

Abstract
Purpose of review
The role of type I IFNs (IFN-I) in the promotion of autoimmunity has been well established. However, its role in the skin fibrosis of systemic sclerosis (SSc) is less clear. IFN-I can participate to tissue repair, and, here, we will consider the extent to which IFN-I’s role in SSc skin fibrosis may reflect in part IFN-I functions during wound healing.
Recent findings
Studies are beginning to delineate whether IFN-I has a protective or pathogenic role and how IFN-I affects tissue biology. Recent support for a pathogenic role came from a study depleting plasmacytoid dendritic cells during bleomycin-induced skin fibrosis. The depletion reduced the bleomycin-induced IFN-I-stimulated transcripts and both prevented and reversed fibrosis. Additionally, two recent articles, one identifying SSc endothelial cell injury markers and one showing repressed IFN signaling in SSc keratinocytes, suggest the possibility of unbalanced IFN-I activities on distinct cells types.
Summary
Recent results support a pathogenic role for IFN-I in skin fibrosis, and recent studies along with others suggest a scenario whereby SSc skin damage results from too much IFN-I-activity driving vasculopathy in combination with too little IFN-I-mediated epidermal integrity and antifibrotic fibroblast phenotype.
Keywords: lupus, scleroderma, skin, type I interferon, wound healing
INTRODUCTION
The contribution of innate immunity to the pathogenesis of systemic sclerosis (SSc) has been well established both using patients’ samples and using multiple mouse models. However, the precise role of innate cells, and in particular macrophages and plasmacytoid dendritic cells (pDCs), in disease and how they regulate the subsequent adaptive response in SSc patients is still unclear. One of the mediators of innate immune activation, type I IFN (IFN-I), has been associated with the development of fibrosis but also with many other autoimmune diseases that are not fibrotic. We will focus on the role that IFN-I can play and will consider the extent to which IFN-I’s role in SSc skin fibrosis may reflect in part IFN-I functions in tissue repair.
IMPACT OF TYPE I INTERFERONS IN IMMUNITY AND DISEASES
The key role of IFN-I in immunity is to participate in the antiviral response and the pleiotropic activity of IFN-I has been well described. The IFN-I are comprised of 17 different proteins including 13 IFN-α subtypes, IFN-β, IFN-ω, IFN-κ, and IFN-ε. The need for such a large number of proteins, all of which share the same receptor, in immunity is unclear and could result from evolutionary pressure on the IFN pathway. It is also possible that these are differentially expressed by different cells, immune or not, and are associated with distinct functions of specific cell types [1].
The contribution by IFN-I to autoimmunity has first been described in systemic lupus erythematosus (SLE) but has since been reported in many other autoimmune diseases, including SSc [2], because of the presence in blood cells of highly expressed genes that are regulated by IFN-I and form the so-called IFN-signature. IFN-I can be produced by most cells in the body and multiple receptors sensing pathogens can induce its production. During an immune response, pathogens are recognized by multiple layers of sensors that will trigger an inflammatory response. However, in autoimmunity, it is likely that the inability to control the response to nucleic acids plays a central role [3], even though the identity of the sensors and the cell types involved is still debated. Recent studies are pointing to a key role for the nucleic acid sensing toll-like receptor (TLR)7, TLR8, and TLR9 in promoting autoimmunity with a particular role for TLR8 in controlling the development of fibrosis [4■■]. Although the activation of pDCs through TLR7 and TLR9 has been well documented in lupus [5–8], the expression of TLR8 by pDCs of SSc patients and the functional consequences on disease using a mouse model of skin fibrosis was unexpected and stresses not only the key role played by nucleic acid recognition in autoimmunity but also the induction of fibrosis.
One of the mediators of nucleic acid recognition and innate cell stimulation is IFN-I. The inflammatory response that follows tissue injury and that is required for tissue repair can, in some cases lead to fibrosis. In particular, macrophages and the nature of their activation can sway the balance between tissue repair and fibrosis [9]. In addition, IFN-I is associated with fibrosis in SSc patients [10,11], but also with many nonfibrotic autoimmune diseases, such as SLE [12–15]. How IFN-I may contribute to SSc skin fibrosis is unclear. Here we will briefly discuss the roles of IFN-I in SLE, wound healing, and SSc, and then highlight recent findings that suggest a role for pDCs and unbalanced IFNI-I activities on skin stromal cells in fibrosis.
