

Abstract
Osteosarcoma is a prominent bone cancer that typically affects adolescents or people in late adulthood. Early recognition of this disease relies on imaging technologies such as x-ray radiography to detect tumor size and location. This paper aims to differentiate osteosarcoma from benign tumors by analyzing both imaging and RNA-seq data through a combination of image processing and machine learning. In experimental results, the proposed method achieved an Area Under the Receiver Operator Characteristic Curve (AUC) of 0.7272 in three-fold cross-validation, and an AUC of 0.9015 using leave-one-out cross-validation.
I. INTRODUCTION
More than 1,900 cancer-related deaths occur in children and adolescents every year in the United States. From 2007–2010, bone cancers accounted for 5% of cancer mortality in children less than 15 years, which increased to 15% in those aged 15–19 years [1]. Osteosarcoma is the most common primary bone malignancy; it has a bimodal distribution in age, with the first peak occurring in adolescence, and the second in older adulthood. [2]. A primary method of tumor diagnosis is x-ray radiography, which is used to locate the tumor and assess its qualities. Some typical features of osteosarcoma tumors in x-ray include a sun-burst appearance, periosteal lifting with the formation of Codman’s triangle, and the creation of new bone in soft tissue, though there are variations depending on the specific type of osteosarcoma [3]. A timely diagnosis is needed in order to increase the chance of patient survival and reduce the risk of metastasis.
Previous work exists related to incorporating multimodal data for osteosarcoma classification. In Shen et al. [4], x-ray imaging features coupled with metabolomic data were used to create a random forest (RF) classification model that achieved an Area Under the Receiver Operator Characteristic Curve (AUC) of 0.94 (standard deviation 0.054). An advantage of RF is that it does not require the number of features to be smaller than the number of samples used for training. Studies such as [5], [6], have used RFs successfully in such instances. Wu et al. [5] compared the performance of RF with other classifiers in discriminating normal from ovarian cancer serum samples with mass spectrometry data, finding that RF produced the least variation in prediction error when compared to other tree-based classifiers. Lee et al. [6] similarly found RF to be the strongest tree-based classifier when using microarray data as model input.
In this paper, we propose an automated method for classification of benign and osteosarcoma tumors using RNA-seq and x-ray images. First, feature extraction methods to obtain salient information from raw RNA-seq data and x-ray images are described. Given the high number of features obtained, chosen methods to reduce the number of features are introduced. Lastly, the performance of the proposed method on an x-ray image and RNA-seq dataset collected by collaborators at Shanghai Jiao Tong University (SJTU) is discussed. The experimental results suggest that automated classification of benign and malignant tumors can be used to enhance computer-aided diagnosis.
II. MATERIALS AND METHODS
A. Dataset
Demographics are provided in Table I. All of the samples contain both RNA-seq data and plain x-ray images by collaborators at SJTU. The RNA sequencing libraries were sequenced in 200bp paired-end mode using the Illumina HiSeq system (Illumina, San Diego, CA, USA). Sample collection for this study was conducted following a Shanghai General Hospital IRB-approved protocol after all participants signed written informed consent. A schematic diagram depicting the overall classification system in presented in Fig. 1.
