

Abstract
Summary: CNS amyloidomas are rare. We describe a 51-year-old man with isolated amyloidomas in the cerebral white matter and in the pons. CT and MR imaging showed a heterogeneous, enhancing mass in the deep cerebral white matter. A second, much smaller linear serpiginous lesion was present in the pons.
CNS amyloidomas are a rare entity with only a few case reports in the literature (1–13). Of these, the most common locations of this disease process in the CNS are in the spinal cord, cerebral white matter, leptomeninges, and gasserian ganglion. These lesions are frequently described as solitary, slow-growing, tumorlike masses. We present a case of multiple cerebral amyloidomas that represented a diagnostic challenge.
Case Report
A 51-year-old man was referred to our institution for neurosurgical consultation for a possible arteriovenous malformation (AVM). He had a 5-year history of right-sided weakness, which had slowly progressed over the last 3 years. The patient also described some word-finding difficulties of 3 years' duration. Past medical history was notable for thyroid disease.
MR imaging performed 3 years before identified two heterogeneous lesions: one in the left corona radiata adjacent to the left lateral ventricle and a second smaller lesion in the central pons (Fig 1). The lesions were stellate and hypointense on T1-weighted images, hyperintense on T2-weighted images, and demonstrated marked enhancement after IV administration of gadopentate dimeglumine. No significant edema was present. At that time, a stereotactic brain biopsy was reportedly nondiagnostic (the slides were not available for our review). MR imaging performed 1 year later showed no changes (Fig 2). Upon referral to our institution, MR scanning again revealed the two lesions, which were stable (Fig 3). A CT scan showed evident heterogeneous calcification of the two lesions (Fig 4). Cerebral angiography was performed to investigate the vascularity of the lesions and they were found to be completely avascular. Prebiopsy diagnosis was atypical neoplasm versus low-flow vascular lesion of unknown type.
fig 1.

T1-weighted postcontrast images.
A, Left parasagittal section at the margin of the lateral ventricle. Heterogeneous, serpiginous, and linear enhancement in the lesion is well demonstrated.
B, Midsagittal image demonstrates a second contrast-enhancing lesion with a linear appearance in the lower pons.
fig 2.

Sagittal (A) and parasagittal (B) fluid-attenuated inversion recovery images obtained 18 months after the study in figure 1. Note the T2 hyperintensity of both lesions. Atrophy of the posterior body of the corpus callosum is also noteworthy
fig 3.

