


Abstract
The paucity of recurrent mutations has hampered efforts to understand and treat neuroblastoma. Alternative splicing and splicing-dependent RNA-fusions represent mechanisms able to increase the gene product repertoire but their role in neuroblastoma remains largely unexplored. Here we investigate the presence and possible roles of aberrant splicing and splicing-dependent RNA-fusion transcripts in neuroblastoma. In addition, we attend to establish whether the spliceosome can be targeted to treat neuroblastoma. Through analysis of RNA-sequenced neuroblastoma we show that elevated expression of splicing factors is a strong predictor of poor clinical outcome. Furthermore, we identified >900 primarily intrachromosomal fusions containing canonical splicing sites. Fusions included transcripts from well-known oncogenes, were enriched for proximal genes and in chromosomal regions commonly gained or lost in neuroblastoma. As a proof-of-principle that these fusions can generate altered gene products, we characterized a ZNF451-BAG2 fusion, producing a truncated BAG2-protein which inhibited retinoic acid induced differentiation. Spliceosome inhibition impeded neuroblastoma fusion expression, induced apoptosis and inhibited xenograft tumor growth. Our findings elucidate a splicing-dependent mechanism generating altered gene products in neuroblastoma and show that the spliceosome is a potential target for clinical intervention.
INTRODUCTION
Neuroblastoma arises within the sympathetic nervous system and is the most frequent extracranial solid childhood cancer, exhibiting a high degree of clinical heterogeneity ranging from spontaneous regression to fatal progression (1). Despite intense sequencing efforts, few recurrently mutated genes have been identified in neuroblastoma (2,3). Instead, high-risk neuroblastoma are characterized by large-scale chromosomal rearrangements such as chromothripsis and loss or gain of chromosomal regions (e.g. loss of 1p36 and 11q or gain of 17q and 2p) with or without MYCN amplification (1,2). In eukaryotic cells, pre-mRNA splicing is essential for intron removal and to increase the number of possible gene products. If not properly regulated, this process can lead to mis-splicing, generating altered and potentially oncogenic proteins. A hyperactivated spliceosome can also provide an avenue of intervention, e.g. in MYC-driven breast cancer, where elevated precursor mRNA synthesis results in an increased burden on the splicing machinery and consequently a vulnerability to spliceosome inhibition (4). In addition, several cancers harbor monoallelic mutations of spliceosome factors (5). Given the lack of recurring mutations and the prominent role of MYCN for high-risk neuroblastoma, understanding a possible role for altered splicing dynamics in high-risk neuroblastoma could provide novel insights on how to target this disease. Notably, in a mouse model of neuroblastoma, intronic splicing motifs were shown to represent hotspots for recurring mutations (6). A potential consequence of aberrant splicing is generation of fusion transcripts through cis-splicing of adjacent genes (7,8). Such fusions differ from those generated by chromosomal translocations, e.g. BCR-ABL in chronic myelogenous leukemia (9). Cis-spliced fusions represent an alternative mechanism, distinct from amino acid changing mutations, able to generate altered gene products with a potential to promote tumor development but also serve as diagnostic markers or novel drug targets (7,10). Besides a fusion resulting from small interstitial genomic deletions at 11q generating either a MLL-FOXR1 or a PAFAH1B2-FOXR2 fusion (11) no intra-chromosomal chimeric transcripts have been described in neuroblastoma. However, they have been shown to be present in other tumor types as well as in non-transformed tissues and be promoted by different types of cellular stress such as infections or mutations (12–16).
In order to explore whether neuroblastoma tumors harbor previously undetected gene fusions, we analyzed a cohort of 172 RNA-sequenced neuroblastoma tumors. We identified several fusion transcripts, of which a significant proportion exhibited a distinct genomic distribution according to tumor risk. As a proof of principle, that fusions can generate novel gene products with alternative properties, we cloned and characterized the ZNF451-BAG2 fusion. The generated protein exhibited distinct protein–protein binding properties compared to wild-type BAG2 and impeded retinoic acid induced differentiation. This reveals how a fusion gene product can influence neuroblastoma response to a drug used in the treatment of high-risk patients (17). Identified fusions were predominantly generated by genes in close proximity and flanked by canonical splicing donors and acceptors. This pattern was distinct to fusions unique for neuroblastoma, whereas fusions identified in normal adrenal gland or in other tumors lacked such a pattern. Furthermore, a subset of identified neuroblastoma specific fusions was hypersensitive to pharmacologic spliceosome inhibition in comparison to their wild type cognates. High expression levels of spliceosome factors were strongly associated with high-risk disease and spliceosome inhibition also selectively promoted apoptosis in neuroblastoma cells in vitro and reduced tumor growth in mouse xenografts.
MATERIALS AND METHODS
Ethical considerations
All animal experiments were performed according to Swedish guidelines and regulations, the ethical permit 6420–2018 was granted by ‘Stockholms Norra djurförsöksetiska nämnd, Sweden’. Neuroblastoma primary tumors came either from the Swedish neuroblastoma Registry (ethical permission (DN03-736) granted by Karolinska Institutets Forskningsetikommitté Nord (clinical information described in Li et al. (18), or from the Irish neuroblastoma cohort, with ethical approval of the Medical and Research Ethics Committee of Our Lady's Children's Hospital, Crumlin, Dublin, Ireland. Informed consent from families of subjects was obtained for samples. Six histologically confirmed normal human adrenal glands were included as controls (covered by existing ethical approvals; Dnr 01-136 and Dnr 01-353 KI forskningsetikkommitté Nord). Four juvenile adrenal glands were from University of Maryland Brain and Tissue Bank; Total RNA of pooled normal adrenal gland was from Clontech (Cat# 636528). Human total RNA from different normal tissues from Clontech (Human Total RNA Master Panel II, Cat#. 636643).
RT-PCR and paired-end RNA-seq
RNA was isolated using the PerfectPure RNA Cultured Cell Kit (cell lines) and PerfectPure RNA Tissue Kit (patient samples) from 5 PRIME. Reverse transcription was performed with iScript cDNA Synthesis Kit (Bio-Rad) and RT-PCR was performed by using iQ SYBR Green Supermix (Bio-Rad). RNA-seq libraries were prepared using TruSeq RNA Library Preparation Kit v2 (Illumina); paired-end RNA sequencing (125 bp) was performed in SciLifeLab (Stockholm).
Data analysis
FusionCatcher (19) was applied to detect the fusion transcripts in paired-end RNA-seq data. Reads were mapped to hg19 and differential expression analysis was performed as described in (20). For enrichment analysis in Figure 1C, only fusions occurring above ∼3% in each risk group were included (1 case in Low/Intermediate-risk, 4 cases in High-risk and five cases for the common fusion transcripts shared by Low/Intermediate-risk and high-risk groups). For the Z-test in 1C the μ and σ values were calculated from genome wide enrichment of fusions at the different chromosomal arms. The analysis presented in Figure 4F–H includes fusion events which occur at a frequency of ≥5 tumors in the NB172 cohort.
Figure 1.

Identification of fusion transcripts in neuroblastoma tumors. (A) Top 25 most frequent fusions identified by FusionCatcher in a cohort of 172 paired end RNA sequenced neuroblastoma patient samples derived from the NCI TARGET project (NB172). (B) Circos plot of genomic distribution of top frequent fusions (>10%) in NB172. (C) Enrichment of fusion transcripts common or unique to low/intermediate-risk or high-risk tumors in chromosomal arms as calculated by a normalized enrichment score = (counts of fusion transcripts in each chromosomal arm / length of chromosomal arm (Mb)) / average enrichment. (D, E) MYCN/LMO1 expression levels are high in neuroblastoma tumors (red) bearing fusion transcripts of which one fusion partner is either MYCN or LMO1, as shown by expression value FPKM (Fragments Per Kilobase per Million mapped reads). P-values in (B,C) were calculated from Z-values, assuming standard normal distribution. *P< 0.05, **P< 0.01, ***P< 0.001.
Figure 4.