Table I:
Characteristics of Patients
| Tumor Type | Mean Age (SD) | Sex |
|---|---|---|
| Benign | 40.33 (12.48) | 5 Male, 1 Female |
| Osteosarcoma | 22.73 (16.14) | 4 Male, 7 Female |
Fig. 1:
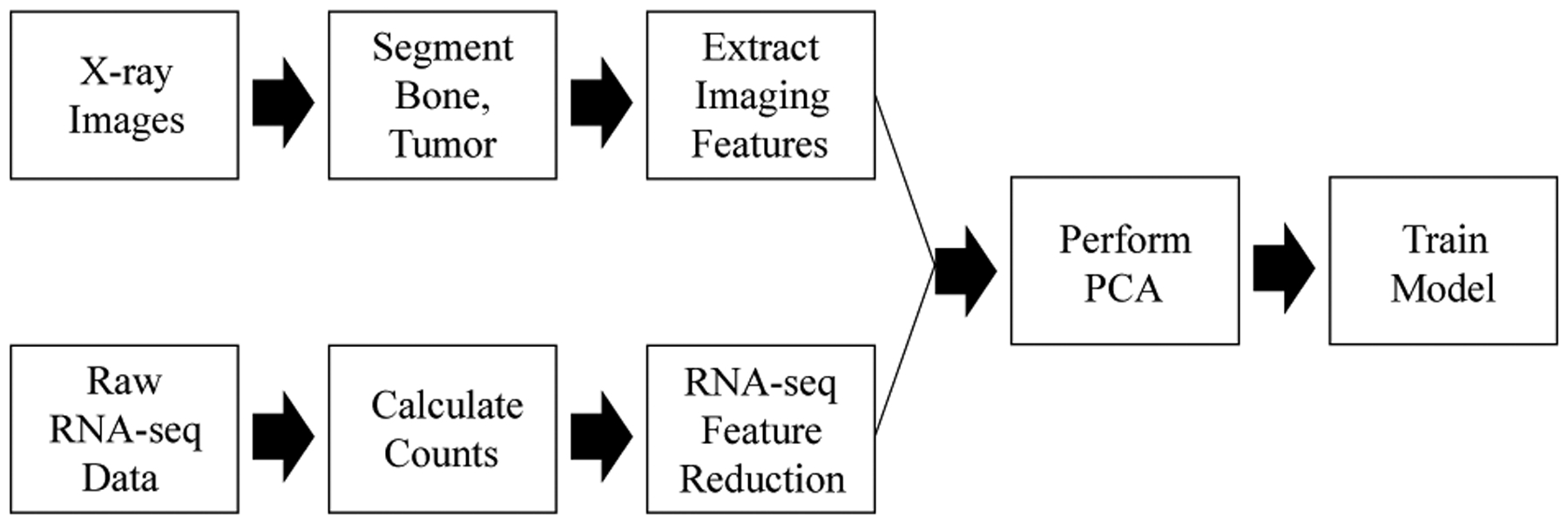
A schematic diagram of the proposed classification system.
B. Image Processing
1). Segmentation:
Imaging data consisted of plain x-ray DICOM images of the tumor and surrounding bone. Images were processed following the methods defined in [4]. A semi-automated segmentation method, Graph Cut with Lazy Snapping [7], was used to segment images into tumor and affected bone regions before feature extraction. With Graph Cut, the user specifies foreground and background seeds to serve as hard constraints for the segmentation. Graph Cut then assigns labels to each node xi (such as 1 for foreground and 0 for background) to minimize the Gibbs Energy E(X) on a graph with respect to the user-defined hard constraints.
Before assigning foreground and background labels, the watershed algorithm pre-segments the graph into superpixels, resulting in a graph , where represents all the small regions, and represents all the arcs that connect them. The energy function E(X) to minimize is defined as
| (1) |
where E1(x), the likelihood energy, measures the cost of labeling node i as xi, and E2(xi,xj), the prior energy, the cost of labeling connected nodes i and j as xi and xj.
Fig. 2 shows the Graph Cut with Lazy Snapping method applied to an x-ray image. Fig. 2a is the original x-ray image of the tumor and bone region. Fig. 2b shows the user-defined foreground seeds (green dots) and background seeds (red dots) to select the tumor regions. Fig. 2c displays the foreground labels that minimize the Gibbs Energy. Fig. 2d is the final mask for the tumor region.
