromatography-mass spectrometry (LC-MS) was performed using an Agilent 1200 Series HPLC interfaced to an Agilent 6130 single quadrupole electrospray mass spectrometer and to a Daly conversion dynode detector with UV detection at 220, 254, and 280 nm. UV-vis spectra were recorded on a SpectraMax M2 spectrophotometer using quartz cuvettes with a 1 cm path length. Cyclic voltammetry measurements were performed using a Nuvant EZstat Pro potentiostat equipped with Ag/Ag+ (3 M NaCl) reference electrode, Pt wire counter electrode, and glassy carbon working electrode. Measurements were performed at room temperature in 0.5 M KNO3 electrolyte. The ferri/ferrocyanide couple was used as an internal standard. The radiochemical purity of the dose to be injected was determined by radio-HPLC (Agilent 1100 Series HPLC unit with a Carroll/Ramsey radiation detector with a silicon PIN photodiode) and radio-TLC (Bioscan AR-2000).
EPR spectroscopy
The EPR experiments were carried out at the University of Arizona EPR Facility, on the X-band EPR spectrometer Elexsys E500 (Bruker Biospin) equipped with the rectangular resonator operating in TE102 mode. In the measurements performed at 77 K and room temperature (~298 K), the microwave frequencies were 9.44 GHz and 9.65 GHz, respectively. The microwave power was 2 mW, and the magnetic field modulation amplitude was 0.5 mT. EPR spectra were fitted using EasySpin software.
Binding studies
Solution 1: 0.2 mM solution of Cu(ClO4)2 was prepared by dissolving Cu(ClO4)2 in milli-Q water (pH = 5.0, adjusted with 60% HClO4). Solution 2: 2.0 mM solution of NSNS2A was prepared by dissolving 2·2HCl in solution 1. Solution 2 was added incrementally to solution 1 in a quartz cuvette and the UV-vis spectrum was recorded after every addition. Absorption at λ = 364 nm was plotted as a function of NSNS2A concentration and fitted in MatLab using the following equation:
where A is the absorbance, ε is the molar attenuation coefficient, is the total concentration of copper, [NSNS2AT] is the total concentration of NSNS2A chelator and KD is the dissociation constant, which is being determined.
Stability tests
Cu(II)(NSNS2A) was dissolved in a MES buffer (0.1M, pH = 5.5), and the absorption spectrum was recorded in a quartz cuvette. EDTA was added, the solution was vigorously shaken, and the absorption spectra were recorded every two minutes until the signal at λ = 364 nm disappeared.
Radiolabelling
64CuCl2 was purchased from the University of Wisconsin-Madison cyclotron lab. All buffers and solutions were prepared using ultrapure water and were chelex-treated for 24h before use. 64CuCl2 (28 mCi) was diluted with 1 mL of citrate buffer (0.3M, pH = 6.0) and an aliquot (50 μL) was added to the ligand. The latter was prepared by mixing 0.3 mL of 1.8 mM NSNS2A in water with 0.2 mL citrate buffer (0.3M, pH = 6.0). The reaction mixture was heated at 80°C for 10 min. The solution was cooled down and was filtered through a sterile syringe filter. The formation of 64Cu(II)(NSNS2A) was confirmed by radio-HPLC. A similar protocol was used for radiolabelling of 64Cu(TE2A), except a higher concentration (25 mM) of the ligand solution was used. The formation of 64Cu(TE2A) was confirmed by radio-TLC, as previously reported.22 Both tracers were diluted with PBS and sterile filtered prior to injection.