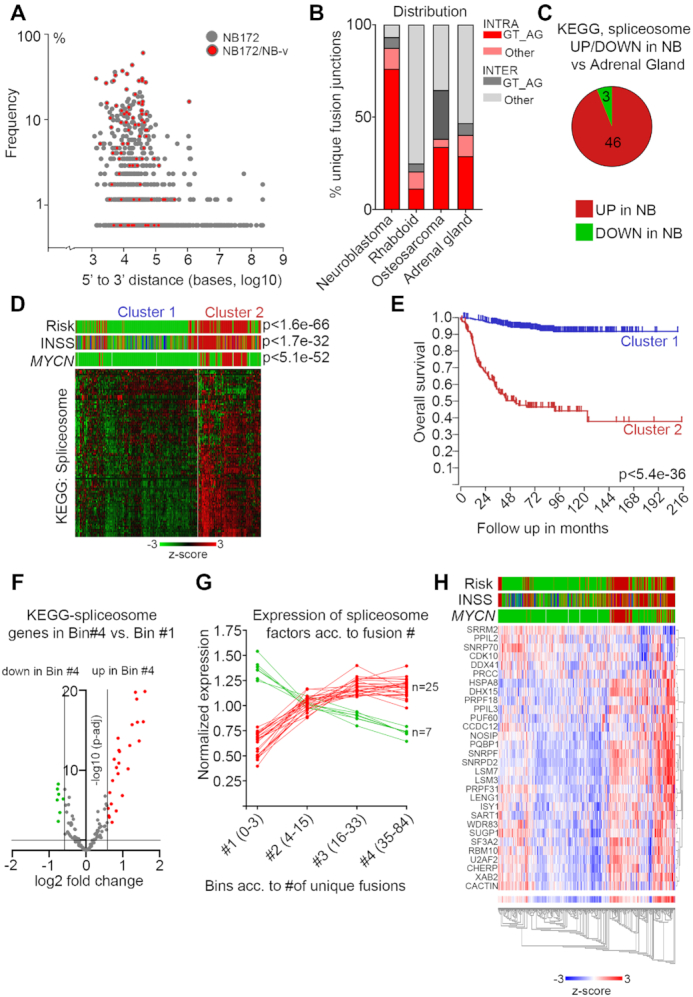
Enrichment of canonical splicing pattern at fusion junctions and aberrant spliceosome activity in high-risk neuroblastoma. (A) Plot of the frequency versus the distance from 5′ to 3′ in the intrachromosomal fusion transcript identified in the NB discovery cohort (NB172); each dot represents a unique fusion transcript; fusion transcripts re-identified in the NB validation cohort (NB-v) were marked as red. (B) Distribution of unique intrachromosomal vs interchromosomal fusion junctions flanked by GT_AG or other nucleotide motifs in neuroblastoma, rhabdoid tumor, osteosarcoma and normal adrenal gland. (C) Differential expression of genes (>1.5×, adj P< 0.05) in the KEGG Spliceosome pathway for neuroblastoma dataset (NBL172) versus human normal adrenal gland dataset. (D) K-means analysis of the 498-SEQC dataset from the R2 database, utilizing the genes in the KEGG spliceosome pathway, generates two clusters with significant differences in Risk, INSS and MYCN-amplification. (E) Overall survival probability of the two clusters identified in (D). (F) Volcano plot showing differential expression of genes belonging to the KEGG Spliceosome category between the tumors with the highest number of unique fusion transcripts (Bin #4) and the tumors with the lowest number (Bin #1). Spliceosome genes with significantly higher expression in Bin #4 are labeled in red and spliceosome genes with significantly lower expression in Bin #4 are labeled green. (G) Normalized expression levels of the genes identified in (F) from Bin #1 to Bin #4. (H) Heatmap and associated clinical classification (Risk, INSS and MYCN status) of the spliceosome genes identified as significantly differentially expressed in (F).
Cloning and expression of wildtype BAG2 and ZNF451-BAG2
cDNA was synthesized from total RNA by the iScript cDNA Synthesis Kit (Bio-Rad). Coding regions of wildtype BAG2 and ZNF451-BAG2 were amplified from SK-N-AS cells for subcloning into p3XFLAG-CMV14 (Sigma) and pLVX-EF1α-IRES-mCherry (Clontech) using primer pair 1 and 2, respectively (Supplementary Table S11), inserted to pCR-Blunt II-TOPO vector via zero Blunt TOPO PCR Cloning Kit (ThermoFisher Scientific) and sequenced. BAG2 and ZNF451-BAG2 were subcloned into p3XFLAG-CMV14 vector and pLVX-EF1α-IRES-mCherry lentiviral vector using EcoRI and BamHI. To generate pLVX-Tetone-puro-IRES-mCherry-empty/BAG2/ZNF451-BAG2 vectors, pLVX-Tetone-puro-empty (Clontech) construct was linearized with AgeI, blunted and digested with EcoRI; pLVX-EF1α-IRES-mCherry-empty/BAG2/ZNF451-BAG2 vectors were linearized with MluI, blunted and digested with EcoRI to release IRES-mCherry-empty/BAG2/ZNF451-BAG2; then linearized and blunted pLVX-Tetone-puro-empty construct was ligated with fragment of IRES-mCherry-empty/BAG2/ZNF451-BAG2 separately.
Lentiviruses expressing pLVX-EF1α-IRES-mCherry-empty/BAG2/ZNF451-BAG2 and pLVX-Tetone-puro-IRES-mCherry-empty/BAG2/ZNF451-BAG2 were produced in 293FT cells using lipofectamine 2000 based protocol.
Cell culture
All eight neuroblastoma cell lines (SH-SY5Y, SK-N-SH, SK-N-FI, SK-N-BE(1), SK-N-AS, LAN-1, CHP-212 and IMR-5) were maintained in RPMI1640 medium supplemented with 10% FBS, 1% penicillin/streptomycin and 1% l-glutamine and grown in 5% CO2 at 37°C.
Differentiation assay, immunofluorescence staining and apoptosis analysis
For short-term retinoic acid (RA) induced differentiation assay, SK-N-FI cells were seeded in six-well plates; after 24 h, cells were infected by lentiviruses expressing pLVX-EF1-IRES-mCherry-empty/BAG2/ZNF451-BAG2 for 48 h; then cells were trypsinized and 20 000 cells were reseeded into 6 well plates with coverslips; 24 h later, cells were treated with 1 μM RA or DMSO as control for 6 days. For doxycycline-inducible system, 20 000 SK-N-BE(1) cells with stable overexpression of pLVX-Tetone-puro-IRES-mCherry-ZNF451-BAG2 were seeded in six-well plates with coverslips, and after 24 h, cells were pre-treated with 1 μM doxycycline or DMSO as a control for one day; then cells received one of the following four different treatment for 4 days: DMSO, 1 μM RA, 1 μM doxycycline or 1 μM RA+1 μM doxycycline.
Cells on coverslips were fixed in RPMI1640 medium containing 2% PFA for 5 min at room temperature (RT), washed once with PBS at RT, fixed with 4% PFA for another 15 min at RT and washed twice with cold PBS; then cells were permeabilized with PBS containing 0.25% Triton X-100 for 15 min at RT and blocked with 3% BSA for 1 h at RT. Immunofluorescent staining was performed with the following primary antibody Tuj1 (Covance, 1:1000) overnight at +4°C.
For apoptosis analysis, cells were seeded on 6 cm dishes 24 h before treatment with Pladienolide B (Santa Cruz) or SPRIN340 (MedChemExpress); DMSO was used as a control; after 48 h treatment, cells, including detached cells in the supernatant, were harvested and total proteins were isolated by lysing cells in lysis buffer containing 100 mM Tris–HCl, pH 7.4, 150 mM NaCl, 1% NP40. Protease and phosphatase cocktail inhibitors (Roche) were added to the lysis buffer before use. Protein levels of apoptosis markers were analyzed by western blotting.
Xenografts
2 × 106 LAN-1 cells in 200 μl PBS:matrigel (BD, 354248) (1:1) were injected s.c. into the right flank of adult female Crl:Nu (Ncr)-Foxn1 nu (nude mice). Treatments started 14 days after transplantation. The control group was injected i.p. with 100 μl vehicle (10% EtOH and 4% Tween-80 in sterile PBS) per mouse per day. Pladienolide B was injected i.p. at a concentration of 10 mg/kg. Tumors were measured externally using a digital caliper; the greatest longitudinal diameter (length) and the greatest transverse diameter (width) were determined, and tumor volume was calculated using the modified ellipsoidal formula [(length × width2)/2]. Overall health was monitored during treatment, and weight was measured weekly. The size of the treated groups was determined based on previous xenograft experiments (20). For tumor growth comparison in Figure 6C mixed-effects analysis with Geisser–Greenhouse's epsilon correction was performed. All statistical analysis was performed in Excel or GraphPad Prism 8.1.1.
Figure 6.
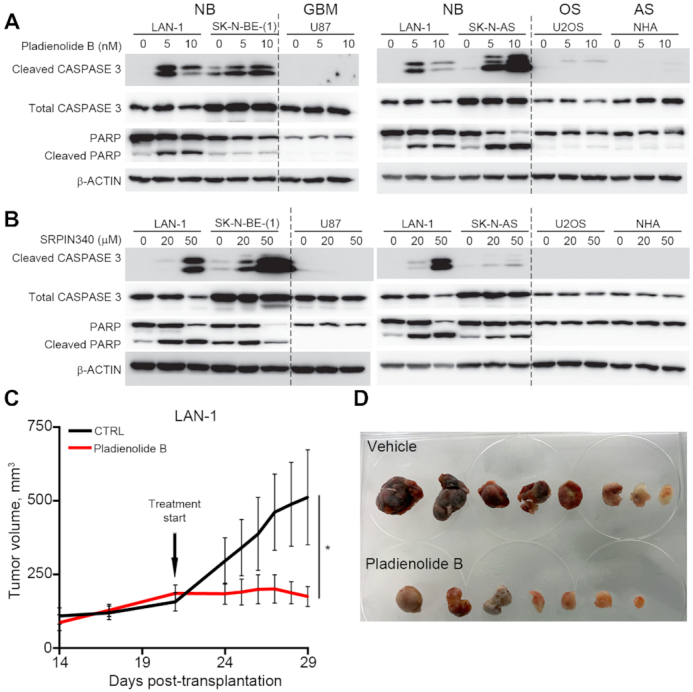
Pharmaceutical inhibition of spliceosome activity selectively induces apoptosis in neuroblastoma cells and impedes neuroblastoma xenograft growth. (A) Induction of apoptosis in LAN-1,SK-N-BE(1) and SK-N-AS neuroblastoma (NB) cells, but not in human glioma (GBM) cells (U87), osteosarcoma (OS) cells (U2OS) or normal human astrocytes (AS), (NHA) upon pladienolide B treatment (0–20 nM) for 48 h, as detected by western blotting of full-length CASPASE-3, cleaved CASPASE-3, full-length PARP and cleaved PARP; β-ACTIN was used as a loading control. (B) Induction of apoptosis in LAN-1,SK-N-BE(1) and SK-N-AS neuroblastoma cells (NB), but not in human glioma (GBM) cells (U87), osteosarcoma cells (U2OS) or normal human astrocytes (NHA) upon SRPIN340 treatment (0–50 mM) for 48 h, as detected by western blotting of full-length CASPASE-3, cleaved CASPASE-3, full-length PARP and cleaved PARP; β-ACTIN was used as a loading control. LAN-1 cells were included in all western blots (A, B) for comparison. (C, D) In mice xenografted with LAN-1 neuroblastoma cells, treatment with Pladienolide B significantly reduced tumor growth (n = 7–8) (C, D). *P<0.05; Mixed-effects model with Geisser-Greenhouse's epsilon correction.
