

Abstract
Background
Epidermolysis bullosa (EB) is a heterogeneous group of rare and incurable genetic disorders characterized by fragility of the skin and mucosae, resulting in blisters and erosions. Several epidemiological studies in other populations have been carried out, reporting varying and sometimes inconclusive figures, highlighting the need for standardized epidemiological analyses in well‐characterized cohorts.
Objectives
To evaluate the epidemiological data on EB in the Netherlands, extracted from the molecularly well‐characterized cohort in the Dutch EB Registry.
Methods
In this observational study all EB‐patients that were based in the Netherlands and captured in the Dutch EB Registry between 1988 and 2018 were included. The epidemiological outcomes were based on complete diagnostic data (clinical features, immunofluorescence, electron microscopy and mutation analysis), with longitudinal follow‐up.
Results
A total of 464 EB‐patients (287 families) were included. The incidence and point‐prevalence of EB in the Netherlands were 41.3 per million live births and 22.4 per million population, respectively. EB Simplex (EBS), Junctional EB (JEB), Dystrophic EB (DEB) and Kindler EB were diagnosed in 45.7%, 18.8%, 34.7% and 0.9% of the EB‐patients, respectively, with an incidence and point‐prevalence of 17.5 and 11.9 (EBS), 9.3 and 2.1 (JEB), 14.1 and 8.3 (DEB), 0.5 and 0.2 (Kindler EB). In 90.5% of the EB‐patients the diagnosis was genetically confirmed. During the investigated time period 73 EB‐patients died, 72.6% of whom as a direct consequence of their EB.
Conclusion
The epidemiological outcomes of EB in the Netherlands are high, attributed to a high detection rate in a well‐organized set‐up, indicating that EB might be more common than previously assumed. These epidemiological data help to understand the extensive need for (specialized) medical care of EB‐patients and is invaluable for the design and execution of therapeutic trials. This study emphasizes the importance of thorough reporting systems and registries worldwide.
Short abstract
Linked Commentary: L. Bruckner‐Tuderman. J Eur Acad Dermatol Venereol 2021; 35: 783–784. https://doi.org/10.1111/jdv.17165.
Introduction
Epidermolysis bullosa (EB) is a heterogeneous group of rare and incurable genetic cutaneous disorders, caused by mutations within the genes encoding critical proteins for the intra‐epidermal cell–cell adhesion and dermo‐epidermal junction (DEJ). 1 , 2 The very recent consensus classification has now divided this heterogeneous group of skin fragility disorders in ‘classical’ EB and ‘EB‐related’ disorders. 2 Depending on the level of skin cleavage, four major types of EB are recognized: EB Simplex (EBS), Junctional EB (JEB), Dystrophic EB (DEB) and Kindler EB. Based on the clinical severity, inheritance pattern and molecular defects, these major types of EB are further divided into many subtypes, that share common features of mechanical fragility and blistering of the skin and mucous membranes. 1 , 2 Most EB‐patients have lifelong blistering and erosions with significant morbidity due to related complications, associated with early mortality. 1
The Center for Blistering Diseases at the University Medical Center Groningen (UMCG) in the Netherlands is a recognized European and the only national expertise centre for EB. Since the registration of its first patient in the Dutch EB Registry (Dutch‐EB‐Reg) in 1988 the centre has evolved and contributed to the diagnosis, research and management of EB‐patients in the Netherlands and other countries for over three decades. 3
Epidemiological data improve our understanding of EB, emphasize the extent and need for (specialized) medical care of EB‐patients and are invaluable for the design and execution of therapeutic trials. Several epidemiological studies in other populations have been carried out, reporting varying and sometimes inconclusive figures (incidences of 1.4–25.0 per million live births and prevalences of 2.82–54.0 per million population), mostly based on clinical features or molecular findings. Through this report, we provide accurate epidemiological figures of each subtype of EB according to the novel consensus report, including subtype‐specific mortality rates, extracted from the molecularly well‐characterized Dutch EB cohort in the Dutch‐EB‐Reg, established over a 31‐year period.