Fig. 2:
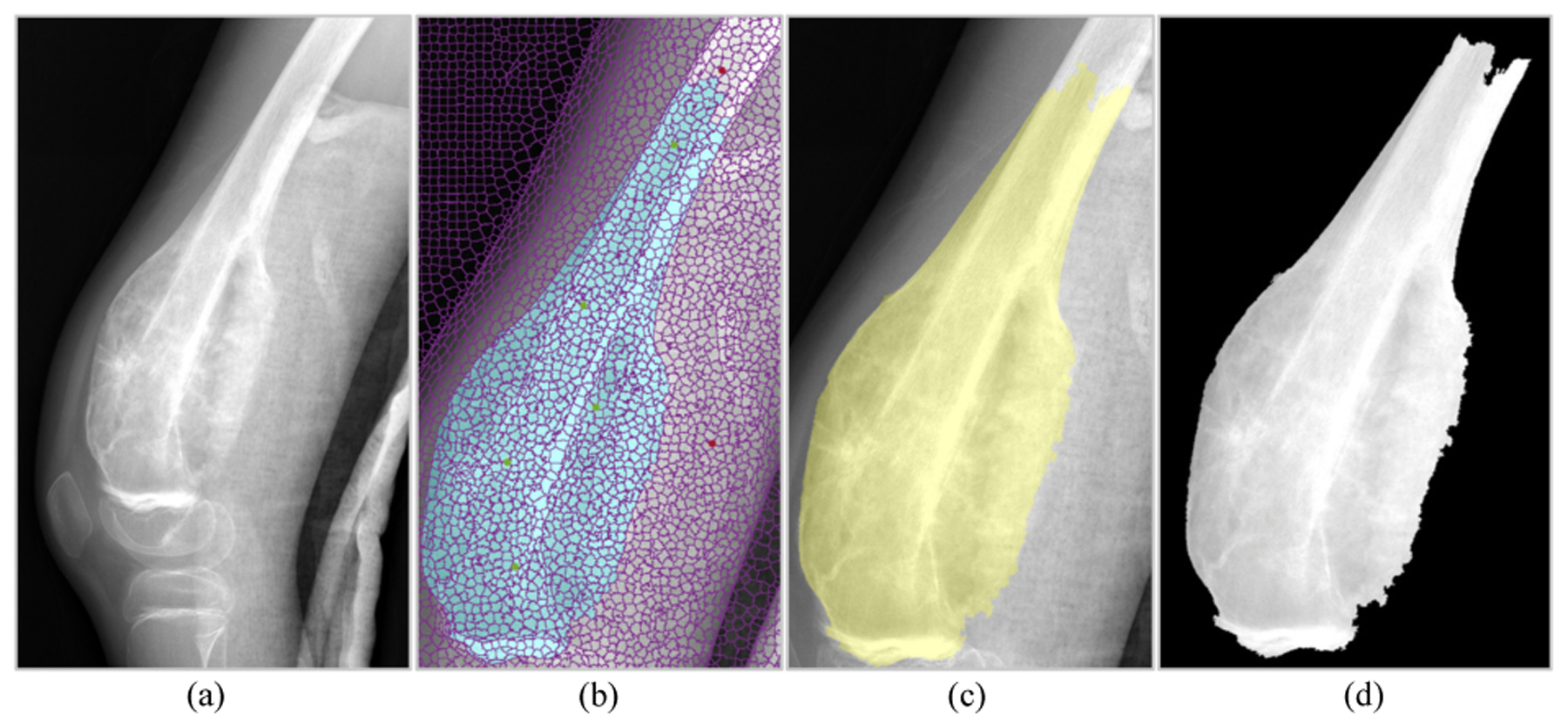
Graph Cut with Lazy Snapping applied to tumor segmentation. [4]
2). Feature Extraction:
The process for extracting features is detailed in previous work [4] and is illustrated in Fig. 3. There are features collected from both the bone and tumor segmentations (tumor border clarity, distance to joint), as well as from the tumor segmentation alone (texture features, morphological features). Table II lists all features extracted from the x-ray images.
Fig. 3:

The image processing techniques utilized for feature extraction.
Table II:
Features from Image Processing
| Morphological Features | Mean, Standard Deviation, Entropy, Kurtosis, Skewness, Convex Area, Eccentricity, Perimeter, Major Axis Length, Minor Axis Length, Solidity |
| Texture Features | Contrast, Correlation, Energy, Homogeneity from gray-level co-occurrence matrix; Mean, Standard Deviation, Kurtosis, Skewness, and Entropy from Approximation, Horizontal Detail, Vertical Detail, and Diagonal Detail Coefficients |
| Other Features | Distance to Joint, Tumor Border Clarity |
C. RNA-seq Processing
A summary of the steps taken to process RNA-seq data for analysis is presented in Fig. 4. First, the quality of all of the samples to be included in the analysis was verified (Section II-C.1). Then, data were aligned to the human reference genome, and raw counts for the aligned reads were obtained (Section II-C.2). After this process, the count data were normalized (Section II-C.3) and reduced (Section II-C.4) before being input to the RF model.