Imaging and biodistribution
All experiments and procedures were performed in accordance with the National Institutes of Health’s “Guide for the Care and Use of Laboratory Animals” and were approved by the Massachusetts General Hospital Institutional Animal Care and Use Committee. 7 Wistar rats (male, 378g-456g) were obtained from Charles River Labs (Wilmington, MA). They were anesthetised with 2% of isoflurane in oxygen and the tracer (120–200 μCi) was administered by IV injection. Quantifications of 64Cu activity in each injected dose was determined using a Capintec CRC-15PET dose calibrator. Rats were imaged on a 4.7 Tesla PET/MRI scanner (Bruker, Billerica MA) and a Bruker PET insert, placed in a holder and heated with warm air. The respiration rate was monitored all through the experiment using Small Animal Instruments (SA Inc.) monitoring equipment and software. MR imaging protocol included 2 T1-weighted 3D FLASH (Fast Low Angle Shot) sequences, covering most of the body, with the following parameters: echo time (TE) = 3.1 ms, 3 repetition time (TR) = 17 ms, field of view (FOV) = 85 × 64 × 40 mm, imaging resolution = 0.221 × 0.327 × 0.625 mm3/voxel, respiratory triggering, and a flip angle = 15 degrees in the first sequence and 30 degrees in the second. All other parameters were identical. A 3D T2-weighted TurboRARE 3 sequence was also acquired with parameters: TE = 2.8 ms, TR = 500 ms, FPV = 80 × 60 × 40 mm, resolution = 0.208 × 0.313 × 0.833 mm3/voxel, RARE factor = 8, and respiratory triggering. To measure PET signal, Amide software was used to draw ROIs on MR images, which were co-registered with the PET images. 3D FLASH images were used for drawing ROIs. Right kidney ROIs were drawn freehand to cover one entire kidney. For the liver and the heart, ellipsoids were placed centered in the liver and the left chamber of the heart, respectively. Time-activity data were acquired from these ROIs. As the data itself was decay corrected during reconstruction, no decay correction was applied to the ROI data. Rats were sacrificed 90 min post injection and the following organs and tissues were collected and weighed: brain, kidneys, liver, heart, spleen, pancreas, urine, blood, lungs, femur, intestines and leg muscle. The counts in each organ were measured using a gamma counter (Wizard, Perkin Elmer) and decay corrected.
Synthesis
TE2A was synthesised as previously reported.22
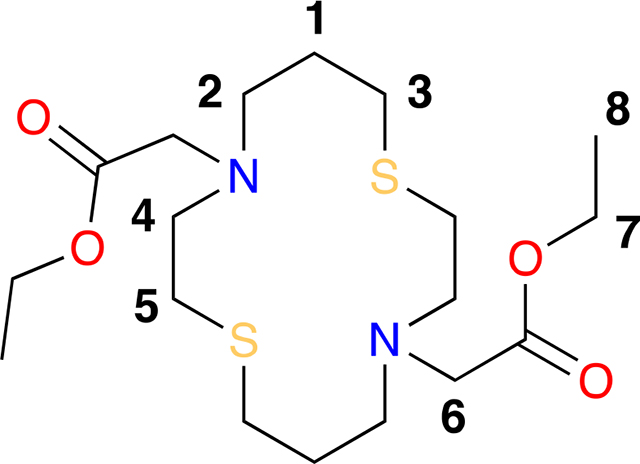
Diethyl 2,2’-(1,8-dithia-4,11-diazacyclotetradecane-4,11-diyl)diacetate (1).