Quantification
Cells were stained as in Figure 3H–M and Supplementary Figure S4G–J. Images were taken by confocal microscopy (10×). For quantification, images were coded and a researcher who had not participated in staining and image acquisition manually counted the ratio of transduced cells extending TUJ1+ neurites/total number of transduced cells/microscopic field. For DOX− SK-N-BE(1) the ratio of cells extending TUJ1+ neurites/total number of cells/microscopic field were counted. Microscopic fields containing less than three transduced cells were discarded. Grubb′s test was performed for outlier detection (alpha P<0.05). For statistical analysis in Figure 3N and Supplementary Figure S4K one-way ANOVA followed by a Tukey's multiple comparison test was performed.
Figure 3.
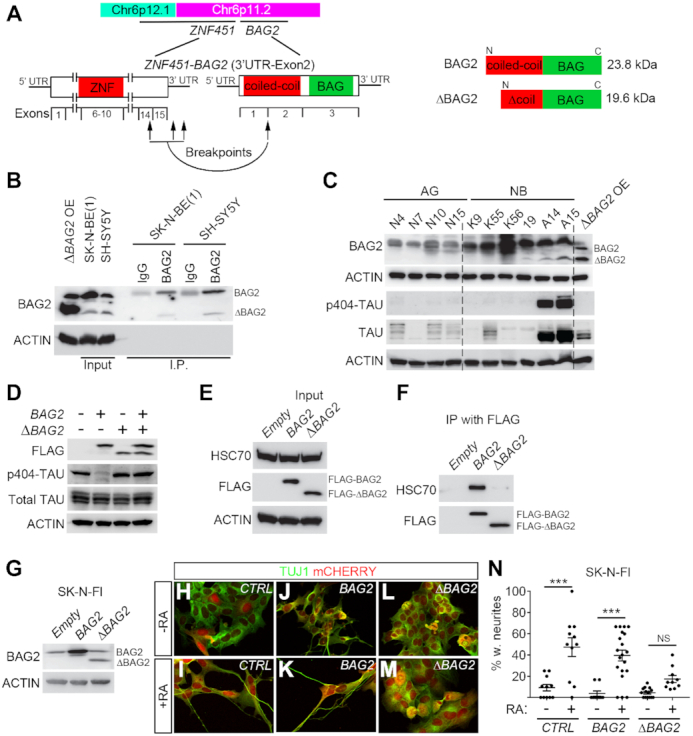
ΔBAG2 impairs clearance of p-TAU, HSC70 binding and inhibits retinoic acid-induced differentiation in neuroblastoma cells. (A) Schematic representation of ZNF451-BAG2 fusion, the resulting truncated BAG2 is referred to as ΔBAG2. (B) BAG2 antibody was able to detect endogenous protein expression of ΔBAG2. Total cell lysates from SK-N-BE(1) and SH-SY5Y was immunoprecipitated with BAG2 antibody (Santa Cruz, catalog sc-390262) and protein levels were detected with the same BAG2 antibody. Whole cell lysate from SK-N-FI cells with lentivirally overexpressed ΔBAG2 was used as a positive control (ΔBAG2 OE). (C) Endogenous tumor ΔBAG2 protein expression is associated with high levels of phosphorylated TAU (p-TAU) in neuroblastoma tumors (NB) as detected by immunoblotting. Whole cell lysate from SK-N-FI cells with lentivirally overexpressed ΔBAG2 was used as a positive control (ΔBAG2 OE). Normal adrenal glands (AG). (D) Overexpression of the ΔBAG2 prevented clearance of pTAU by wt BAG2. SK-N-FI cells were transfected with p3XFLAG-CMV14-empty/BAG2/ΔBAG2/BAG2+ΔBAG2 for 48 h. Cells were harvested and proteins were extracted; whole-cell lysates were used to detect FLAG, p-TAU on Ser404, total TAU and ACTIN. (E, F) Wild type BAG2 but not ΔBAG2 binds to HSC70. FLAG-tagged proteins were immunoprecipitated from whole-cell lysates as prepared in (D) using Anti-FLAG M2 magnetic beads and eluted. Immunoprecipitated proteins were western blotting to detect HSC70, FLAG. (G–N) Constitutive lentiviral overexpression of ΔBAG2, but not wildtype BAG2 (backbone pLVX-EF1α-IRES-mCherry) inhibited RA-induced differentiation (6 days of treatment) in SK-N-FI cells. Data presented in (G) is western blot analysis of protein levels. Data in N is represented as mean of transduced cells with TUJ1+ neurites/total number of transduced cells ± SEM, each data-point represents this ratio in a single 10× microscopic field (n = 10–20). ***P< 0.001, one-way ANOVA with Tukey's multiple comparison test.
Western blotting
Immunoblotting was performed using standard protocols. Following primary antibodies were used: BAG2(sc-390262, 1:200), HSC70(sc-7298, 1:200) from Santa Cruz; phosphor-ser404-Tau(44–758G, 1:200) from ThermoFisher Scientific; total Tau(A0024, 1:1000) from Dako; beta-actin(AC-15, 1:3000), FLAG(F1804, 1:1000) from Sigma; PARP (9542s), Caspase-3 (14220s) and cleaved caspase-3 (9664s) from Cell Signaling.
Co-immunoprecipitation
SK-N-FI cells were seeded in 15 cm dishes 24 h before transfection. Transfection of p3XFLAG-CMV14-empty/BAG2/ZNF451-BAG2 constructs was performed using lipofectamine 2000. Forty-eight hours post-transfection, cells were harvested and proteins were extracted using lysis buffer containing 10 mM Tris–HCl (pH7.4), 150 mM NaCl, 0.5% NP40, complete protease and phosphatase inhibitors (Roche). FLAG-tagged fusion proteins were immunoprecipitated using Anti-FLAG M2 magnetic beads (Sigma) according to manufacturer's instructions. Bound proteins were examined by silver staining or western blotting using standard protocols.
RESULTS
Fusion transcripts are a common feature of neuroblastoma
To reveal novel gene fusions in neuroblastoma we analyzed a data set (National Cancer Institute TARGET, dbGap Study Accession: phs000218.v16.p6) comprising 172 paired-end RNA sequenced neuroblastoma tumors (referred to as ‘NB172’), out of which 139 were diagnosed as high-risk, 19 as intermediate-risk and 14 as low-risk according to the Children's Oncology Groups staging (COG), (Supplementary Table S1). We applied the fusion detection tool FusionCatcher (19) and identified chimeric transcripts in 163 out of 172 cases with an average of 31 distinct fusion transcripts per tumor (Supplementary Table S2). Short homologous sequences (SHS) have been suggested to serve as templates for reverse transcriptase dependent false positive chimeras/fusions (21). In order to avoid potential false positive fusions, we removed any fusion that incorporated genes with SHSs of five or more nucleotides, which reduced the number of identified fusions from 1073 to 924. Structural consequences of the fusions ranged from truncated proteins through bona fide fusion proteins to deletion of genes. The majority of fusions (786/924) revealed by our analysis were intra-chromosomal fusion transcripts (Supplementary Tables S2 and S3), many of which consisted of adjacent genes. Importantly, 114 fusions occurred at a frequency of 5% or more (Top 25 in Figure 1A, all >10% in Figure 1B and full list in Supplementary Table S3) and all of these were intrachromosomal. There was a significant enrichment of fusion junctions at chromosomes 17 and 22 (Supplementary Figure S1A). Furthermore, there was a significant enrichment of fusions that occurred in >10% of tumors at the same chromosomes (Figure 1B).