TYPE I INTERFERONS IN LUPUS
The first observation that IFN-I may contribute to the pathogenesis of SLE were described about 40 years ago [16,17]. Patients can now be characterized by measuring the levels of IFN-induciblegenes, which are chronically elevated and can correlate with some of the clinical features of patients [12–15]. The cells responsible for the high presence of IFN-I are believed to be the pDCs, although it is likely that other cells may participate to the overall IFN response over time in patients [8,18]. The role played by pDCs and IFN-I is complex as the key findings rely on measuring interferon-stimulated genes (ISGs) in the blood, not tissues. Blood constitute a heterogeneous population of cells and the dependence of ISGs on certain IFN is still unclear and whether specific ISGs can be linked to specific symptoms is still undefined. Recent findings show that depleting or attenuating pDCs can prevent disease in lupus-prone mice [19,20] and can reduces the IFN-I response in the skin of SLE patients with encouraging signs of clinical efficacy in the skin [21■■]. These data suggest that IFN-I produced by pDCs may have a critical role in generating lesions in the skin whereas its contribution to the overall disease is less clear. IFN-I can also be produced by other cells as well such like epithelial cells [22], which adds complexity to our understanding of how IFN-I impacts the disease. Cutaneous diseases including lichen planus, dermatomyositis, lichen sclerosis, and cutaneous GVHD that share the common pathological feature named ‘interface dermatitis’ [23] all have IFN-signature in the skin but are not associated primarily with fibrosis. These findings may reflect the different tissue microenvironmental contexts of each disease.
Although SLE is not generally considered a fibrosing disease, Putterman and colleagues recently published that nonresponder lupus nephritis patients can have a high IFN-I signature and a fibrotic signature in kidney tubular epithelial cells [24■]. The fibrotic signature was not associated with frank, histological fibrosis, suggesting that these signatures are pointing to an early event and is a harbinger of things to come. The results suggest the possibility of a functional interaction between IFN-I and fibrosis, especially at early stages, and support a potential role for type I IFN in driving fibrosis.
TISSUE REPAIR
Type I IFN can promote wound healing in the skin [25,26]. Repair of epidermal tissues, such as skin and gut involves epithelial closure to reform a protective barrier, and the protective role of IFN-I in wound healing in part reflects its contributions to epithelial barrier formation or maintenance. In a tape stripping injury model whereby tape is applied and then pulled off the skin a number of times, IFN-α, likely from pDCs [27], was upregulated within a day after injury, and mice deficient in the IFN alpha/beta receptor (IFNAR) were less able to induce a protective keratinocyte activation response [25]. Similarly, mice lacking TLR3 failed to close skin wounds [28], and, in the gut, IFNAR has a similar function in promoting barrier integrity of the intestinal epithelium [29]. Consistent with these observations, overexpression of IFN-I promoted, via myeloid cells, epithelial proliferation, and repair [26]. Together, the data point to a protective role for IFN-I in promoting epithelial integrity during wound healing.
How exactly IFN-I promotes wound healing and epithelial closure is less clear. With tape stripping, IFNAR−/− mice showed reduced IL-6, IL-17, and IL-22 responses, suggesting that a lack of inflammatory responses contribute to reduced wound healing [25,27]. However, the importance of the reduced inflammatory cytokines was not directly tested. IFN-I also had a protective role upon skin injury with subacute ultraviolet radiation (UVR) [30], as IFNAR−/− mice showed greater skin inflammation compared with controls. Although these studies were apparently at odds with each other with regard to the effect on skin inflammation, IFNAR clearly had a beneficial role in both models. Elkon and colleagues proposed that differences in levels, source, and inducers of IFN (higher, by pDCs, and dependent on TLR7 and TLR9 for tape stripping, whereas UVR produced lower levels of IFN in the absence of pDCs and the IFN was dependent on TLR3 and STING) could potentially account for differences in inflammation. Potentially, some of the differences could be because of direct dermal damage by UVR but not tape stripping, that leads to inflammation that is controlled by IFNAR, perhaps via effects on epidermal integrity. IFNAR can also act on myeloid cells to promote their ability to stimulate healing. IFNAR on myeloid cells has been shown to promote keratinocyte proliferation [26], and IFNAR will modulate pDC expression of cytokines, potentially contributing to the inflammatory cytokines that were lost in IFNAR−/− mice with tape stripping [25].
In addition to a role for IFNAR in epithelial barrier function, IFN-I can affect immunity at the skin level, which could have implications for wound healing. For example, IFN-I produced by keratinocytes activates dendritic cells in the skin [31]. This causes them to be better antigen-presenting cells and presumably to migrate toward the draining lymph node to activate the immune system. IFNAR can also activate accumulation of effector T cells in a tissue by activating macrophages [32] and can initiate tissue inflammation by promoting macrophage necroptosis [33]. Regulatory T cells, which have tissue repair properties [34], are also directly regulated by IFNAR to promote their development and function [35]. Cumulatively, the data suggest that IFNAR can promote wound healing by acting on local cells to repair tissue and also by stimulating immune cells to control infectious agents.
In some settings involving an excess of cytokine, however, IFN-I can also impede wound healing. For example, injection of IFN-α into wound edges delayed wound closure [36]. This was associated with an antiproliferative effect of IFN-α on the capillaries that normally grow into the wound, as well as on fibroblasts and keratinocytes. These results are in line with the sensitivity of endothelial cells to IFN-α in vitro [37], an ehrlichial infection model whereby infection-induced IFN-I signaling on nonhematopoietic cells is detrimental to the host [38] and a skin viral infection model that was associated with lymphatic dysfunction [39]. Whether excessive IFN combined with an unrepaired wound (versus excessive IFN in the absence of a wound) leads eventually to fibrosis would be interesting to investigate.