Materials and methods
Study design and data collection
In this population‐based and observational (cross‐sectional and longitudinal) study, all EB‐patients registered at our centre from 01‐Jan‐1988 until 31‐Dec‐2018 were included. EB‐patients referred to our expertise centre by specialists from foreign countries were excluded in order to provide accurate epidemiological outcomes of EB in the Netherlands. The diagnosis of EB was established based on clinical features, skin biopsies for immunofluorescence antigen mapping (IFM) and transmission electron microscopy (TEM), and in particular mutation analysis, which over the years evolved from sanger sequencing of single candidate genes to next‐generation sequencing of multiple EB‐related genes in parallel. 4 The subclassification of EB was conducted according to the latest consensus report. 2 The medical records of the EB‐patients were systematically collected during the investigated time period and retrospectively examined. This study was carried out in accordance with the principles of the Declaration of Helsinki.
EB‐related skin fragility disorders
The 2020 EB‐consensus reclassification separated the former suprabasal EBS phenotypes as ‘EB‐related skin fragility disorders’ from the classical EB‐phenotypes (acral peeling skin syndrome, lethal acantholytic EB, skin fragility‐woolly hair syndrome and skin fragility‐ectodermal dysplasia syndrome). 1 , 2 Therefore, patients with such phenotypes were excluded in the calculations and described in the Appendix S1 (Supporting Information).
Data analysis
The quantitative and qualitative characteristics of the EB‐patients were summarized as absolute numbers, averages together with standard deviations (SD) and proportions (%). Incidences were calculated per million live births [total no. of new EB‐patients born (1‐Jan‐1988 until 31‐Dec‐2018)/total no. of live births (1988–2018)*1 000 000], when calculated per year the incidences were presented as rates. Prevalences were calculated per million population [total no. of EB‐patients alive at a time point (31‐Dec‐2018)/total population at a time point (31‐Dec‐2018)*1 000 000]. The number of live births, average life expectancy of the Dutch population and population size of the Netherlands were collected from the national Central Agency for Statistics (https://opendata.cbs.nl/#/CBS/nl/). The epidemiological data were analysed using SPSS 23.0 (SPSS Inc., Chicago, IL, USA). There was no correction for multiplicity.
Results
Incidence and point‐prevalence
From 1 January 1988 until 31 December 2018, 544 EB‐patients (358 families) were registered at our centre, of which 80 EB‐patients (71 families; 14.7%) referred by specialists from foreign countries. The remaining 464 EB‐patients (287 families; 247 males and 217 females) were based in the Netherlands, of which 346 patients were of Dutch descent (74.6%), 27 patients had a mixed ethnic background (5.8%), 78 patients were immigrants (16.8%) (n = 13 unknown, 2.8%). The incidence and point‐prevalence of EB in the Netherlands were calculated to be 41.3 per million live births (5 830 469 live births, 1988–2018, StatLine, CBS, The Hague, (South Holand), Netherlands) and 22.4 per million population (17 282 163 total population, 31‐Dec‐2018, StatLine, CBS).
Overall, a genetic diagnosis was made in 420 patients (254 families; 90.5%). In 22 patients (18 families; 4.7%) no DNA was available for mutation analysis. In another 22 patients (15 families; 4.7%) no pathogenic mutation could be found (or just a single pathogenic mutation in case of recessive EB) and hence classified as genetically unsolved (Fig. 2b). However, the level of blister formation could be established by IFM and TEM on available skin biopsies in most of these patients, and therefore the major type of EB (n = 10 unknown). 4 Dominant inheritance was present in 304 patients (149 families; 65.5%) and recessive inheritance in 160 patients (138 families; 34.5%). Only in 16 patients with recessive EB it remained uncertain whether the parents were consanguineous or not.
Figure 2.