Gain of 17q is the most frequently occurring genomic alteration in high-risk neuroblastoma and a marker for adverse clinical outcome (1), whereas 22q alterations have been reported to be involved in the transition to metastatic and more aggressive neuroblastoma (22). We analyzed the fusion transcripts occurring exclusively in low/intermediate-risk and exclusively in high-risk tumors as well as fusion transcripts common to low/intermediate-risk and high-risk levels (Supplementary Tables S4 and S5). Chimeric transcripts unique to low/intermediate-risk tumors exhibited significant enrichment at several chromosomal arms including 11q, a region commonly lost in high-risk neuroblastoma (Figure 1C). In contrast, both common and high-risk unique fusion transcripts were enriched at 17q and 22q but not at 11q (Figure 1C), with a pronounced increase in frequency of 17q fusion transcripts in high-risk tumors (Figure 1C). Thus, with increased risk the frequency of 17q located fusion junctions also increases and was more than seven times higher than the average fusion rate per chromosomal arm. Several fusion transcripts encompassed factors involved in neuroblastoma pathogenesis, including ARID1B, CASZ1, HDAC8, LMO1, MYCN, BRCA1, TERT and PDE6G (Supplementary Table S6). Notably, tumors harboring fusion transcripts of well-known neuroblastoma oncogenes (e.g. MYCN and LMO1) also exhibited high expression levels of their wild-type cognates (Figure 1D and E). The high-risk susceptibility locus in LMO1 is significantly associated with MYCN-non amplified high-risk neuroblastoma but not with MYCN-amplified high-risk neuroblastoma (23), interestingly the LMO1-RIC3 fusion (Supplementary Figure S1B) was exclusively detected in MYCN-non amplified high-risk neuroblastoma (Supplementary Table S6). The BRCA1-VAT1 fusion was also only detected in MYCN-non amplified high-risk cases; previously it has been shown that copy number amplification of BRCA1 in neuroblastoma is restricted to cases lacking MYCN-amplification (24) (Supplementary Table S6).
Validation of fusion transcripts specific for neuroblastoma
To corroborate the fusion transcripts observed in the NB172 dataset in an independent cohort, we performed paired-end RNA-sequencing in an additional cohort containing 14 neuroblastoma patient samples, together with eight neuroblastoma cell lines, NB-validation (NB-v, Materials and Methods). We identified 139 fusions, of which 82 (∼59%) were present in the NB172 dataset (Figure 2A and Supplementary Tables S7 and S8). To investigate whether the identified fusions were neuroblastoma specific, we analyzed a cohort of 161 sequenced tissue samples from human normal adrenal glands (25). Out of 342 detected fusions in the adrenal gland cohort, only 23 (∼6.7%) were present in the NB172 dataset and only 4 (∼1.2%) of these were present in the NB-v cohort (Figure 2A and Supplementary Table S9). This enrichment of common fusion transcripts in the neuroblastoma cohorts vs. the adrenal gland dataset was highly significant (chi-square test with Yate's correction, P-value < 0.0001). Fusion transcript associated genes unique to and shared by the two neuroblastoma datasets were enriched at 17q and 2p, two chromosomal regions where gains are closely associated with high-risk neuroblastoma (Figure 2B). Parametric analysis of gene set enrichment (PAGE) (26) comparing high-risk grade 4 tumors with low-risk grade 4s tumors (according to the International Neuroblastoma Staging System, INSS) in the R2 498-SEQC data base (27) showed that the NB172/NB-v common genes identified in Figure 2A are enriched in the grade 4 high-risk tumors (Supplementary Figure S2A). To further investigate whether the identified fusions are distinct for neuroblastoma we analyzed a set of 65 sequenced rhabdoid tumors (National Cancer Institute TARGET, dbGap Study Accession: phs000470.v17.p7) wherein 2055 unique fusion transcripts were detected. However, the overlap with the fusions detected in NB172 dataset was limited to 44 transcripts (∼2.1%) (Figure 2C). In a cohort of 177 sequenced osteosarcoma tumors (National Cancer Institute TARGET, dbGap Study Accession: phs000468.v17.p7; Figure 2C) we could detect 1650 unique fusion transcripts but there was no overlap with the NB172 cohort (Figure 2C). In contrast to fusions detected in neuroblastoma, the majority of detected fusions in rhabdoid tumors and osteosarcoma were inter-chromosomal (Supplementary Figure S2B). Detected fusions that occur at higher frequencies than 5% are more abundant in neuroblastoma (in 12.3% of tumors) than in rhabdoid tumor (5.7%) and osteosarcoma (0.4%) (Supplementary Figure S2C). In osteosarcoma there was a considerable number of tumors (∼24.9%, 44/177) harboring fusions predicting substantial deletion of the TP53 tumor suppressor, disruption of the gene or a truncation at the C-terminal (Figure 2D, Supplementary Table S10). Notably, TP53 is one of the most frequently altered genes in osteosarcoma (28). As a comparison we could not detect any fusions containing TP53 in the neuroblastoma tumors (0/172) (Figure 2D).
Figure 2.
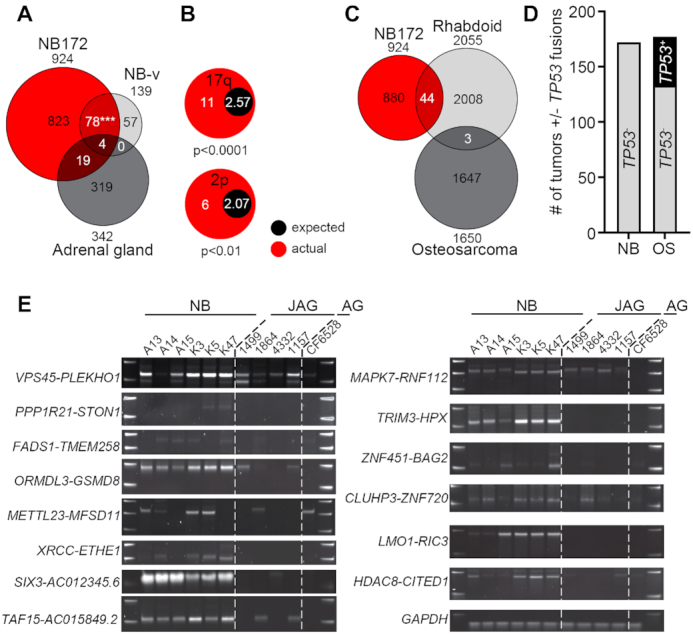
Validation of fusion transcripts. (A) Venn diagram of identified fusions in three datasets, NB172 (NCI TARGET), Validation-NB (NB-v, 14 neuroblastoma patients plus 8 neuroblastoma cell lines) and adrenal gland (161 samples from normal human adrenal gland). A significant number of fusions detected in NB-v were present in the NB172 cohort, chi-square with Yate's correction P-value <0.001. (B) Enrichment of fusion transcript genes unique to the NB172 and the NB-v cohorts on chromosomal arms 2p and 17q. (C) Venn diagram of identified number of fusions in three datasets, NB172 (NCI TARGET), Rhabdoid (65 Rhabdoid tumor patient samples) and Osteosarcoma (177 Osteosarcoma patient samples). (D) Comparison of the number of neuroblastoma (NB) and osteosarcoma (OS) tumors with at least one fusion transcript containing TP53. (E) Validation of fusion transcripts by RT-PCR in neuroblastoma tumors (NB), juvenile adrenal glands (JAG) and adult adrenal gland (AG). GAPDH was used for normalization. P-values in (A) were calculated by chi-square test with Yate's correction and in (B) by Fisher's exact test. ***P< 0.001.
Several programs are available for the identification of fusion transcripts from RNA-seq data. However, results vary considerably between different algorithms (29) why validation with independent methods is warranted. We designed primers spanning the fusion junctions of selected chimeric transcripts which occurred at high frequency or encompassed genes previously shown to be important for neuroblastoma biology. We initially designed junction spanning primers for the selected chimeric transcripts VPS45-PLEKHO1, METTL23-MFSD11, HDAC8-CITED1, ZNF451-BAG2, TRIM3-HPX and PRR11-SMG8 (Supplementary Table S11). We performed PCR in 10 neuroblastoma tumor samples and in a panel consisting of cDNA from 14 untransformed human tissues. All selected candidates, except PRR11-SMG8, were expressed in a neuroblastoma specific manner (Supplementary Figure S2D). For validation, these PCR-products including ten additional fusions, PPP1R21-STON1, FADS1-TMEM258, ORMDL3-GSDM8, XRCC1-ETHE1, SIX3-AC012354.6, MAPK7-RNF112, CLUHP3-ZNF720, LMO1-RIC3, CHCHD10-VPREB3 and TAF15-AC015849.2, were excised, inserted into the pCR-Blunt II-TOPO vector and subsequently sequenced. All the sequenced PCR-products exhibited an identical sequence of nucleotides to that of the fusions identified by FusionCatcher, except the SIX3-AC012354.6 chimeric transcript, which harbors a point mutation in SK-N-BE(1) cell line (Supplementary Table S12). To understand how well the transcripts predicted by FusionCatcher corresponds to successfully PCR-validated candidates we designed primers targeting the fusion junctions of all unique transcripts detected by FusionCatcher in the LAN-1 and SK-N-BE(1) neuroblastoma cell lines. Out of 17 predicted fusions in the LAN-1 cells we could validate 15 (88%) with PCR followed by Sanger sequencing whereas in the SK- N-BE(1) cells 9 (75%) out of 12 fusions were validated (Supplementary Figure S3A, B and Supplementary Table S13). A previous report identified the VPS45-PLEKHO1 fusion in non-transformed tissue (16), but it was not detected in our panel of non-transformed tissues. Neuroblastoma commonly appear in and around the adrenal gland (1). To further investigate specificity of identified fusion transcripts in neuroblastoma compared to healthy juvenile adrenal glands we performed PCR for 14 fusion transcripts on six neuroblastoma tumors, four healthy juvenile adrenal glands (age 0–5 years) and a pool of normal adult adrenal glands (Figure 2E). The PCR analysis shows that there is an enrichment of fusions in neuroblastoma but also that a subset of normal juvenile adrenal glands expresses certain fusions, albeit at lower levels. Importantly, the PCR validation shows that fusions detected at high frequency with FusionCatcher have a high probability of being true positives. In addition to identifying several fusions incorporating well known neuroblastoma oncogenes (Figure 1D, E and Supplementary Table S6) we also analyzed selected fusion transcripts occurring at high frequency for the predicted effect on functional domains of wild type transcripts (Supplementary Table S14).
