

Abstract
The evolution of coenzymes, or their impact on the origin of life, is fundamental for understanding our own existence. Having established reasonable hypotheses about the emergence of prebiotic chemical building blocks, which were probably created under palaeogeochemical conditions, and surmising that these smaller compounds must have become integrated to afford complex macromolecules such as RNA, the question of coenzyme origin and its relation to the evolution of functional biochemistry should gain new impetus. Many coenzymes have a simple chemical structure and are often nucleotide‐derived, which suggests that they may have coexisted with the emergence of RNA and may have played a pivotal role in early metabolism. Based on current theories of prebiotic evolution, which attempt to explain the emergence of privileged organic building blocks, this Review discusses plausible hypotheses on the prebiotic formation of key elements within selected extant coenzymes. In combination with prebiotic RNA, coenzymes may have dramatically broadened early protometabolic networks and the catalytic scope of RNA during the evolution of life.
Keywords: coenzymes, origin of life, prebiotic chemistry, protometabolism, RNA world theory
Although coenzymes play key roles in biotic metabolism, they have not been in the focus as elements in protometabolism nor has their possible prebiotic generation been discussed in detail. This Review provides a conceptual view of the role of coenzymes with respect to known theories of protometabolism and their incorporation into the RNA world hypothesis.
1. Introduction
“The origin of Life cannot be discovered, it has to be reinvented” (Albert Eschenmoser). [1]
How did life start on planet Earth? The most significant yet unanswered question of the natural sciences. [2] Studying the origin of life is made tangible to our own experience when we consider that extant life still carries the imprint of its origin and that it is possible to extract ancestral concepts from the realm of extant mechanism. Nowadays, this question of origin has been linked to that of nucleotides and proteins, but little focus has been spent on the evolution of coenzymes. [3] This is rather surprising as coenzymes are typically small and simple organic non‐protein compounds, such as pyridoxal phosphate (PLP, 1), that specifically bind to enzyme macromolecules and actively participate in catalytic biotransformations (Scheme 1). [4] In many cases, it has been shown that protein structures (apoenzymes) are inactive without a coenzyme partner and that this association is synergistic. This alliance is very productive in a biological context, promoting site‐specific oxidation and reduction, group transfer reactions such as acylation, phosporylation, methylation, and formal acyl‐anion transfer, many of which cannot be affected by enzymes that are purely based on a protein scaffold. Consequently, at some stage during the evolution of life, coenzymes must have played a key role in the creation of complex metabolic networks. [5]
Scheme 1.
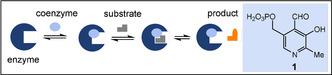
Coenzymes in enzyme catalysis and structure of the coenzyme pyridoxal phosphate (PLP, 1).
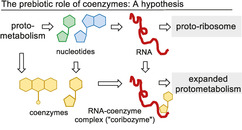
The question remains: At which stage during this evolutionary process did coenzymes first appear? Coenzymes may have evolved in an early primitive prebiotic metabolism, well before enzymes or other polymers existed, and prior to the existence of molecular replication. [6] This early metabolism might have been capable of self‐propagation through autocatytic pathways and cycles, [7a] which would have depended on a constant flux of organic building blocks, including those with coenzyme‐like properties. In this context, a number of small‐molecule metabolic cycles have already been discussed as probable prebiotic mechanisms. [7]
However, assuming this hypothesis, means speculating about a protometabolism of small coenzyme‐like molecules, for which hardly any vestiges or evidence can be found today. In general, coenzymes themselves exert poor catalytic properties compared to coenzyme–enzyme complexes and it is reasonable to assume that this would also be the case for their role in a primitive metabolic network. Their full potential must, therefore, have been realized with the arrival of macromolecular templates, thereby resulting in coenzyme‐like molecules with catalytic properties and/or the ability to promote a greater diversity of chemical transformations. If such an association were to have developed at the same time that RNA macromolecules first emerged,[ 8 , 9 ] the coenzymes, or early simpler analogues, could very well have acted as “holoribozymes” (in analogy to the term holoenzyme) by binding to nucleic acid fragments either through electrostatic or hydrogen‐bonding interactions, [10] or through covalent linkage to the RNA terminus through a phosphate ester (Scheme 2 A). Indeed, the latter concept has been discussed for the nucleotide‐derived coenzymes nicotinamide adenine dinucleotide, riboflavin, and S‐adenosylmethionine. [11] In a broader sense, it is perhaps also conceivable that this association or templating of nucleotide bases with primitive coenzyme‐like molecules in itself might have supported and catalyzed the first RNA syntheses.
Scheme 2.
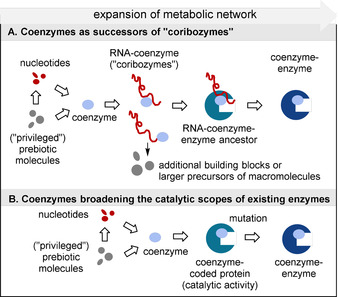
Hypotheses on coenzyme‐dependent enzyme evolution; A) as part of RNA‐world theory; B) evolution in a coded protein world (in this scheme, the term coenzyme includes small molecules as well as metal cations or metal complexes).
Eventually, single amino acids would become associated with catalytic RNA templates, enabling their condensation and giving rise to the first peptide structures. The combination of these new peptides with RNA is likely to have expanded the pool of catalytic macromolecular binding sites and yielded a greater diversity and complexity of organic reactions. As molecular evolution progressed, protein structures would then displace the role of nucleic acid subunits, thereby creating enzymes that still bear the catalytic core of their active sites—including those that are connected with today's coenzymes. Further molecular evolution would then have created vast families of homologous enzymes capable of performing complex self‐regulating biochemical networks (Scheme 2 A). In view of the presumed advantages of proteins over RNA as catalysts, such as chemical stability and structural diversity of the available monomeric units, this nucleotide/amino acid replacement scenario is more plausible than the reverse. Alternatively, it remains possible that coenzymes became involved in the evolution of peptides at a later stage, directly broadening the catalytic scope of protein‐based biotransformations (Scheme 2 B) and merely participating as bystanders during the hypothesized RNA world.
Deciphering the evolution of life is one of the few highly speculative and hypothesis‐driven scientific pursuits [12] in which chemists have and need to play a central role. [1] The Miller–Urey spark discharge experiment [13] is a key example of how chemical sciences offer a unique contribution to unraveling such a fundamental scientific mystery. The experiment showed that amino acids and lower carboxylic/fatty acids can all result from four components: methane, ammonia, diatomic hydrogen, and carbon monoxide under strongly reducing conditions and with an electric spark discharge (see Figure 4 in Section 3.1). This was the starting point for chemists to “reinvent” the molecular basis of the origin of life; by theorizing how protometabolic networks may have emerged from principal building blocks that are assumed to be formed under prebiotic conditions. Notably, these ideas were not only conceived as theories purely based on thought; they were also supported by new experimental evidence. Although coenzymes are themselves chemical entities that perform transformations through distinct mechanisms, they have mostly escaped the focus of our contemporary discussion on abiogenesis or the origin of life.
Figure 4.

Overview on prebiotic molecules formed according to experimental evidence under different conditions. * The list is not comprehensive; products listed are regarded to have major relevance for current hypotheses on prebiotic evolution; for quantities and ratios refer to the original publications. # not practically demonstrated yet.
In this account, I consider how coenzyme‐like molecules may have formed under prebiotic conditions and discuss the possible roles of these structures in the development of primitive metabolism. I explore how the presence of coenzymes in a prebiotic and pre‐RNA world may align with or further contribute to existing theories of chemical abiogenesis. Further, I contemplate: which of the principal features or analogues of modern coenzymes could have formed under typical prebiotic conditions? Are these entities able to bind to nucleotides or RNA fragments, thereby fostering their catalytic activity? In the attempt to answer these questions, I firstly summarize established theories and concepts relating to prebiotic chemistry, especially those that describe the emergence of privileged organic building blocks and early protometabolic networks. Then, the potential for these rudimentary elements to have contributed to the assembly of prebiotic coenzymes is critically evaluated also using known present‐day mechanisms of biosynthesis and current trends in biotechnology as a guide. Throughout this discussion, it is my intention to suggest new avenues of research relating to prebiotic chemistry and the origin of life, and to advocate future efforts to better understand the historical role of coenzyme molecules using modern experimentation. At this point it should not be forgotten that prebiotic simulation experiments should be compatible with the plausible geochemical conditions that likely existed on early Earth.
2. The RNA World, Ribozymes, and Riboswitches
In the evolutionary RNA scenario, nucleic acids of defined sequence precede proteins of defined sequence. This is premised on the fact that extant RNA molecules, unlike proteins, have the potential for self‐replication and also function to transcribe protein structures. Interestingly, the ability of RNA molecules to catalyze their own synthesis from activated monomers has been demonstrated in a controlled laboratory environment. [15] Accordingly, RNA is thought to have played a key role in the evolution of prebiotic replication. [8]
The RNA world theory suggests that RNA fragments first emerged from simple prebiotic molecules (Scheme 3). These early ribozymes are then believed to have formed complexes with amino acids, promoting their condensation, and leading to formation of the first peptides. Over time, the peptides grew to develop a complex tertiary structure that enhanced catalytic activity and selectivity. The diversification of advanced peptides coupled with their greater chemical stability allowed them to replace RNA as biocatalysts, eventually leading to the modern‐day enzymes. Indeed, ribozymes typically have turnover times of several minutes, whereas protein‐catalyzed reactions are often ten to one hundred thousand times faster. It is, therefore, unlikely that RNA would have continued to function primarily as a biocatalyst after the rise of functional proteins. Instead, RNA began to assume different roles in the evolution of modern biology, such as transcription and expression of the genetic code. Further specialization gave rise to double‐helix DNA and, consequently, any key role as an essential biological catalyst was relinquished. Therefore, it is nowadays assumed that RNA preceded proteins, which preceded DNA.
Scheme 3.
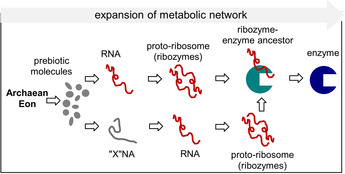
Facettes of the RNA world hypothesis (from prebiotic molecules via ribozymes and protoribosome to enzymes). The possible role of coenzymes is not depicted (for this refer to Scheme 2).
Another theory suggests that structurally simpler pre‐RNA or “XNA” molecules may have emerged from prebiotic building blocks as a forerunner to the RNA world. This alternative pre‐biological (or nonbiological) system may have supported some of the basic functions of life, acting as a road map for the evolution of RNA and peptide structures. A primordial self‐replicating “XNA” world would presumably have had some degree of chemical compatibility with respect to RNA building blocks and base pairing to facilitate transition from one to the other. However, the theory is speculative and there are no extant structural artefacts that appear to suggest such a pre‐RNA world. The closest idea of a pre‐RNA world has been discussed for inosine, the first biosynthetic intermediate with subsequent conversion into the other purine nucleotides. Based on this and the pairing ability of hypoxanthine, a role in pre‐RNA variants has been suggested. [16a] As we are bound to develop a retrospective view of the evolution of life, it is very challenging to verify or falsify the “XNA” hypothesis through experimentation. [16]
Despite being largely replaced by more robust protein structures, RNA has maintained its specialized biocatalytic role as a synthesizer of peptides. As Steitz and co‐workers put it very briefly: “The ribosome is a ribozyme”. [17] Mechanistically, Yonath and co‐workers postulated that ancient proto‐ribosomes may have catalyzed the formation of early peptides through phosphoanhydride‐activation of amino acid monomers. [14a] This hypothesis, which was critically commented, [14b] stems from the sequence similarities between key catalytic elements found in the peptidyl transferase active site of the ribosome and sequences of in vitro selected RNAs having related catalytic activity. The proto‐ribosomes were supposedly of dimeric nature, with an active symmetrical cavity for accommodating two substrates oriented face‐to‐face.
Consistent with the notion that RNA gave rise to prebiotic catalysis, ribozymes could have principally been responsible for the synthesis of coenzyme molecules or simpler functional analogues of the extant coenzymes (Figure 2, top left). However, from a chemical viewpoint, the diversity of known ribozyme‐catalyzed transformations is rather small, thus limited to acid/base‐promoted hydrolytic reactions and selected group‐transfer reactions of nucleotidic and peptidic elements. More complex structural transformations, such as redox chemistry, alkylations, and C−C bond‐forming reactions depend on co‐catalysts, including the known coenzymes FMN and FAD (flavins) (2 a,b), S‐adenosylmethionine (SAM, 3), thiamine pyrophosphate (TPP, 4) and tetrahydrofolate (THF, 5; Figure 1). To construct more elaborate organic molecules, these co‐catalytic small molecules (or simpler analogues thereof) must have already existed so that functional co‐ribozymes could arise (Scheme 2 A and Figure 2 top right). [18] Presumably, this association, pivotal to the evolution of biological catalysis, was achieved in a prebiotic world through small‐molecule binding to the RNA template through hydrogen bonding and/or electrostatic interactions that resemble most extant coenzyme/protein complexes (Figure 2, top right). [19a] Alternatively, coenzymes may have covalently linked to the 5′‐terminus of a ribozyme (Figure 2, bottom). Indeed, this strategy of expanding the metabolic scope of RNA molecules may be evidenced by the fact that the coenzymes FAD (2 b), a derivative of FMN (2 a), nicotinamide (NAD, 8), and coenzyme A (CoA, 10) all include the adenosine monophosphate handle (AMP handle) as a structural element. This may indicate the association of these coenzymes with RNA molecules in a very ancient RNA world.[ 8a , 11c ]
Figure 2.
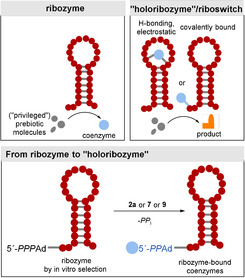
Top: Ribozyme and “holoribozyme”/riboswitch concepts for the role of coenzymes in the RNA world. Bottom: Covalent attachment of FMN (2 a), NMN (7), and pantetheine phosphate (9) to ribozyme by “self‐catalysis”.[ 3d , 19 ]
Figure 1.
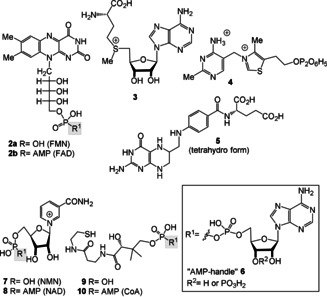
Coenzymes FMN/FAD (2 a,b), SAM (3), and TPP (4), THF (5), NMN/NAD (7/8), and pantetheine phosphate/coenzyme A (9/10). The “AMP‐handle” 6 is found in FAD, NAD, and CoA and FMN, NMN, and pantetheine phosphate (note: phosphate groups are presented fully protonated throughout the text).
Recently, Huang et al. used in vitro selection starting with 5′‐ATP initiation to isolate RNA sequences that are capable of synthesizing the ribozyme‐bound coenzymes FAD (2 b), NAD (8), and CoA (10) from their precursors FMN (2 a), nicotinamide monophosphate (7), and 4′‐phosphopantetheine (9; Figures 1 and 2, top right and lower half). [19b] This result demonstrates that modern coenzyme molecules can be accessed from their core structural elements by ribozymes. In a prebiotic world, this chemical elaboration would have led to an expansion of the metabolic portfolio of RNA and eventually the proteins and enzymes that replaced them. However, it does not show that the essential core structures themselves arise through biocatalytic pathways.
Today, the ability of small molecules, including coenzymes, to bind to RNA is manifested in riboswitches that are responsible for regulating gene translation. [20] Riboswitches are short, relatively simple sequences in mRNAs that bind metabolites directly, without the need for intermediary proteins. This interaction alters the secondary RNA structure, which can have the effect of activating or deactivating gene expression. There are many small molecules that regulate gene expression, including TPP (4), [21] FMN (2 a),[ 22a , 22b ] SAM (3),[ 22c , 22d ] THF (5), [22e] adenosylcobalamine (AdoCbl), [22f] and other related metabolites. [22g] Currently, more than a dozen classes or subclasses of riboswitches respond to nine nucleotide‐like coenzymes. In selected cases, it was shown that binding is linked to π‐stacking of nucleobases with the aromatic elements of coenzymes as well as Mg2+ complexation (Figure 2, top right).
Besides coenzymes, amino acids such as lysine and nucleobases such as guanine can also bind to riboswitches for gene regulation. [23] Lysine is central to the operation of aldolase class 1 enzymes, as an imine progenitor with dihydroxyacetone phosphate, which is a basic building block in the catalytic assembly of monosaccharides, and as a participant in many related acid/base‐catalyzed biotransformations. [24] Through its many roles as a co‐catalyst, one can envisage the importance of lysine complexation, or that of lysine‐containing small peptides, with polynucleotide fragments in an RNA world. The notion that riboswitch receptors have persisted through evolution, fulfilling a similar catalytic role throughout, is highly speculative. It is yet to be shown through experimentation that the complexes of riboswitches and coenzymes show catalytic activity that is different from ribozymes. [25]
Coenzyme molecules may also have participated in the evolution of RNA fragments to DNA, a more secure method of preserving nucleotide sequences that transcribe the synthesis of proteins. In the search for possible prebiotic models for dehydrogenases, Visser suggested that nicotinamide (as well as 5‐deaza‐FMN, see Figure 7 in Section 4.2) could fit in most double‐helix conformations, thus promoting ribonucleotide reduction (Figure 3). [3d] Depending on the orientation of the amide substituent, nicotinamide might act as an adenine analogue that forms a base pair with uracil. Alternatively, it may also serve as a guanine analogue that forms hydrogen bonds with cytosine. The theory was modeled on the known activation of nicotinamide through the hydrogen‐bonding interactions of extant dehydrogenases.
Figure 7.
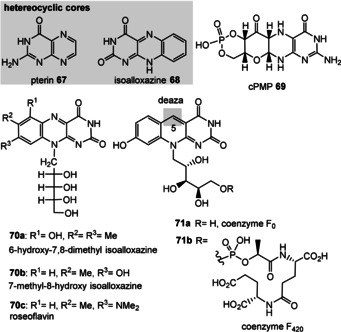
Structures of pterin (67) and isoaloxazine (68), natural redox flavin derivatives 69, 70, and the archaea coenzymes F0 and F420 (71 a,b); for structures of FMN (2 a) and FAD (2 b) see Figure 1.
Figure 3.
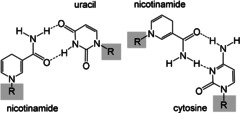
Nicotinamide (reduced form) as a possible dehydrogenase from a pregenetic code . [3d]
3. Evolution of Life: On Prebiotics and Protometabolism
3.1. Overview
Some evidence has been collected that suggests nucleotide‐derived coenzymes or related heterocycles being a component of the RNA world hypothesis. However, could the conditions on early Earth have favored the formation of such molecular entities?