Fig. 4:
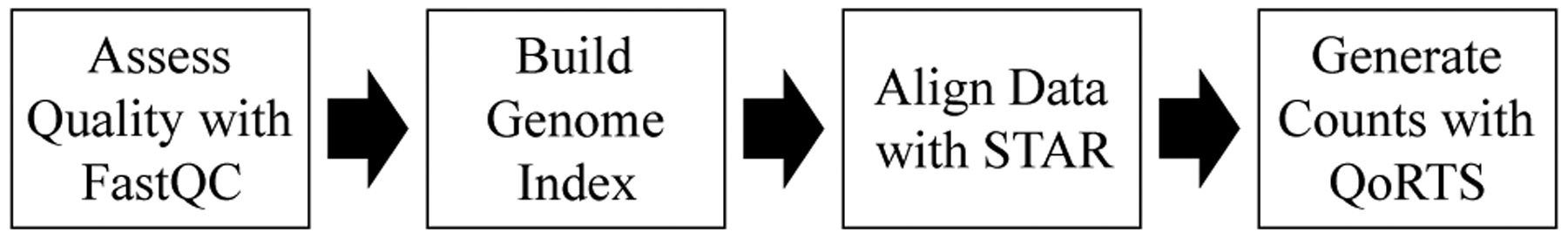
Processing RNA-seq data to obtain counts.
1). Data Quality Assessment:
Before aligning RNA-Seq data against the reference genome, FastQC version 0.11.8 [8] was used to assess the quality of the raw RNA-seq data. The raw FASTQ files were used as input to FastQC, which was run using default parameters. For each sample, FastQC generated an HTML report, showing scores and graphics that illustrated quality metrics such as per-base sequence quality, GC content, duplicate, and over-represented sequence. We examined these reports before proceeding to the alignment step. The overall sequence quality was deemed acceptable, and thus, no samples were removed from the analysis, nor was adapter trimming applied.
2). Genome Alignment and Generation of Count Data:
Using STAR [9] with default parameters, a genome index was generated from the current human reference genome (hg38) and its corresponding General Transfer Format (GTF) annotation file. The reference genome and GTF files were obtained from the UCSC website [10], using Gencode V29 [11]. STAR was then used to map the raw RNA-Seq reads against the indexed reference genome. The maximum number of multiple alignments allowed for a read was set to 20. The resultant BAM file was sorted by coordinate as required by QoRTs [12]. All samples achieved a ratio of uniquely mapped reads greater than 76%. The aligned RNA-Seq data were processed via the Java utility in QoRTs [12]. The processed data were then read into R using the QoRTs library to generate count files. These counts, after performing the normalization and feature reduction described in the following sections, serve as RNA-seq features for the classification model.
3). Normalization of RNA-seq Count Data:
With RNA-seq data, counts can vary from sample to sample, and should therefore be normalized. The normalization method used in this paper follows the example of [13], which relies on DESeq2’s [14] normalization method. DESeq2 takes raw counts as input, then uses a median ratio equation [14], [15] to normalize the counts across samples. For each sample, DESeq2 calculates the size factor, then divides the sample’s raw counts by its size factor. For n transcripts and m samples, the size factor for sample j is calculated as
| (2) |
with kij the raw count for transcript i in sample j. Any transcript having a count of zero was excluded from this calculation, following the method from DESeq2 [15], [16].
The training data were normalized using the size factors from (2), which were also used to derive the size factors for each sample j* in the test set, shown in (3).
| (3) |
In this manner, the test set is also normalized by the data in the training set. This approach is justified, for if the original equation from [15] was employed, i.e., using all of the data available for normalization, the training samples would be normalized by both themselves as well as the test samples, which would lead to data leakage. Likewise, the test samples would also be normalized by themselves, leading to overfitting and data leakage.
4). RNA-seq Feature Reduction:
Due to the large number of features (counts of transcripts) in our RNA-seq dataset, we produced a list of genes of interest. We limited our inclusion of RNA-seq transcript counts to those which could be associated with the genes of interest. A previous literature search identified 211 genes either whose upregulation is associated with tumorigenesis and metastasis, or whose downregulation is associated with tumorigenesis and metastasis. This feature reduction step was performed to reduce potential noise being added to the classification model, and to limit the RNA-seq features to those which could be biologically relevant.
D. Machine Learning
After performing image processing and selecting the RNA-seq features of interest, the two data types were combined into one dataset. In this manner, every observation consisted of one individual, containing both features from image processing and counts from transcripts of interest from RNA-seq.
To create the classification of osteosarcoma or benign tumor, RF [17] was used. In each model, the number of trees used was 50, the square root of the total number of features was selected for each decision split, and the minimum leaf size was 1. RF models were constructed using both three-fold cross-validation and leave-one-out cross-validation.