The ZNF451-BAG2 fusion generates a truncated BAG2 protein, present in a subset of neuroblastoma tumors
One of the 25 most frequently occurring fusion transcripts encompassed the BCL2 associated athanogene (BAG2) which encodes a co-chaperone, BAG2, involved in targeting misfolded proteins for degradation through an ubiquitin independent pathway (30). BAG2 levels have previously been shown to increase upon neuronal differentiation in neuroblastoma cells (31). In addition, BAG2 clears phosphorylated TAU from neuronal microtubule (30), potentially promoting stabilization of axons, an important component of neuronal differentiation. We thus selected to investigate ZNF451-BAG2 to understand whether a protein with alternative properties could be generated as a result of these fusion transcripts.
The ZNF451-BAG2 fusion spans the 3′ UTR or exon 14 of ZNF451 and the second exon of BAG2, potentially generating a truncated BAG2 transcript lacking the first exon (Figure 3A). The first exon of BAG2 encodes part of a coiled-coil domain that is absent in the ZNF451-BAG2 fusion (Figure 3A). Full length BAG2 encodes a 23.8 kDa protein, whereas the ZNF451-BAG2 fusion transcript encodes a smaller 19.6 kDa protein (ΔBAG2) (Figure 3A). The ZNF451-BAG2 chimera was present in 31 of the 172 sequenced tumors (18%). Alignment of wild-type BAG2 protein (BAG2) across different species showed that in ΔBAG2 the highly conserved N-terminal coiled-coil domain was truncated (Supplementary Figure S4A), implying functional relevance of the truncated region for BAG2. For validation of the ZNF451-BAG2 fusion transcript we performed PCR of SK-N-BE(1) cells utilizing primers covering the fusion junction and specific for distinct regions predicted by the FusionCatcher, followed by Sanger sequencing, see Supplementary Figure S4B, C and Supplementary Tables S11 and S12. We then cloned the ΔBAG2 transcript (Supplementary Figure S4B and C) for lentiviral overexpression in SK-N-FI neuroblastoma cells, wherein it was not detected by FusionCatcher. Firstly, we validated the capacity of a BAG2 antibody to detect the ΔBAG2 protein when expressed in SK-N-FI neuroblastoma cells. In addition, we probed its capacity to detect endogenous ΔBAG2 protein as well as immunoprecipitated ΔBAG2 in both SK-N-BE(1) and SH-SY5Y neuroblastoma cells (Figure 3B). To understand if tumors wherein ZNF451-BAG2 was identified (Supplementary Figure S2D) also had detectable levels of ΔBAG2 protein, we then performed immunoblotting with the validated BAG2 antibody. All probed (n = 13) tumors contained BAG2 protein at varying levels however only five tumors also co-expressed detectable levels of ΔBAG2, while no detectable levels of ΔBAG2 were observed in tissue from six human normal adrenal glands (Figure 3C and Supplementary Figure S4D).
ΔBAG2 impairs clearance of phosphorylated TAU and binding to HSC70
BAG2 has been shown to be important for clearance of phosphorylated forms of the TAU protein (pTAU) and thus been implicated as a stabilizer of microtubules (30). To test if this capacity was attenuated by the presence of ΔBAG2 we probed the levels of pTAU in a panel of neuroblastoma tumors and normal adrenal glands. This showed a clear association between the presence of pTAU and endogenous ΔBAG2 (Figure 3C and Supplementary Figure S4D). To validate that this was caused by ΔBAG2 protein expression we expressed a FLAG-tagged version of ΔBAG2 and BAG2 alone or in combination in SK-N-FI neuroblastoma cells. Upon BAG2 overexpression, the levels of pTAU were significantly reduced whereas total TAU was present in amounts similar to those in control-transduced cells (Figure 3D). ΔBAG2 overexpressing cells retained pTAU levels and more importantly, upon co-expression BAG2 failed to clear pTAU (Figure 3D), implying that ΔBAG2 can act as a negative regulator of BAG2 function. BAG2 has been shown to bind the heat shock cognate 70 (HSC70) (32), a chaperone protein important for pTAU ubiquitination (33) and axon outgrowth (34). Overexpression of BAG2 and ΔBAG2 in SK-N-FI neuroblastoma cells followed by BAG2 immunoprecipitation via FLAG revealed that BAG2 but not ΔBAG2 binds a 70 kDa protein (Supplementary Figure S4E). Since BAG2 has been reported to bind HSC70 (32), we performed additional immunoprecipitation followed by immunoblotting with a HSC70 specific antibody. This revealed that BAG2 binds HSC70, whereas ΔBAG2 does not (Figure 3E and F).
ΔBAG2 impedes differentiation of neuroblastoma in response to retinoic acid
To investigate whether ΔBAG2 impinges on the capacity of neuroblastoma cells to differentiate, we treated CTRL (empty vector), BAG2 orΔBAG2 expressing SK-N-FI cells (Figure 3G) with retinoic acid (RA), a compound used in adjuvant therapy of high-risk neuroblastoma patients (17). After six days of RA treatment, cells transduced with either CTRL (Figure 3H–I, N) or BAG2 (Figure 3J–K, N) acquired neuronal morphology with long neurites. In contrast, ΔBAG2 transduced cells exhibited a weaker response to RA, with significantly less neurite formation (Figure 3L–N). To validate this effect we transduced SK-N-BE(1) neuroblastoma cells with a doxycycline-inducible version of the ΔBAG2 fusion genes (Supplementary Figure S4F). Upon doxycycline induction, ΔBAG2 expressing cells exhibited a reduced capacity to respond to RA (Supplementary Figure S4G–K).
Fusion transcripts with canonical splicing motifs are enriched in neuroblastoma and elevated expression of spliceosome factors is a strong predictor of high-risk tumors
Previously it has been suggested that cis-splicing between adjacent genes can generate fusion transcripts in prostate cancer (7,10). To elucidate whether there was a correlation between distance and frequency we plotted the distance between 5′ and 3′ of the fusion junction in the intrachromosomal fusion transcript versus the fusion frequency in the NB172 and NB-v data sets (Figure 4A). Fusions occurring at high frequency in both data sets were enriched between 1 and 100 kb, whereas transcripts separated by more than 100 kb occurred at lower frequencies and almost exclusively in the larger NB172 data set (Figure 4A). The genomic proximity of identified transcripts suggests that these fusions could be a result of cis-splicing (7,12). Inspection of nucleotide sequences located 5′ and 3′ of the fusion junctions for canonical splicing donors (GT) and acceptors (AG) revealed that 81.9% carried GT and AG at the 5′ and 3′ fusion sites (GT*AG) (Figure 4B). Notably, 87% of intra-chromosomal fusions had GT*AG at the fusion junction whereas only 45.9% of inter-chromosomal fusions contained canonical splicing sites at the junctions. Thus, in total there was ∼3 times more intrachromosomal fusions with canonical splicing sites than expected by chance in neuroblastoma, (P-value < 0.0001, chi-square test with Yate's correction). This pattern was unique to neuroblastoma as fusions detected in normal adrenal gland, osteosarcoma and rhabdoid tumor exhibited no enrichment of intrachromosomal fusions with canonical splicing sites (Figure 4B). Aberrant RNA splicing has been suggested as a driving event for several cancers and mutations in genes coding for components of the spliceosome have been identified in several types of tumors (35). Notably, in MYC-driven breast cancer, it has been shown that spliceosome is a therapeutic vulnerability (4), whereas in small cell lung cancer spliceosome inhibition was reported to be effective regardless of MYC status (36). We compared expression levels of genes of the KEGG, ‘Spliceosome’ gene category between neuroblastoma and normal adrenal gland. Out of 134 genes 46 had significantly higher expression levels in the neuroblastoma, whereas only three had lower levels of expression (Figure 4C). To investigate whether there were clinically relevant differences in expression of spliceosome genes between low- and high-risk neuroblastoma we performed k-means analysis of the 498-SEQC neuroblastoma cohort (Figure 4D). Together with previous observations (37) our analysis elucidates how the differential expression of spliceosome factors clearly identifies tumors of different clinical outcome, with high expression levels of splicing factors predicting high-risk tumors with bleak clinical outcome and substantially shorter overall survival (Figure 4D and E). The group of genes belonging to the KEGG ‘Spliceosome ‘category able to predict clinical outcome (Figure 4D and E) are listed in Supplementary Table S15. To investigate whether this association was dependent on MYCN status, we redid the k-means analysis on non-MYCN amplified tumors within the same dataset. After removal of all MYCN-amplified tumors there was a slightly reduced albeit still highly significant correlation between high-risk neuroblastoma, worse outcome and high expression levels of spliceosome factors (Supplementary Figure S5A and B). To elucidate whether the number of identified fusions were correlated with spliceosome factor expression levels we divided the NB172 neuroblastoma cohort into four bins according to the number of detected unique fusion transcripts/tumor. Analysis of differentially expressed genes between bin #4, containing the highest number of fusions, and bin #1 with the lowest number of fusions, was performed. Among the differentially expressed genes, 32 belonged to the KEGG ‘Spliceosome’ category and of these 25 exhibited significantly higher expression levels in Bin #4 whereas seven were expressed at higher levels in Bin# 1 (Figure 4F). To further understand whether this correlation was also present in Bin #2 and Bin #3, we plotted the average expression of each of the 32 differentially expressed KEGG ‘Spliceosome’ factors in each bin. This analysis showed that there was a continuous trend with increased expression of KEGG ‘Spliceosome’ factors with increased numbers of fusions (Figure 4G). Reflecting the k-means analysis (Figure 4D and E) there was a correlation between high levels of these spliceosome factors and high-risk tumors (Figure 4H). Furthermore, inspection of previous genome sequencing studies of neuroblastoma patients revealed mutations in several spliceosome factors in primary tumors and de novo mutations in spliceosome factors occurred in relapsed tumors (2,3,38,39) (summarized in Supplementary Table S16). In tumors wherein the ZNF451-BAG2 fusion had been detected, three tumors also had mutations in spliceosome factors (Supplementary Table S17). There was however no differentially expressed spliceosome genes in the ZNF451-BAG2 expressing tumors in comparison to the ZNF451-BAG2 negative tumors (Supplementary Table S18).