A 25 mL round bottom flask equipped with a septum and magnetic stir bar was charged with 1,8-dithia-4,11-diazacyclotetradecane (0.100 g, 0.427 mmol), anhydrous potassium carbonate (0.295 g, 2.14 mmol), dichloromethane (4.3 mL) and bromo ethyl acetate (0.1 mL, 0.901 mmol) in the indicated order. The reaction was stirred at room temperature for 12 h and filtered through a pad of celite. The light yellow solution was concentrated in vacuo, recrystallized in minimal amounts of ethanol and filtered affording diethyl 2,2’-(1,8-dithia-4,11-diazacyclotetradecane-4,11-diyl)diacetate (0.134 g, 0.329 mmol) in a 77% yield as a white crystalline solid. MP = 103–105 °C. 1H NMR (400 MHz, CDCl3) δ 4.15 (H7, q, J = 7.1 Hz, 4H), 3.36 (H6, s, 4H), 2.86 (H4, t, J = 7.8, 6.2 Hz, 4H), 2.76 (H2, t, J = 6.4 Hz, 4H), 2.66 (H3, H5, dt, J = 8.7, 7.0 Hz, 8H), 1.77 (H1, p, J = 6.6 Hz, 4H), 1.26 (H8, t, J = 7.1 Hz, 6H). 13C NMR (101 MHz, CDCl3) δ 171.13 (COOEt), 60.3 (C7), 55.3 (C6), 52.9 (C4), 52.5 (C2), 29.9 (C5), 29.2 (C3), 27.7 (C1), 14.3 (C8). IR νmax 2979, 2923, 2847, 1727, 1458, 1415, 1186, 1135, 1033 cm−1. HRMS (ESI) m/z [M+Na+] calcd. for C18H34N2NaS2 429.1852, found 429.1854.
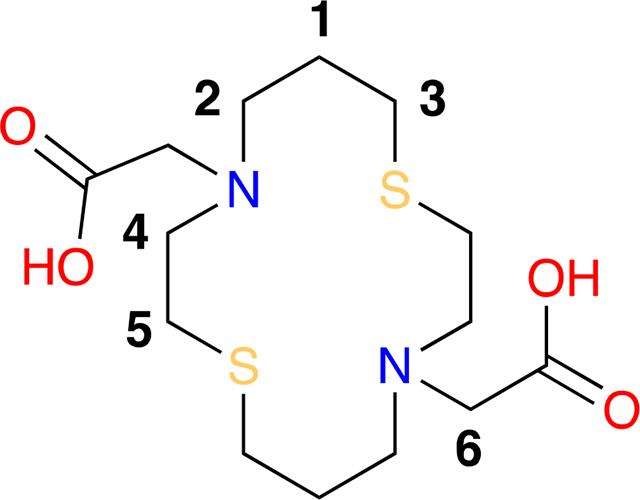
2,2’-(1,8-dithia-4,11-diazacyclotetradecane-4,11-diyl)diacetic acid dihydrochloride (2).
A 5 mL round bottom flask equipped with a magnetic stir bar and septum was charged with 1 M HCl (1.2 mL) and diethyl 2,2’-(1,8-dithia-4,11-diazacyclotetradecane-4,11-diyl)diacetate (0.050 g, 0.123 mmol) and refluxed for 6 hours. The solution was concentrated in vacuo to provide 2,2’-(1,8-dithia-4,11-diazacyclotetradecane-4,11-diyl)diacetic acid dihydrochloride (0.052 g, 0.123 mmol) as a white solid in quantitative yield. 1H NMR (400 MHz, D2O) δ 4.00 (H6, s, 4H), 3.44 (H4, m, 4H), 3.35 (H2, m, 4H), 2.89 (H5, dd, J = 8.9, 6.8 Hz, 4H), 2.61 (H3, t, J = 7.4 Hz, 4H), 1.97 (H1, p, J = 7.0 Hz, 4H). 13C NMR (101 MHz, D2O) δ 168.1 (COOH), 55.0 (C6), 51.9 (C4), 51.6 (C2), 26.9 (C3), 23.1 (C5), 22.7 (C1). IR νmax 3375, 2918, 2498, 2426, 2363, 1737, 1635, 1480, 1467, 1439, 1410, 1241, 1214, 1025, 971, 923 cm−1. HRMS (ESI) m/z [M-2HCl+H+]+ calcd for C14H27O4N2S2 351.1407, found 351.1407.
2,2’-(1,8-dithia-4,11-diazacyclotetradecane-4,11-diyl)diacetate copper(II) complex, Cu(II)(NSNS2A).