Several different environments have been proposed as plausible sites for the origin of life on planet Earth, and among these, hydrothermal vents and hydrothermal fields have been most intensively discussed. [26] Five conditions in terrestrial environments (the Miller–Urey spark discharge experiment, the iron‐sulfur hypothesis, the formose reaction, HCN and formamide oligomerizations, and cyanosulfidic protometabolism) will briefly be discussed in this section because it is plausible to assume that under these conditions privileged building blocks for the prebiotic formation of coenzymes may have formed (Figure 4).
It is generally acknowledged that the origin of life did not occur at a single setting because of the range of possible pathways that require specific conditions (heat, light, catalytic surfaces, reductive environment, and coupling–cooling cycles etc.). [27] These had to be linked for intratransportation of products and reactants. It has to be noted that none of the scenarios discussed below are able to provide a general explanation for the appearance of chirality in life. [28]
3.2. The Miller–Urey Spark Discharge and Related Experiments
Miller's spark discharge experiment is regarded as the first attempt at an efficient abiotic synthesis of organic compounds under simulated primitive Earth conditions. [29] The classic experiment used a reducing gas mixture composed of H2, H2O, CH4, and NH3. It yielded different carboxylic acids including several proteinogenic amino acids (Figure 4, experiment 1). Under these conditions, HCN [30] and formaldehyde [31] as well as acetylene (19) [32] and cyanoacetylene (20) are also formed.
One of the early experiments conducted in Miller's laboratories that contained H2S and CO2 was recently re‐analyzed (Figure 4, experiment 2). [33] These conditions could have been prevalent on the early Earth before extensive continents formed. The analysis revealed the presence of additional amino acids, including ones that contained sulfur, carboxylic acids, and small‐molecular‐weight molecules with nitrogen and sulfur as heteroatoms, which were postulated to have resulted from cysteine degradation.
The limited structural diversity of molecules led to doubts on the relevance of the Miller–Urey experiment. In recent repetitions of this experiment, both electric discharge and laser‐driven plasma impact simulations were carried out in a reducing atmosphere containing NH3 and CO (Figure 4, experiment 3). [34] These experiments provided all four canonical nucleobases of RNA [adenine (A), guanine (G), cytosine (C), uracil (U)], supposedly formed after the generation of small radicals (H, CN, NH2) and formamide followed by 2‐amino‐2‐hydroxyacetonitrile (15), 2‐amino‐2‐hydroxymalononitrile (16), 2,3‐diaminomaleonitrile (17), 4‐amino‐1H‐imidazole‐5‐carbonitrile (18), and cyanoacetylene (20; Figure 5). [35]
Figure 5.
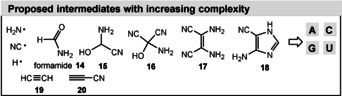
Proposed nucleobase precursors formamide and intermediates 14–18 of Miller–Urey experiments (purines: adenine (A) and guanine (G) from 18; pyrimidines: cytosine (C) and uracil (U) from 20 via cyanoacetaldehyde and urea.
The yields and compositions of the nucleobases, amino acids, and other products formed under different spark discharge and related irradiation and shock heating conditions strongly depend on the conditions such as pH value, dissolved species, and the redox state of the environment. [26] This also applies for the equilibrium between α‐hydroxy‐ and α‐aminonitriles formed, which depends on the presence of NH3 and is itself pH‐dependent. Higher pH‐values favour the presence of aminonitriles. Interestingly, formaldehyde strongly accelerates the hydrolysis of amino nitriles compared to hydroxynitriles and thus favors the formation of amino acids. [36]
3.3. Wächtershäuser's Iron–Sulfur Hypothesis
Hydrothermal vents provide a sustained source of chemical energy and, therefore, have widely been discussed as a site for the formation of prebiotic molecules. [37] First proposed by Günter Wächtershäuser, the iron–sulfur hypothesis suggests that deposits of iron sulfide minerals near deep‐sea hydrothermal vents are able to provide the reductive medium and energy to catalyze complex reaction sequences from simple precursors such as carbon monoxide and carbon dioxide, hydrogen cyanide, and hydrogen sulfide. It was shown that ammonia is generated from nitrate under these reducing conditions (FeS, H2S). [38] Methanethiol (CH3‐SH) and carbon oxysulfide (COS) are formed from CO2 and FeS/H2S, [39] or from CO and H2 in the presence of NiS. [38] This scenario provides methanethiol and, hence in the presence of nickel sulfide and iron sulfide, also S‐methyl ethanethioate (CH3C(O)SCH3; Figure 4). It was postulated that this thioester acts as a starting point for subsequent exergonic transformations [40] and is also involved in endergonic reactions, notably the formation of (phospho)anhydride compounds (see also Section 3.8). [41]
When HCN is present, which may have formed as a result of meteorite bombardments in the Archean Eon, [42] it became involved in the formation of pairs of α‐hydroxy and α‐amino acids such as glycolate/glycine, lactate/alanine, and glycerate/serine as well as pyruvic acid. This nickel‐catalyzed reaction proceeds after the formation of nickel cyanide in the presence of CO. [43] Furthermore, it is known that α‐keto acids including pyruvic acid react with ammonia in the presence of ferrous hydroxide or ferrous sulfide and H2S to furnish α‐amino acids, including alanine. [44] The reaction of α‐amino acids with COS or with CO and H2S in an aqueous medium leads to dipeptides and tripeptides (see also Scheme 14 in Section 3.8). [45]
Scheme 14.
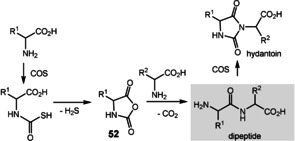
COS‐mediated peptide formation.
The attractive aspect of this theory is associated with the role of H2S, which also plays a key role as a reductant in Sutherland's cyanosufidic protometabolism (see Section 3.7). Furthermore, FeS is present in 16 classes of enzymes. [46] Conceptually, H2S has to be favored over H2 as an energy source. Typically, more H2S than H2 is exhausted from hydrothermal vents. The oxidation of H2S produces more energy than H2 oxidation and the former generates additional equivalents of H2. Indeed, the complete oxidation of H2S to H2SO4 releases eight electrons compared to the two electrons released by H2 oxidation. [47] It was noted, however, that the salty character of the sea hampers condensation reactions to form oligomers, and protocells are unlikely to form due to the huge osmotic pressure. Therefore, hydrothermal fields that are still present today (e.g. Yellowstone National Park, USA) have also been discussed as favorable prebiotic environments for the formation of protometabolic networks. [26]
However, the discussion in this Review is not focused on the issue of which specific environment coenzyme formation may have likely occurred in, but on principal chemical pathways that could have operated during the early days of our planet. In this way, Eschenmoser's quote made at the beginning will be a recurring theme.
3.4. The Formose Reaction
The formose reaction, discovered as early as 1861 by the Russian chemist Butlerow, [48] is thought to have played a key role in the prebiotic formation of simple sugars from the C1 building block formaldehyde. Commonly, the reaction proceeds under basic conditions (pH 10–11, 60–80 °C) in the presence of a divalent metal salt such as the chelator calcium hydroxide. The multistep formose reaction is iterative in nature and is based on aldol reactions, reverse aldol reactions, and aldose‐ketose isomerizations (Scheme 4). [49] It is initiated by the dimerization of formaldehyde to yield glycolaldehyde (11).
Scheme 4.
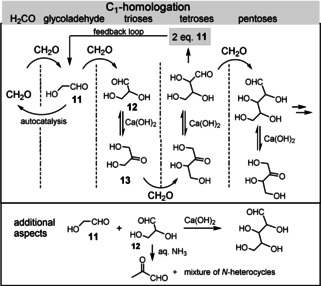
Schematic presentation of the formose reaction.
The mechanism of this rate‐determining first step is unknown and might be radically triggered. The next homologation step yields glyceraldehyde (12), which is equilibrated to dihydroxyacetone (13). From there, tetrulose is available which again can undergo ketose‐aldose isomerization in the presence of Ca(OH)2.
The resulting aldotetrose is homologated to pentoses and finally hexoses. [50] It is noteworthy that the retroaldol reaction of tetroses results in the formation of two equivalents of glycolaldehyde (11) and this step can act as a feedback loop. Importantly, glycolaldehyde itself exerts autocatalytic activity on the dimerization of formaldehyde, the rate‐determining step. However, the formose reaction yields complex mixtures, with ribose being formed in rather low yield (<1 %). [51] Furthermore, high concentrations (>0.1 m) of formaldehyde are required, thus raising doubts whether these would reflect prebiotic conditions. Another disadvantage is the chemical lability of ribose under the reaction conditions employed (t 1/2=73 min; 100 °C, pH 7). [52]
Additional aspects involve the direct synthesis of pentoses by the aldol reaction between glycolaldehyde (11) and glyceraldehyde (12) in the presence of Ca(OH)2. When 12 and ammonia are mixed, pyruvaldehyde and a complex mixture of N‐heterocycles are formed. Transferring this product mixture to a fresh aliquot of a glyceraldehyde solution leads to a gradual enhancement in the rate of pyruvaldehyde formation. So far, the autocatalytically active components are unknown. [53] Some of these major drawbacks were shown to be circumvented by the presence of borate minerals, as these preferentially promote the formation of ribose and two epimeric pentuloses. Indeed, the borate complex of ribose is particularly stable compared to all other products of the formose reaction. [54]
3.5. HCN Oligomerization
In 1961, Joan Oró pointed out that the nucleotide bases are oligomers of hydrogen cyanide. It was found that adenine is formed from concentrated aqueous ammonium cyanide (1–15 m) under refluxing conditions (Figure 4). [55] The process proceeds via the dimer 21, trimer 23, and tetramers 17 and 18, and was verified for single transformations under aqueous conditions (HCN→17 (11 %); [56] 23→17 (79 %); [56] 17→18 (80 %); [57] Scheme 5). [58] Two tetrameric constitutional isomers were postulated, of which 4‐amino‐1H‐imidazole‐5‐carbonitrile (18) directly leads to adenine (A) in the presence of formamidine or directly through a photochemically induced short track, or alternatively in the presence of guanidine (24) to 2,6‐diamino purine (18→adenine (3 %); [57] HCN/NH3→adenine (22 %) [56] ).
Scheme 5.
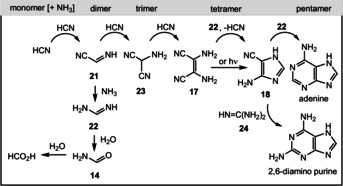
Oligomers of HCN and of ammonia adduct formamidine (22) as well as hydrolysis products formamide (14) and formic acid.
Additionally, several amino acids are generated from HCN and ammonia under these conditions, although their structures have not been proven unequivocally. Other RNA and DNA nucleobases can be obtained through simulated prebiotic chemistry in a reducing atmosphere. [56] The tetramerization of HCN is accelerated by the presence of formaldehyde. [60] However, it was reported that nucleobases have relatively short lifetimes in aqueous solution (t 1/2: 1 year for adenine, 19 days for cytosine, 0.8 years for guanine, 12 years for uracil at 100 °C, pH 7), which is of relevance when considering that high concentrations are required for the following oligo‐ or polymerization.
Dimer 21 can also undergo aminolysis to formamidine (22) and, under aqueous conditions, to formamide (14) and further to formic acid, with the latter being discussed as a possible prebiotic reductant. And formamide is an alternative to HCN for oligomerizations as discussed in Section 3.8.
Related to the oligomerization of HCN is a protometabolic network based on the cyanide cyanogen (25) and cyanoacetylene (20), which was mainly established by the groups of Orgel and Eschenmoser (Scheme 6).[ 61 , 62 , 63 , 64 ] This extension of options allows the number of accessible pyrimidines to increase, a necessity for proposing prebiotic pathways towards the heterocyclic cores of pterin‐containing coenzymes (see Section 4.2). Cyanogen (25) is the starting point for cyanamide (26), guanidine (2), and urea (27), as well as ammonium cyanate. Cyanoacetylene (20) is a suitable precursor for the malono derivative 28 and plays a role in the protometabolic scenarios proposed by Sutherland and Carell (see Section 3.7).
Scheme 6.
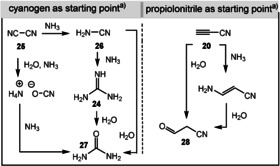
Protometabolic networks leading to cyanamide (26), guanidine (24), urea (27), and cyanoacetaldehyde (28). a) Formed under spark discharge conditions. Some transformations have only been proven under high‐temperature conditions (>200 °C).
3.6. Formamide Oligomerization
Closely related to the C1 building block HCN is its formal hydrate formamide (Figure 4). [65] Compared to HCN, formamide is less reactive, more soluble in water, and has a high boiling point. A series of studies under high‐energy conditions, the first one published as early as 1980, [66] were conducted with formamide, which is formed from ammonia and CO or formic acid at high temperature using different heterogeneous catalysts such as calcium carbonate, silica, alumina, silver metal oxides, and others or upon UV irradiation as well as proton and heavy‐particle radiation (Figure 4 and Scheme 7). [67]
Scheme 7.
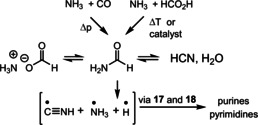
Prebiotic formamide and nucleobase formation.
The group of Saladino also detected the formation of carboxylic acids and amino acids and, importantly, also urea and carbodiimide using different meteroite‐derived heterogeneous additives (Figure 4). [68] In addition, scenarios have been discussed in which formamide serves as a geochemically plausible solvent for the formation of prebiotic building blocks. [69]
3.7. Sutherland's Cyanosulfidic Protometabolism
The prebiotic conditions described so far provide privileged prebiotic building blocks such as amino acids, nucleobases, and sugars which are of relevance for supporting the RNA‐world theory. However, except for the possibility of creating simple peptides under the influence of COS (see Scheme 14 in Section 3.8), these routes do not provide larger and functionally complex building blocks, especially not nucleosides or nucleotides, respectively. The formation of the N‐glycosidic bond in RNA monomers has been a topic of critical discussion for several decades. It is the key issue to be solved, especially in confirming the RNA‐world hypothesis, but in the absence of phosphate activation its formation is principally endergonic.[ 11 , 13 ] Conceptually, the biotic nucleoside formation approach separates sugar from nucleobase syntheses and merges both elements in the final step.
Powner, Sutherland, and co‐workers suggested an unconventional and rather comprehensive theory (Figure 4). It bypasses the need for the direct formation of an N‐glycosidic bond. The route to activated ribonucleotides is based on what has been termed the cyanosulfidic chemical homologation process.[ 11c , 16 ] This theory provides the key building blocks of RNA, proteins, and lipids only from hydrogen cyanide as the sole carbon and nitrogen source. Hydrogen sulfide serves as a reductant under UV‐irradiation conditions in the presence of CuI/CuII catalysis. In fact, this combination acts as a photoredox system. [70] As a result, glycolaldehyde (11), glyceraldehyde (12), and dihydroxyacetone (13) are generated, being formed from the corresponding cyanohydrins after H2S reduction (Figure 4 and Scheme 8).
Scheme 8.
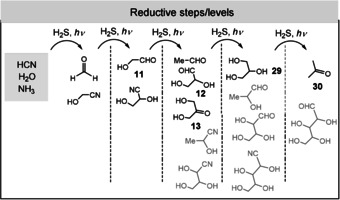
Sutherland's cyanosulfidic protometabolism based on the photochemical Kiliani–Fischer reaction, which can be accelerated by copper(I/II) photoredox cycling (structures that are drawn in gray were not reported by Sutherland and co‐workers, but could principally form).
Importantly, the reductive properties also allow “reduced sugars” such as glycerol (29; lipid building block) and acetone (30) to be generated. [11c] Higher sugars such as tetroses, pentoses, and hexoses should principally be accessible following this reductive homologation logic, but these are not relevant in Sutherland's prebiotic approach to ribonucleotides. In fact, glycolaldehyde (11) and glyceraldehyde (12) and not ribose are the sugar components in their prebiotic ribonucleotide synthesis. Moreover, several reactions were found to be promoted by inorganic phosphate. In a sequential process, 2‐aminooxazole (31) is first formed from glycolaldehyde (17) and cyanamide (25). In the presence of glyceraldehyde (12), 31 furnishes arabinoside 32 and the corresponding ribo‐derivative, bearing an oxazole ring at C1 and C2 (carbohydrate numbering; Scheme 9). Then, the pyrimidine ring is formed upon reaction with cyanoacetylene (20) and the phosphate‐mediated ring opening with inversion of the configuration at C2 to yield the phosphorylated cytidine derivative 33. Finally, photochemical tautomerization and hydrolysis furnishes the corresponding uridine derivative 34. [16b]
Scheme 9.
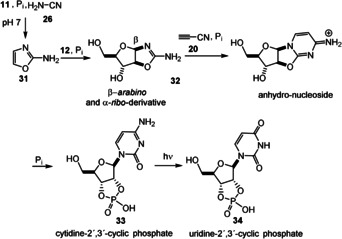
Synthesis of cytidine‐2’,3’‐cyclic phosphate (33) and uridine‐2’,3’‐cyclic phosphate (34) via 2‐aminooxazole (31).
Unlike pyrimidine ribosides, finding prebiotic routes towards purine ribosides turned out to be challenging, when directly coupling, for example, adenine with ribose under extreme conditions. [71] This observation is associated with the lack of regiocontrol and reactivity. First attempts to access purine‐based ribonucleotides yielded advanced intermediate 35 (Scheme 10). [16b] It was achieved by mixing the three established building blocks 12, 18, and 31 at pH 4–5 and this mixture underwent a Mannich‐type multicomponent reaction. From here, the prebiotic conversion of pyrimidine and purine anhydronucleosides into Watson–Crick base‐pairing arabino‐furanosyl nucleosides in water was demonstrated. [72] Sutherland's theory circumvents the formation of N‐glycoside formation by exploiting heterocyclic reactions. In other words, the theory separates the C‐C/C‐O chemistry of sugars from the C‐N chemistry of nucleobases. Interestingly, the possible formation of myriads of products is tamed to a few key intermediates by the presence of phosphate.
Scheme 10.
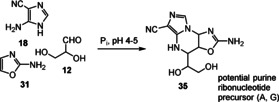
Multicomponent Mannich reaction towards potential purine precursor 35.