Spliceosome inhibition impedes expression of fusion transcripts, induces neuroblastoma apoptosis and inhibits growth of neuroblastoma xenografts
To investigate whether inhibition of spliceosome activity would selectively impede the generation of fusion transcripts but not their wild type cognates, we treated neuroblastoma cells (LAN-1 and SK-N-BE(1)) with the spliceosome inhibitor, Pladienolide B, targeting SF3B1 and PHF5, two key components of the splicing machinery (40,41). Upon treatment, there was a loss of expression for a majority of selected and sequenced high frequency fusion transcripts whereas expression of wild type genes constituting the fusions were largely unaffected at these concentrations (Figure 5A, B and Supplementary Figure S6A). Furthermore, in both cell lines there was a specific reduction of the ΔBAG2 protein upon Pladienolide B treatment (Supplementary Figure S6B and C). To substantiate the impact of spliceosome inhibition we also treated LAN-1 cells with SRPIN340 (42), an inhibitor of SRPK1 which phosphorylates proteins containing serine-arginine-rich (SR) domains, a function necessary for proper splicing. SRPIN340 treatment also impeded the expression of fusion transcripts in LAN-1 cells, however not as potent as Pladienolide B (Supplementary Figure S6D). In order to ensure that this spliceosome inhibitory effect was not just a reflection of different initial expression levels of wild type genes we exploited the VPS45 and LMO1 wild type transcripts that both had a reduction in expression upon Pladienolide B treatment. If reduced expression of transcripts was a mere reflection of initial expression levels it would be expected that the genes affected by Pladienolide B treatment to be among the lowliest expressed genes. This is however not the case for VPS45 nor LMO1 which both are expressed at higher levels than affected transcripts (Supplementary Figure S6E). To elucidate if selective loss of fusion transcripts upon spliceosome inhibition was associated with increased apoptosis we treated the neuroblastoma cell lines LAN-1, SK-N-BE(1) and SK-N-AS with increasing concentrations of Pladienolide B. As controls we utilized a panel of transformed and non-transformed human cells consisting of U87 (glioma), U2OS (osteosarcoma) and NHA (normal human astrocytes). Already at 5nM the neuroblastoma cell lines exhibited increased levels of cleaved caspase-3 and cleaved PARP, whereas control cells showed no signs of cell death. At 10nM cell death in both neuroblastoma cell lines was further increased but control cells were still non-responsive (Figure 6A). As with Pladienolide B, albeit not as efficiently, SRPIN340 selectively induced apoptosis in neuroblastoma cells, but not in the other cell types tested (Figure 6B). To test whether inhibition of the spliceosome could affect neuroblastoma growth in vivo we transplanted LAN-1 cells into the flanks of nude mice. When xenografts had reached a volume of ∼200 mm3 we initiated treatment with Pladienolide B. At the endpoint, Pladienolide B treated tumors exhibited significantly reduced growth in comparison to vehicle treated tumors (Figure 6C and D). To test if systemic delivery of Pladienolide B also selectively affected fusion transcript levels, we generated cDNA from the dissected tumors and performed PCR with primers specific for selected wild type transcripts and fusion transcripts. Partly reflecting the in vitro effects (Figure 5A), the levels of several fusion transcripts, among them ZNF451-BAG2, were reduced in the treated animals whereas the wild type transcripts were largely unaffected (Supplementary Figure S6F). Despite a correlation between selectively reduced fusion levels and apoptosis or abrogated xenograft growth upon spliceosome inhibition, our data cannot conclusively infer a causal link between these effects as they could represent parallel but independent events.
Figure 5.
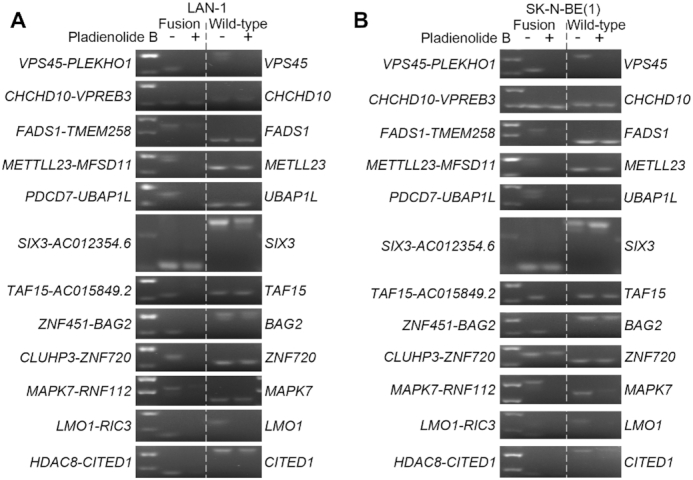
Pharmaceutical inhibition of spliceosome activity selectively reduces expression of fusion transcripts in neuroblastoma cells. (A, B) Splicing-dependent generation of several high frequent fusion transcripts in LAN-1 (A) and SK-N-BE(1) (B) neuroblastoma cells. SK-N-BE(1) and LAN1 cells were treated with splicing inhibitor Pladienolide B at 100 nM (SK-N-BE(1)) and 5–50 nM (LAN-1) for 6 h; RNA was isolated from harvested cells and reversely transcribed to cDNA; RT-PCR were performed with primers spanning the fusion junctions (fusion transcript) or exon–exon boundary (wild-type cognate).
DISCUSSION
Our analysis revealed that high expression levels of splicing associated factors is a distinguishing feature of high-risk neuroblastoma, representing a strong predictor of poor clinical outcome. Furthermore, pharmacological inhibition of the spliceosome selectively induced apoptosis in neuroblastoma cells and abrogated tumor growth in vivo. Our study shows that a high frequency of neuroblastoma specific fusion transcripts could constitute an overlooked process through which altered transcripts are generated. It has been shown that upon cellular stress (e.g. viral infection, replicative or osmotic stress and mutational events) transcriptional termination can be blocked, increasing the probability of generating ‘downstream of genes’- transcripts (15). Thus, a proportion of fusions could represent passengers that occur as a response to cellular stress combined with neuroblastoma associated events such as gain or loss of chromosomal regions (e.g. 17q and 2p). In contrast to such passengers, certain fusions could be early events preceding and promoting other transformative events. Pharmacologic inhibition of splicing selectively repressed expression of several top frequent fusion transcripts but not of their wild-type cognates and there was a high frequency of splicing donor/acceptor sites in neuroblastoma specific fusions but not in fusions detected in normal adrenal gland, osteosarcoma or rhabdoid tumors. This pattern implies that a substantial proportion of the detected fusions are of the same cis-splicing type as reported in prostate cancer (7). Regardless of the mechanisms underlying the generation of these fusions, they are not dependent on amino acid changing mutations but can still provide a source of modified gene products with a potential to promote neuroblastoma but also reveal novel drug targets. Given the low frequency of recurrent mutations in neuroblastoma, such a pool of altered gene products could indeed be relevant for tumor pathophysiology. A background of expressed fusion transcripts could potentially augment the effect of oncogenic drivers. Notably, our analysis shows that when established drivers of neuroblastoma (e.g. MYCN and LMO1) are part of the fusion transcripts the expression levels of wild type cognates are elevated. In addition, a panel of neuroblastoma specific fusions occurring at high frequency could serve as biomarkers for diagnosis and the presence of risk-specific fusions could sub-divide neuroblastoma patients for precision therapy. A previous study indeed shows how the SLC45A3-ELK4 read-through fusion transcript is elevated in prostate cancer tissue, is androgen-regulated and can be detected in an non-invasive assay from biopsies of men at risk of having prostate cancer (13).