A 5 mL round bottom flask was equipped with a magnetic stir bar, nitrogen inlet, and rubber septum was charged with 2,2’-(1,8-dithia-4,11-diazacyclotetradecane-4,11-diyl)diacetic acid dihydrochloride salt (0.054 g, 0.128 mmol), MilliQ water (1 mL), acetonitrile (1 mL) and copper(II) acetate (0.030 g, 0.128 mmol). The dark red solution was stirred at 23 °C for 10 min and sonicated at 23 °C for 5 min. The solution was concentrated in vacuo providing a red solid in quantitative yield. HRMS (ESI) m/z [M-2H++Na+]+ calcd for C14H24O4N2CuNaS2 434.0366, found 434.0366.
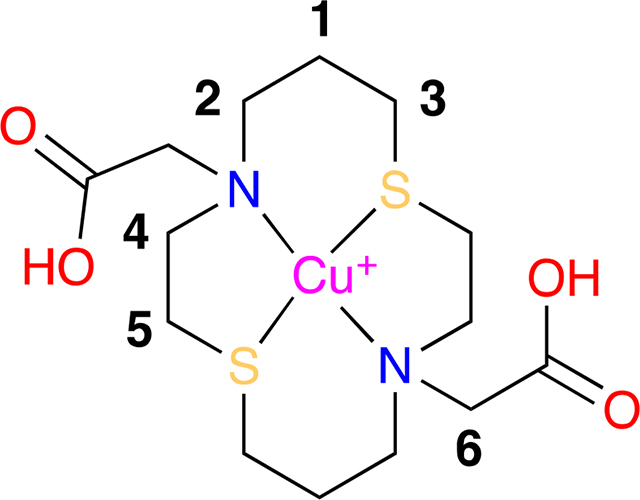
copper(I) hexafluorophosphate-2,2’-(1,8-dithia-4,11-diazacyclotetradecane-4,11-diyl)diacetic acid, Cu(I)(H2NSNS2A).
A 5mm NMR tube, flushed with nitrogen, was charged with 2,2’-(1,8-dithia-4,11-diazacyclotetradecane-4,11-diyl)diacetic acid ·2HCl (0.020 g, 0.047 mmol) and DMSO-d6 (1.5 mL, 0.031 M). The solution was then degassed (3x freeze-pump-thaw cycles), using Schlenk techniques, and charged with freshly prepared tetrakis(acetonitrile)copper(I) hexafluorophosphate (0.018 g, 0.047 mmol). The solution/NMR tube was warmed for 5 min at 50 °C via external water bath, then sonicated at 23 °C for 20 s. The clear solution was then subjected to NMR experiments. 1H NMR (400 MHz, DMSO-d6) δ 4.08 (H6, s, 4H), 3.34 (H4, m, 4H), 3.17 (H2, m, 4H), 2.87 (H5, m, 4H), 2.58 (H3, m, 4H), 1.81 (H1, m, 4H). 13C NMR (101 MHz, DMSO-d6) δ 168.2 (COOH), 54.5 (C6), 50.7 (C4), 50.6 (C2), 25.4 (C3), 23.0 (C5), 21.5 (C1).
X-ray Crystallographic Data Collection and Structure Solution and Refinement
Crystals were obtained from the slow evaporation of a saturated solution of corresponding solvent at room temperature and stored under atmospheric conditions. Single crystal data for all structures were collected on a Bruker CCD-based diffractometer with dual Cu/Mo ImuS microfocus optics (Cu Kα radiation, λ =1.54178 Å or Mo Kα radiation, λ = 0.71073). Crystals were mounted on a cryoloop using Paratone oil and placed under a steam of nitrogen at 100 K (Oxford Cryosystems). The data were corrected for absorption with the SADABS program. The structures were refined using Bruker SHELXTL Software Package (Version 6.1) and were solved using direct methods until the final anisotropic full-matrix least squares refinement of F2 converged.28 Electronic Supplementary Information (ESI) available: CCDC 2003358, 2003359 and 2003361 contain the supplementary crystallographic data for this paper. These data can be obtained free of charge from The Cambridge Crystallographic Data Centre via www.ccdc.cam.ac.uk/data_request/cif.