The recent work by Carell, Becker and co‐workers is in line with such thinking, in that formamidopyrimidines 36, accessible from formic acid and aminopyrimidines, are excellent precursors to form N‐9‐purine ribosides 38 with sugars such as ribose 37 (Scheme 11 A). [73a] The aminopyridines themselves derive from the reaction of the trimer of HCN (23) and formamidine (22). [74] The underlying concept follows the idea that the N‐glycosidic bond is generated first through imine formation and the imidazole ring is constructed afterwards by nucleophilic attack of the O,N‐acetal on the formamide followed by elimination of water. Thus, the timing of the glycoside and nucleobase formation is reversed compared to classical chemical glycosylation approaches. Although not expressed explicitly by the authors, this work provides a direct link to the biosynthesis of the coenzyme thiaminepyrophosphate (TPP, 4) in bacteria and plants, particularly with respect to the ring closure that yields 5‐aminoimidazole ribonucleotide (AIR, 38; Scheme 11 B). [18]
Scheme 11.
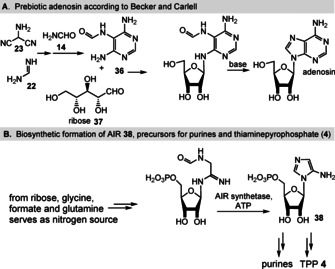
A) Formamidopyrimide (36) is a precursor for purine ribosides, such as adenosine, according to Carell and Becker. [73a] B) Comparison with the biotic formation of AIR (38), [18] the precursor of purines and TPP (4).
Very recently, the same group extended their studies to the formation of pyrimidine nucleosides. By using a similar prebiotic milieu as disclosed for the purine nucleosides they achieved a unified approach towards nucleotides (Scheme 12). [73b] Cyanoacetylene (20) readily reacts with hydroxylamine (39; formed from nitrite, which is partially reduced by sulfite) to 3‐aminoisoxazole (40). In the presence of urea (27), isoxazolylurea (41) was shown to be the next intermediate that reacts to form urea adduct 42 in the presence of ribose (37). Finally, an iron(II)‐catalyzed reduction of the N−O bond of the isoxazole moiety initiates formation of the pyrimidine ring and consequently nucleoside 43.
Scheme 12.
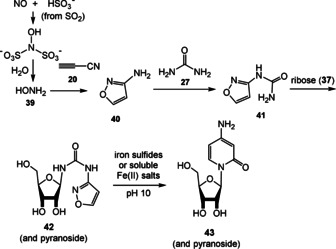
Prebiotic plausible synthesis of pyrimidine‐RNA building blocks according to Becker and Carell. [73b]
The chemistry of Becker and Carell requires so‐called wet‐dry cycling, in which the evaporation and concentration of the reactants favor condensation reactions. This is followed by redissolving the concentrates. Such cycles are of much broader relevance for the creation of complex protometabolisms and are found in ponds and hydrothermal fields. [26]
Returning back to Sutherland's cyanosulfidic protometabolism, one has to discuss a second feature. It commences from acetylene (19), which was shown to react with HCN to yield acrylonitrile (44) or cyanoacetylene (20) depending on the oxidation state of the copper catalyst (Scheme 13). One strand leads to aminopropionitrile (45), which can either serve as a precursor for arginine after condensation with cyanamide (26), [75] or allows access to proline via 3‐aminopropanal, 4‐amino‐2‐hydroxybutanenitrile (46), and pyrrolidine‐2‐carbonitrile (47). Cyanoacetylene (20) is the starting point for the second strand, which can furnish either glutamine and glutamate (via maleonitrile (48) or succinonitrile and asparagine and aspartate (via 2‐aminosuccinonitrile (49)). Eschenmoser and Koch [74b] found mild conditions to partially hydrolyze α‐aminonitriles to the corresponding amides. Importantly, formaldehyde promotes amide formation after cyclization of the O,N‐acetal and formation of oxazolidin‐5‐imine (Scheme 13, bottom). [76] Prolonged reaction times and access of formaldehyde led to further reactions mainly resulting in aldol products.
Scheme 13.
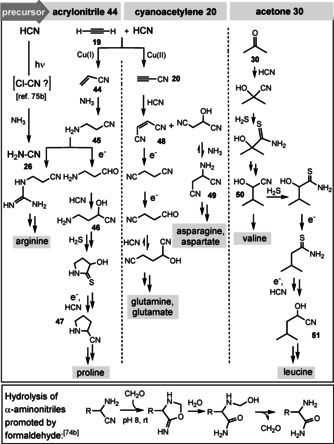
Prebiotic formation of selected amino acids according to the cyanosulfidic protometabolism (top) and formaldehyde‐promoted nitrile hydrolysis (bottom).
Acetone (30; see also Scheme 8) was shown to serve as a precursor for the amino acids valine and leucine, with 2‐hydroxy‐3‐methylbutanenitrile (50) as an intermediate; an alternative route proceeds via 2‐hydroxy‐3‐methylbutanethioamide and 2‐hydroxy‐4‐methylpentanenitrile (51). [11c] Finally, within the concept of cyanosulfidic protometabolism, carbohydrate precursors can serve as a starting point for the formation of amino acids (2‐hydroxyacetonitrile→glycine; glycolaldehyde (11)→alanine, and threonine; 2,3‐dihydroxypropanenitrile→serine. [11c]
3.8. Functional Group Activations and Group Transfer Reactions
The formation of esters and amides are key reactions in the biotic and prebiotic worlds. These are formed by oligo‐ and polymerizations of amino acids to form peptides [77] or nucleotides that result in nucleic acids and require either the activation of the carboxy group typically as a mixed anhydride or the alcohol function as a phosphate ester. Functional group activation could be of relevance when considering coenzymes such as SAM and ATP or simpler analogues being likely created under palaeogeochemical conditions. Several synthetic scenarios and solutions for obtaining these high‐energy intermediates have been reported.
Mixed anhydrides: COS (see also hydrothermal vents in Figure 4), formed from the condensation of CO and H2S, is regarded as being a prebiotic key player in the activation of amino acids and, hence, has been made responsible for peptide formation (Scheme 14). Thus, α‐amino acids are activated via the corresponding thiocarbamates as carboanhydrides (Leuchs anhydride) 52. This process is kinetically accelerated in the presence of metal cations, including Fe2+ and an oxidizing agent. Moreover, K3Fe(CN)6 was found to be an efficient promoter. [78] Finally, the oxidative dimerization of thioacids also provide good acylating species for the formation of peptides. [79]
Phosphate esters: Finding answers to the question of how organic phosphates could have been formed under palaeogeochemical conditions should pave the way to expand protometabolic networks. Phosphate esters are important as structural elements (e.g. nucleic acids) as well as for the chemical activation of organic molecules, including coenzymes. However, the quest for abundant phosphate in a prebiotic world was found to be a delicate topic. Orthophosphate is only able to phosphorylate alcohols when utilized in deep eutectic solvents (urea and choline chloride). [80] In addition, inorganic phosphate is sequestered by calcium in the unreactive mineral hydroxyapatite, only soluble at a pH value of around 4.5, so that free inorganic phosphate is thought to have been scarce on early Earth. Mg2+ ions and borate are able to sequester phosphate from calcium to form the mineral lüneburgite. Ribonucleosides that are stabilized by borate mobilize borate and phosphate from lüneburgite, which leads to regioselective phosphorylation of nucleosides by the mineral. [81] Furthermore, formamide and phosphate minerals also promote the phosphorylation of nucleosides. [82]
Besides mineral activation, several abiotically formed small molecules were found to activate inorganic phosphate with subsequent phosphorylation of alcohols and carboxylic acids. These are a) COS, b) cyanamide (H2NCN, 26), a tautomer of carbodiimide, c) urea, [83] the hydrate of diimide, and d) diamidophosphate (DAP, 57) and amidotriphosphate (AmTP, 58) (Scheme 15).
Scheme 15.
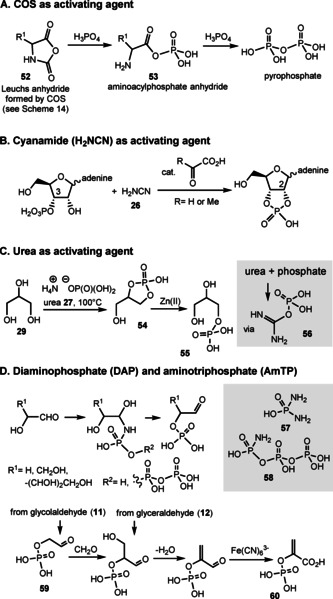
Examples of phosphorylations under prebiotic conditions (reagent systems A–D).
COS and orthophosphate in the presence of an α‐amino acid yield the corresponding aminoacyl phosphate anhydride 53 along with pyrophosphate (Scheme 15 A). Importantly, pyrophosphate is a candidate for being a primary chemical energy donor in prebiotic systems or even early forms of life. [84] Again, the Leuchs anhydrides 52 can serve as key intermediates for this transformation. [85] Cyanamide (26) rapidly activates phosphate in the presence of glyoxylate or pyruvate. These act as catalysts in the phosphorylation of adenosine‐3′‐phosphate and the generation of adenosine‐2′,3′‐cyclic phosphate (Scheme 15 B). [86] Urea (27) forms adducts 56 with ammonium dihydrogen phosphate. In the presence of glycerol (29) this reagent system yields cyclic phosphate diester 54, preferentially affording regioisomeric monoester 55 after treatment with zinc(II) salts (Scheme 15 C). This process was claimed to be an entry into the world of lipoidic glycerate esters. [87]
Eschenmoser und co‐workers showed that the product complexity of the formose reaction is greatly suppressed by using glycolaldehyde phosphate (55). Glycol aldehyde (11) is phosphorylated in the presence of diamidophosphate (57) or amidotriphosphate (58), which are known to be formed by ammonolysis of cyclotriphosphate in water (Scheme 15 D). Cyclotriphosphate itself is not effective as a phosphorylating agent under such conditions, due to the ability of amidotriphosphate to reversibly form a carbonyl addition product.[ 87a , 87b , 87c ] Another important example is the preferred 2‐phosphorylation of d‐ribose. This selectivity pattern blocks entry of the key aldehyde components glycolaldehyde (11) and glyceraldehyde (12) into ribonucleotide syntheses. [88] The versatility of this approach was further demonstrated by Powner et al. using a modification of Eschenmoser's phosphorylation procedure. High‐energy phosphate phosphoenol pyruvate (60) was obtained from prebiotic nucleotide precursors glycolaldehyde (11) and glyceraldehyde (12). The sequence also included the oxidation of the enal intermediate to pyruvate 60, for which hexacyanoferrate(III) was found to be a suitable oxidant. [89] Related to these amino‐activated phosphates 57/58, is thiophosphate (HSPO3H2). Within the cyanosulfidic world it served to phosphorylate adenosine, however, without regioselectivity. [87d]
The formation of high‐energy phosphoanhydrides such as ATP (62) and adenosine diphosphate was reported by Yamagata. [90] These are formed from adenosine monophosphate (AMP) and calcium phosphate in the presence of cyanate as the condensing agent in a mildly acidic environment. Finally, formamide, has also been discussed to promote phosphate donor formation with phosphate mineral sources. [91] Phosphorylation is not only a key issue for prebiotic nucleotide chemistry but also for coenzymes such as pyridoxal phosphate (1). Most of these have retained a phosphoester group as a structural element that is not directly involved in individual steps of the catalytic process, but often exerts anchoring properties.
Other group transfer reactions: Isocyanic acid (HNCO) is considered to have been present on the archaean Earth as a result of the decomposition of urea (27). This assumption is further supported by the observation that HCNO was detected in interstellar gases and on comet 67P/Churyumov‐Gerasimenko. [92] The reaction of this highly reactive C1 building block with methylamine [93] provides N‐methylurea, which can decompose to methylcyanate and ammonia (Scheme 16). Methylcyanate provides methylurea derivatives in reactions with amino groups, including adenosine. Alternatively, nitrosylation of methylurea (61) would provide diazomethane, a strong methylating agent. Nitrate, required for nitrosylation, could have been available from nitrogen via NO and NO2 during lightning. [94] It has to be noted that these types of methylation are abiotically triggered and not mediated by small molecules such as the coenzyme SAM (3).
Scheme 16.
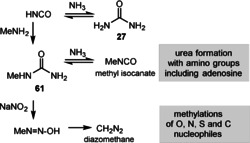
Possible routes for prebiotic methylations and carbamoylations. [86]
4. On the Prebiotic Evolution of Coenzymes
4.1. General Considerations
The prebiotic networks of Sutherland and Carell combined with the functional group activation procedures are particularly powerful as they bypass one endergonic step, namely the N‐glycosylation to nucleotides. Furthermore, feasable solutions for the phosphorylation of alcohols that bypass the need for adenosine triphosphate (ATP, 62) were reported. Having elementary prebiotic building blocks such as nucleobases, α‐amino acids, and sugars at hand that are formed from only a very few achaean molecules (NH3, HCN, H2S, CH2O, HC(O)NH2, COS, CO2, H2, N2) from which protometabolic networks emerged,[ 16 , 61 ] we return to the question whether coenzymes or simpler analogues derived therefrom might have been part of this intrinsic structural propinquity of biomolecules and prebiotic building blocks.[ 3 , 5 ] The poor (catalytic) activity of such prebiotically formed coenzymes could be overcome by binding to short RNA fragments. Such a scenario should have dramatically expanded the range of possible protometabolic transformations. A list of organic transformations promoted by biotic coenzymes are found in Table 1.
Table 1.
Most important organic reactions promoted by biotic coenzymes.
|
Chemical Transformation |
Coenzymes |
|---|---|
|
activation of alcohols |
ATP (62) |
|
activation of carboxylic acids |
ATP (62), coenzyme A (10) |
|
C1 transfer reactions |
biotin (64), THF (5), SAM (3) |
|
reductions, oxidations |
NAD (8), FAD/FMN (2), lipoic acid (63), PQQ (65) |
|
chlorinations, brominations |
FAD/FMN (2) (requires O2) |
|
aminations, racemization, eliminations, decarboxylations, Michael additions, retro aldol reactions, |
PLP (1) |
|
acylanions and transformations (benzoin‐type reactions) |
TPP (4) |
|
S‐ylid chemistry |
SAM (3) |
|
radical chemistry |
SAM (3) (with [FeS] cluster), heme, cobalamines, and others all derived from urogen III (66). |
Important coenzymes 1–5, 8, and 10 have been listed earlier and these are complemented in Figure 6 with coenzymes 62–65 as well as urogen III (66), a biosynthetic precursor of heme, coenzyme F430, cobalamines, and siroheme.[ 3a , 18 , 95 ] Structurally, four coenzymes (SAM (3), NAD (8), coenzyme A (10), and ATP (62)) are composed of ribonucleoside or nucleotide units. Moreover, several other members are biosynthetically formed from nucleotides. These include the riboflavins FMN/FAD (2) and tetrahydrofolic acid (THF, 5) that are derived from guanosine triphosphate (GTP). These facts have been taken as an argument suggesting that coenzymes and the RNA world very likely evolved simultaneously. [11b] In contrast, lipoic acid (63) is a fatty acid derivative, biotin (65) a hybrid composed of a fatty acid and the amino acid glycine, while PQQ (66) originates from a dipeptide (glutamic acid and tyrosine). PLP (1) and the pyridine ring in NAD (8) are either derived from sugars or from a sugar and the amino acid aspartic acid, whereas possible prebiotic building blocks leading to thiamine pyrophosphate (TPP, 4) are ambiguous, because three different, chemically rather complex biosynthetic routes evolved in plants, bacteria, and fungi. [96]
Figure 6.
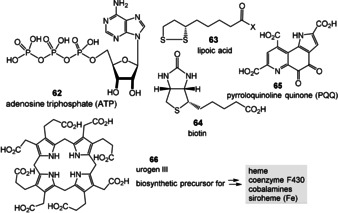
Coenzymes ATP (62), lipoic acid (63), biotin (64), PQQ (65), and urogen III (66), with the last being a precursor for key iron and cobalt‐dependent cofactors.
Eschenmoser claimed that the origin of life has to be reinvented. [1] Nevertheless, can one raise the question, how sensible is it to extend this reinvention step to small molecules that are thought to exist in the RNA world and serve as a raw model for structurally related coenzymes whose evolutionary foundations and biosynthesis lie in the protein world? An important link between the prebiotic and the biotic world are privileged building blocks discussed in the previous section because these serve as the basis of the transitional RNA world. Based on the assumption that privileged prebiotic building blocks are not solely linked to RNA but also at least to some coenzymes, possible scenarios are discussed for the formation of selected members or simpler analogues that experienced structural alterations once their formation was under the control of the protein regime. The structural changes during evolution were likely directed towards chemical reactivity and binding properties for the macromolecular template. As many coenzymes are derived from nucleotides, proposals on their prebiotic formation should exploit the flexibility suggested by Sutherland (mixing small carbohydrate building blocks with HCN oligomers) and Carell (imine formation instead of N‐glycosidation).
Beyond the question of how to make coenzymes part of the RNA world theory as discussed in Sections 1 and 2, their possible formation under palaeogeochemical conditions has to considered too, and this will be covered in the following.
4.2. Considerations on the Prebiotic Formation of Pterin‐ and Flavin‐Derived Coenzymes
When experimentally pursuing possible prebiotic scenarios that give coenzymes or simplified analogues, one is entitled to choose harsher geochemical conditions than commonly exist for their biosynthesis. [97] Several extant coenzymes such as FMN (2 a) and FAD (2 b) as well as related derivatives 70, coenzyme F420 (71) and remarkably also folic acid (5) are based on heterocyclic elements that are associated with pterin (67) and the tricyclic isoalloxazine (68) cores.
Flavins: Riboflavins (2) are coenzymes that promote a diverse number of redox reactions. In their reduced form (e.g. FMNH2 and FADH2), they commonly perform hydrogen transfer reactions in a single‐electron transfer mode. In combination with oxygen, peroxyflavin species are generated that provide a single atom of molecular oxygen or halogen. [98] However, for the reason that these types of oxidations require molecular oxygen, this additional reactivity very likely appeared later in the evolution of life. [99] Other known natural flavin derivatives 70 with redox properties have different substitution patterns in the benzene ring (Figure 7).