One concern with previously reported fusions is that relatively few cases have been independently validated. In 2015, only 3% of fusions identified by deep sequencing could be reproducibly detected (9). Arguably, the ‘non-genomic’ characteristic of this type of intrachromosomal fusions potentially augments the detection of false positives. The risk that a proportion of fusion transcripts constitute false positives as the result of spurious transcription or of sequencing errors is reduced by our crosswise analysis with other tumors and healthy tissues. There is a clear enrichment of common fusions unique for the neuroblastoma data sets that do not appear in any of the other tumors nor in adult normal adrenal glands. Neuroblastoma specific fusions are enriched for genes located at chromosomal regions (2p or 17q) which are commonly gained in high-risk neuroblastoma. PCR with primers spanning the fusion points and selective loss of several fusion transcripts upon spliceosome inhibition provide support, independent of bioinformatic tools, that splicing dependent fusion transcripts indeed are present. Interestingly, in a few of the examined juvenile adrenal glands there was low but detectable expression of certain fusion transcripts. Thus, our data suggest that certain fusion transcripts can be lowly expressed at the most common site of neuroblastoma and then be enriched for upon tumor formation. Tumor specific distribution of fusions is also evident in the osteosarcoma tumors where the TP53 tumor suppressor is a fusion partner in an disproportional amount of the detected fusions, reflecting the importance of inactivating mutations in TP53 for this disease (28). It has previously been reported that the presence of short homology sequences (SHS) can generate false RNA-chimeras due to template switching during the reverse transcriptase reaction (21). Such a mechanism would presumably generate random fusions between transcripts containing matching SHSs with no preference for any particular chromosomal location nor for intrachromosomal vs interchromosomal fusion transcripts. The non-random enrichment of fusions at chromosomal locations that mirror the disease as well as the enrichment of intrachromosomal fusions between closely located genes suggests that reverse transcriptase induced template switching is not the cause of these fusions. Moreover, our study shows that an altered protein, ΔBAG2, with novel properties can be generated as a consequence of an intrachromosomal RNA-fusion. Although, the altered ΔBAG2 protein influences the response to a drug (RA) commonly used to treat high-risk neuroblastoma patients, further studies are required to evaluate the full function of this particular fusion product in neuroblastoma.
Our study provides novel insights of the role of splicing and splicing-dependent RNA-fusions in neuroblastoma, but additional studies are necessary to determine whether the selective loss of RNA-fusion expression and impeded tumor growth upon spliceosome inhibition are causally linked. In addition, the functional role of specific fusions as well as the total impact of collected fusions or distinct combinations of fusions remain to be investigated.
Taken together, our data show that high expression levels of splicing associated factors are characteristic of high-risk neuroblastoma and that the spliceosome can serve as a target for pharmacological intervention. In addition, splicing dependent RNA-fusions provide a mechanism, distinct from amino acid changing mutations, through which altered gene products can be generated. This could potentially compensate for the lack of recurrent mutations in neuroblastoma. Enrichment of fusions at chromosomal regions associated with high-risk neuroblastoma and the specificity of expression imply that this type of altered gene products is relevant for the biology of the disease and could serve as potential novel drug targets and diagnostic markers.
DATA AVAILABILITY
Data needed to evaluate the conclusions in the paper are present in the paper and/or the Supplementary Materials. RNA-sequences from neuroblastoma available at: National Cancer Institute TARGET, dbGap Study Accession: phs000218.v16.p6. RNA-sequences from rhabdoid tumors available at: National Cancer Institute TARGET, dbGap Study Accession: phs000470.v17.p7. RNA-sequences from osteosarcoma available at: National Cancer Institute TARGET, dbGap Study Accession: phs000468.v17.p7. Sequenced neuroblastoma cell lines, GEO#: GSE145075.
Supplementary Material
ACKNOWLEDGEMENTS
We thank Johan Ericson and members of the Holmberg and Schlisio groups for valuable comments.
Contributor Information
Yao Shi, Department of Cell and Molecular Biology, Karolinska Institutet, Solnavägen 9, SE-171 65 Stockholm, Sweden.
Juan Yuan, Department of Cell and Molecular Biology, Karolinska Institutet, Solnavägen 9, SE-171 65 Stockholm, Sweden.
Vilma Rraklli, Department of Cell and Molecular Biology, Karolinska Institutet, Solnavägen 9, SE-171 65 Stockholm, Sweden.
Eva Maxymovitz, Department of Cell and Molecular Biology, Karolinska Institutet, Solnavägen 9, SE-171 65 Stockholm, Sweden.
Miriam Cipullo, Department of Medical Biochemistry and Biophysics, Karolinska Institutet, Solnavägen 9, SE-171-65 Solna, Sweden.
Mingzhi Liu, Department of Cell and Molecular Biology, Karolinska Institutet, Solnavägen 9, SE-171 65 Stockholm, Sweden.
Shuijie Li, Department of Microbiology, Tumor- and Cellbiology, Karolinska Institutet, Solnavägen 9, SE-171 65 Solna, Sweden.
Isabelle Westerlund, Department of Cell and Molecular Biology, Karolinska Institutet, Solnavägen 9, SE-171 65 Stockholm, Sweden.
Oscar C Bedoya-Reina, Department of Microbiology, Tumor- and Cellbiology, Karolinska Institutet, Solnavägen 9, SE-171 65 Solna, Sweden.
Petra Bullova, Department of Microbiology, Tumor- and Cellbiology, Karolinska Institutet, Solnavägen 9, SE-171 65 Solna, Sweden.
Joanna Rorbach, Department of Medical Biochemistry and Biophysics, Karolinska Institutet, Solnavägen 9, SE-171-65 Solna, Sweden.
C Christofer Juhlin, Department of Oncology-Pathology, Karolinska Institutet, Cancer Center Karolinska (CCK), Karolinska University Hospital, SE-171 76 Stockholm, Sweden.
Adam Stenman, Department of Oncology-Pathology, Karolinska Institutet, Cancer Center Karolinska (CCK), Karolinska University Hospital, SE-171 76 Stockholm, Sweden.
Catharina Larsson, Department of Oncology-Pathology, Karolinska Institutet, Cancer Center Karolinska (CCK), Karolinska University Hospital, SE-171 76 Stockholm, Sweden.
Per Kogner, Department of Women's and Children's Health, Karolinska Institutet, Karolinska University Hospital, SE-171 76 Stockholm, Sweden.
Maureen J O’Sullivan, Department of Histopathology, Our Lady's Children's Hospital, Dublin, Ireland; Trinity Translational Medicine Institute, Trinity College, Dublin, Ireland.
Susanne Schlisio, Department of Microbiology, Tumor- and Cellbiology, Karolinska Institutet, Solnavägen 9, SE-171 65 Solna, Sweden.
Johan Holmberg, Department of Cell and Molecular Biology, Karolinska Institutet, Solnavägen 9, SE-171 65 Stockholm, Sweden.
SUPPLEMENTARY DATA
Supplementary Data are available at NAR Online.
FUNDING
Johan Holmberg lab was supported by the Swedish Children Cancer Foundation, the Swedish Cancer Foundation, Knut and Alice Wallenberg Foundation, Swedish Research Council (to V.R.); Strategic Research Programme in Cancer (StratCan, SFO); Swedish Brain Foundation. Funding for open access charge: Swedish Research Council.
Conflict of interest statement. None declared.