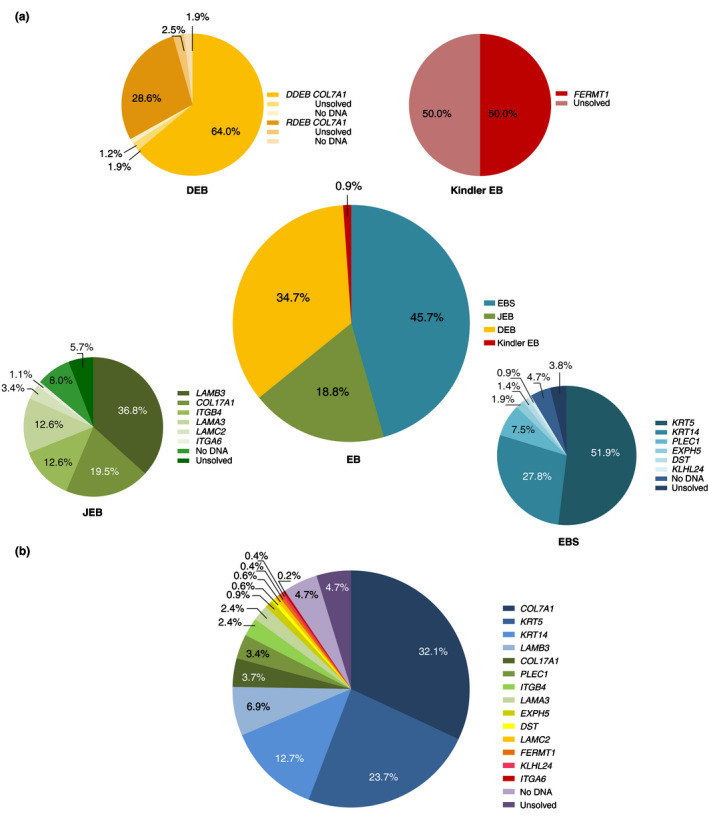
Distribution of the major types and genes involved in patients with Epidermolysis Bullosa in the Dutch Epidermolysis Bullosa Registry for the time period 1988–2018, n = 464. (a) The total distribution and genes involved in the major EB types in the cohort of the Dutch Epidermolysis Bullosa Registry (Dutch‐EB‐Reg). (b) The total distribution of the 14 genes involved in the Dutch Epidermolysis Bullosa Registry (Dutch‐EB‐Reg). Patients without mutation analysis performed were classified as ‘No DNA’; patients in which no pathogenic mutation(s) could be found with mutation analysis were genetically classified as ‘Unsolved’.
EB simplex
In the Dutch‐EB‐Reg, EBS was most frequently diagnosed (212 patients; 108 families; 45.7%) with an incidence of 17.5 per million live births and a point‐prevalence of 11.9 per million population (Figs. 1 and 2a, Table 1). Dominant EBS was more common than recessive EBS (191 patients; 94 families; 90.1% vs. 21 patients; 14 families; 9.9%). In 81.0% of the patients with recessive EBS a homozygous mutation was found, of which 52.9% associated with known parental consanguinity. Mutation analysis implicated six genes in the EBS cohort. In 8.5% of the EBS‐patients (n = 18) the causative gene could not be discovered.
Figure 1.