Cyclic pyranopterin monophosphate (69, cPMP) is a biosynthetic precursor of the dithiolene group bearing molybdopteridin. This binds molybdenum to furnish the molybdenum cofactor (MoCo). Molybdenum‐containing enzymes are regarded as being ancient redox enzymes. [100] As a consequence of their privileged role in life, methanogens, which belong to prokaryotic archaea, rely on several unique coenzymes. For example, coenzymes F0 and F420 (71 a,b), 5‐deazaflavin derivatives of FMN (2), and others are involved in the formation of methane, which is a metabolic by‐product under hypoxic conditions. [101]
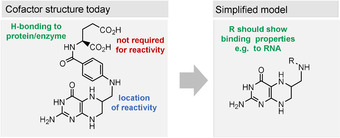
Like many other coenzymes, riboflavins contain structural elements, for example, the ribityl substituent, that is not essential for specific redox properties (Figure 8). These elements are important for binding to the macromolecular template—proteins in the biotic world. A first hint that simplified pterins may be of relevance as prebiotic redox models is the oxidation product of guanine 8‐oxo‐7,8‐dihydroguanine (72). It still shows redox properties similar to FMN (2), as was shown when being covalently incorporated into DNA or RNA strands. In this environment, 72 is able to promote photorepair reactions (Scheme 17). However, 72 has not been tested as a reductant or oxidant for organic substrates yet. [102]
Figure 8.
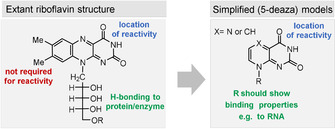
Structural and functional elements of riboflavins (oxidized form) and proposed simplified model.
Scheme 17.
8‐Oxo‐7,8‐dihydroguanine (72), the redox active derivative of guanine, and the flavinium cation (73; both are presented in their oxidized form).
Contemporary trends in biotechnology demonstrate that simplified and cheaper analogues of coenzymes show similar chemical properties to extant coenzymes, and that these can be employed in vitro as well as in vivo. The flavinium cation (73) accelerates the rate of NAD(P)H oxidation by three orders of magnitude. [103] It is worth noting that such flavinium cations were shown to also operate in intracellular redox regulations of E. coli.
First efforts to prepare pterin derivatives from prebiotic molecules were conducted by Ried and co‐workers 40 years ago. [104] Pyrolysis of solid compositions of three different amino acids at 160–200 °C for several hours provided complex product mixtures from which pterin derivatives 74 and 75 were detected by mass spectrometric analysis. However, further structural assignments, including stereochemical aspects of the products, were not carried out (Scheme 18). In fact, it is arguable whether such conditions are truly prebiotic, except for hydrothermal vents or fields. [26]
Scheme 18.
Pyrolytic formation of pterin derivatives 74 and 75 from amino acids.
A closer look at the biosynthesis of FMN/FAD (2) provided an advanced model for its prebiotic formation. The amazing aspect of riboflavin biosynthesis is the disproportionation of two molecules of the bicyclic 6,7‐dimethyl‐8‐d‐ribityllumazine (76) that results in the formation of one molecule of the tricyclic riboflavin (77) and one monocyclic 5‐amino‐6‐d‐ribitylaminouracil (78; Scheme 19). [98] The latter and ribulose‐5‐phosphate (80) can serve as biosynthetic precursors for d‐ribytillumazine (76) in a two‐step process via 3,4‐dihydroxybutanone‐4‐phosphate (82). [98] Mechanistically, both enzyme‐catalyzed transformations are extraordinary. Even more remarkable, however, is the fact that this disproportionation is also achieved in vitro in the absence of any enzymes as was discovered by Wood and co‐workers.[ 99 , 100 , 105 , 106 ] Both the chemical as well as the enzyme‐catalyzed transformations proceed with the same regiocontrol. In addition, the initial biotransformation was also mimicked in the absence of enzymes when ribulose 1,5‐bisphosphate (81) and pyrimidine (79) were heated and lumazine derivative 76 was formed.[ 3b , 107 ] Likewise, the condensation of 74 and 3,4‐dihydroxy‐2‐butanone 4‐phosphate (82) yields ribityllumazine derivative 76 in the absence of lumazine synthase. [108]
Scheme 19.
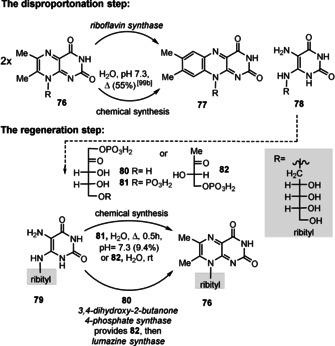
Late‐stage biosynthetic steps towards riboflavins 77 and corresponding biomimetic chemical transformations.
However, a prebiotic route to 5‐amino‐6‐d‐ribitylaminouracil (79) has not been fully established so far. 5,6‐Diaminouracil (84) could be generated from guanidine (24) and 2‐aminomalononitrile (23), which yields pyrimidine‐2,4,5,6‐tetraamine (83). Attempts to transform this pyrimidine derivative into 84 turned out to be a challenge under basic conditions. [74b] However, under acidic conditions, two partially hydrolyzed products 86 and 87 as well as the uracil derivative 84, a minor product, were formed (Scheme 20). [74b] It is debatable whether the reaction conditions resemble a prebiotic environment. Even more importantly, the formation of the ribityl derivative 78 from 84 under such conditions has not been reported yet. Principally, this should be a straightforward task if one considers the regioselective reductive amination between 78 and ribose (37), for which formic acid was proposed as a reductant. [74b] As an alternative, photocatalytic reduction with H2S as a hydrogen donor could also be envisaged. It was found, however, that the lack of nucleophilicity of the amino group at position 6 compared to position 5 is a major obstacle of this step, as the former amino group is in principal a vinylogous amide, so that imine formation was found to preferentially occur at position 5. [74b]
Scheme 20.
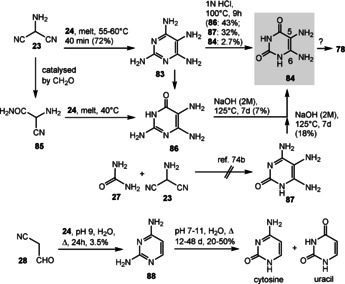
Prebiotic synthesis of different pyrimidine derivatives 84, 86–88, cytosine, and uracil.
A variation of this approach starts with the partial hydrolysis of α‐aminonitrile (23) and synthesis of amide 85 (see also Scheme 13, bottom field). In a similar manner, the reaction with guanidine (24) yields pyrimidine derivative 86, which can also be hydrolyzed under basic conditions to furnish uracil derivative 84. It was noticed that guanidine cannot be exchanged by urea (27) in these condensation reactions, so that 4,5,6‐triamino‐2‐pyrimidinone (87) could not directly be accessed. [74b] Related to these results are studies reported about five decades ago by Orgel and co‐workers. [62] Guanidine (24) and cyanoacetaldehyde (28; see Scheme 6) react in a dilute aqueous solution to afford 2,4‐diaminopyrimidine (88), which in turn is hydrolyzed to cytosine and uracil under rather mild conditions.
Recently, Carell and co‐workers suggested a prebiotic alternative to a series of pyrimidines (uracil, cytosine, 4‐thiouracil, and aminomethylated cytosines) [109] which could have played a role as building blocks for prebiotically relevant simple flavin analogues. The sequence is initiated by adding dimethylamine to cyanoacetylene (20), followed by thiolysis of the cyanide, S‐methylation of the resulting thioamide, and condensation with HNCO.[ 92 , 93 ]
All these studies support, but not unequivocally prove, the hypothesis that flavin‐type molecules could have been formed under plausible prebiotic conditions.
5‐Deazaflavins: It has been argued that coenzyme F420 (71) instead of the riboflavins resembles the truly ancient redox factor.[ 110 , 111 ] It is taxonomically restricted, but functionally it facilitates various two‐electron redox reactions in methanogenic, sulfate‐reducing, and likely methanotrophic archaea. Coenzyme F420 is also found in a wider range of bacteria, such as filamentous actinomycetes and mycobacteria. [113] Chemically, coenzymes F0 and F420 more resemble nicotinamide (see Section 4.3) than flavins so that it has occasionally been termed a “nicotinamide in a flavin's clothing” (Scheme 21). [112]
Scheme 21.
Coenzyme F420 (71), a “nicotinamide in a flavin's clothing”.
Finally, when suggesting that redox‐active coenzymes based on the pterin or isoalloxazine core were part of the prebiotic world, one has to consider the issue of regeneration in both directions between the reduced and oxidized forms. This issue has not been addressed so far, but the chemistry discussed above can potentially serve as a solution. Fe(CN)6 3− is able to oxidize the reduced form of flavins, while photoredox catalysis (H2S, hν) might be suitable for the regeneration of the reduced form of flavins.
Folic acid: Folic acid is a coenzyme that consists of a pteridine portion linked through p‐aminobenzoic acid to l‐glutamic acid (Figure 9). The reduced form of folic acid, tetrahydrofolate (5), serves as a donor of C1 units (methyl, hydroxymethyl, and formyl transfer) in a wide range of biosynthetic processes. Important examples are the formation of methionine, purines, and thymine. [113] Plants and most microorganisms obtain folates by de novo biosynthesis from guanosine triphosphate (GTP), which leads to the pterin ring of GTP cyclohydrolase in the intermediate 6‐hydroxymethyl‐7,8‐dihydropterin (91; Figure 10).
Figure 9.
Structural elements of THF (5) and a simplified model.
Figure 10.
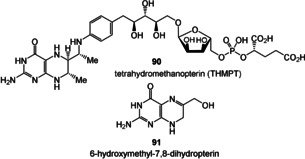
Structures of the archaea tetrahydromethanopterin (THMPT, 90) and the biosynthetic key precursor 6‐hydroxymethyl‐7,8‐dihydropterine (91).
Structurally and functionally related to tetrahydrofolic acid is tetrahydromethanopterin (90). It is a coenzyme in the methanogenesis of archaea and is responsible for methyl transfer to coenzyme M. [114] Unlike 5, THMPT does not contain an electron‐withdrawing substituent in the para position of the phenyl ring and, as a consequence, the reactivity of the formaldehyde adducts towards reductants is reduced compared to that of 5.
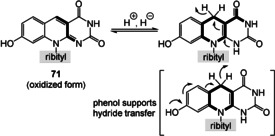
A biomimetic chemical synthesis of the pterin derivative 95 was pursued by Koch and Eschenmoser (Scheme 22). [74b] The condensation between 4‐cyanoaniline (for which a possible prebiotic synthesis was described in Ref. [74b]) and glyceraldehyde (12) provides an imine 93 a and the tautomeric aminohydroxyketone, which further reacted to the dehydrofolate derivative 94 in the presence of aminopyrimidine 83. The latter was chosen instead of derivative 86 to bypass the lack of reactivity of the vinylic amido group at position 6 in 86 (see Scheme 20)
Scheme 22.
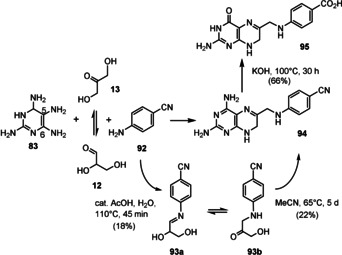
A prebiotic route to simplified dihydrofolate derivate 95. [74b]
The reduction of dihydrotetrahydrofolate, for example, using prebiotic reductants H2S (under photocatalytic conditions) or formic acid, would lead to the active coenzyme analogue for which, however, a procedure has not been reported so far. As tetrahydrofolate is a mediator for C1‐transfer, a concept of transfer and reloading has to be found. Under biotic conditions, THF (5) receives formaldehyde from a pyridoxal phosphate mediated release from serine and transfer. In the prebiotic world, this step does not need to be considered, as formaldehyde was present anyway. So why place folic acid in an early RNA world, if its main role would be the trapping and transfer of formaldehyde? It can be argued that folic acid acts as a vehicle that could bind to the template RNA, from where formaldehyde transfer onto a substrate could occur in a more controlled fashion. However, conditions of formaldehyde release or its reduction to N6‐methyl‐THF, that is, a methyl transfer agent similar to SAM (3) have not been studied so far.
4.3. Considerations on the Prebiotic Formation of NAD
Related to the deazaflavins 71 is nicotinamide adenine dinucleotide (NAD(P), 8). It acts as a hydride acceptor or hydride donor in its reduced form (NAD(P)H) and plays a key role in biotic redox reactions, for example, catalyzed by dehydrogenases, reductases, and hydroxylases. Typically, NAD(P)H‐dependent enzymes are involved in the reduction of C=O and electron‐deficient C=C bonds. The reactivity is located in the electronically “frustrated” aromatic ring—the pyridinium moiety (Figure 11). Besides hydride transfer, dihydropyridines can also undergo electron‐transfer reactions under photoredox conditions, which is another interesting chemical feature. [115]
Figure 11.
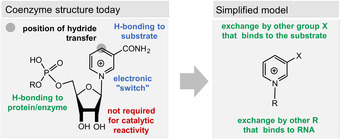
Structural and functional elements of nicotinamides (oxidized form) and proposed simplified model.
The first attempts to prepare the pyridine moiety in NAD(P) under prebiotic conditions were initiated by Orgel and co‐workers, who found that cyanoacetylene (20), propiolaldehyde (96), and ammonia yield nicotinamide (99; Scheme 23).[ 116a , 116b ] Later, Cleaves and Miller [116c] demonstrated that the biosynthetically simpler of two known pathways to nicotinamide‐derived coenzymes [117] can be mimicked by mixing dihydroxyacetone phosphate with aspartic acid [118] to yield nictotinic acid (97) and quinolic acid (98) in respectable yields. When glyceraldehyde‐3‐phosphate was employed, the yields dropped (for 97: 0.8 %; for 98: 6.0). However, to have a redox functional moiety in hand, a prebiotically sensible method for the quaternization of the ring nitrogen atom and formation of the pyridinium moiety had to be found.
Scheme 23.
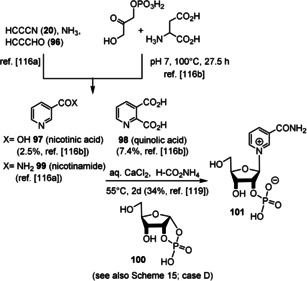
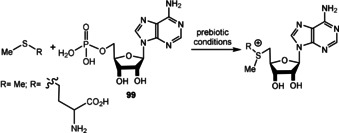
Biomimetic de novo synthesis of nicotinic acid (97), quinolic acid (98), and nicotinamide (99), as well as the generation of nicotinamide nucleotide 101.
Recently, the results of studies by Orgel [116a] and Eschenmoser [87c] (see also Scheme 15 D) were combined for the generation of the pyridinium moiety in nicotinamide nucleotide 101. Kim and Benner demonstrated the formation of 101 by reacting nicotinamide (99) with ribose‐1,2‐cyclic phosphate (100) under prebiotically plausible conditions. [119] Although, this nucleotide is structurally closely related to NAD(P) (8), it can be questioned whether the pyridinium nitrogen atom has to be part of an O,N‐acetal, or whether structurally simplified analogues also show redox properties like 8. An answer to that question has been obtained by recent advances in the field of biotechnology on the topic of “artificial” coenzymes. [120] Synthetic alkyl analogues (Figure 12, right; X=CONH2, CO2H, CN and R=Bu or Bn) still promote enolate reductase catalyzed biotransformations without compromising the activity or stereoselectivity of the bioreduction process. [121] Although not directly transferable to the prebiotic world, these findings are indicative that one can search for structurally more basic, prebiotically formed analogues of nicotinamide‐based coenzymes. In addition, regeneration procedures for both redox forms under palaeogeochemical conditions have not been investigated so far.
Figure 12.
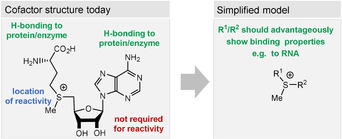
Structural elements of SAM (3) and a simplified model.
4.4. Considerations on the Prebiotic Formation of S‐Adenosylmethionine (SAM)
S‐Adenosylmethionine (3) is involved in methyl, adenosyl, and aminopropyl transfer reactions, the first being the most important one. [122] Cosubstrates of SAM such as the “ancient” iron‐sulfur clusters or vitamin B12, which are able to promote single‐electron transfer, facilitate radical chemistry after homolytic cleavage between the sulfur atom and the ribosyl group. [123] Sulfur‐ylide chemistry has also been reported for SAM‐dependent enzymes. Biosynthetically, it is derived from adenosine triphosphate (ATP, 62) and methionine. Indeed, in bacteria, the biosynthesis of SAM is regulated by SAM riboswitches.[ 22c , 22d ]
The chemical reactivity is located in the sulfonium group, while the polar functionalities of the adenosine group can orient the coenzyme within the macromolecular template (Figure 12). The first experimental studies on possible prebiotic models of SAM utilized the trimethylsulfonium ion. [124] Indeed, the Me3S+ cation 98 shows chemoselective methylating properties under dilute, aqueous conditions when exposed to 2‐aminoethan‐1‐ol (Scheme 24). However, the possible prebiotic origin of the trimethylsulfonium moiety is currently not clear. [124]
Scheme 24.
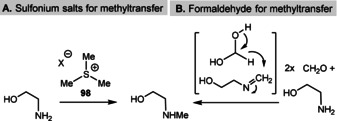
Models of prebiotic methyl transfer reactions.
Alternatively, formaldehyde was studied as a possible prebiotic methylating agent. Indeed, methylation principally works if imine formation is accompanied by hydride transfer from formaldehyde hydrate in a Cannizzaro‐type mode. Thus, glycine was transformed into N‐methylglycine (sarcosine), with the alcohol function remaining untouched (Scheme 25). [125] Along this line, one has to return to Carell's recent work on a possible prebiotic route to diazomethane (Scheme 14). In this case, it is also unclear whether this small molecule lives long enough to initiate methyl transfer onto a nucleophile under plausible geochemical conditions (see Scheme 14). [93]
Scheme 25.
Prebiotic formation of SAM (3) and analogues.
These alkylating methods do not seem to be suited to achieve regioselective methyl transfer, foremost because of the absence of a macromolecular template, such as proteins or RNA. Such templates are essential for controlling the reactivity, stability, and selectivity of the methyl transfer.
Under RNA world conditions, SAM could have formed if a prebiotically generated methyl thioether, such as methionine [33b] or dimethyl sulfide directly reacted with adenosine monophosphate (AMP, 99; Scheme 25). Laurino and Tawfik demonstrated that ATP spontaneously reacts with methionine to yield SAM, which could also work for the prebiotically more relevant AMP. [126] Importantly, the resulting sulfonium cations, whether SAM (3) or another derivative, could bind to the macromolecular template RNA. As discussed for riboflavins and nicotinamides, the regeneration of the methylating agents, be it SAM or simpler analogues, is the second key issue, if such methylating agents were of prebiotic relevance. Under biotic conditions, SAM receives its methyl group by a C1‐transfer sequence that is initiated by the pyridoxal phosphate promoted release of formaldehyde from serine and transfer onto tetrahydrofolic acid (5). Next, the resulting N,N‐acetal is reduced, typically by NADH (8). [117] So far, simple regeneration concepts for thioethers have not been probed.