1). Three-Fold Cross-Validation:
For three-fold cross-validation (CV), three folds of observations were created, with each fold containing 2 benign observations and 3–4 of osteosarcoma. The observations in each fold were selected randomly, and there was no overlap among each of the folds. Two folds were used to train the RF model, and the third fold was used to test performance. This was repeated, utilizing each fold once as the test fold. Before data were input to the model, principal component analysis (PCA) was applied to the training folds to reduce the feature space. The test fold was scaled by the training folds, and the coefficients from PCA applied to the training folds were used to yield the principal components (PCs) for the test fold, in order to prevent data leakage.
2). Leave-One-Out Cross-Validation:
For this method, all observations except one were used as a training set for the RF model, and the last observation was used to test performance. This was repeated so that each observation was used once for testing. Before data were input to the model, PCA was applied to the training set to reduce the feature space. The test observation was scaled by the training data, and the coefficients from PCA applied to the training set were used to yield the PCs for the test set.
III. RESULTS & DISCUSSION
In this study, a RF model was created using features extracted from RNA-seq and x-ray image data to classify a given tumor as benign or osteosarcoma. A dataset was developed, containing imaging features as well as RNA-seq counts from transcripts associated with genes from a relevant literature search. Before training each model, PCA was performed to reduce the feature space of the imaging features combined with transcript counts in the training set. Three-fold and leave-one-out CV were used to assess performance. Performing three-fold CV with RF trained on only one PC achieved an AUC of 0.7272. The performance results of three-fold CV with one PC are displayed in Table III. Performing leave-one-out CV achieved AUC of 0.9015 when 2 PCs were used to train the model. The performance results are displayed in Table IV.
Table III:
Average Performance of the Random Forest Model with Three-Fold Cross-Validation
| PCs | AUC | F1 | Sensitivity | Specificity | Accuracy |
|---|---|---|---|---|---|
| 1 | 0.7272 | 0.8462 | 1 | 0.3333 | 0.9647 |
Table IV:
Average Performance of the Random Forest Model with Leave-One-Out Cross-Validation
| PCs | AUC | F1 | Sensitivity | Specificity | Accuracy |
|---|---|---|---|---|---|
| 1 | 0.4470 | 0.8148 | 1 | 0.1667 | 0.7059 |
| 2 | 0.9015 | 0.9091 | 0.9091 | 0.8333 | 0.8824 |
One major concern when performing this study was limiting the number of variables to train the model. Thousands of transcripts were aligned to the genome. Thus, PCA was used to capture the variance explained by those features, while limiting the number of features exposed to the model.
Increasing the number of PCs increases the amount of information being provided to the model, so it is expected that additional PCs lead to higher AUC, as shown in Table IV. However, performance in both three-fold and leave-one-out CV did not significantly improve after adding more PCs than those displayed, which may indicate that a large amount of information introduced is noise.
Limitations of this study include small sample size and class imbalance. The small number of observations in this study reduces the ability to assess the generalizability of this classification method. For example, because there were only 6 benign cases, a difference of 1 incorrect classification could result in large changes in specificity. Additionally, there are almost twice as many osteosarcoma cases as benign tumor cases in our dataset. Despite these limitations, the results suggest that the models created in this study may be used to assist clinicians in accurately distinguishing between benign and malignant tumors in target patient populations.
ACKNOWLEDGMENT
Olivia Alge, Lu Lu, and Zhi Li are part of the University of Michigan Bioinformatics Graduate Program.
Work by the authors was supported by The University of Michigan-Shanghai Jiao Tong University Joint Institute.
Olivia Alge is supported by the University of Michigan NIH NIGMS Bioinformatics Training Grant of the National Institutes of Health under award number T32GM070449. The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Institutes of Health.