The idea that IFN-I effects are context-dependent is supported in considering the role of IFN-I in bacterial infection secondary to viral infections [40]. Here, the timing of type I IFN seems to be a critical parameter that determines its exact effect. IFN expressed at high levels prior to secondary infection can suppress the host response, including neutrophil recruitment, Th17 generation, and expression of antibacterial peptides, leaving the host less able to control the secondary infection. This is similar to the role of IFN-I in suppressing a protective Th1 response with Mycobacterium tuberculosis and Mycobacterium leprae. On the other hand, IFN at 14 days after the start of viral infection reduces the risk of secondary infections. The reasons for a protective role later after a viral infection is unclear, but it is interesting to speculate that IFN-I -mediated remodeling of the affected tissue or of the secondary lymphoid organs where immune responses are initially generated after the initial infection contributed to better protection.
In wound healing then, IFN-I can have different functions in different contexts and has effects in addition to effects on immunity. Overall, IFN-I is protective in promoting epidermal barrier integrity during would healing, but excessive IFN-I can disrupt vascular function.
TYPE I INTERFERONS IN SYSTEMIC SCLEROSIS
In SSc, there is an association between IFN-I and disease. An IFN signature is found in the blood of about 50% of SSc patients, and it is apparent even early in disease [41–43]. Importantly, an IFN signature is also found in skin, again in association with early disease [44–46]. Polymorphisms of IFN regulatory genes IRF-4,5,7,8 and STAT4 have been found to be associated with SSc [41], further suggesting the possibility of a pathogenic role for IFN-I. As in SLE, there is also anecdotal evidence that IFN-I used for treating other diseases was associated with development of SSc [41]. Intriguingly, anti-IFNAR (Anifrolumab) in phase I trials led to suppression of IFN signature and TGFβ signaling in SSc skin [47] and further trials to assess clinical efficacy could help us understand the extent to which IFN-I contributes to pathogenesis.
A pathogenic role for IFN-I in skin fibrosis was further supported by two recent studies. Delaney et al. [49] treated a graft-versus-host disease (GVHD) model that is characterized by skin inflammation and fibrosis and used as an SSc model [48] with anti-IFNAR. The GVHD model showed upregulated IFN-I stimulated gene expression in the skin and anti-IFNAR reduced the IFN-I signature as well as fibrosis development, suggesting that IFN-I drove fibrosis development. In 2018, Ah Kioon et al. [4■■] examined the role of pDCs in scleroderma that can express very large quantities of IFN-I. They found that pDCs accumulate in SSc skin and express higher levels of IFN-α than cells from healthy controls spontaneously in culture. Although the pDCs also expressed additional cytokines, such as CXCL4, pDC depletion reversed the upregulated IFN-associated genes seen in the bleomycin-induced skin fibrosis model and improved fibrosis. Both these studies in different models [4■■,49], then, supported the idea that IFN-I overall promotes skin fibrosis, presumably via a local mechanism.
How IFN-I affects skin biology during fibrosis is critical for insight into the role of IFN-I, but understanding is currently limited. Interferon alpha expression in human scleroderma skin has been attributed to pDCs or other types of immune cells that are located near blood vessels and associated with capillary rarefaction [50]. This suggests a scenario whereby high levels of IFN-I and its antiangiogenic effects on endothelial cells may contribute to the pathogenesis of scleroderma skin fibrosis. The Delaney et al. [49] study treating the GVHD model with anti-IFNAR also showed that the reduced fibrosis development was associated with prevention of endothelial cell injury, supporting the idea that IFN-I -mediated vascular injury contributed to fibrosis. Single-cell RNA sequencing of SSc skin has not reported the finding of an IFN-I signature per se in endothelial cells [51■■], but endothelial cells were found to express higher levels of vWF [51■■], a marker of endothelial cell damage, and vWF has been shown to have downregulated anifrolomab in SLE patients [52]. Potentially, then, excessive IFN-I contributes to the endothelial cell damage in scleroderma skin.
In contrast, however, short-term primary cultures of keratinocytes from SSc skin have shown repressed IFN-I signaling when compared with transcriptome of healthy control keratinocytes [53■■]. This echoes the repressed IFN-I signaling found in cultured SSc skin and lung fibroblasts [46,54]. Whether the repression in keratinocytes and, potentially, fibroblasts reflects the in-vivo fibroblast state or is a result of culturing remains to be further sorted out. Interesting, recent work by Browning and colleagues showing some increased VCAM-1 expression by perivascular but not other fibroblasts in SSc skin [55], which could potentially reflect increased IFN-I signaling [49,56]. This study points to the heterogeneity of fibroblasts in all tissues that is becoming better understood [57,58■–61■,62] and demonstrates the potential for specific regulation of a subset of cells that may not be detected in bulk sequencing of a particular cell type or that may be lost upon culturing. In the study of scleroderma, the results from single-cell RNA sequencing analyses of fibroblasts and keratinocytes directly from SSc skin will likely be illuminating.