4.5. Considerations on the Prebiotic Formation of Pyridoxal Phosphate.
Vitamin B6 comprises a group of structurally related pyridine derivatives namely pyridoxal, pyridoxine, and pyridoxamine, from which the active 5‐phosphorylated forms such as pyridoxal phosphate (PLP, 1) are derived. [127]
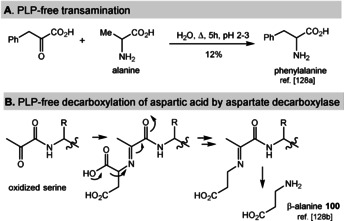
PLP promotes a large and diverse number of biotransformations in the arena of amino acid metabolism as well as in modifying secondary metabolites. Transamination as well as decarboxylations of α‐amino acids can be regarded as being the two key reactions performed by PLP (1) and its amino derivative 101. It was shown that these two reactions can also occur between amino acids and α‐keto acids without PLP, preferably at low pH values of 2–3 (Scheme 26 A). [128a] In addition, enzyme‐catalyzed decarboxylations of aspartic acid that yield β‐alanine (100) are known to occur without the presence of PLP as coenzyme (Scheme 26 B). [128b] Instead, protein‐bound pyruvate (derived from a terminal serine) takes over the role of PLP (1). This clearly is an indication that there are alternative and simpler concepts for decarboxylations available that could have also operated under prebiotic conditions. [129] However, as a consequence of the central role of PLP in the formation of amino acids and its role to access other small molecules, it is worth considering a protometabolic scenario for PLP.
Scheme 26.
A) Transamination between a α‐keto acid and alanine and B) PLP‐free decarboxylation of aspartic acid by aspartate decarboxylase.
Thus, one can envisage a simplified model of PLP (1) that retains its principal reactivities (Figure 13). The methyl substituent is likely to be unnecessary, while substituents that bind to a macromolecular template such as proteins or to prebiotic RNA (OH and Y) preferentially should be kept. In fact, the prebiotic formation of such functionalized pyridine rings have been calculated using cyanoacetylene (20), diacetylene, and CO in aqueous phosphoric acid, [130] but experimentally this computer‐assisted model has not been verified so far.
Figure 13.
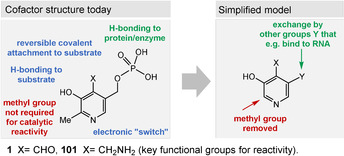
Functional and less necessary elements in pyridoxal and pyridoxamine phosphates (101) and core (blue: necessary functional elements; green: required for binding to a macromolecular template, but structural flexibility is presumed to be allowed; red: less necessary structural elements).
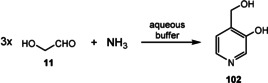
The first example of a simplified pyridoxine derivative formed under supposedly prebiotic conditions is based on the trimerization of glycolaldehyde (11) in a heated buffered solution that contained ammonia. This Chichibabin‐type pyridine synthesis provided 4‐(hydroxymethyl)pyridine‐3‐ol (102; Scheme 27)[ 131 , 132 ] and is related to one of the two known biosynthetic pathways to PLP (1), specifically the one that does not require any additional coenzymes (Scheme 28). [133] The condensation of ribose (37; or any other pentose; Scheme 4) with glyceraldehyde‐3‐phosphate (from 58; its formation is described in Scheme 15 D) in the presence of ammonia could principally yield pyridoxine phosphate (103), which presumably should be oxidized to PLP (1) by Fe(CN)6 3.
Scheme 27.
De novo synthesis of the simplified pyridoxine derivative 102.
Scheme 28.
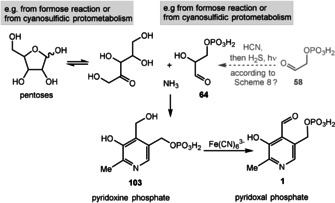
Hypothetical prebiotic formation of pyridoxine phosphate (103) and PLP (1) from pentoses and glyceraldehyde‐3‐phosphate (64).
4.6. Considerations on the Prebiotic Formation of Lipoic Acid (63), Biotin (64), and Coenzyme A (10)
A discussion on the possible prebiotic role of lipoic acid (63) and biotin (64) must follow two lines of consideration: a) their possible prebiotic formation and b) their chemical usefulness in a protometabolic scenario. The first point is associated with the role of fatty acids in the evolution of life because these two coenzymes contain linear hydrocarbon chains (lipoic acid: octanoic acid, biotin: pimelic acid). It is astounding to note that fatty acids are almost absent in Figure 4, although the six concepts discussed there are regarded as key routes for the creation of basic prebiotic molecules and privileged building blocks. However, fatty acids are important for the formation of lipid membranes and these are essential for compartment generation, thereby separating metabolic activities from the non‐self outside world. The creation of compact spaces leads to higher concentrations of reactants and consequently to an accelerated expansion of protometabolic networks. [134] Surprisingly, little evidence has been accumulated so far of how fatty acids first appeared on the scene. [135] The most widely discussed proposal of prebiotic fatty acid formation is the Fischer–Tropsch process. [136] It provides linear hydrocarbons from CO and H2 under high‐temperature gas‐phase conditions. Fatty acids and fatty alcohols are formed as by‐products.
Another important study is based on one‐pot reactions of acetylene and CO in the presence of nickel sulfide in an aqueous environment mimicking hydrothermal subseafloor vents. [137] The primary products detected are unsaturated short‐ to medium‐chain carboxylic acids with uneven carbon numbers that can undergo further reduction in the presence of CO.
Finally, Prieur hypothesized that sulfur ylide chemistry may have been responsible for the formation of fatty acids, but this suggestion has not been verified experimentally so far. [138]
To solve this overall dilemma it was speculated that instead of fatty acid based compartmentation, alternative amphiphilic building blocks such as alkyl phosphates, alkyl sulfates, and polyprenyl chains created the first micelles. [136] Archaea provide a clue of such molecular alternatives as their membranes are not composed of fatty acid esters, but are based on polyprenolic ethers. Indeed, 2‐methylbut‐3‐en‐2‐ol oligoprenols and 2‐methylbut‐3‐en‐2‐ol polyprenols are formed from 3‐methylbut‐2‐en‐1‐ol in the presence of the mineral clay montmorillonite K10 at room temperature. [139] It was suggested that these C5‐building blocks may have formed from isobutene and formaldehyde by a Prins‐type reaction. Phosphorylation of the final allyl alcohols and ether formation with glycerol (29; see Scheme 8) could have constituted membranes already known from archaea.
To sum up, no compelling story on the prebiotic formation of amphiphilic building blocks that result in the generation of micelle‐type architectures can currently be told. This statement is of consequence when considering a possible prebiotic synthesis of fatty acid derived lipoic acid (63) and biotin (64) [140] and to a lesser degree of coenzyme A (10). For this reason, it may very well be that these coenzymes did not exist in the emerging RNA world. The second line of consideration mentioned above asks whether their absence would have had any consequences for the expanding protometabolic network. Lipoic acid (63) promotes redox reactions (thiols⇌disulfide). [141] However, other redox coenzymes that operate in the absence of oxygen are nicotin amides (as NAD (8) or simpler analogues), and the riboflavins are known alternatives. More importantly, the oldest redox systems discussed to have existed in an oxygen‐free atmosphere are FeS clusters. These act as single‐electron‐transfer cofactors and form spontaneously in hydrothermal vents (see Figure 4). Further evidence was recently collected when the prebiotic synthesis of iron‐sulfur clusters driven by UV light was reported. [142]
Likewise, several controversial arguments on the prebiotic role of biotin have been discussed. [143] Biotin (64) promotes the trapping and the transfer of carbon dioxide onto nucleophiles, predominantly the α‐position of thioesters. The resulting malonates can serve as enol precursors in decarboxylative Claisen reactions. Malonates are a highly labile species and the reversible decarboxylation is a facile process with protonation of the enol, a somehow useless process. To use malonates in C−C coupling reactions, the two reaction partners need to be protected from protic media and this is commonly guaranteed in the active site of enzymes, such as fatty acid synthases or ketosynthases.
Unlike most other coenzymes, biotin alone is chemically rather inert and only exerts its coenzymatic role in multienzyme systems with carboxyphosphate (HO2C‐OPO3H2) as the substrate. Biotically, this anhydride is presumed to form from bicarbonate and ATP prior to transfer onto biotin but it has not been isolated nor observed directly by experiment. [144] However, biotin‐free transfer is also well‐established, for example, the use of carbamoyl phosphate (H2N(O)C‐OPO3H2) in pyrimidine biosynthesis. Furthermore, several alternative C1 building blocks that are more electrophilic than carbon dioxide and that are of prebiotic relevance have been covered in this account. These include guanidine ((H2N)2C=NH, 24), cyanic acid (HCNO), cyanamide (H2NCN, 26), and urea (27).
Collecting all these arguments and facts it is reasonable to assume that this coenzyme likely did not play a role in the early evolution towards life. Furthermore, biotin‐mediated carboxylation is not the only biotic option to activate and introduce CO2 into carbon backbones. The enzyme RuBisCo [145] plays a key role in CO2 fixation. It uses magnesium as a cofactor and ATP as an energy source for this endergonic process. However, the hypothesis that a RuBisCo‐like mechanism of carbon dioxide fixation might have been part of a protometabolic network has not yet been tested experimentally.
Coenzyme A (10) [146] is a structurally complex thiol composed of adenosine diphosphate (ADP), pantoic acid, β‐alanine, and cysteamine (Figure 14). It is commonly present in the form of thioester derivatives including those based on fatty acids. Thioesters show enhanced carbonyl activity as well as α‐acidity. Coenzyme A serves as an acyl group carrier, which is formed from the corresponding acylated phosphates, a highly reactive acyl transfer species (see also 53 in Scheme 15 A). Compared to these mixed anhydrides, thioesters are less prone to hydrolysis and can survive in water for long periods of time under prebiotically plausible conditions. [147]
Figure 14.
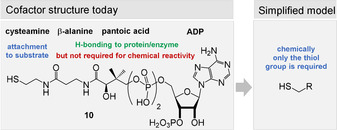
Structure of coenzyme A (10) and a simplified model (blue: necessary functional elements; green: required for binding to a macromolecular template, but structural flexibility is presumed to be allowed; red: less necessary structural elements with respect to chemical reactivity).
From a chemical standpoint, the relevant chemical reactivity in coenzyme A (10) is located in the thiol group so that simplified coenzyme A analogues such as methanethiol, cysteamine (104), or the water‐soluble coenzyme M (mercaptoethanesulfonate, 108) were considered to have played a role in the prebiotic world. Remarkably, coenzyme M is found in methanogens, where it is responsible for methyl transfer reactions during methanogenesis. [148] The abundance of methanethiol was detected in diverse hydrothermal fluids emanating from ultramafic, mafic, and sediment‐covered midocean ridge settings. [149]
In the case of cysteamine or larger fragments of coenzyme A, several reactions under supposedly prebiotic conditions that lead to β‐alanine (100), [29] cysteamine (104), [33b] and pantoic acid (105) [150] have been studied (Scheme 29). [151] However, it has to be stressed that cysteamine (104) is not ideally suited for thioester formation because of a preferential chemoselective acyl transfer to the amino instead of the thiol group, which may also explain the low yield of diamide 106 under thermal conditions. Attempts to phosphorylate pantetheine with ADP or ATP at 120 °C provided only trace amounts of coenzyme A (10) (<0.01 %). As an alternative route, Jadhav and Yarus proposed its formation through a ribozyme‐catalyzed reaction. [152]
Scheme 29.
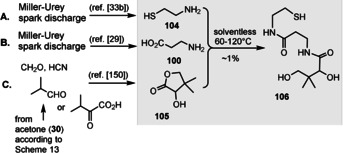
Three experimental studies (A–C) for the formation of cysteamine (104), β‐alanine (100), and pantoic acid (105) as well as the subsequent formation of pantetheine (106).
As early as 1993, Miller and Schlesinger searched for prebiotic conditions to form coenzyme M (108). [153] Ethene, formed during prebiotic processes in planetary atmospheres, [154] is transformed into ethylene sulfide (107), presumably in the presence of 3p sulfur atoms (Scheme 30). [155] These sulfur atoms are generated upon photolysis of prebiotic molecules COS and CS2. The reaction of ethylene sulfide (107) with ammonia yields cysteamine (104). [153] Likewise, coenzyme M (108) is formed in the presence of sulfite.
Scheme 30.
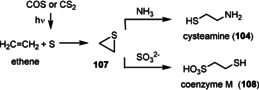
Experimental evidence for the prebiotic formation of cysteamine (104) and coenzyme M (108) from ethene and sulfur.
If these thioalcohols are presumed to activate carboxylic acids in the form of the corresponding thioesters, a plausible activation of these prior to thioester formation has to be considered. For the formation of amino acid based thioesters 109, anhydride 52 and/or aminoacylphosphate anhydride 53 can be regarded as sensible candidates (Scheme 31).
Scheme 31.
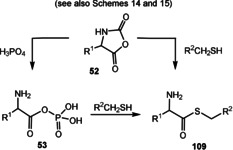
Plausible formation of thioesters 109 from amino acid derived anhydride 52 and mixed anhydride 53.
4.7. Considerations on the Prebiotic Formation of Thiamine Pyrophosphate (4)
Thiamine pyrophosphate (4, TPP) promotes acylanion transfer reactions. [96] Surprisingly, it was recently reported that the biologically active form of 4 is not required by Borrelia burgdorferi, the bacterium that causes Lyme disease. This observation challenges the paradigm that this coenzyme is essential for all living organisms. [156]
Nevertheless, from a metabolic point of view, 4 promotes a large variety of different C−C coupling reactions, which is clearly very beneficial for substantially broadening a possible protometabolism on prebiotic Earth. The chemical reactivity for catalysis is located in the thiazolium unit of TPP (Figure 15). The existence of TPP riboswitches (see Section 2) could be taken as an argument that TPP or simpler carbene analogues may indeed be “old” coenzymes. However, their prebiotic formation is still unclear, and inspirations borrowed from the known biosynthetic routes are not helpful. [157] Two biosynthetic routes are currently known for thiazole formation that utilize different sets of building blocks. In bacteria and plants, these are ribulose‐5‐phosphate, cysteine as a source for sulfur, and glycine or alternatively tyrosine (both are converted into the common precursor dehydroglycine). Alternatively, in yeast, eukaryotes, and archaea, biosynthesis relies on glycine, nicotine amide (8, NAD+; note that NAD+ provides atoms here and not a redox environment), and a sulfur source. Both routes furnish hydroxyethylthiazole (HET, 110) as a key precursor. The biosynthesis is terminated by quaternization of the thiazole nitrogen atom in 110 with 4‐amino‐2‐methyl‐5‐hydroxymethylpyrimidine pyrophosphate (HMP‐PP, 111); Figure 16). In bacteria and plants, 111 is biosynthesized from 5‐aminoimidazole ribonucleotide (38; see also its role in the biosynthesis of purines in Scheme 11 B), while in yeast, histidine and PLP (serving as an atom source and not as a coenzyme) are the principal building blocks.
Figure 15.
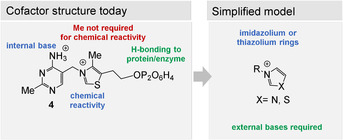
Structure of thiamine pyrophosphate (4) and a simplified analogue (blue: necessary functional elements; green: required for binding to a macromolecular template, but structural flexibility is presumed to be allowed; red: less necessary structural elements with respect to chemical reactivity).
Figure 16.
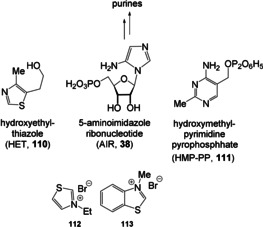
Intermediates of TPP biosyntheses: HET (110), AIR (111), HMP‐PP (112), and examples of simplified thiazolium salts 113 and 114.
None of these biosynthetic routes have inspired scientists to develop prebiotic syntheses so far, because in several of them coenzymes are required as building blocks. One could, however, consider simpler analogues that bear the key chemical reactivity of TPP (Figure 15). Indeed, purely synthetic studies on the self‐condensation of formaldehyde with simpler TPP derivatives 112 and 113 revealed that the first step of the formose reaction (see Scheme 4) selectively yields dihydroxyacetone (13), but not glycolaldehyde (11). [158] This could imply that once TPP (4) or a simpler analogue appeared on the prebiotic scene, the formose reaction as a source for small carbohydrate‐type prebiotic building blocks could have become increasingly important.
In essence very little research on TPP in the context of this Review has been conducted so far so that a discussion on the prebiotic role of TPP (4) or simplified carbene analogues is still highly speculative.
4.8. Considerations on the Prebiotic Formation of Porphyrins, Urogen III (66), and 5‐Aminolevulanic Acid
Porphyrin‐type cofactors are composed of a macrocyclic tetrapyrrole ligand (see urogen II, 66) that tightly binds metal ions. Biotically, they perform group transfer and redox reactions that include the transfer of electrons. [159] Porphyrin‐containing proteins are ubiquitously distributed in all kingdoms of life and among those heme and chlorophylls are the most important ones. Remarkably, cytochrome P450 enzymes, which bear the heme core, are thought to have existed for more than 3.5 billion years, [160] so that a role in the prebiotic world has been discussed to be likely. [161] Porphyrin abiogenesis from pyrrole and formaldehyde under simulated geochemical conditions was reported as early as 1967. [162] Later, Strasdeit and co‐workers collected simple pyrroles 114 under abiotic conditions when seawater, containing amino acids, was exposed to molten lava. [161] They suggested that, on primordial volcanic islands, the volatile pyrroles and HCl must have condensed at cooler locations and pyrrole oligomerization may have occurred. Thus, treatment of 2,4‐diethylpyrrole and HCl with formaldehyde and nitrite yielded highly stable octaethylporphyrin (115) as well as other oligomers [163] (Scheme 32 A). An approach closely related to the biosynthesis of the more water soluble urogen III (66) was pursued by Lindsey and co‐workers. [164] They showed that porphyrinogens such as urogen (66) can be generated by self‐condensation from α‐aminoketones 116 as well as β‐diketones or β‐ketoesters in water under supposedly prebiotic conditions (Scheme 32 B). As was established in biosynthetic studies on urogen (66), dimer 117 is the key intermediate of this process.
Scheme 32.
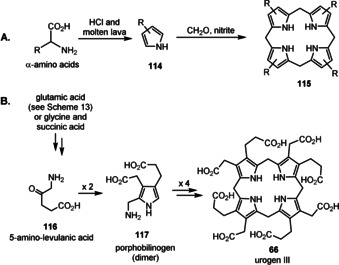
A) Abiotic and B) biomimietic formation of porphyrin core structures.