Computational details
Geometry optimisation and harmonic frequencies calculations were performed using Gaussian16 software package40 with hybrid B3LYP functional and triple-zeta def2-TZVP basis set41 with empirical account of dispersion interactions using D342 parametrisation, and SMD43 continuum model of water. EPR parameters were calculated with ORCA 4.144, 45 for optimised geometries using B3LYP/def2-TZVP and core polarised CP(PPP)46 basis set on Cu with refined grid. Spin-orbit coupling was calculated using SOMF(1X)47 mean-field approximation. TD-DFT calculations of UV-Vis spectra were performed with ORCA 4.1 using B3LYP/def2-TZVP basis set for optimised structures.
Supplementary Material
Scheme 1.

Synthetic pathways for copper complexes discussed in the paper. Crystal structures of 1 and 2·2HCl can be found in ESI.
Acknowledgements
S.S. wants to thank Dr. Mariane Le Fur for her help with radiolabelling and fruitful discussions. P.C. acknowledges support of the National Institutes of Health Office of the Director for instrumentation support (OD010650, OD023503, OD025234). C.J.Z thanks Ohio Board of Regents for funds used to purchase the Bruker-Nonius Apex CCD X-ray diffractometer.
Footnotes
Electronic Supplementary Information (ESI) available: [details of any supplementary information available should be included here]. See DOI: 10.1039/x0xx00000x
Notes and references
- 1.Kardos J, Heja L, Simon A, Jablonkai I, Kovacs R and Jemnitz K, Cell Commun Signal, 2018, 16, 71. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Bass LA, Wang M, Welch MJ and Anderson CJ, Bioconjug Chem, 2000, 11, 527–532. [DOI] [PubMed] [Google Scholar]
- 3.Boswell CA, Sun X, Niu W, Weisman GR, Wong EH, Rheingold AL and Anderson CJ, J Med Chem, 2004, 47, 1465–1474. [DOI] [PubMed] [Google Scholar]
- 4.Cai Z and Anderson CJ, J Labelled Comp Radiopharm, 2014, 57, 224–230. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Zia NA, Cullinane C, Van Zuylekom JK, Waldeck K, McInnes LE, Buncic G, Haskali MB, Roselt PD, Hicks RJ and Donnelly PS, Angew Chem Int Ed Engl, 2019, 58, 14991–14994. [DOI] [PubMed] [Google Scholar]
- 6.Kelly JM, Ponnala S, Amor-Coarasa A, Zia NA, Nikolopoulou A, Williams C Jr., Schlyer DJ, DiMagno SG, Donnelly PS and Babich JW, Mol Pharm, 2020, 17, 1954–1962. [DOI] [PubMed] [Google Scholar]
- 7.Woodin KS, Heroux KJ, Boswell CA, Wong EH, Weisman GR, Niu W, Tomellini SA, Anderson CJ, Zakharov LN and Rheingold AL, European Journal of Inorganic Chemistry, 2005, 2005, 4829–4833. [Google Scholar]
- 8.Le Fur M, Beyler M, Le Poul N, Lima LM, Le Mest Y, Delgado R, Platas-Iglesias C, Patinec V and Tripier R, Dalton Trans, 2016, 45, 7406–7420. [DOI] [PubMed] [Google Scholar]
- 9.Brown OC, Baguna Torres J, Holt KB, Blower PJ and Went MJ, Dalton Trans, 2017, 46, 14612–14630. [DOI] [PubMed] [Google Scholar]
- 10.Bodio E, Boujtita M, Julienne K, Le Saec P, Gouin SG, Hamon J, Renault E and Deniaud D, ChemPlusChem, 2014, 79, 1284–1293. [Google Scholar]