REFERENCES
- [1].Smith Malcolm A., Altekruse Sean F., Adamson Peter C., Reaman Gregory H., and Seibel Nita L.. Declining childhood and adolescent cancer mortality. Cancer, 120(16):2497–2506, August 2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [2].Ottaviani Giulia and Jaffe Norman. The Epidemiology of Osteosarcoma. In Jaffe Norman, Bruland Oyvind S., and Bielack Stefan, editors, Pediatric and Adolescent Osteosarcoma, volume 152, pages 3–13. Springer US, Boston, MA, 2009. [DOI] [PubMed] [Google Scholar]
- [3].Kundu Zile Singh. Classification, imaging, biopsy and staging of osteosarcoma. Indian Journal of Orthopaedics, 48(3):238–246, May 2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [4].Shen Rebecca, Li Zhi, Zhang Linglin, Hua Yingqi, Mao Min, Li Zhicong, Cai Zhengdong, Qiu Yunping, Gryak Jonathan, and Najarian Kayvan. Osteosarcoma Patients Classification Using Plain X-Rays and Metabolomic Data. In 2018 40th Annual International Conference of the IEEE Engineering in Medicine and Biology Society (EMBC), pages 690–693, Honolulu, HI, July 2018. IEEE. [DOI] [PubMed] [Google Scholar]
- [5].Wu B, Abbott T, Fishman D, McMurray W, Mor G, Stone K, Ward D, Williams K, and Zhao H. Comparison of statistical methods for classification of ovarian cancer using mass spectrometry data. Bioinformatics, 19(13):1636–1643, September 2003. [DOI] [PubMed] [Google Scholar]
- [6].Jae Won Lee Jung Bok Lee, Park Mira, and Song Seuck Heun. An extensive comparison of recent classification tools applied to microarray data. Computational Statistics & Data Analysis, 48(4):869–885, April 2005. [Google Scholar]
- [7].Li Yin, Sun Jian, Tang Chi-Keung, and Shum Heung-Yeung. Lazy snapping. ACM Transactions on Graphics (ToG), 23(3):303–308, 2004. [Google Scholar]
- [8].Andrews Simon. FastQC A Quality Control tool for High Throughput Sequence Data, 2010.
- [9].Dobin Alexander, Davis Carrie A., Schlesinger Felix, Drenkow Jorg, Zaleski Chris, Jha Sonali, Batut Philippe, Chaisson Mark, and Gingeras Thomas R.. STAR: ultrafast universal RNA-seq aligner. Bioinformatics, 29(1):15–21, January 2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [10].Karolchik Donna, Hinrichs Angela S., Furey Terrence S., Roskin Krishna M., Sugnet Charles W., Haussler David, and Kent W. James. The UCSC Table Browser data retrieval tool. Nucleic Acids Res, 32(Database issue):D493–496, January 2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [11].Frankish Adam, Diekhans Mark, Ferreira Anne-Maud, Johnson Rory, Jungreis Irwin, Loveland Jane, Jonathan M Mudge Cristina Sisu, Wright James, Armstrong Joel, Barnes If, Berry Andrew, Bignell Alexandra, Silvia Carbonell Sala Jacqueline Chrast, Cunning-ham Fiona, Toms Di Domenico Sarah Donaldson, Ian T Fiddes Carlos Garca Girn, Jose Manuel Gonzalez Tiago Grego, Hardy Matthew, Hourlier Thibaut, Hunt Toby, Osagie G Izuogu Julien Lagarde, Fergal J Martin Laura Martnez, Mohanan Shamika, Muir Paul, Navarro Fabio C P, Parker Anne, Pei Baikang, Pozo Fernando, Ruffier Magali, Bianca M Schmitt Eloise Stapleton, Suner Marie-Marthe, Sycheva Irina, Barbara Uszczynska-Ratajczak Jinuri Xu, Yates Andrew, Zerbino Daniel, Zhang Yan, Aken Bronwen, Jyoti S Choudhary Mark Gerstein, Guig Roderic, Hubbard Tim J P, Kellis Manolis, Paten Benedict, Reymond Alexandre, Tress Michael L, and Flicek Paul. GENCODE reference annotation for the human and mouse genomes. Nucleic Acids Research, 47(D1):D766–D773, January 2019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [12].Hartley Stephen W. and Mullikin James C.. QoRTs: a comprehensive toolset for quality control and data processing of RNA-Seq experiments. BMC Bioinformatics, 16(1), December 2015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [13].Goksuluk Dincer, Zararsiz Gokmen, Korkmaz Selcuk, Eldem Vahap, Gozde Erturk Zararsiz Erdener Ozcetin, Ozturk Ahmet, and Ahmet Ergun Karaagaoglu. MLSeq: Machine learning interface for RNA-sequencing data. Computer Methods and Programs in Biomedicine, 175:223–231, July 2019. [DOI] [PubMed] [Google Scholar]
- [14].Love Michael I., Huber Wolfgang, and Anders Simon. Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2. Genome Biology, 15(12):550, December 2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [15].Anders Simon and Huber Wolfgang. Differential expression analysis for sequence count data. Genome Biol, 11(10):R106, 2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [16].Love Michael, Anders Simon, and Huber Wolfgang. Package ‘DE-Seq2’, June 2019.
- [17].Breiman Leo. Random Forests. Machine Learning, 45(1):5–32, October 2001. [Google Scholar]