REFERENCES
- 1. Brodeur G.M. Neuroblastoma: biological insights into a clinical enigma. Nat. Rev. Cancer. 2003; 3:203–216. [DOI] [PubMed] [Google Scholar]
- 2. Pugh T.J., Morozova O., Attiyeh E.F., Asgharzadeh S., Wei J.S., Auclair D., Carter S.L., Cibulskis K., Hanna M., Kiezun A.et al.. The genetic landscape of high-risk neuroblastoma. Nat. Genet. 2013; 45:279–284. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Molenaar J.J., Koster J., Zwijnenburg D.A., van Sluis P., Valentijn L.J., van der Ploeg I., Hamdi M., van Nes J., Westerman B.A., van Arkel J.et al.. Sequencing of neuroblastoma identifies chromothripsis and defects in neuritogenesis genes. Nature. 2012; 483:589–593. [DOI] [PubMed] [Google Scholar]
- 4. Hsu T.Y., Simon L.M., Neill N.J., Marcotte R., Sayad A., Bland C.S., Echeverria G.V., Sun T., Kurley S.J., Tyagi S.et al.. The spliceosome is a therapeutic vulnerability in MYC-driven cancer. Nature. 2015; 525:384–388. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Yoshida K., Ogawa S.. Splicing factor mutations and cancer. Wiley Interdiscip. Rev. RNA. 2014; 5:445–459. [DOI] [PubMed] [Google Scholar]
- 6. Chen J., Hackett C.S., Zhang S., Song Y.K., Bell R.J., Molinaro A.M., Quigley D.A., Balmain A., Song J.S., Costello J.F.et al.. The genetics of splicing in neuroblastoma. Cancer Discov. 2015; 5:380–395. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Qin F., Song Z., Babiceanu M., Song Y., Facemire L., Singh R., Adli M., Li H.. Discovery of CTCF-sensitive cis-spliced fusion RNAs between adjacent genes in human prostate cells. PLoS Genet. 2015; 11:e1005001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Akiva P., Toporik A., Edelheit S., Peretz Y., Diber A., Shemesh R., Novik A., Sorek R.. Transcription-mediated gene fusion in the human genome. Genome Res. 2006; 16:30–36. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Mertens F., Johansson B., Fioretos T., Mitelman F.. The emerging complexity of gene fusions in cancer. Nat. Rev. Cancer. 2015; 15:371–381. [DOI] [PubMed] [Google Scholar]
- 10. Zhang Y., Gong M., Yuan H., Park H.G., Frierson H.F., Li H.. Chimeric transcript generated by cis-splicing of adjacent genes regulates prostate cancer cell proliferation. Cancer Discov. 2012; 2:598–607. [DOI] [PubMed] [Google Scholar]
- 11. Santo E.E., Ebus M.E., Koster J., Schulte J.H., Lakeman A., van Sluis P., Vermeulen J., Gisselsson D., Ora I., Lindner S.et al.. Oncogenic activation of FOXR1 by 11q23 intrachromosomal deletion-fusions in neuroblastoma. Oncogene. 2012; 31:1571–1581. [DOI] [PubMed] [Google Scholar]
- 12. Jia Y., Xie Z., Li H.. Intergenically spliced chimeric RNAs in cancer. Trends Cancer. 2016; 2:475–484. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Rickman D.S., Pflueger D., Moss B., VanDoren V.E., Chen C.X., de la Taille A., Kuefer R., Tewari A.K., Setlur S.R., Demichelis F.et al.. SLC45A3-ELK4 is a novel and frequent erythroblast transformation-specific fusion transcript in prostate cancer. Cancer Res. 2009; 69:2734–2738. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Maher C.A., Kumar-Sinha C., Cao X., Kalyana-Sundaram S., Han B., Jing X., Sam L., Barrette T., Palanisamy N., Chinnaiyan A.M.. Transcriptome sequencing to detect gene fusions in cancer. Nature. 2009; 458:97–101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Proudfoot N.J. Transcriptional termination in mammals: stopping the RNA polymerase II juggernaut. Science. 2016; 352:aad9926. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Babiceanu M., Qin F., Xie Z., Jia Y., Lopez K., Janus N., Facemire L., Kumar S., Pang Y., Qi Y.et al.. Recurrent chimeric fusion RNAs in non-cancer tissues and cells. Nucleic Acids Res. 2016; 44:2859–2872. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Matthay K.K., Reynolds C.P., Seeger R.C., Shimada H., Adkins E.S., Haas-Kogan D., Gerbing R.B., London W.B., Villablanca J.G.. Long-term results for children with high-risk neuroblastoma treated on a randomized trial of myeloablative therapy followed by 13-cis-retinoic acid: a children's oncology group study. J. Clin. Oncol. 2009; 27:1007–1013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Li S., Fell S.M., Surova O., Smedler E., Wallis K., Chen Z.X., Hellman U., Johnsen J.I., Martinsson T., Kenchappa R.S.et al.. The 1p36 tumor suppressor KIF 1Bbeta is required for calcineurin activation, controlling mitochondrial fission and apoptosis. Dev. Cell. 2016; 36:164–178. [DOI] [PubMed] [Google Scholar]
- 19. Nicorici D., Şatalan M., Edgren H., Kangaspeska S., Murumägi A., Kallioniemi O., Virtanen S., Kilkku O.. FusionCatcher – a tool for finding somatic fusion genes in paired-end RNA-sequencing data. 2014; bioRxiv doi:19 November 2014, preprint: not peer reviewed 10.1101/011650. [DOI]
- 20. Westerlund I., Shi Y., Toskas K., Fell S.M., Li S., Surova O., Sodersten E., Kogner P., Nyman U., Schlisio S.et al.. Combined epigenetic and differentiation-based treatment inhibits neuroblastoma tumor growth and links HIF2alpha to tumor suppression. Proc. Natl. Acad. Sci. U.S.A. 2017; 114:E6137–E6146. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Xie B., Yang W., Ouyang Y., Chen L., Jiang H., Liao Y., Liao D.J.. Two RNAs or DNAs May artificially fuse together at a short homologous sequence (SHS) during reverse transcription or polymerase chain reactions, and thus reporting an shs-containing chimeric RNA requires extra caution. PLoS One. 2016; 11:e0154855. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Khan F.H., Pandian V., Ramraj S., Natarajan M., Aravindan S., Herman T.S., Aravindan N.. Acquired genetic alterations in tumor cells dictate the development of high-risk neuroblastoma and clinical outcomes. BMC Cancer. 2015; 15:514. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Hungate E.A., Applebaum M.A., Skol A.D., Vaksman Z., Diamond M., McDaniel L., Volchenboum S.L., Stranger B.E., Maris J.M., Diskin S.J.et al.. Evaluation of genetic predisposition for MYCN-Amplified neuroblastoma. J. Natl. Cancer Inst. 2017; 109:djx093. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Lee S.H., Kim J.S., Zheng S., Huse J.T., Bae J.S., Lee J.W., Yoo K.H., Koo H.H., Kyung S., Park W.Y.et al.. ARID1B alterations identify aggressive tumors in neuroblastoma. Oncotarget. 2017; 8:45943–45950. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Consortium, G.T. The Genotype-Tissue Expression (GTEx) project. Nat. Genet. 2013; 45:580–585. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Kim S.Y., Volsky D.J.. PAGE: parametric analysis of gene set enrichment. BMC Bioinformatics. 2005; 6:144. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Koster J. R2: Genomics Analysis and Visualization Platform. 2008; http://r2.amc.nl.
- 28. Martin J.W., Squire J.A., Zielenska M.. The genetics of osteosarcoma. Sarcoma. 2012; 2012:627254. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Haas B.J., Dobin A., Li B., Stransky N., Pochet N., Regev A.. Accuracy assessment of fusion transcript detection via read-mapping and de novo fusion transcript assembly-based methods. Genome Biol. 2019; 20:213. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Carrettiero D.C., Hernandez I., Neveu P., Papagiannakopoulos T., Kosik K.S.. The cochaperone BAG2 sweeps paired helical Filament-Insoluble tau from the microtubule. J. Neurosci. 2009; 29:2151–2161. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Santiago F.E., Almeida M.C., Carrettiero D.C.. BAG2 is repressed by NF-kappaB signaling, and its overexpression is sufficient to shift Abeta1-42 from neurotrophic to neurotoxic in undifferentiated SH-SY5Y neuroblastoma. J. Mol. Neurosci. 2015; 57:83–89. [DOI] [PubMed] [Google Scholar]
- 32. Qin L., Guo J., Zheng Q., Zhang H.. BAG2 structure, function and involvement in disease. Cell Mol. Biol. Lett. 2016; 21:18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Shimura H., Schwartz D., Gygi S.P., Kosik K.S.. CHIP-Hsc70 complex ubiquitinates phosphorylated tau and enhances cell survival. J. Biol. Chem. 2004; 279:4869–4876. [DOI] [PubMed] [Google Scholar]
- 34. DeGeer J., Kaplan A., Mattar P., Morabito M., Stochaj U., Kennedy T.E., Debant A., Cayouette M., Fournier A.E., Lamarche-Vane N.. Hsc70 chaperone activity underlies Trio GEF function in axon growth and guidance induced by netrin-1. J. Cell Biol. 2015; 210:817–832. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Wang B.D., Lee N.H.. Aberrant RNA splicing in cancer and drug resistance. Cancers (Basel). 2018; 10:458. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Suda K., Rozeboom L., Yu H., Ellison K., Rivard C.J., Mitsudomi T., Hirsch F.R.. Potential effect of spliceosome inhibition in small cell lung cancer irrespective of the MYC status. PLoS One. 2017; 12:e0172209. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Guo X., Chen Q.R., Song Y.K., Wei J.S., Khan J.. Exon array analysis reveals neuroblastoma tumors have distinct alternative splicing patterns according to stage and MYCN amplification status. BMC Med. Genet. 2011; 4:35. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Eleveld T.F., Oldridge D.A., Bernard V., Koster J., Daage L.C., Diskin S.J., Schild L., Bentahar N.B., Bellini A., Chicard M.et al.. Relapsed neuroblastomas show frequent RAS-MAPK pathway mutations. Nat. Genet. 2015; 47:864–871. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Sausen M., Leary R.J., Jones S., Wu J., Reynolds C.P., Liu X., Blackford A., Parmigiani G., Diaz L.A. Jr, Papadopoulos N.et al.. Integrated genomic analyses identify ARID1A and ARID1B alterations in the childhood cancer neuroblastoma. Nat. Genet. 2013; 45:12–17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Mizui Y., Sakai T., Iwata M., Uenaka T., Okamoto K., Shimizu H., Yamori T., Yoshimatsu K., Asada M.. Pladienolides, new substances from culture of Streptomyces platensis Mer-11107. III. In vitro and in vivo antitumor activities. J. Antibiot. (Tokyo). 2004; 57:188–196. [DOI] [PubMed] [Google Scholar]
- 41. Teng T., Tsai J.H., Puyang X., Seiler M., Peng S., Prajapati S., Aird D., Buonamici S., Caleb B., Chan B.et al.. Splicing modulators act at the branch point adenosine binding pocket defined by the PHF5A-SF3b complex. Nat. Commun. 2017; 8:15522. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Fukuhara T., Hosoya T., Shimizu S., Sumi K., Oshiro T., Yoshinaka Y., Suzuki M., Yamamoto N., Herzenberg L.A., Herzenberg L.A.et al.. Utilization of host SR protein kinases and RNA-splicing machinery during viral replication. Proc. Natl. Acad. Sci. U.S.A. 2006; 103:11329–11333. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
Data needed to evaluate the conclusions in the paper are present in the paper and/or the Supplementary Materials. RNA-sequences from neuroblastoma available at: National Cancer Institute TARGET, dbGap Study Accession: phs000218.v16.p6. RNA-sequences from rhabdoid tumors available at: National Cancer Institute TARGET, dbGap Study Accession: phs000470.v17.p7. RNA-sequences from osteosarcoma available at: National Cancer Institute TARGET, dbGap Study Accession: phs000468.v17.p7. Sequenced neuroblastoma cell lines, GEO#: GSE145075.