Despite the likely heterogeneity there is in any given cell type, however, the surprising results of Kahlenberg and colleagues raise the interesting possibility that the high levels of IFN-I in the skin in SSc may not signal to all cell types in a balanced way; some cell types, such as keratinocytes and fibroblasts, may be ‘nonresponders’ whereas others, such as endothelial cells, may be very sensitive to IFN-I [53■■]. Interestingly, Varga and colleagues have shown that IFN-β induces an antifibrotic phenotype in cultured fibroblasts, raising the possibility that reduced IFN-I signaling in SSc fibroblasts may be pathogenic [46,63]. Similarly, given the role of IFN-I in promoting epidermal barrier integrity, repressed IFN-I responses in keratinoctyes in SSc has the potential to disrupt barrier integrity, thus contributing to the wound healing phenotype of the epidermis [64] and skin damage in SSc. Potentially, the high levels of IFN-I in SSc is meant to be a protective response to a perceived ‘wound’, but the unbalanced response to IFN-I results in damage to the skin, thereby contributing to the fibrotic phenotype.
CONCLUSION
Although high IFN-I levels are associated with SSc and recent data support the idea that high IFN-I is overall pathogenic in SSc skin fibrosis, we propose here that understanding how IFN-I is pathogenic may require us to consider that an imbalance in the nonhematopoietic ‘stromal’ response to IFN-I may be a contributing factor. Potentially, the sensitive endothelial cell response to IFN-I is driving a vasculopathy whereas the repressed responses in keratinocytes and fibroblasts drives epidermal dysfunction and a profibrotic fibroblast phenotype (Fig. 1). In an attempt to repair the ‘wound’ that is not healing, IFN-I levels are driven up, further exacerbating the skin damage. It is also possible that the stromal responses reflect also indirect responses whereby IFN-I acts on myeloid and other immune cells to mediate the distinct endothelial, keratinocyte, and fibroblast responses. Here, more detailed understanding of the effects of IFN-I on the stromal elements in SSc patients or scleroderma models and of the direct and indirect IFN-I-responding cells will help us better understand how IFN-I should be therapeutically targeted in SSc skin fibrosis, and potentially in wound healing and other autoimmune and inflammatory conditions.
FIGURE 1.
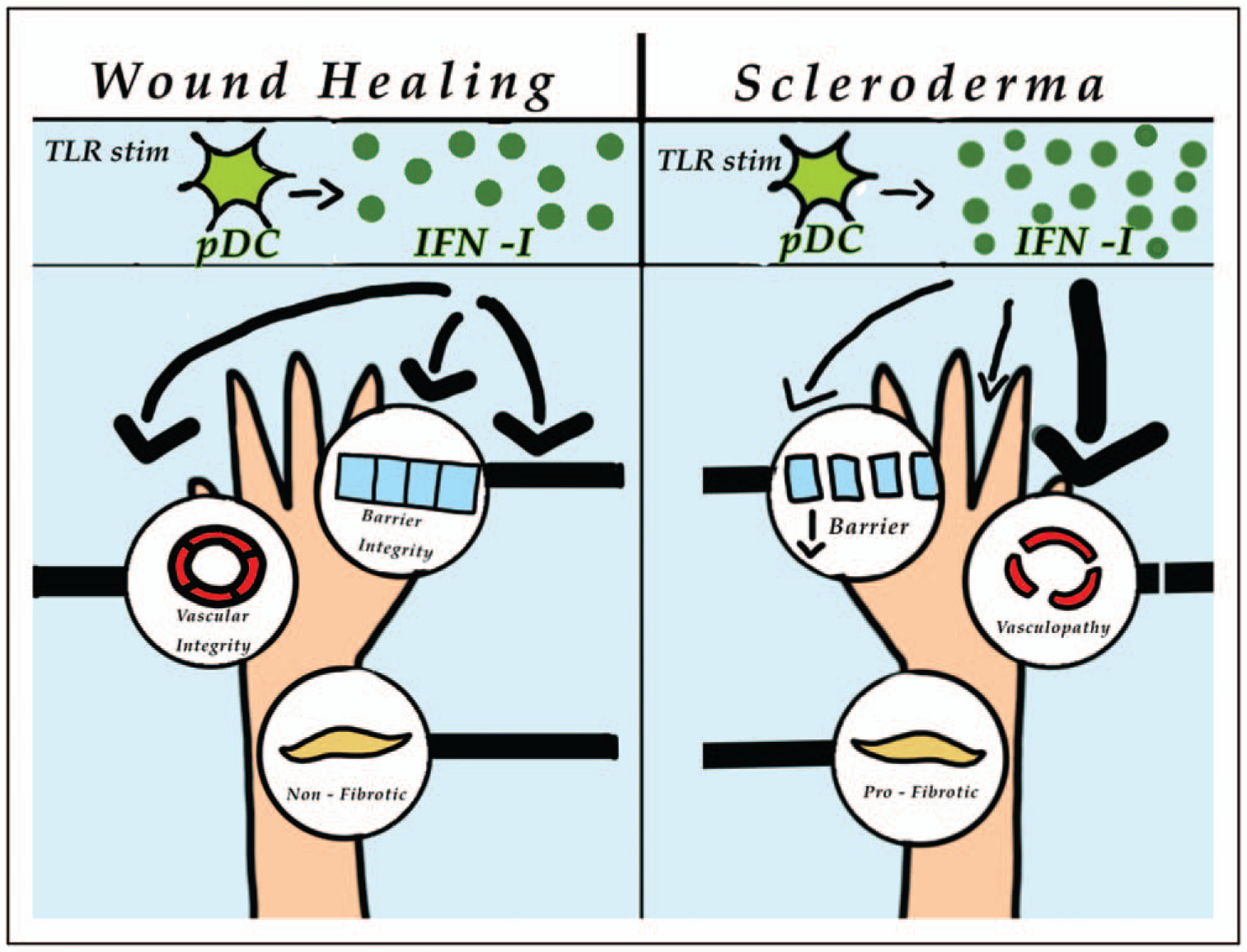