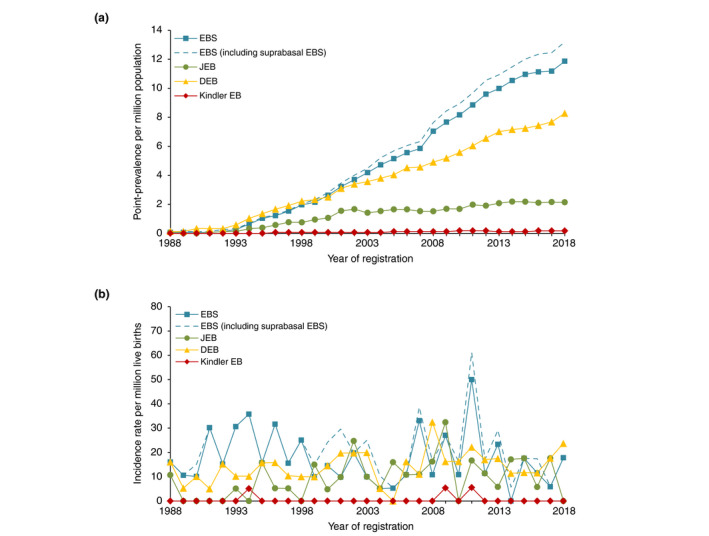
Epidemiological outcomes of each major type of Epidermolysis Bullosa in the Netherlands for the time period 1988–2018,n = 490. (a) Annual point‐prevalence (per million population) of each major type of EB. (b) Annual incidence rates (per million live births). Based on the Dutch Epidermolysis Bullosa Registry (Dutch‐EB‐Reg). DEB, dystrophic epidermolysis bullosa; EB, epidermolysis bullosa; EBS, epidermolysis bullosa simplex; JEB, junctional epidermolysis bullosa.
Table 1.
Epidemiological data and distribution of Epidermolysis Bullosa Simplex in the Netherlands for the time period 1988–2018, n = 212
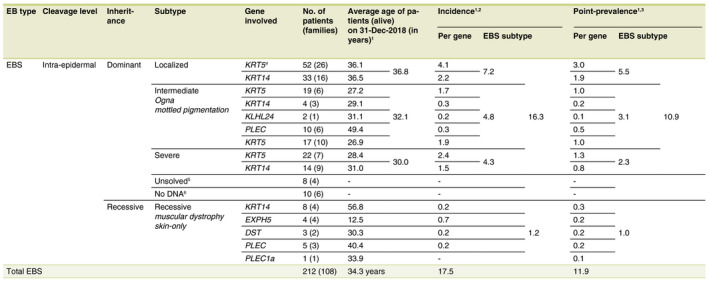
†If no mutation analysis was performed or pathogenic mutation(s) could be found, the patients were still included in the calculation of the incidence and point‐prevalence for the concerned subtype of EBS. ‡Incidence (per million live births) = total no. of new EBS‐patients born (1988–2018)/total no. of live births (1988–2018)*1 000 000. Total live births in the Netherlands from 01‐Jan‐1988 until 31‐Dec‐2018: 5 830 469 (StatLine, CBS). §Point‐prevalence (per million population) = total no. of EBS‐patients alive at a time point (31‐Dec‐2018)/population at a time point (31‐Dec‐2018)*1 000 000. Total Dutch population, 31‐Dec‐2018: 17 282 163 (StatLine, CBS). ¶Two patients with EBS‐localized, without mutation analysis performed, were included because a pathogenic mutation in KRT5 was found in a family member, with a comparable clinical presentation. ††The group of ‘Unsolved’ EBS‐patients consisted of the following: n = 7 EBS‐localized and n = 1 EBS‐intermediate. ‡‡The group of ‘No DNA’ EBS‐patients consisted of the following: n = 4 EBS‐localized, n = 3 EBS‐intermediate and n = 3 EBS‐severe.
EB, epidermolysis bullosa; JEB, junctional epidermolysis bullosa.
Junctional EB
Junctional EB was diagnosed in 87 patients (72 families; 18.8%) with an incidence of 9.3 per million live births and a point‐prevalence of 2.1 per million population (Figs. 1 and 2a, Table 2). JEB was inherited autosomal recessively in 94.3% of the cases, of which 43.9% carried a homozygous mutation (of which 58.3% associated with known parental consanguinity). In five patients (one family; 5.7%) a dominant form of JEB was diagnosed, with a mutation in the ITGB4 gene. 5 Six genes were implicated in the pathogenesis of JEB in our cohort. In 13.8% of the JEB‐patients (n = 12) the diagnosis could not be confirmed genetically.