However, the link between basic prebiotic molecules and the starting building blocks, especially with succinic acid and further downstream with 5‐aminolevulanic acid (116), has not been established experimentally yet.
5. Conclusions
The foundations of the RNA world theory are self‐replication and catalysis. In this world, RNA preceded a possible protoribosome, [14a] the ribosome, DNA, and hence the protein world. [165] The existence of coenzymes that are structurally closely related to nucleotides, for example, manifested in the “AMP‐handle” 6 or being hidden in the heterocyclic core structures of selected coenzymes, strongly supports the hypothesis that these were already present when the first RNA monomers appeared on the scene and before protoribosomes existed (Scheme 33). [14a]
Scheme 33.
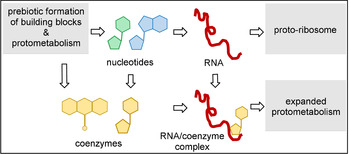
Hypothetic prebiotic formation of coenzymes (here flavin‐ and nicotinamide‐type representatives are sketched) and their possible role within the RNA world.
If RNA served as a template for coenzymes, a complex catalytic protometabolism could have emerged. From a reverse view, one can argue that the prebiotic existence of coenzymes would also enforce the RNA‐world theory.
Table 2 gives a condensed summary of the (experimental) status of coenzyme syntheses under plausible prebiotic conditions, as has been discussed in this Review. Nevertheless, the role of coenzymes as catalytically important small molecules in an RNA world can still be debated because a) only larger, but not complete, parts of several jigsaw puzzles on their prebiotic syntheses have been studied experimentally and b) RNA/coenzyme conjugates or complexes have not been structurally studied nor have chemical transformations been carried with such architectures.
Table 2.
The status of coenzyme syntheses under assumed prebiotic conditions (coenzymes are listed that have been discussed in detail).
|
Coenzyme (ordered by numbering) |
Prebiotic analogue obtained by synthesis |
Comments |
|---|---|---|
|
PLP (1) |
|
glycolaldehyde and ammonia provide (102) a simplified derivative of pyridoxine. Oxidation to aldehyde likely occurs under plausible prebiotic conditions. |
|
flavins (2) |
|
pyrolytic formation of 75 from glycine, alanine, and lysine, but only proven by mass spectrometry. Formation of flavin derivatives 77 under plausible prebiotic conditions reported in part. Several early steps not established yet. |
|
SAM (3) |
|
substitution of AMP with thioethers reported. Methionine can be exchanged by dimethylsulfide (R=Me), but prebiotically relevant thioethers are not well studied. |
|
TPP (4) |
|
no prebiotically plausible synthetic route reported. Evidence was collected that simpler analogues such as 112 are catalytically active (e.g. in the formose reaction). |
|
folic acid (5) |
|
formation of simplified folic acid derivative 95 reported. Plausible prebotic conditions for some steps are questionable (e.g. 94→95). |
|
NMN, NAD(P) (7/8) |
|
formation of NAD+ derivative 101 under plausible prebiotic conditions reported.
|
|
coenzyme A (10) |
|
pantetheine derivative 106 prepared in trace amounts. Simpler thiols such as methanethiol or coenzyme M could have played a role under prebiotic conditions instead. |
|
ATP (62) |
– |
formation of nucleotide monophosphates (NMPs) under plausible prebiotic conditions reported. ATP itself is very likely not stable to exist in a prebiotic environment. |
|
lipoic acid (63) |
– |
its presence and catalytic role in a prebiotic is world questionable. |
|
biotin (64) |
– |
its presence and catalytic role in a prebiotic world questionable. Besides CO2, other electrophilic derivatives likely existed so that biotin for CO2 transfer is not essentially required. |
|
porphyrins and urogen III (66) |
|
formation of porphyrin core occurs on molten lava starting from α‐amino acids, HCl, and formaldehyde (R originates from the α‐amino acid side chain). |
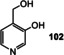
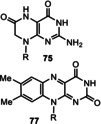
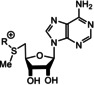
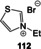
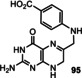
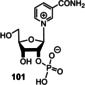
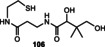
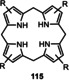
6. Outlook
As early as 1976, White III proposed that coenzymes are the surviving vestiges of nucleic acid enzymes. [11d] He claimed that their occurrence preceded the evolution of ribosomal protein synthesis. However, at that time, catalytic active ribozymes were unknown. Indeed, these were not discovered until 1982 by Cech and co‐workers. [9a] Many years later, the existence of RNA riboswitches were reported, which are able to bind coenzymes and thus could have served as templates under prebiotic conditions [166] before proteins and enzymes appeared on the stage of molecular evolution (Scheme 33). However, it should be kept in mind that hypotheses other than the RNA‐world theory have been proposed. These include the lipid, metabolism, protein, and thioester worlds. [167] For these prebiotic worlds a protometabolism also has to be reinvented that would provide all other bio(macro)molecules that make up life. In all cases, the emergence of catalytic processes had to be of key importance, and small molecules such as coenzymes or simpler analogues and metal cations could serve these requirements.
Eschenmoser's initial statement [1] positions chemistry in the center of such an endeavor. Recent advances in unraveling plausible prebiotic metabolic networks not only provide experimental support for the RNA‐world hypothesis, but they now pave the way to focus more vividly on prebiotic scenarios that lead to coenzymes, especially those that are structurally and (bio)synthetically linked with nucleotides. This Review is intended to refocus on conezymes and inspire chemical groups to pursue research programs that expand the protometabolic networks with coenzymes including simpler analogues. This, however, needs to be accompanied by:
studies on the (catalytic) reactivity of such moieties under prebiotic conditions,
their ability to possibly bind to RNA templates, and
a chemistry that allows for their regeneration, especially in the cases of coenzymes that promote redox and group transfer reactions.
There is a quest for “reinventing” a protometabolism of coenzymes.
Conflict of interest
The authors declare no conflict of interest.
Biographical Information
Andreas Kirschning studied chemistry at the University of Hamburg and at Southampton University (UK). In Hamburg, he joined the group of Prof. Ernst Schaumann and received his PhD in 1989 working in the field of organosilicon chemistry. After a postdoctoral stay at the University of Washington (Seattle, USA) with Prof. Heinz G. Floss, he started his independent research at the Clausthal University of Technology in 1991, where he finished his habilitation in 1996. In 2000 he moved to Leibniz University Hannover. His research interests cover structure elucidation as well as the semi‐ and total synthesis of natural products.

Acknowledgements
I thank Dr. Matthew D. Norris for helpful discussions. Open access funding enabled and organized by Projekt DEAL.
A. Kirschning, Angew. Chem. Int. Ed. 2021, 60, 6242.
References
- 1. Eschenmoser A., Tetrahedron 2007, 63, 12821–12844. [Google Scholar]
- 2.First conceptual thoughts on prebiotic chemistry were published by A. I. Oparin, J. B. S. Haldane:
- 2a. Oparin A. I., Proischogdenie Zhizni, Moscovsky Robotschii, Moscow, 1924; translated by A. Synge into English in: [Google Scholar]; Bernal J. D., The Origin of Life, Weidenfeld & Nicolson, London, 1967, pp. 199–234; [Google Scholar]
- 2b. Haldane J. B. S., Ration. Annu. 1929, 148, 3 reprinted in Bernal's book (see Ref. [1a], pp. 242–249. [Google Scholar]
- 3.
- 3a. Holliday G. L., Thornton J. M., Marquet A., Smith A. G., Rebeille F., Mendel R., Schubert H. L., Lawrence A. D., Warren M. J., Nat. Prod. Rep. 2007, 24, 972–987; [DOI] [PubMed] [Google Scholar]
- 3b. Eschenmoser A., Loewenthal E., Chem. Sov. Rev. 1992, 21, 1–16; [Google Scholar]
- 3c. King G. A. M., BioSystems 1980, 13, 2345; [Google Scholar]
- 3d. Visser C. M., Origins Life 1982, 12, 165–179. [DOI] [PubMed] [Google Scholar]
- 4.
- 4a. Richter M., Nat. Prod. Rep. 2013, 30, 1324–1345; [DOI] [PubMed] [Google Scholar]
- 4b. Fischer J. D., Holliday G. L., Rahman S. A., Thornton J. M., J. Mol. Biol. 2010, 403, 803–824. [DOI] [PubMed] [Google Scholar]
- 5. Handler P., Evolution of the coenzymes, in: Proceed. 5th Intern. Congr. Biochem. (Ed.: Oparin A. I.), Macmillan, New York, 1961, pp. 149–157. [Google Scholar]
- 6.
- 6a. Wächtershäuser G., Chem. Biodiversity 2007, 4, 584–601; [DOI] [PubMed] [Google Scholar]
- 6b. Johnston W. K., Unrau P. J., Lawrence M. S., Glasner M. E., Bartel D. P., Science 2001, 292, 1319–1325. [DOI] [PubMed] [Google Scholar]
- 7.
- 7a. Orgel L. E., Proc. Natl. Acad. Sci. USA 2000, 97, 12503–12507; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7b. Orgel L. E., PLoS Biol. 2008, 6, 5–13; [Google Scholar]
- 7c. Sharov A. A., Biosystems 2016, 144, 8–17;26968100 [Google Scholar]
- 7d. Ross D. S., Origins Life Evol. Biospheres 2007, 37, 61–65; [DOI] [PubMed] [Google Scholar]
- 7e. Smith E., Morowitz H. J., Proc. Natl. Acad. Sci. USA 2004, 101, 13168–13173; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7f. Morowitz H. J., Kostelnik J. D., Yang J., Cody G. D., Proc. Natl. Acad. Sci. USA 2000, 97, 7704–7708; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7g. Kauffman S. A., J. Theor. Biol. 1986, 119, 1–24. [DOI] [PubMed] [Google Scholar]
- 8.
- 8a. Gilbert W., Nature 1986, 319, 618; [Google Scholar]
- 8b. Joyce G. F., Nature 2002, 418, 214–221; [DOI] [PubMed] [Google Scholar]
- 8c. Robertson M. P., Joyce G. F., Cold Spring Harbor Perspect. Biol. 2012, 4, a003608; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8d. Penny D., Biol. Philos. 2005, 20, 633–671. [Google Scholar]
- 9.
- 9a. Kruger K., Grabowski P.-J., Zaug A. J., Sands J., Gottschling D. E., Cech T. R., Cell 1982, 31, 147–157; [DOI] [PubMed] [Google Scholar]
- 9b. Chen X., Li N., Ellington A. D., Chem. Biodiversity 2007, 4, 633–655. [DOI] [PubMed] [Google Scholar]
- 10.
- 10a. Eakin R. E., Proc. Natl. Acad. Sci. USA 1963, 49, 360–366; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10b. Sassanfar M., Szostak J. W., Nature 1993, 364, 550–553; [DOI] [PubMed] [Google Scholar]
- 10c. Burgstaller P., Famulok M., Angew. Chem. 1994, 33, 1084–1087; [Google Scholar]
- 10d. Pross A., Origins Life Evol. Biospheres 2004, 34, 307–321; [DOI] [PubMed] [Google Scholar]
- 10e. Saladino R., Botta G., Pino S., Costanzo G., Di Mauro E., Chem. Soc. Rev. 2012, 41, 5526–5565; [DOI] [PubMed] [Google Scholar]
- 10f. Patel B. H., Percivalle C., Ritson D. J., Duffy C. D., Sutherland J. D., Nat. Chem. 2015, 7, 301–307. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.
- 11a. Yarus M., Cold Spring Harbor Perspect. Biol. 2011, 3, a003590; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11b. Yarus V. R., Biochimie 2002, 84, 877–888; [DOI] [PubMed] [Google Scholar]
- 11c. Connell G. J., Christian E. L., Orig Life 1993, 23, 291–297; [DOI] [PubMed] [Google Scholar]
- 11d. H. B. White III , J. Mol. Evol. 1976, 7, 101–104. [DOI] [PubMed] [Google Scholar]
- 12.
- 12a. Szostak J. W., Bartel D. P., Luisi P. L., Nature 2001, 409, 387–390; [DOI] [PubMed] [Google Scholar]
- 12b. Ruiz-Mirazo K., Briones C., de la Escosura A., Chem. Rev. 2014, 114, 285–366; [DOI] [PubMed] [Google Scholar]
- 12c. Sutherland J. D., Angew. Chem. Int. Ed. 2016, 55, 104–121; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2016, 128, 108–126. [Google Scholar]
- 13. Bada J. L., Chem. Soc. Rev. 2013, 42, 2186–2196. [DOI] [PubMed] [Google Scholar]
- 14.
- 14a. Davidovich C., Belousoff M., Wekselman I., Shapira T., Krupkin M., Zimmerman E., Bashan A., Yonath A., Isr. J. Chem. 2010, 50, 29–35; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14b. Meyer S. C., Nelson P. A., Bio-complexity 2011, 1–10. [Google Scholar]
- 15.
- 15a. Cech T. R., Uhlenbeck C., Nature 1994, 372, 39–40; [DOI] [PubMed] [Google Scholar]
- 15b. Fedor M. J., Williamson J. R., Nat. Rev. Mol. Cell Biol. 2005, 6, 399–412. [DOI] [PubMed] [Google Scholar]
- 16.
- 16a. Anastasi C., Buchet F. F., Crowe M. A., Parkes A. L., Powner M. W., Smith J. M., Sutherland J. D., Chem. Biodiversity 2007, 4, 721–739; [DOI] [PubMed] [Google Scholar]
- 16b. Islam S., Powner M. W., Chem 2017, 2, 470–501; [Google Scholar]
- 16c. Kim S. C., O'Flaherty D. K., Zhou L., Lelyveld V. S., Szostak J. W., Proc. Natl. Acad. Sci. USA 2018, 115, 13318–13323. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.
- 17a. Nissen P., Hansen J., Ban N., Moore P. B., Steitz T. A., Science 2000, 289, 920–930; [DOI] [PubMed] [Google Scholar]
- 17b. Steitz T. A., Moore P. B., Trends Biochem. Sci. 2003, 28, 411–418. [DOI] [PubMed] [Google Scholar]
- 18.
- 18a. Webb M. E., Marquet A., Mendel R. R., Rébeillé F., Smith A. G., Nat. Prod. Rep. 2007, 24, 988–1008. [DOI] [PubMed] [Google Scholar]
- 19.
- 19a. Tsukiji S., Pattnaik S. B., Suga H., J. Am. Chem. Soc. 2004, 126, 5044–5045; [DOI] [PubMed] [Google Scholar]
- 19b. Huang F., Bugg C. W., Yarus M., Biochemistry 2000, 39, 15548–15555; [DOI] [PubMed] [Google Scholar]
- 19c. Breaker R. R., Joyce G. F., J. Mol. Evol. 1995, 40, 551–558. [DOI] [PubMed] [Google Scholar]
- 20.
- 20a. McCown P. J., Corbino K. A., Stav S., Sherlock M. E., Breaker R. R., RNA 2017, 23, 995–1011; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20b. Sherwood A. V., Henkin T. M., Annu. Rev. Microbiol. 2016, 70, 361–374; [DOI] [PubMed] [Google Scholar]
- 20c. Serganov A., Patel D. J., Annu. Rev. Biophys. 2012, 41, 343–370; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20d. Serganov A., Polonskaia A., Tuan Phan A., Breaker R. R., Patel D. J., Nature 2006, 441, 1167–1171. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.
- 21a. Winkler W., Nahvi A., Breaker R. R., Nature 2002, 419, 952–955; [DOI] [PubMed] [Google Scholar]
- 21b. Croft M. T., Moulin M., Webb M. E., Smith A. G., Proc. Natl. Acad. Sci. USA 2007, 104, 20770–20775. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.
- 22a. Serganov A., Huang L., Patel D. J., Nature 2009, 458, 233–237; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22b. Pedrolli D. B., Matern A., Wang J., Ester M., Siedler K., Breaker R., Mack M., Nucleic Acids Res. 2012, 40, 8662–8673; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22c. Grundy F. J., Henkin T. M., Mol. Microbiol. 1998, 30, 737–749; [DOI] [PubMed] [Google Scholar]
- 22d. Winkler W. C., Nahvi A., Sudarsan N., Barrick J. E., Breaker R. R., Nat. Struct. Biol. 2003, 10, 701–707; [DOI] [PubMed] [Google Scholar]
- 22e. Ames T. D., Rodionov D. A., Weinberg Z., Breaker R. R., Chem. Biol. 2010, 17, 681–685; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22f. Regulski E. E., Moy R. H., Weinberg Z., Barrick J. E., Yao Z., Ruzzo W. L., Breaker R. R., Mol. Microbiol. 2008, 68, 918–932; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22g. Nahvi A., Sudarsan N., Ebert M. S., Zou X., Brown K. L., Breaker R. R., Chem. Biol. 2002, 9, 1043–1049. [DOI] [PubMed] [Google Scholar]
- 23.
- 23a. Serganov A., Huang L., Patel D. J., Nature 2008, 455, 1263–1267; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23b. Mandal M., Boese B., Barrick J. E., Winkler W. C., Breaker R. R., Cell 2003, 113, 577–586. [DOI] [PubMed] [Google Scholar]
- 24. Damblon C., Raquet X., Lian L. Y., Lamotte-Brasseur J., Fonze E., Charlier P., Roberts G. C., Frère J. M., Proc. Natl. Acad. Sci. USA 1996, 93, 1747–1752. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Nelson J. W., Breaker R. R., Sci Signal 2017, 10, eaam8812. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Deamer D., Assembling Life “How can Life Begin on Earth and Other Habitable Planets?, Oxford University Press, Oxford, 2019. [Google Scholar]
- 27. Kitadai N., Maruyama S., Geosci. Front. 2018, 9, 1117–1153. [Google Scholar]
- 28.
- 28a. Plasson R., Kondepudi D. K., Hugues H. B., Commeyras A., Asakura K., Chirality 2007, 19, 589–600; [DOI] [PubMed] [Google Scholar]
- 28b. Jafarpour F., Biancalani T., Goldenfeld N., Phys. Rev. E 2017, 95, 032407. [DOI] [PubMed] [Google Scholar]
- 29.
- 29a. Miller S. L., Science 1953, 117, 528–529; [DOI] [PubMed] [Google Scholar]
- 29b. Miller S. L., Urey H. C., Science 1959, 130, 245–251. [DOI] [PubMed] [Google Scholar]
- 30. Tian F., Kasting J. F., Zahnle K., Earth Planet. Sci. Lett. 2011, 308, 417–423. [Google Scholar]
- 31.