Model of type I interferon activities on skin stromal cells during wound healing and in scleroderma. Left panel: during wound healing, IFN-I from toll-like receptor (TLR)-stimulated pDCs acts on the epithelium to promote skin barrier integrity, the vasculature to promote normal function, and fibroblasts to maintain an antifibrotic phenotype. Right panel: in scleroderma skin fibrosis, the excess IFN-I has an imbalanced effect on different stromal compartments. The vasculature is sensitive to the IFN-I, leading to vasculopathy, whereas the epidermis and fibroblasts have repressed IFN-I signaling, leading to disrupted barrier function and a profibrotic phenotype, respectively. IFN-1, type I interferon.
KEY POINTS.
IFN-I can both promote wound healing by promoting epithelial barrier integrity and disrupt wound healing by inhibiting proliferation or survival of endothelial cells, and its activities may be dose-dependent and context-dependent.
Recent results support a pathogenic role for IFN-I in skin fibrosis.
Recent results also show endothelial injury that could be IFN-I-mediated and suppressed IFN-I-stimulated genes in SSc keratinocytes.
We speculate that the skin damage of SSc skin fibrosis may reflect, in part, excessive IFN-I effects on endothelial combined with too little on keratinocytes and fibroblasts, resulting in vascular damage, disruption of the epidermal integrity, and profibrotic fibroblasts.
Acknowledgements
The authors thank Charlotte Black for the figure illustration.
Financial support and sponsorship
This work was supported by grants from the NIH (1R01AI132447 to F.J.B.; NIH R01AI079178 to T.T.L.), from the Scleroderma Research Foundation (F.J.B.), Scleroderma Foundation (T.T.L.), the Lupus Research Alliance (T.T.L.), St. Giles Foundation (T.T.L.), and A Lasting Mark Foundation (T.T.L.).
Footnotes
Conflicts of interest
F.J.B. has been acting as a consultant for Astra Zeneca, Jansen, Biogen, and EMD Serono but otherwise has no other conflicts. T.T.L. has no conflicts of interest.
REFERENCES AND RECOMMENDED READING
Papers of particular interest, published within the annual period of review, have been highlighted as:
■ of special interest
■■ of outstanding interest
- 1.Fensterl V, Chattopadhyay S, Sen GC. No love lost between viruses and interferons. Annu Rev Virol 2015; 2:549–572. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Crow MK, Olferiev M, Kirou KA. Type I interferons in autoimmune disease. Annu Rev Pathol 2019; 14:369–393. [DOI] [PubMed] [Google Scholar]
- 3.Barrat FJ, Elkon KB, Fitzgerald KA. Importance of nucleic acid recognition in inflammation and autoimmunity. Annu Rev Med 2016; 67:323–336. [DOI] [PubMed] [Google Scholar]
- 4.■■.Ah Kioon MD, Tripodo C, Fernandez D, et al. Plasmacytoid dendritic cells promote systemic sclerosis with a key role for TLR8. Sci Transl Med 2018; 10:423. [DOI] [PMC free article] [PubMed] [Google Scholar]; This article demonstrates the pathogenic role of plasmacytoid dendritic cells as a key source of IFN-I in SSc skin fibrosis.