Table 2.
Epidemiological data and distribution of Junctional Epidermolysis Bullosa in the Netherlands for the time period 1988–2018, n = 87
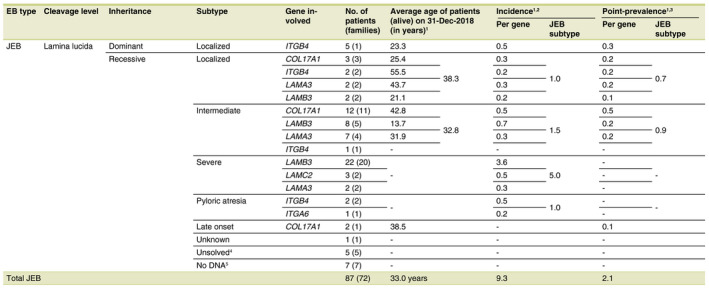
†If no mutation analysis was performed or pathogenic mutation(s) could be found, the patients were still included in the calculation of the incidence and point‐prevalence for the concerned subtype of JEB (two stillborn patients, an induced partus of a foetus and a patient who was born and has died before 1988, were excluded in the calculations of the incidence and point‐prevalence, but included in the number of affected patients). ‡Incidence (per million live births) = total no. of new JEB‐patients born (1988–2018)/total no. of live births (1988–2018)*1 000 000. Total live births in the Netherlands from 01‐Jan‐1988 until 31‐Dec‐2018: 5 830 469 (StatLine, CBS). §Point‐prevalence (per million population) = total no. of JEB‐patients alive at a time point (31‐Dec‐2018)/population at a time point (31‐Dec‐2018)*1 000 000. Total Dutch population, 31‐Dec‐2018: 17 282 163 (StatLine, CBS). ¶The group of ‘Unsolved’ JEB‐patients consisted of the following; n = 3 JEB‐localized (n = 2 one pathogenic mutation in LAMA3, n = 1 one pathogenic mutation in ITGB4) and n = 2 JEB‐pyloric atresia (n = 1 one pathogenic mutation in ITGB4). ††The group of ‘No DNA’ JEB‐patients consisted of the following; n = 4 JEB‐severe, n = 2 JEB‐subtype unknown (two stillborn patients) and n = 1 JEB‐pyloric atresia.
EB, epidermolysis bullosa; JEB, junctional epidermolysis bullosa.
Dystrophic EB
Dystrophic EB was identified in 161 patients (103 families; 34.7%) with recurrent and unique mutations in the COL7A1 gene (Fig. 2a). An incidence of 14.1 per million live births was calculated and a point‐prevalence of 8.3 per million population (Fig. 1, Table 3), with dominant DEB (DDEB) in 108 patients (54 families; 67.1%) and recessive DEB (RDEB) in 53 patients (49 families; 32.9%), of which 35.8% carried a homozygous mutation (94.7% of which had consanguineous parents). In 7.5% of the DEB‐patients (n = 12) the genotype remained unknown.
Table 3.