- 31a. Lazcano A., Bada J. L., Origins Life Evol. Biospheres 2003, 33, 235–242; [DOI] [PubMed] [Google Scholar]
- 31b. Bada J. L., Chem. Soc. Rev. 2013, 42, 2186–2196. [DOI] [PubMed] [Google Scholar]
- 32. Guillemin J. C., Bouyahyi M., Riague E. H., Adv. Space Res. 2004, 33, 81–87. [Google Scholar]
- 33.
- 33a. Parker E. T., Cleaves H. J., Dworkin J. P., Glavin D. P., Callahan M., Aubrey A., Lazcano A., Bada J. L., Proc. Natl. Acad. Sci. USA 2011, 108, 5526–5531; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33b. Parker E. T., Cleaves H. J., Callahan M. P., Dworkin J. P., Glavin D. P., Lazcano A., Bada J. L., Origins Life Evol. Biospheres 2011, 41, 201–212; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33c. Johnson A. P., Cleaves H. J., Dworkin J. P., Glavin D. P., Lazcano A., Bada J. L., Science 2008, 322, 404; [DOI] [PubMed] [Google Scholar]
- 33d. Cleaves H. J., Chalmers J. H., Lazcano A., Miller S. L., Bada J. L., Origins Life Evol. Biospheres 2008, 38, 105–115. [DOI] [PubMed] [Google Scholar]
- 34.Ammonia formation by meteorite impact: Shimamura K., Shimojo F., Nakano A., Tanaka S., Sci. Rep. 2016, 6, 38953. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.
- 35a. Robertson M. P., Miller S. L., Nature 1995, 375, 772–774; [DOI] [PubMed] [Google Scholar]
- 35b. Ferus M., Pietrucci F., Saitta A. M., Knížek A., Kubelíka P., Ivanek O., Shestivska V., Civiš S., Proc. Natl. Acad. Sci. USA 2017, 114, 4306. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Taillades J., Beuzelin I., Garrel L., Tabacik V., Bied C., Commeyras A., Origins Life Evol. Biospheres 1998, 28, 61–77. [DOI] [PubMed] [Google Scholar]
- 37. Martin W., Russell M. J., Philos. Trans. R. Soc. London Ser. B 2007, 362, 1887–1925. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Blöchl E., Keller M., Wächtershäuser G., Stetter K. O., Proc. Natl. Acad. Sci. USA 1992, 89, 8117–8120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Heinen W., Lauwers A. M., Origins Life Evol. Biospheres 1996, 26, 131–150. [DOI] [PubMed] [Google Scholar]
- 40. Huber C., Wächtershäuser G., Science 1997, 276, 245–247. [DOI] [PubMed] [Google Scholar]
- 41. Wächtershäuser G., Adams M. W. W., in Thermophiles: The Keys to Molecular Evolution and the Origin of Life (Ed.: Wiegel J.), 1998, pp. 47–57. [Google Scholar]
- 42.By late heavy bombardement, HCN is generated from carbon and N2: Kurosawa K., Sugita S., Ishibashi K., Hasegawa S., Sekine Y., Ogawa N. O., Kadono T., Ohno S., Ohkouchi N., Nagaoka Y., Matsui T., Origins Life Evol. Biospheres 2013, 43, 221–245. [DOI] [PubMed] [Google Scholar]
- 43. Huber C., Wächtershäuser G., Science 2006, 314, 630–632. [DOI] [PubMed] [Google Scholar]
- 44. Huber C., Wächtershäuser G., Tetrahedron Lett. 2003, 44, 1695–1697. [Google Scholar]
- 45.
- 45a. Huber C., Wächtershäuser G., Science 1998, 281, 670–672; [DOI] [PubMed] [Google Scholar]
- 45b. Huber C., Eisenreich W., Hecht S., Wächtershäuser G., Science 2003, 301, 938–940; [DOI] [PubMed] [Google Scholar]
- 45c. Leman L., Orgel L., Ghadiri M. R., Science 2004, 306, 283–286. [DOI] [PubMed] [Google Scholar]
- 46. Russell M. J., Martin W., Trends Biochem. Sci. 2002, 29, 348–363. [DOI] [PubMed] [Google Scholar]
- 47. Olson K. R., Straub K. D., Physiology 2016, 31, 60–72. [DOI] [PubMed] [Google Scholar]
- 48.
- 48a. Butlerow A. M., Comptes rendus 1861, 53, 145–147 in Justus Liebigs Ann. Chem. 1861, 120, 295–298; structure elucidation by [Google Scholar]
- 48b. Fischer E., Ber. Dtsch. Chem. Ges. 1888, 21, 988–992; [Google Scholar]
- 48c. Fischer E., Ber. Dtsch. Chem. Ges. 1890, 23, 370–394. [Google Scholar]
- 49.
- 49a. Breslow R., Tetrahedron Lett. 1959, 1, 22–26; [Google Scholar]
- 49b. Appayee C., Breslow R., J. Am. Chem. Soc. 2014, 136, 3720–3723. [DOI] [PubMed] [Google Scholar]
- 50.
- 50a. Mizuno T., Weiss A. H., Adv. Carbohydr. Chem. Biochem. 1974, 29, 173–227; [Google Scholar]
- 50b. Socha R. F., Weiss A. H., Sakharov M. M., J. Catal. 1981, 67, 207–217; [Google Scholar]
- 50c.review on the prebiotic geochemistry of formaldehyde see H. J. Cleaves II , Precambrian Res. 2008, 164, 111–118. [Google Scholar]
- 51. Shapiro R., Origins Life Evol. Biospheres 1988, 18, 71–85. [DOI] [PubMed] [Google Scholar]
- 52. Larralde R., Robertson M. P., Miller S. L., Proc. Natl. Acad. Sci. USA 1995, 92, 8158–8160. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Weber A. L., Origins Life Evol. Biospheres 2007, 37, 105–111. [DOI] [PubMed] [Google Scholar]
- 54.
- 54a. Ricardo A., Carrigan M. A., Olcott A. N., Benner S. A., Science 2004, 303, 196; [DOI] [PubMed] [Google Scholar]
- 54b. Kim H.-J., Ricardo A., Illangkoon H. I., Kim M. J., Carrigan M. A., Frye F., Benner S. A., J. Am. Chem. Soc. 2011, 133, 9457–9468. [DOI] [PubMed] [Google Scholar]
- 55.
- 55a. Oró J., Biochem. Biophys. Res. Commun. 1960, 2, 407–412; [Google Scholar]
- 55b. Oró J., Kimball A. P., Arch. Biochem. Biophys. 1961, 94, 217–227; [DOI] [PubMed] [Google Scholar]
- 55c. Oró J., Kamat S. S., Nature 1961, 190, 442–443. [DOI] [PubMed] [Google Scholar]
- 56. Sanchez R. A., Ferris J. P., Orgel L. E., J. Mol. Biol. 1967, 30, 223–253. [PubMed] [Google Scholar]
- 57. Ferris J. P., Orgel L. E., J. Am. Chem. Soc. 1966, 88, 1074. [DOI] [PubMed] [Google Scholar]
- 58. Roy D., Najafian K., von Ragué Schleyer P., Proc. Natl. Acad. Sci. USA 2007, 104, 17272–17277. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. Origins of Prebiological Systems and of Their Molecular Matrices (Eds.: Fox S. W.), Academic Press, New York, 1967. [Google Scholar]
- 60.
- 60a. Schwartz A. W., Goverde M., J. Mol. Evol. 1982, 18, 351–353; [DOI] [PubMed] [Google Scholar]
- 60b. Schwartz A. W., Joosten H., Voet A. B., Biosystems 1982, 15, 191–193. [DOI] [PubMed] [Google Scholar]
- 61. Prebiotic Chemistry and Chemical Evolution of Nucleic Acids (Ed.: Menor-Salván C.), Springer Nature, Switzerland GmbH, 2018. (ISBN 978-3-319-93583-6). [Google Scholar]
- 62.
- 62a. Ferris J. P., Zamek O. S., Altbuch A. M., Freimann H., J. Mol. Evol. 1974, 3, 301–309; [DOI] [PubMed] [Google Scholar]
- 62b. Ferris J. P., Sanchez R. A., Orgel L. E., J. Mol. Biol. 1968, 33, 693–704; [DOI] [PubMed] [Google Scholar]
- 62c. Sanchez R. A., Ferris J. P., Orgel L. E., Science 1966, 154, 784–785. [DOI] [PubMed] [Google Scholar]
- 63. Lohrmann R., J. Mol. Evol. 1972, 1, 263–269. [DOI] [PubMed] [Google Scholar]
- 64.
- 64a.G. Bold (A. Eschenmoser), Ph.D. thesis, ETH Zürich 7702, 1984;
- 64b.H. P. Buser (A. Eschenmoser), Ph.D. thesis, ETH Zürich 7867, 1985;
- 64c.Y. B. Xiang (A. Eschenmoser), Ph.D. thesis, ETH Zürich 7993, 1986.
- 65.
- 65a. Biver N., Bockelée-Morvan D., Debout V., Crovisier J., Boissier J., Lis D., Russo N. D., Moreno R., Colom P., Paubert G., Astron. Astrophys. 2014, 566, L5; [Google Scholar]
- 65b. Saitta M., Saija F., Proc. Natl. Acad. Sci. USA 2014, 111, 13768–13773; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65c. Saladino R., Di Mauro E., García-Ruiz J. M., Chem. Eur. J. 2019, 25, 3181–3189. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66. Yamada H., Hirobe M., Okamoto T., Yakugaku Zasshi 1980, 100, 489–492. [Google Scholar]
- 67. Bizzarri B. M., Botta L., Pérez-Valverde M. I., Saladino R., Di Mauro E., García-Ruiz J. M., Chem. Eur. J. 2018, 24, 8126–8132 and references cited therein. [DOI] [PubMed] [Google Scholar]
- 68. Saladino R., Botta G., Delfino M., Di Mauro E., Chem. Eur. J. 2013, 19, 16916–16922. [DOI] [PubMed] [Google Scholar]
- 69. Bada J. L., Chalmers J. H., H. J. Cleaves II , Phys. Chem. Chem. Phys. 2016, 18, 20085–20095. [DOI] [PubMed] [Google Scholar]
- 70.
- 70a. Sagan C., Khare B. N., Science 1971, 173, 417–420; [DOI] [PubMed] [Google Scholar]
- 70b. Khare B. N., Sagan C., Nature 1971, 232, 577–579. [DOI] [PubMed] [Google Scholar]
- 71. Fuller W. D., Sanchez R. A., Orgel L. E., J. Mol. Biol. 1972, 67, 25–33. [DOI] [PubMed] [Google Scholar]
- 72. Roberts S. J., Szabla R., Todd Z. R., Stairs S., Sasselov D. K. J. D. D., Powner M. W., Nat. Commun. 2018, 9, 4073. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.
- 73a. Becker S., Thoma I., Deutsch A., Gehrke T., Mayer P., Zipske H., Carell T., Science 2016, 352, 833–836; [DOI] [PubMed] [Google Scholar]
- 73b. Becker S., Feldmann J., Wiedemann S., Okamura H., Schneider C., Iwan K., Crisp A., Rossa M., Amatov T., Carell T., Science 2019, 366, 76–82. [DOI] [PubMed] [Google Scholar]
- 74.
- 74a.U. P. Trinks (A. Eschenmoser) Ph.D. thesis, ETH Zürich, 1987, ETH 2-collection under 10.3929/ethz-a-000413538; [DOI]
- 74b.K. E. Koch (A. Eschenmoser) Ph.D. thesis, ETH Zürich, 1992, ETH e-collection under 10.3929/ethz-a-000694124; [DOI]
- 74c. Traube W., Liebigs Ann. Chem. 1904, 331, 64–88. [Google Scholar]
- 75.
- 75a. Schimpl A., Lemmon R. M., Calvin M., Science 1965, 147, 149–150. The dimer of cyanamide, dicyandiamide, is formed upon irradiation of dilute cyanide solutions with ultraviolet light, and by irradiation of a mixture of methane, ammonia, and water with electrons. Recently, evidence was collected that chloride present in sea water promotes the formation of cyanamide via cyanogen chloride (ClCN, 36) under radiolytic conditions.17790694 [Google Scholar]
- 75b. Yi R., Hongo Y., Yoda I., Adam Z. R., Fahrenbach A. C., ChemistrySelect 2018, 3, 10169–10174. [Google Scholar]
- 76.Liebig was the first to find that dicyane is hydrolyzed to the oxamide in water, catalyzed by acetaldehyde: von Liebig J., Liebigs Ann. 1860, 113, 246–247. [Google Scholar]
- 77.Depsipeptides have also been considered as prebiotic polymers:
- 77a. Frenzel-Pinter M., Haynes J. W., Martin C., Petrov A. S., Burcar B. T., Krishnamurthy R., Hud N. V., Leman L. J., Williams L. D., Proc. Natl. Acad. Sci. USA 2019, 116, 16338–16346; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77b. Forsythe J. G., Yu S.-S., Mamajanov I., Grover M. A., Krishnamurthy R., Fernández F. M., Hud N. V., Angew. Chem. Int. Ed. 2015, 54, 9871–9875; [DOI] [PMC free article] [PubMed] [Google Scholar]; Angew. Chem. 2015, 127, 10009–10013. [Google Scholar]
- 78. Leman L., Orgel L., Ghadiri M. R., Science 2004, 306, 283–286. [DOI] [PubMed] [Google Scholar]
- 79. Liu R., Orgel L., Nature 1997, 389, 52–54. [DOI] [PubMed] [Google Scholar]
- 80. Gull M., Zhou M., Fernández F. M., Pasek M. A., J. Mol. Evol. 2014, 78, 109–117. [DOI] [PubMed] [Google Scholar]
- 81. Kim H. J., Furukawa Y., Kakegawa T., Bita A., Scorei R., Benner S. A., Angew. Chem. Int. Ed. 2016, 55, 15816–15820; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2016, 128, 16048–16052. [Google Scholar]
- 82. Saladino R., Carota E., Botta G., Kapralov M., Timoshenko G. N., Rozanov A. Y., Krasavin E., Di Mauro E., Proc. Natl. Acad. Sci. USA 2015, 112, E2746–E2755. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.
- 83a. Nuevo M., Bredehöft J. H., Meierhenrich U. J., d'Hendecourt L., Thiemann W. H. P., Astrobiology 2010, 10, 245–256; [DOI] [PubMed] [Google Scholar]
- 83b. Robertson M., Miller S. L., Nature 1995, 375, 772–774. [DOI] [PubMed] [Google Scholar]
- 84. Baltscheffsky H., Acta Chem. Scand. 1967, 21, 1973–1974. [DOI] [PubMed] [Google Scholar]
- 85.
- 85a. Leman L. J., Orgel L., Ghadiri M. R., J. Am. Chem. Soc. 2006, 128, 20–21; [DOI] [PubMed] [Google Scholar]
- 85b. Biron J. P., Pascal R., J. Am. Chem. Soc. 2004, 126, 9198–9199; [DOI] [PubMed] [Google Scholar]
- 85c. Biron J. P., Parkes A. L., Pascal R., Sutherland J. D., Angew. Chem. Int. Ed. 2005, 44, 6731–6734; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2005, 117, 6889–6892. [Google Scholar]
- 86. Tsanakopoulou M., Sutherland J. D., Chem. Commun. 2017, 53, 11893–11896. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.
- 87a. Müller D., Pitsch S., Kittaka A., Wagner E., Wintner C. E., Eschenmoser A., Helv. Chim. Acta 1990, 73, 1410–1468; [Google Scholar]
- 87b. Krishnamurthy R., Arrhenius G., Eschenmoser A., Origins Life Evol. Biospheres 1999, 29, 333–354; [DOI] [PubMed] [Google Scholar]
- 87c. Krishnamurthy R., Guntha S., Eschenmoser A., Angew. Chem. Int. Ed. 2000, 39, 2281–2285; [PubMed] [Google Scholar]; Angew. Chem. 2000, 112, 2369–2373; [Google Scholar]
- 87d. Ritson D. J., Zu J., Sutherland J. D., Synlett 2017, 28, 64–67; [Google Scholar]
- 87e. Gibard C., Gorell I. B., Jimémez E. I., Klee T. P., Pasek M. A., Krishnamurthy R., Angew. Chem. 2019, 131, 8235–8239. [Google Scholar]
- 88. Powner M. W., Gerland B., Sutherland J. D., Nature 2009, 459, 239–242. [DOI] [PubMed] [Google Scholar]
- 89. Coggins A. J., Powner M. W., Nat. Chem. 2017, 9, 310–317. [DOI] [PubMed] [Google Scholar]
- 90. Yamagata Y., Origins Life Evol. Biospheres 1999, 29, 511–520. [DOI] [PubMed] [Google Scholar]
- 91. Costanzo G., Saladino R., Crestini C., Ciciriello F., Di Mauro E., J. Biol. Chem. 2007, 282, 16729–16735. [DOI] [PubMed] [Google Scholar]
- 92.
- 92a. Goesmann F., Rosenbauer H., Bredehöft J. H., Cabane M., Ehrenfreund P., Gautier T., Giri C., Krüger H., le Roy L. L., MacDermott A. J., Science 2015, 349, aab0689; [DOI] [PubMed] [Google Scholar]
- 92b. Altwegg K., Balsiger H., Bar-Nun A., Berthelier J.-J., Bieler A., Bochsler P., Briois C., Calmonte U., Combi M. R., Cottin H., Sci. Adv. 2016, 2, e1600285; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92c. Ehrenfreund P., Chanley S. B., Annu. Rev. Astron. Astrophys. 2000, 38, 427–483. [Google Scholar]
- 93. Schneider C., Becker S., Okamura H., Crisp A., Amatov T., Stadlmeier M., Carell T., Angew. Chem. Int. Ed. 2018, 57, 5943–5946; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2018, 130, 6050–6054. [Google Scholar]
- 94. Yung Y. L., Mcelroy M. B., Science 1979, 203, 1002–1004. [DOI] [PubMed] [Google Scholar]
- 95.
- 95a. Begley T. D., Chatterjee A., Hanes J. W., Hazra A., Ealick S. E., Curr. Opin. Chem. Biol. 2008, 12, 118–125; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95b. Begley T. P., Nat. Prod. Rep. 2006, 23, 15–25. [DOI] [PubMed] [Google Scholar]
- 96.Three main biosynthetic routes towards TPP have been described in plants, fungi, and bacteria:
- 96a. Lin S., Cronan J. E., Mol. BioSyst. 2011, 7, 1811–1821; [DOI] [PubMed] [Google Scholar]
- 96b. Mann S., Ploux O., Biochim. Biophys. Acta Proteins Proteomics 2011, 1814, 1459–1466; [DOI] [PubMed] [Google Scholar]
- 96c. Lin S., Hanson R. E., Cronan J. E., Nat. Chem. Biol. 2010, 6, 682–688; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96d. Pohl M., Sprenger G. A., Müller M., Curr. Opin. Biotechnol. 2004, 15, 335–342. [DOI] [PubMed] [Google Scholar]
- 97.It has to be noted that hyperthermophiles such as the species Nanoarchaeota are regarded as one of the earliest members which thrive in submarine hot vents: Stetter K. O., Philos. Trans. R. Soc. London Ser. B 2006, 361, 1837–1843. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.