- 5.Barrat FJ, Meeker T, Gregorio J, et al. Nucleic acids of mammalian origin can act as endogenous ligands for toll-like receptors and may promote systemic lupus erythematosus. J Exp Med 2005; 202:1131–1139. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Vollmer J, Tluk S, Schmitz C, et al. Immune stimulation mediated by auto-antigen binding sites within small nuclear RNAs involves Toll-like receptors 7 and 8. J Exp Med 2005; 202:1575–1585. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Means TK, Latz E, Hayashi F, et al. Human lupus autoantibody-DNA complexes activate DCs through cooperation of CD32 and TLR9. J Clin Invest 2005; 115:407–417. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Guiducci C, Gong M, Xu Z, et al. TLR recognition of self nucleic acids hampers glucocorticoid activity in lupus. Nature 2010; 465:937–941. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Wynn TA, Vannella KM. Macrophages in tissue repair, regeneration, and fibrosis. Immunity 2016; 44:450–462. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Wu M, Assassi S. The role of type 1 interferon in systemic sclerosis. Front Immunol 2013; 4:266. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Laurent P, Sisirak V, Lazaro E, et al. Innate immunity in systemic sclerosis fibrosis: recent advances. Front Immunol 2018; 9:1702. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Bennett L, Palucka AK, Arce E, et al. Interferon and granulopoiesis signatures in systemic lupus erythematosus blood. J Exp Med 2003; 197:711–723. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Baechler EC, Batliwalla FM, Karypis G, et al. Interferon-inducible gene expression signature in peripheral blood cells of patients with severe lupus. Proc Natl Acad Sci U S A 2003; 100:2610–2615. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Kirou KA, Lee C, George S, et al. Coordinate overexpression of interferon-alpha-induced genes in systemic lupus erythematosus. Arthritis Rheum 2004; 50:3958–3967. [DOI] [PubMed] [Google Scholar]
- 15.Banchereau R, Hong S, Cantarel B, et al. Personalized immunomonitoring uncovers molecular networks that stratify lupus patients. Cell 2016; 165:551–565. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Hooks JJ, Moutsopoulos HM, Geis SA, et al. Immune interferon in the circulation of patients with autoimmune disease. N Engl J Med 1979; 301:5–8. [DOI] [PubMed] [Google Scholar]
- 17.Ytterberg SR, Schnitzer TJ. Serum interferon levels in patients with systemic lupus erythematosus. Arthritis Rheum 1982; 25:401–406. [DOI] [PubMed] [Google Scholar]
- 18.Rodero MP, Tesser A, Bartok E, et al. Type I interferon-mediated autoinflammation due to DNase II deficiency. Nat Commun 2017; 8:2176. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Rowland SL, Riggs JM, Gilfillan S, et al. Early, transient depletion of plasmacytoid dendritic cells ameliorates autoimmunity in a lupus model. J Exp Med 2014; 211:1977–1991. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Sisirak V, Ganguly D, Lewis KL, et al. Genetic evidence for the role of plasmacytoid dendritic cells in systemic lupus erythematosus. J Exp Med 2014; 211:1969–1976. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.■■.Furie R, Werth VP, Merola JF, et al. Monoclonal antibody targeting BDCA2 ameliorates skin lesions in systemic lupus erythematosus. J Clin Invest 2019; 129:1359–1371. [DOI] [PMC free article] [PubMed] [Google Scholar]; This study depletes plasmacytoid dendritic cells in human SLE and shows associated skin improvement.
- 22.Sarkar MK, Hile GA, Tsoi LC, et al. Photosensitivity and type I IFN responses in cutaneous lupus are driven by epidermal-derived interferon kappa. Ann Rheum Dis 2018; 77:1653–1664. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Wenzel J, Tuting T. An IFN-associated cytotoxic cellular immune response against viral, self-, or tumor antigens is a common pathogenetic feature in ‘interface dermatitis’. J Invest Dermatol 2008; 128:2392–2402. [DOI] [PubMed] [Google Scholar]
- 24.■.Der E, Suryawanshi H, Morozov P, et al. Tubular cell and keratinocyte single-cell transcriptomics applied to lupus nephritis reveal type I IFN and fibrosis relevant pathways. Nat Immunol 2019; 20:915–927. [DOI] [PMC free article] [PubMed] [Google Scholar]; This article shows by single-cell RNA sequencing that kidney epithelial cells from lupus nephritis patients have IFN-I and fibrosis signatures that are reflected in the skin.