Epidemiological data and distribution of Dystrophic Epidermolysis Bullosa in the Netherlands for the time period 1988–2018, n = 161
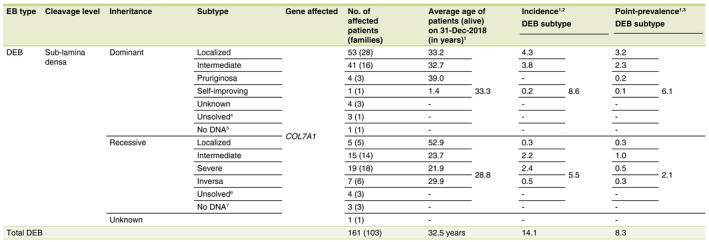
†If no mutation analysis was performed or pathogenic mutation(s) could be found, the patients were still included in the calculation of the incidence and point‐prevalence for the concerned subtype of DEB. ‡Incidence (per million live births) = total no. of new patients born (1988–2018)/total no. of live births (1988–2018)*1 000 000. Total live births in the Netherlands from 01‐Jan‐1988 until 31‐Dec‐2018: 5 830 469 (StatLine, CBS). §Point‐prevalence (per million population) = total no. of patients alive at a time point (31‐Dec‐2018)/population at a time point (31‐Dec‐2018)*1 000 000. Total Dutch population, 31‐Dec‐2018: 17 282 163 (StatLine, CBS). ¶The group of ‘Unsolved’ DDEB‐patients consisted of a family of three patients with DDEB‐localized. ††In this patient the subtype of DDEB remained uncertain. ‡‡The group of ‘Unsolved’ RDEB‐patients consisted of the following; n = 3 RDEB‐intermediate and n = 1 RDEB‐localized (in all patients one pathogenic mutation in COL7A1 could be found). §§The group of ‘No DNA’ RDEB‐patients consisted of the following; n = 2 RDEB‐severe (both deceased patients were included in the calculation of the average age at death for RDEB‐severe) an n = 1 RDEB‐intermediate.
DEB, dystrophic epidermolysis bullosa; EB, epidermolysis bullosa.
Kindler EB
Kindler EB was diagnosed in only four patients (four families; 0.9%) with an incidence of 0.5 per million live births and a point‐prevalence of 0.2 per million population (Figs. 1 and 2a, Table 4). In two patients with Kindler EB compound heterozygous mutations in FERMT1 were found, in the remaining two patients a clinical phenotype of Kindler EB was present, but no pathogenic mutations could be found.
Table 4.
Epidemiological data and distribution of Kindler Epidermolysis Bullosa in the Netherlands for the time period 1988–2018, n = 4

†In two patients no pathogenic mutation(s) could be found. Those patients were still included in the calculation of the incidence and point‐prevalence of Kindler EB. ‡Incidence (per million) = total no. of new Kindler EB‐patients born (1988–2018)/total no. of live births (1988–2018)*1 000 000. Total live births in the Netherlands from 01‐Jan‐1988 until 31‐Dec‐2018: 5 830 469 (Statline, CBS). §Point‐prevalence (per million) = total no. of Kindler EB‐patients alive at a time point (31‐Dec‐2018)/population at a time point (31‐Dec‐2018)*1 000 000. Total Dutch population, 31‐Dec‐2018: 17 282 163 (Statline, CBS).
EB, epidermolysis bullosa; Kindler EB, Kindler epidermolysis bullosa.
Epidemiological trends
Based on the increasing number of registered EB‐patients at our centre, the point‐prevalence of EBS and DEB has progressively increased over the years, while it plateaued for JEB after an initial period of increase (Fig. 1a). On the other hand, the yearly incidence rates showed a fluctuating pattern (Fig. 1b).
EB and mortality
For the Netherlands, the average life expectancy was 81.8 years (80.2 years for males and 83.3 years for females; 31‐Dec‐2018, StatLine, CBS). During the investigated time period, 73 EB‐patients (15.7%) in the Dutch‐EB‐Reg died, with an average age at death (AAD) of 22.9 years (n = 72; SD: 31.0; n = 1 AAD unknown). Of the 73 deceased EB‐patients, 68 patients had a recessive type of EB (47 JEB‐patients, 16 DEB‐patients, four EBS‐patients and one patient with Kindler EB) and five patients a dominant type. A total of 53 patients with recessive EB (77.9%) died as a direct consequence of known complications of their EB; 72.6% of the total group of 73 deceased EB‐patients. In another six patients with recessive EB it was not possible to find out whether their death was related to EB (8.8%). The remaining nine patients with recessive EB died from EB‐unrelated causes (13.2%), which was also true for the five patients with dominant EB (Table 5).