- 98a. Fischer M., Bacher A., Nat. Prod. Rep. 2005, 22, 324–350; [DOI] [PubMed] [Google Scholar]
- 98b. Bacher A., Eberhardt S., Fischer M., Kis K., Richter G., Annu. Rev. Nutr. 2000, 20, 153–167; [DOI] [PubMed] [Google Scholar]
- 98c. Mack M., Grill S., Appl. Microbiol. Biotechnol. 2006, 71, 265–275. [DOI] [PubMed] [Google Scholar]
- 99. Visser C. M., Origins Life 1982, 12, 165–179. [DOI] [PubMed] [Google Scholar]
- 100. Iobbi-Nivol C., Leimkühler S., Biochim. Biophys. Acta Bioenerg. 2013, 1827, 1086–1101. [DOI] [PubMed] [Google Scholar]
- 101. Sato T., Atomi H., Curr. Opin. Microbiol. 2011, 14, 307–314 and references cited therein. [DOI] [PubMed] [Google Scholar]
- 102. van Nguyen K., Burrows C. J., J. Am. Chem. Soc. 2011, 133, 14586–14589. [DOI] [PubMed] [Google Scholar]
- 103.
- 103a. Tan Z., Zhu C., Fu J., Zhang X., Li M., Zhuang W., Ying H., Angew. Chem. 2018, 130, 16702–16706; [DOI] [PubMed] [Google Scholar]
- 103b. Zhu C., Li Q., Pu L., Kan Z., Guo K., Ying H., Ouyang P., ACS Catal. 2016, 6, 4989–4994. [Google Scholar]
- 104. Heinz B., Ried W., Dose K., Angew. Chem. Int. Ed. 1979, 18, 478–483; [Google Scholar]; Angew. Chem. 1979, 91, 510–511. [Google Scholar]
- 105.
- 105a. Rowan T., Wood H. C. S., Proc. Chem. Soc. 1963, 21–22; [Google Scholar]
- 105b. Rowan T., Wood H. C. S., J. Chem. Soc. C 1968, 452–458; [DOI] [PubMed] [Google Scholar]
- 105c. Paterson T., Wood H. C. S., Chem. Commun. 1969, 290–291; [Google Scholar]
- 105d. Paterson T., Wood H. C. S., J. Chem. Soc. Perkin Trans. 1 1972, 1051–1056; [DOI] [PubMed] [Google Scholar]
- 105e. Crout D. H. G., Hatfield J. A., J. Chem. Soc. Chem. Commun. 1987, 742–744. [Google Scholar]
- 106.
- 106a. Beach R. L., Plaut G. W. E., Biochemistry 1970, 9, 760–770; [DOI] [PubMed] [Google Scholar]
- 106b. Plaut G. W. E., Beach R. L., in Flavins and Flavoproteins (Ed.: Singer T. P.), Elsevier, Amsterdam, 1976, pp. 737–746. [Google Scholar]
- 107.C. J. Strupp, Ph.D. thesis, Nr. 9832, ETH Zürich, 1992.
- 108. Kis K., Kugelbrey K., Bacher A., J. Org. Chem. 2001, 66, 2555–2559. [DOI] [PubMed] [Google Scholar]
- 109. Okamura H., Becker S., Tiede N., Wiedemann S., Feldmann J., Carell T., Chem. Commun. 2019, 55, 1939–1942. [DOI] [PubMed] [Google Scholar]
- 110. Reuke B., Korn S., Eisenreich W., Bacher A., J. Bacteriol. 1992, 174, 4042–4049. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111. Greening C., Ahmed F. H., Mohamed A. E., Lee B. M., Pandey G., Warden A. C., Scott C., Oakeshott J. G., Taylor M. C., Jackson C. J., Microbiol. Mol. Biol. Rev. 2016, 80, 451–493. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112.
- 112a. Walsh C., Acc. Chem. Res. 1986, 19, 216–221; [Google Scholar]
- 112b. Friedrich W., Vitamins, Walter de Gruyter, Berlin, 1988. [Google Scholar]
- 113. Bermingham A., Derrick J. P., BioEssays 2002, 24, 637–648. [DOI] [PubMed] [Google Scholar]
- 114.
- 114a. Acharya P., Warkentin E., Ermler U., Thauer R. K., Shima S., J. Mol. Biol. 2006, 357, 870–879; [DOI] [PubMed] [Google Scholar]
- 114b. White R. H., Vitam. Horm. 2001, 61, 299–337. [DOI] [PubMed] [Google Scholar]
- 115. Wang P.-Z., Chen J.-R., Xiao W.-J., Org. Biomol. Chem. 2019, 17, 6936–6951. [DOI] [PubMed] [Google Scholar]
- 116.
- 116a. Dowler M. J., Fuller W. D., Orgel L. E., Sanchez R. A., Science 1970, 169, 1320–1321; [DOI] [PubMed] [Google Scholar]
- 116b. Friedmann N., Miller S. L., Sanchez R. A., Science 1971, 171, 1026–1027; [DOI] [PubMed] [Google Scholar]
- 116c. Cleaves H. J., Miller S. L., J. Mol. Evol. 2001, 52, 73–77. [DOI] [PubMed] [Google Scholar]
- 117. Begley T. P., Kinsland C., Mehl R. A., Osterman A., Dorrestein P., Vitam. Horm. 2001, 61, 103–119. [DOI] [PubMed] [Google Scholar]
- 118.Abiotic formation of aspartic acid: Civiš S., Juha L., Babánková D., Cvačka J., Frank O., Jehlička J., Králiková B., Krása J., Kubát P., Muck A., Pfeifer M., Skála J., Ullschmied J., Chem. Phys. Lett. 2004, 386, 169–173. [Google Scholar]
- 119. Kim H., Benner S. A., Chem. Eur. J. 2018, 24, 581–584. [DOI] [PubMed] [Google Scholar]
- 120. Guarneri A., van Berkel W. J. H., Paul C. E., Curr. Opin. Biotechnol. 2019, 60, 63–71. [DOI] [PubMed] [Google Scholar]
- 121.
- 121a. Paul C. P., Hollemann F., Appl. Microbiol. Biotechnol. 2016, 100, 4773–4778; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 121b. Paul C. E., Arends I. W. C. E., Hollemann F., ACS Catal. 2014, 4, 788–797; [Google Scholar]
- 121c. Paul C. E., Gargiulo S., Opperman D. J., Lavandera I., Gotor-Fernández V., Gotor V., Taglieber A., Arends I. W. C. E., Hollmann F., Org. Lett. 2013, 15, 180–183. [DOI] [PubMed] [Google Scholar]
- 122.
- 122a. Liscombe D. K., Louie G. V., Noel J. P., Nat. Prod. Rep. 2012, 29, 1238–1250; [DOI] [PubMed] [Google Scholar]
- 122b. O'Hagan D., Schmidberger J. W., Nat. Prod. Rep. 2010, 27, 900–918. [DOI] [PubMed] [Google Scholar]
- 123.
- 123a. Sofia H. J., Chen G., Hetzler B. G., Reyes-Spindola J. F., Miller N. E., Nucleic Acids Res. 2001, 29, 1097–1106; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 123b. Zhang Q., van der Donk W. A., Liu W., Acc. Chem. Res. 2012, 45, 555–564; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 123c. Frey P. A., Annu. Rev. Biochem. 2001, 70, 121–148. [DOI] [PubMed] [Google Scholar]
- 124. Prieur B. E., J. Biol. Phys. 1995, 20, 301–312. [Google Scholar]
- 125. Waddell T. G., Eilders L. L., Patel B. P., Sims M., Origins Life Evol. Biospheres 2000, 30, 539–548. [DOI] [PubMed] [Google Scholar]
- 126. Laurino P., Tawfik D. S., Angew. Chem. Int. Ed. 2017, 56, 343–345; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2017, 129, 349–351. [Google Scholar]
- 127.
- 127a. Du Y.-L., Ryan K. S., Nat. Prod. Rep. 2019, 36, 430–457; [DOI] [PubMed] [Google Scholar]
- 127b. Eliot A. C., Kirsch J. F., Annu. Rev. Biochem. 2004, 73, 383–415; [DOI] [PubMed] [Google Scholar]
- 127c. Drewke C., Leistner E., Vitam. Horm. 2001, 61, 121–155. [DOI] [PubMed] [Google Scholar]
- 128.
- 128a. Bishop J. C., Cross S. D., Waddell T. G., Origins Life Evol. Biospheres 1997, 27, 319–324. For the first report on transaminations, see [DOI] [PubMed] [Google Scholar]; Herbst R. M., J. Am. Chem. Soc. 1936, 58, 2239; [Google Scholar]
- 128b. Williamson J. M., Brown G. M., J. Biol. Chem. 1979, 254, 8074–8082. [PubMed] [Google Scholar]
- 129. Raushel F. M., Thoden J. B., Holden H. M., Acc. Chem. Res. 2003, 36, 539–548. [DOI] [PubMed] [Google Scholar]
- 130.
- 130a. Aylward N., Bofinger N., Biophys. Chem. 2006, 123, 113–121; [DOI] [PubMed] [Google Scholar]
- 130b.a de novo pyridine synthesis related to this approach was recently reported: Zhou G., Zhao X., Dan W., Tetrahedron Lett. 2017, 58, 3085–3088. [Google Scholar]
- 131. Austin S. M., Waddell T. G., Origins Life Evol. Biospheres 1999, 29, 287–296. [DOI] [PubMed] [Google Scholar]
- 132. Chichibabin A. E., J. Russ. Phys. Chem. Soc. 1906, 37, 1229. [Google Scholar]
- 133.
- 133a. Fitzpatrick T. B., Amrhein N., Kappes B., Macheroux P., Tews I., Raschle T., Biochem. J. 2007, 407, 1–13; [DOI] [PubMed] [Google Scholar]
- 133b. Mukherjee T., Hanes J., Tews I., Ealick S. E., Begley T. P., Biochim. Biophys. Acta Proteins Proteomics 2011, 1814, 1585–1596; [DOI] [PubMed] [Google Scholar]
- 133c. Drewke C., Leistner E., Vitam. Horm. 2001, 61, 121–155. [DOI] [PubMed] [Google Scholar]
- 134.
- 134a. Szostak J. W., Bartel D. P., Luisi P. I., Nature 2001, 409, 387–390; [DOI] [PubMed] [Google Scholar]
- 134b. Deamer D., Life 2017, 7, 5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 135.
- 135a. Thomas J. A., Rana F. R., Origins Life Evol. Biospheres 2007, 37, 267–285; [DOI] [PubMed] [Google Scholar]
- 135b. McCollom T. M., Rev. Minerol. Geochem. 2013, 75, 467–494; [Google Scholar]
- 135c. McCollom T. M., Seewald J. S., Chem. Rev. 2007, 107, 382–401. [DOI] [PubMed] [Google Scholar]
- 136.
- 136a. McCollom T. M., Ritter G., Simoneit B. R. T., Origins Life Evol. Biospheres 1999, 29, 153–166; [DOI] [PubMed] [Google Scholar]
- 136b. Nooner D. W., Oro J., Proc. Adv. Chem. 1980, 178, 159–171; [Google Scholar]
- 136c. Rushdie A. I., Simoneit B. R. T., Origins Life Evol. Biospheres 2001, 31, 103; [DOI] [PubMed] [Google Scholar]
- 136d. Simoneit B. R. T., Rushdie A. I., Deamer D. W., Adv. Space Res. 2007, 40, 1649–1656. [Google Scholar]
- 137. Scheidler C., Sobotta J., Eisenreich W., Wächtershäuser G., Huber C., Sci. Rep. 2016, 6, 27595. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 138. Prieur B. E., in Chemical Evolution: Physics of the Origin and the Evolution of Life (Eds.: Chela-Flores J., Raulin F.), Kluver, Dordrecht, Boston, London, 1996, pp. 171–176. [Google Scholar]
- 139.
- 139a. Désaubry L., Nakatani Y., Ourisson G., Tetrahedron Lett. 2003, 44, 6959–6961; [Google Scholar]
- 139b. Cafferty B. J., Yuan L., Baghbanzadeh M., Rappoport D., Beyzavi N. H., Whitesides G. M., Angew. Chem. Int. Ed. 2019, 58, 8097–8102; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2019, 131, 8181–8186. [Google Scholar]
- 140. Marquet A., Bui B. T. S., Florentin D., Vitam. Horm. 2001, 61, 51–101. [DOI] [PubMed] [Google Scholar]
- 141.Evidence was collected that carbon disulfide can serve as source for organic di- and oligosulfides under hydrothermal reducing conditions. These mimic the redox properties of lipoic acid in a prebioic world: Rushdi I., Simonet B. R. T., Astrobiology 2005, 5, 749–769. [DOI] [PubMed] [Google Scholar]
- 142. Bonfio C., Valer L., Scintilla S., Shah S., Evans D. J., Jin L., Szostak J. W., Sasselov D. D., Sutherland J. D., Mansy S. S., Nat. Chem. 2017, 9, 1229–1234. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 143. Visser C. M., Kellogg R. M., J. Mol. Evol. 1978, 11, 171–187. [DOI] [PubMed] [Google Scholar]
- 144. Kochanek S. E., Clymer T. M., Pakkala V. S., Hebert S. P., Reeping K., Firestine S. M., Evanseck J. D., J. Phys. Chem. B 2015, 119, 1184–1191. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 145. Tabita F. R., Hanson T. E., Li H., Satagopan S., Singh J., Tabita S. C., Microbiol. Mol. Biol. Rev. 2007, 71, 576–599. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 146. Begley T. B., Kinsland C., Strauss E., Vitam. Horm. 2001, 61, 157–171. [DOI] [PubMed] [Google Scholar]
- 147. Bracher P. J., Snyder P. W., Bohall B. R., Whitesides G. M., Origins Life Evol. Biospheres 2011, 41, 399–412. [DOI] [PubMed] [Google Scholar]
- 148. Enzmann F., Mayer F., Rother M., Holtmann D., AMB Express 2018, 8, 1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 149. Reeves E. P., McDermott J. M., Seewald J. S., Proc. Natl. Acad. Sci. USA 2014, 111, 5474–5479. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 150. Miller S. L., Schlesinger G., J. Mol. Evol. 1993, 36, 308–314. [DOI] [PubMed] [Google Scholar]
- 151. Keefe A. D., Newton G. L., Miller S. L., Nature 1995, 373, 683–685. [DOI] [PubMed] [Google Scholar]
- 152. Jadhav R., Yarus M., Biochemie 2002, 84, 877–888. [DOI] [PubMed] [Google Scholar]
- 153. Miller S. L., Schlesinger G., J. Mol. Evol. 1993, 36, 302–307. [DOI] [PubMed] [Google Scholar]
- 154. Raulin F., Frère C., Grana 1991, 30, 510–513. [Google Scholar]
- 155. Breckenridge W. H., Taube H., J. Chem. Phys. 1970, 53, 1750–1766. [Google Scholar]
- 156. Downs D., Nat. Microbiol. 2016, 2, 16252. [DOI] [PubMed] [Google Scholar]
- 157. Maupin-Furlow J. P., B Group Vitamins—Current Uses and Perspectives (Eds.: LeBlanc J. G., de Giori G. S.), IntechOpen Limited, London, 2018, Chapter 2, 10.5772/intechopen.77170. [DOI] [Google Scholar]
- 158. Matsumoto T., Yamamoto H., Inoue S., J. Am. Chem. Soc. 1984, 106, 4829–4832. [Google Scholar]
- 159. Leeper F. J., Nat. Prod. Rep. 1989, 6, 171–203. [DOI] [PubMed] [Google Scholar]
- 160. Nelson D. R., Kamataki T., Waxman D. J., Guengerich F. P., Estabrook R. W., Feyereisen R., Gonzalez F. J., Coon M. J., Gunsalus I. C., Gotoh O., Okuda K., Nebert D. W., DNA Cell Biol. 1993, 12, 1–51. [DOI] [PubMed] [Google Scholar]
- 161.
- 161a. Fox S., Strasdeit H., Astrobiology 2013, 13, 578–595; [DOI] [PubMed] [Google Scholar]
- 161b. Pleyer H. L., Strasdeit H., Fox S., Origins Life Evol. Biospheres 2018, 48, 347–371. [DOI] [PubMed] [Google Scholar]
- 162. Hodgson G. W., Baker B. L., Nature 1967, 216, 29–32. [DOI] [PubMed] [Google Scholar]
- 163. Callot H. J., Ocampo R., in The porphyrin handbook, Vol. 1 (Eds.: Kadish K. M., Smith K. M., Guilard R.), Academic Press, San Diego, 2000, pp. 349–398. [Google Scholar]
- 164.
- 164a. Lindsey J. S., Ptaszek M., Taniguchi M., Origins Life Evol. Biospheres 2009, 39, 495–515; [DOI] [PubMed] [Google Scholar]
- 164b. Lindsey J. S., Chandrashaker V., Taniguchi M., Ptaszek M., New J. Chem. 2011, 35, 65–75; [Google Scholar]
- 164c. Soares A. R. M., Taniguchi M., Chandrashaker V., Lindsey J. S., Chem. Sci. 2012, 3, 1963–1974; [Google Scholar]
- 164d. Soares A. R. M., Taniguchi M., Chandrashaker V., Lindsey J. S., New J. Chem. 2013, 37, 1073–1086; [Google Scholar]
- 164e. Soares A. R. M., Anderson D. R., Chandrashaker V., Lindsey J. S., New J. Chem. 2013, 37, 2716–2732. [Google Scholar]
- 165. Pohorille A. Early ancestors of existing cells. in: Protocells: bridging nonliving and living matter (Eds.: Rasmussen S., Bedau M. A., Chen L., Deamer D., Krakauer D. C., Packard N. H., Stadler P. F.), MIT Press, Cambridge, 2009, pp. 563–581. [Google Scholar]
- 166. Breaker R. R., Cold Spring Harbor Perspect. Biol. 2012, 4, a003566. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 167. Krishnamurthy R., Acc. Chem. Res. 2017, 50, 455–459. [DOI] [PubMed] [Google Scholar]