- 11.Xiao Z, Donnelly PS, Zimmermann M and Wedd AG, Inorg Chem, 2008, 47, 4338–4347. [DOI] [PubMed] [Google Scholar]
- 12.Barnard PJ, Holland JP, Bayly SR, Wadas TJ, Anderson CJ and Dilworth JR, Inorg Chem, 2009, 48, 7117–7126. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Holm RH, Kennepohl P and Solomon EI, Chem Rev, 1996, 96, 2239–2314. [DOI] [PubMed] [Google Scholar]
- 14.Warren JJ, Lancaster KM, Richards JH and Gray HB, J Inorg Biochem, 2012, 115, 119–126. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Clark KM, Yu Y, Marshall NM, Sieracki NA, Nilges MJ, Blackburn NJ, van der Donk WA and Lu Y, J Am Chem Soc, 2010, 132, 10093–10101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Dockal ER, Jones TE, Sokol WF, Engerer RJ, Rorabacker DB and Ochrymowycz LA, J Am Chem Soc, 1976, 98, 4322–4324. [DOI] [PubMed] [Google Scholar]
- 17.Ambundo EA, Deydier M-V, Grall AJ, Aguera-Vega N, Dressel LT, Cooper TH, Heeg MJ, Ochrymowycz LA and Rorabacher DB, Inorganic Chemistry, 1999, 38, 4233–4242. [Google Scholar]
- 18.Papini G, Alidori S, Lewis JS, Reichert DE, Pellei M, Gioia Lobbia G, Biddlecombe GB, Anderson CJ and Santini C, Dalton Trans, 2009, DOI: 10.1039/b808831d, 177–184. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Walker TL, Mula S, Malasi W, Engle JT, Ziegler CJ, van der Est A, Modarelli J and Taschner MJ, Dalton Trans, 2015, 44, 20200–20206. [DOI] [PubMed] [Google Scholar]
- 20.Taschner IS, Aubuchon E, Schrage BR, Ziegler CJ and van der Est A, Dalton Trans, 2020, 49, 3545–3552. [DOI] [PubMed] [Google Scholar]
- 21.Chapman J, Ferguson G, Gallagher JF, Jennings MC and Parker D, Journal of the Chemical Society, Dalton Transactions, 1992, DOI: 10.1039/dt9920000345. [DOI] [Google Scholar]
- 22.Pandya DN, Kim JY, Park JC, Lee H, Phapale PB, Kwak W, Choi TH, Cheon GJ, Yoon YR and Yoo J, Chem Commun (Camb), 2010, 46, 3517–3519. [DOI] [PubMed] [Google Scholar]
- 23.Walker TL, Malasi W, Bhide S, Parker T, Zhang D, Freedman A, Modarelli JM, Engle JT, Ziegler CJ, Custer P, Youngs WJ and Taschner MJ, Tetrahedron Letters, 2012, 53, 6548–6551. [Google Scholar]
- 24.Nikles DE, Powers MJ and Urbach FL, Inorganic Chemistry, 1983, 22, 3210–3217. [Google Scholar]
- 25.Silversides JD, Allan CC and Archibald SJ, Dalton Trans, 2007, DOI: 10.1039/b615329a, 971–978. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Rybak-Akimova EV, Nazarenko AY, Chen L, Krieger PW, Herrera AM, Tarasov VV and Robinson PD, Inorganica Chimica Acta, 2001, 324, 1–15. [Google Scholar]
- 27.Sakaguchi U and Addison AW, Journal of the Chemical Society, Dalton Transactions, 1979, DOI: 10.1039/dt9790000600. [DOI] [Google Scholar]
- 28.Addison AW, in Copper Coordination Chemistry: Biochemical and Inorganic Perspectives, ed. Karlin JZKD, Adenine Press, New York, 1983. [Google Scholar]