Table 5.
Mortality data of each major type and subtype of Epidermolysis Bullosa in the Netherlands for the time period 1988–2018, n = 73
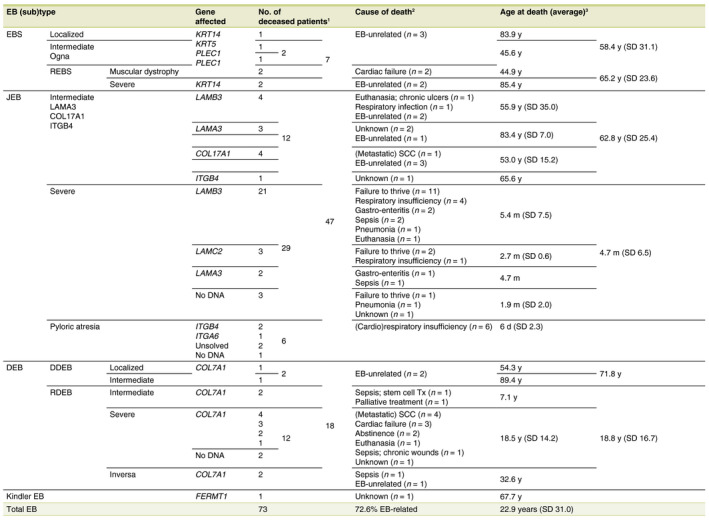
†Excluding two stillbirths with an unknown subtype of JEB, an induced partus of a foetus with JEB‐severe and a patient with JEB‐severe who died before 1988 but was captured in the Dutch‐EB‐Reg during the investigated time period. ‡Every cause of death other than ‘unknown’ or ‘EB‐unrelated’ are considered to be EB‐related. §In one patient with JEB‐intermediate the age at death remained unknown (n = 72).
EB, epidermolysis bullosa; EBS, epidermolysis bullosa simplex; DDEB, dominant dystrophic epidermolysis bullosa; DEB, dystrophic epidermolysis bullosa; JEB, junctional epidermolysis bullosa; Kindler EB, Kindler epidermolysis bullosa; RDEB, recessive dystrophic epidermolysis bullosa; REBS, recessive epidermolysis bullosa simplex; SCC, squamous cell carcinoma; SD, standard deviation.
Mortality and EBS
The average age of the alive EBS‐patients in the Dutch‐EB‐Reg was 34.3 years (n = 205; SD: 20.9) (31‐Dec‐2018). Seven EBS‐patients died during the investigated time period. Two patients with REBS‐muscular dystrophy died at the age of 43.7 and 46.0 years from cardiac failure. Two other patients with REBS died at the age of 88.6 and 82.3 years, both considered being EB‐unrelated. Three patients with dominant EBS died at the age of 83.9, 67.3 and 23.8 years, all three considered being EB‐unrelated (Tables 1 and 5).
Mortality and JEB
The average age of the alive JEB‐patients in the Dutch‐EB‐Reg was 33.0 years (n = 36; SD: 24.3) (31‐Dec‐2018). A total of 47 JEB‐patients died during the investigated time period (excluding two stillbirths with an unknown subtype of JEB, an induced partus of a foetus with JEB‐severe and a patient with JEB‐severe who died before 1988 but was captured in the Dutch‐EB‐Reg during the investigated time period). For JEB‐pyloric atresia an AAD of 6 days was calculated (n = 6; SD: 2.3). All of whom died shortly after (cardio)respiratory problems occurred and an abstaining policy was initiated. For JEB‐severe an AAD of 4.7 months (n = 29; SD: 6.5) was calculated, with an exceptional outlier of 2.7 years, of which 26 patients (90%) died within the first year of life. All patients with JEB‐severe died from EB‐related complications, in order of frequency: failure to thrive, respiratory insufficiency, infectious diseases and euthanasia (n = 1 unknown). For JEB‐intermediate an AAD of 62.8 years (n = 11; SD: 25.4) was calculated. Those patients died from: infectious diseases, euthanasia, metastatic squamous cell carcinoma and EB‐unrelated causes (n = 3 unknown) (Table 5).