- 25.Gregorio J, Meller S, Conrad C, et al. Plasmacytoid dendritic cells sense skin injury and promote wound healing through type I interferons. J Exp Med 2010; 207:2921–2930. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Sun L, Miyoshi H, Origanti S, et al. Type I interferons link viral infection to enhanced epithelial turnover and repair. Cell Host Microbe 2015; 17:85–97. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Guiducci C, Tripodo C, Gong M, et al. Autoimmune skin inflammation is dependent on plasmacytoid dendritic cell activation by nucleic acids via TLR7 and TLR9. J Exp Med 2010; 207:2931–2942. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Lin Q, Fang D, Fang J, et al. Impaired wound healing with defective expression of chemokines and recruitment of myeloid cells in TLR3-deficient mice. J Immunol 2011; 186:3710–3717. [DOI] [PubMed] [Google Scholar]
- 29.Fischer JC, Bscheider M, Eisenkolb G, et al. RIG-I/MAVS and STING signaling promote gut integrity during irradiation- and immune-mediated tissue injury. Sci Transl Med 2017; 9:eaag2513. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Sontheimer C, Liggitt D, Elkon KB. Ultraviolet B irradiation causes stimulator of interferon genes–dependent production of protective type I interferon in mouse skin by recruited inflammatory monocytes. Arthritis Rheumatol 2017; 69:826–836. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Zhang LJ, Sen GL, Ward NL, et al. Antimicrobial peptide LL37 and MAVS signaling drive interferon-β production by epidermal keratinocytes during skin injury. Immunity 2016; 45:119–130. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Marro BS, Legrain S, Ware BC, Oldstone MB. Macrophage IFN-I signaling promotes autoreactive T cell infiltration into islets in type 1 diabetes model. JCI Insight 2019; 4; pii: 125067 [Epub ahead of print] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.McComb S, Cessford E, Alturki NA, et al. Type-I interferon signaling through ISGF3 complex is required for sustained Rip3 activation and necroptosis in macrophages. Proc Natl Acad Sci U S A 2014; 111:E3206–E3213. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Li J, Tan J, Martino MM, Lui KO. Regulatory T-cells: potential regulator of tissue repair and regeneration. Front Immunol 2018; 9:585. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Metidji A, Rieder SA, Glass DD, et al. IFN-alpha/beta receptor signaling promotes regulatory T cell development and function under stress conditions. J Immunol 2015; 194:4265–4276. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Stout AJ, Gresser I, Thompson WD. Inhibition of wound healing in mice by local interferon alpha/beta injection. Int J Exp Pathol 1993; 74:79–85. [PMC free article] [PubMed] [Google Scholar]
- 37.Kaiser WJ, Kaufman JL, Offermann MK. IFN-α sensitizes human umbilical vein endothelial cells to apoptosis induced by double-stranded RNA. J Immunol 2004; 172:1699–1710. [DOI] [PubMed] [Google Scholar]
- 38.Zhang Y, Thai V, McCabe A, et al. Type I interferons promote severe disease in a mouse model of lethal ehrlichiosis. Infect Immun 2014; 82:1698–1709. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Loo CP, Nelson NA, Lane RS, et al. Lymphatic vessels balance viral dissemination and immune activation following cutaneous viral infection. Cell Rep 2017; 20:3176–3187. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Boxx GM, Cheng G. The roles of type I interferon in bacterial infection. Cell Host Microbe 2016; 19:760–769. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Skaug B, Assassi S. Type I interferon dysregulation in systemic sclerosis. Cytokine 2019. pii: S1043–4666(19)30006–7. doi: 10.1016/j.cyto.2018.12.018. [DOI] [PubMed] [Google Scholar]
- 42.Ciechomska M, Skalska U. Targeting interferons as a strategy for systemic sclerosis treatment. Immunol Lett 2018; 195:45–54. [DOI] [PubMed] [Google Scholar]
- 43.Brkic Z, van Bon L, Cossu M, et al. The interferon type I signature is present in systemic sclerosis before overt fibrosis and might contribute to its pathogenesis through high BAFF gene expression and high collagen synthesis. Ann Rheum Dis 2016; 75:1567–1573. [DOI] [PubMed] [Google Scholar]
- 44.Johnson ME, Mahoney JM, Taroni J, et al. Experimentally-derived fibroblast gene signatures identify molecular pathways associated with distinct subsets of systemic sclerosis patients in three independent cohorts. PLoS One 2015; 10:e0114017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Assassi S, Swindell WR, Wu M, et al. Dissecting the heterogeneity of skin gene expression patterns in systemic sclerosis. Arthritis Rheumatol 2015; 67:3016–3026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Gardner H, Shearstone JR, Bandaru R, et al. Gene profiling of scleroderma skin reveals robust signatures of disease that are imperfectly reflected in the transcript profiles of explanted fibroblasts. Arthritis Rheum 2006; 54:1961–1973. [DOI] [PubMed] [Google Scholar]
- 47.Guo X, Higgs BW, Bay-Jensen AC, et al. Suppression of T cell activation and collagen accumulation by an anti-IFNAR1 mAb, anifrolumab, in adult patients with systemic sclerosis. J Invest Dermatol 2015; 135: 2402–2409. [DOI] [PubMed] [Google Scholar]
- 48.Ruzek MC, Jha S, Ledbetter S, et al. A modified model of graft-versus-host-induced systemic sclerosis (scleroderma) exhibits all major aspects of the human disease. Arthritis Rheum 2004; 50:1319–1331. [DOI] [PubMed] [Google Scholar]
- 49.Delaney TA, Morehouse C, Brohawn PZ, et al. Type I IFNs regulate inflammation, vasculopathy, and fibrosis in chronic cutaneous graft-versus-host disease. J Immunol 2016; 197:42–50. [DOI] [PubMed] [Google Scholar]
- 50.Fleming JN, Nash RA, McLeod DO, et al. Capillary regeneration in scleroderma: stem cell therapy reverses phenotype? PLoS One 2008; 3:e1452. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.■■.Apostolidis SA, Stifano G, Tabib T, et al. Single cell RNA sequencing identifies HSPG2 and APLNR as markers of endothelial cell injury in systemic sclerosis skin. Front Immunol 2018; 9:2191. [DOI] [PMC free article] [PubMed] [Google Scholar]; This article analyzes SSc skin endothelial cells by single-cell RNA sequencing and delineates the markers of injury expressed by these cells.