- 29.Bernardo MM, Heeg MJ, Schroeder RR, Ochrymowycz LA and Rorabacher DB, Inorganic Chemistry, 1992, 31, 191–198. [Google Scholar]
- 30.Anderson CJ and Ferdani R, Cancer Biother Radiopharm, 2009, 24, 379–393. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Benesi HA and Hildebrand JH, Journal of the American Chemical Society, 1949, 71, 2703–2707. [DOI] [PubMed] [Google Scholar]
- 32.Gagne RR, Allison JL and Lisensky GC, Inorganic Chemistry, 1978, 17, 3563–3571. [Google Scholar]
- 33.Cheung PM, Berger RF, Zakharov LN and Gilbertson JD, Chem Commun (Camb), 2016, 52, 4156–4159. [DOI] [PubMed] [Google Scholar]
- 34.Shee NK, Das D, Oluwafunmilayo Adekunle FA, Drew MGB and Datta D, Inorganica Chimica Acta, 2011, 366, 198–202. [Google Scholar]
- 35.Dahl EW and Szymczak NK, Angew Chem Int Ed Engl, 2016, 55, 3101–3105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Gagne RR, Allison JL and Ingle DM, Inorganic Chemistry, 1979, 18, 2767–2774. [Google Scholar]
- 37.Funkemeier D and Mattes R, Chemische Berichte, 1991, 124, 1357–1362. [Google Scholar]
- 38.Pandya DN, Kim JY, Kwak W, Park JC, Gawande MB, An GI, Ryu EK and Yoo J, Nucl Med Mol Imaging, 2010, 44, 185–192. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Mathias CJ, Welch MJ, Green MA, Diril H, Meares CF, Gropler RJ and Bergmann SR, J. Nucl. Med, 1991, 32, 475–480. [PubMed] [Google Scholar]
- 40.Frisch MJ, Trucks GW, Schlegel HB, Scuseria GE, Robb MA, Cheeseman JR, Scalmani G, Barone V, Petersson GA, Nakatsuji H, Li X, Caricato M, Marenich AV, Bloino J, Janesko BG, Gomperts R, Mennucci B, Hratchian HP, Ortiz JV, Izmaylov AF, Sonnenberg JL, Williams F. Ding, Lipparini F, Egidi F, Goings J, Peng B, Petrone A, Henderson T, Ranasinghe D, Zakrzewski VG, Gao J, Rega N, Zheng G, Liang W, Hada M, Ehara M, Toyota K, Fukuda R, Hasegawa J, Ishida M, Nakajima T, Honda Y, Kitao O, Nakai H, Vreven T, Throssell K, Montgomery JA Jr., Peralta JE, Ogliaro F, Bearpark MJ, Heyd JJ, Brothers EN, Kudin KN, Staroverov VN, Keith TA, Kobayashi R, Normand J, Raghavachari K, Rendell AP, Burant JC, Iyengar SS, Tomasi J, Cossi M, Millam JM, Klene M, Adamo C, Cammi R, Ochterski JW, Martin RL, Morokuma K, Farkas O, Foresman JB and Fox DJ, Journal, 2016. [Google Scholar]
- 41.Weigend F and Ahlrichs R, Physical Chemistry Chemical Physics, 2005, 7, 3297–3305. [DOI] [PubMed] [Google Scholar]
- 42.Grimme S, Antony J, Ehrlich S and Krieg H, The Journal of Chemical Physics, 2010, 132, 154104. [DOI] [PubMed] [Google Scholar]
- 43.Marenich AV, Cramer CJ and Truhlar DG, The Journal of Physical Chemistry B, 2009, 113, 6378–6396. [DOI] [PubMed] [Google Scholar]
- 44.Neese F, WIREs Computational Molecular Science, 2012, 2, 73–78. [Google Scholar]
- 45.Neese F, WIREs Computational Molecular Science, 2018, 8, e1327. [Google Scholar]
- 46.Neese F, Inorganica Chimica Acta, 2002, 337, 181–192. [Google Scholar]
- 47.Neese F, The Journal of Chemical Physics, 2005, 122, 034107. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.