Noncontrast axial CT scan obtained 2.5 years after the initial imaging study. There is extensive heterogeneous calcification of the left periventricular lesion. Note the minimal mass effect, which is unchanged from previous studies.
fig 4. Axial T1-weighted postcontrast image obtained at the time of the second stereotactically guided biopsy. The lesion is similar in appearance to the earliest study
A stereotactic brain biopsy of the lesion in the left corona radiata was performed. This revealed abundant spherical and irregular amorphous masses of pale eosinophilic hyaline material adjacent to and interspersed between small fragments of brain parenchyma. The material was Congo red positive and displayed green birefringence when polarized. Scattered reactive astrocytes, minimal number of plasma cells, and small foci of calcifications were also noted. The walls of several small- and medium-sized blood vessels within the mass also showed amyloid deposition. No vascular malformation could be identified (Fig 5). Immunoperoxidase stains for lambda and kappa light chains showed a high background and were difficult to interpret. There was partial immunoreactivity to transthyretin, an amyloid protein that has been implicated in rare familial leptomeningeal amyloidosis (2). A diagnosis of primary cerebral amyloidoma was reached.
fig 5.
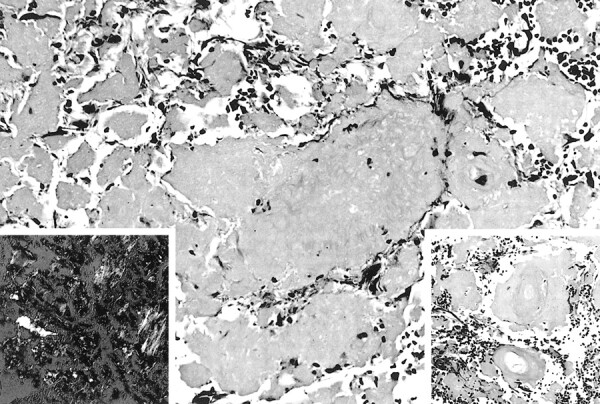
Masses of amyloid interspersed between fragments of reactive brain tissue (hematoxylin & eosin [H&E], magnification ×200). Right insert shows involvement of blood vessel walls with amyloid (H&E, magnification ×300), while the left shows the characteristic apple-green birefringence (Congo red, polarized ×300)
Discussion
This report describes the radiographic appearance of multifocal cerebral amyloidomas involving the cerebral deep white matter and the pons. Isolated cerebral amyloidomas are rare. Sometimes called primary CNS amyloidomas, they are a localized form of amyloid deposit that is distinguished from systemic amyloidosis. In the CNS, isolated amyloidomas are also much rarer than the more common congophilic amyloid angiopathy. However, the amyloid deposits in this group of diseases are physiochemically similar (3). What determines their distribution, localized or systemic, is unknown.
The radiographic findings of amyloidoma in the CNS are typically patchy lesions with intense enhancement following contrast administration. The lesions have been predominantly described as heterogeneous on T2-weighted images and isointense to slightly hyperintense on T1-weighted images. The heterogeneous signal is felt to represent nonuniform deposits of amyloid protein. Lee et al (3) stated that areas of more dense amyloid deposition are more likely to have brighter signal. The marked enhancement following contrast administration is felt to be secondary to the disruption of the blood-brain barrier, attributed to the amyloid involvement in the blood vessel walls seen microscopically. This same phenomenon is described in scintigraphic studies using technetium 99M pertechnetate (8). Two prior reports described MR findings similar to ours, with irregular radial enhancement at the periphery of the lesions that corresponded to amyloid deposition in the vascular walls as demonstrated by biopsy. These imaging findings may resemble a vascular lesion or an infiltrative glioma (3, 5). Diagnosis of the majority of reported cases was made by excisional biopsy. Stereotactic biopsy can be diagnostic, as demonstrated in our case and as reported in earlier cases (3, 11).
The few reported cases of CNS amyloidomas have predominantly occurred in adults in their late 40s and older. The most common locations have been in the thoracic spinal cord and meninges, cerebral white matter, and gasserian ganglion. Only one previous report described a lesion within the brain stem (5, 6). In our case, the pontine lesion was not proven by biopsy to be an amyloidoma; however, its signal characteristics and appearance were identical to the biopsy of a proven cerebral amyloidoma, making this the most likely diagnosis.
Cerebral amyloidomas are most commonly described as occurring in isolation. Three prior case reports describe multiple (two) CNS lesions (5–8). In our case, the multiplicity of the lesions, the lack of surrounding edema, and the imaging characteristics were suggestive of a vascular lesion.
Cerebral angiography was performed because of the imaging findings. AVMs may present with CT and MR imaging findings of serpiginous, sometimes calcified, lesions with enhancement, but usually have prominent signal voids, which were absent in the present case. Telangiectasias have some MR features similar to our case, such as contrast enhancement, but occur almost exclusively in the pons. The negative angiogram excluded a high-flow AVM but not a neoplastic process.
CNS amyloidomas have been shown to be slow-growing lesions and demonstrate similar clinical characteristics to indolent tumors. Therefore, although benign entities, they present with the same treatment difficulties as slow-growing tumors, depending on their location within the CNS. Notably, CNS amyloidomas are a different entity from systemic amyloidosis, or from amyloid angiopathy. They occur in isolation and have no known etiologic factors.
Conclusion
CNS amyloidoma is a rare entity. Clinically and radiographically, it is similar to a slow-growing neoplasm (3). Cerebral angiography was an important diagnostic study in the workup of this case because it helped to exclude a vascular lesion and, therefore, facilitated the decision to perform a stereotactic biopsy. Cerebral amyloidomas, although rare, should be considered in the differential diagnosis of slow-growing tumors and heterogeneous enhancing intracranial lesions.
Footnotes
Address reprint requests to Barton Lane, MD, FACR, Chief of Radiology (114), Palo Alto Veterans Administration Medical Center, 3801 Miranda Ave., Palo Alto, CA 94304.
References
- 1.Vorster SJ, Lee JH, Ruggieri P. Amyloidoma of the gasserian ganglion. AJNR Am J Neuroradiol 1998;19:1853-1855 [PMC free article] [PubMed] [Google Scholar]
- 2.Herrick MK, DeBruyne K, Horoupian DS, et al. Massive leptomeningeal amyloidosis associated with a Val30Met transthyretin gene. Neurology 1996;47:988-992 [DOI] [PubMed] [Google Scholar]
- 3.Lee J, Krol G, Rosenblum M. Primary amyloidoma of the brain: CT and MR presentation. AJNR Am J Neuroradiol 1995;16:712-714 [PMC free article] [PubMed] [Google Scholar]
- 4.Caerts B, Mol V, Sainte T, et al. CT and MRI of amyloidoma of the CNS. Eur Radiol 1997;7:474-476 [DOI] [PubMed] [Google Scholar]
- 5.Cohen M, Lanska D, Roessmann U, et al. Amyloidoma of the CNS. I. Clinical and pathologic study. Neurology 1992;42:2019-2023 [DOI] [PubMed] [Google Scholar]
- 6.Vidal RG, Ghiso J, Gallo G, et al. Amyloidoma of the CNS. II. Immunohistochemical and biochemical study. Neurology 1992;42:2024-2028 [DOI] [PubMed] [Google Scholar]
- 7.Spaar FW, Goebel HH, Volles E, Wickboldt J. Tumor-like amyloid formation (amyloidoma) in the brain. J Neurol 1981;224:171-182 [DOI] [PubMed] [Google Scholar]
- 8.Moreno AJ, Brown JM, Brown TJ, et al. Scintigraphic findings in a primary cerebral amyloidoma. Clin Nucl Med 1983;8:528-530 [DOI] [PubMed] [Google Scholar]
- 9.Hori A, Kitamoto T, Tateishi J, et al. Focal intracerebral accumulation of a novel type of amyloid protein. An early stage of cerebral amyloidoma? Acta Neuropathol (Berl) 1988;76:212-215 [DOI] [PubMed] [Google Scholar]
- 10.O'Brien TJ, McKelvie PA, Vrodos N. Bilateral trigeminal amyloidoma: an unusual case of trigeminal neuropathy with a review of the literature. Case report. J Neurosurg 1994;81:780-783 [DOI] [PubMed] [Google Scholar]
- 11.Schroder R, Linke RP, Voges J, et al. Intracerebral A lambda amyloidoma diagnosed by stereotactic biopsy. Clin Neuropathol 1995;14:347-350 [PubMed] [Google Scholar]
- 12.Bornemann A, Bohl J, Hey O, et al. Amyloidoma of the gasserian ganglion as a cause of symptomatic neuralgia of the trigeminal nerve: report of three cases. J Neurol 1993;241:10-14 [DOI] [PubMed] [Google Scholar]
- 13.Matsumoto T, Tani E, Fukami M, et al. Amyloidoma in the gasserian ganglion: case report. Surg Neurol 1999;52:600-603 [DOI] [PubMed] [Google Scholar]