- 52.Casey KA, Guo X, Smith MA, et al. Type I interferon receptor blockade with anifrolumab corrects innate and adaptive immune perturbations of SLE. Lupus Sci Med 2018; 5:e000286. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.■■.McCoy SS, Reed TJ, Berthier CC, et al. Scleroderma keratinocytes promote fibroblast activation independent of transforming growth factor beta. Rheumatology (Oxford) 2017; 56:1970–1981. [DOI] [PMC free article] [PubMed] [Google Scholar]; As part of a study underscoring the potential role of epidermal dysfunction in modulating dermal fibrosis, this article points out that cultured keratinocytes from SSc skin show repressed IFN-I signalling.
- 54.Lindahl GE, Stock CJ, Shi-Wen X, et al. Microarray profiling reveals suppressed interferon stimulated gene program in fibroblasts from scleroderma-associated interstitial lung disease. Respir Res 2013; 14:80. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Barron AMS, Mantero JC, Ho JD, et al. Perivascular adventitial fibroblast specialization accompanies T cell retention in the inflamed human dermis. J Immunol 2019; 202:56–68. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Kobayashi T, Takaku Y, Yokote A, et al. Interferon-β augments eosinophil adhesion-inducing activity of endothelial cells. Eur Respir J 2008; 32:1540–1547. [DOI] [PubMed] [Google Scholar]
- 57.Nazari B, Rice LM, Stifano G, et al. Altered dermal fibroblasts in systemic sclerosis display podoplanin and CD90. Am J Pathol 2016; 186:2650–2664. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.■.Tabib T, Morse C, Wang T, et al. SFRP2/DPP4 and FMO1/LSP1 define major fibroblast populations in human skin. J Invest Dermatol 2018; 138:802–810. [DOI] [PMC free article] [PubMed] [Google Scholar]; Single-cell RNA sequencing analysis of normal human skin underscores the multiple subtypes of fibroblasts.
- 59.■.Croft AP, Campos J, Jansen K, et al. Distinct fibroblast subsets drive inflammation and damage in arthritis. Nature 2019; 570:246–251. [DOI] [PMC free article] [PubMed] [Google Scholar]; The authors use a combination of techniques including CyTOF and single-cell RNA sequencing analyses of murine arthritis model synovial fibroblasts to demonstrate functionally distinct subtypes of cells.
- 60.■.Zhang F, Wei K, Slowikowski K, et al. Defining inflammatory cell states in rheumatoid arthritis joint synovial tissues by integrating single-cell transcriptomics and mass cytometry. Nat Immunol 2019; 20:928–942. [DOI] [PMC free article] [PubMed] [Google Scholar]; The authors use a combination of techniques including CyTOF and single-cell RNA sequencing to identify immune and stromal cell populations, including four fibroblast subtypes, in human rheumatoid arthritis synovium.
- 61.■.Rodda LB, Lu E, Bennett ML, et al. Single-cell RNA sequencing of lymph node stromal cells reveals niche-associated heterogeneity. Immunity 2018; 48:1014 e1016–1028.e1016. [DOI] [PMC free article] [PubMed] [Google Scholar]; The authors use single-cell RNA sequencing to delineate nine different types of lymph node stromal fibroblasts that appear to have specific niche-restricted functions.
- 62.Malhotra D, Fletcher AL, Astarita J, et al. , Immunological Genome Project Consortium. Transcriptional profiling of stroma from inflamed and resting lymph nodes defines immunological hallmarks. Nat Immunol 2012; 13:499–510. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Fang F, Ooka K, Sun X, et al. A synthetic TLR3 ligand mitigates profibrotic fibroblast responses by inducing autocrine IFN signaling. J Immunol 2013; 191:2956–2966. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Aden N, Shiwen X, Aden D, et al. Proteomic analysis of scleroderma lesional skin reveals activated wound healing phenotype of epidermal cell layer. Rheumatology (Oxford) 2008; 47:1754–1760. [DOI] [PubMed] [Google Scholar]