Mortality and DEB
The average age of the alive DEB‐patients in the Dutch‐EB‐Reg was 32.5 years (n = 143; SD: 23.0) (31‐Dec‐2018), divided into an average age of 28.8 years for patients with RDEB (n = 37; SD: 20.7) and 33.3 years for patients with DDEB (n = 105; SD: 23.5). A total of 16 RDEB‐patients died during the investigated time period with an average AAD of 18.8 years (SD: 16.7), further subdivided into an average AAD of 18.5 years for RDEB‐severe (n = 12; SD: 14.2). All RDEB‐severe patients died from EB‐related complications: (metastatic) SCC, cardiac failure, abstinence, euthanasia and sepsis (n = 1 unknown). Two patients with RDEB‐intermediate died during the investigated time period at a young age from EB‐related complications (AAD 8.4 months and 13.5 years). Two patients with RDEB‐inversa died during the investigated time period at the age of 8.0 and 57.3 years, from sepsis caused by chronic wounds and an EB‐unrelated cause, respectively. Both patients with DDEB died from EB‐unrelated causes (AAD of 54.3 and 89.4 years; Table 5).
Mortality and Kindler EB
One patient with Kindler EB died during the investigated time period at the age of 67.7 years. The cause of death remained unknown and therefore a potential correlation with EB uncertain (Table 5). The other patients with Kindler EB in the Dutch‐EB‐Reg were still alive, with an average age of 13.8 years (n = 3; SD 9.3) (31‐Dec‐2018) (Table 4).
Discussion
Point‐prevalence and incidence
This observational study presents epidemiological data on each subtype of EB, extracted from the molecularly well‐characterized Dutch EB cohort in the Dutch‐EB‐Reg. In recent decades, several epidemiological studies have been carried out in different countries, reporting varying and sometimes inconclusive incidences and prevalences (1.4–25.0 per million live births and 2.82–54.0 per million population, respectively), mostly based on clinical features or molecular findings. 6 , 7 , 8 , 9 , 10 , 11 , 12 , 13 , 14 , 15 , 16 , 17 , 18 , 19 In 90.5% of the EB‐patients in our cohort the diagnosis was genetically confirmed, allowing accurate epidemiological analyses.
Both the calculated incidence of 41.3 per million live births and point‐prevalence of 22.4 per million population are high compared to that reported in other countries, especially if the 26 patients with an EB‐related skin fragility disorder would be included (incidence of 44.9 per million live births; point‐prevalence of 23.7 per million population) (Fig. 1, Table S1, Supporing Information). Over the years, increased knowledge, advanced diagnostics and the reputation of our centre ensured that an increasing number of EB‐patients were seen at our centre and captured in the Dutch‐EB‐Reg. The well‐organized set‐up in a small and highly populated country, a robust healthcare system with a good insurance policy and collaboration between specialists in the Netherlands ensured that most EB‐patients were referred to our centre (near‐complete ascertainment), especially those with severe subtypes of EB. Furthermore, being the only national expertise centre for EB, all mutation analysis for EB takes place in the UMCG. Therefore, we believe that the Dutch‐EB‐Reg documents most of the EB‐patients in the Dutch population and our calculated figures come close to the actual numbers, even though they are higher than previously reported. Our epidemiological data from the well‐characterized Dutch EB cohort thus indicate that EB may be more common than considered so far.
Risk of underestimation
Despite the expertise, knowledge and reputation of our centre, it is assumed that still not every patient is referred to our national centre, especially those with rather mild subtypes of EB. Those patients may be treated by their general practitioner or local specialist, as earlier noticed by Horn et al. 14 Besides, patients with milder subtypes of EB who were born in the last few years are expected to be registered in the following years, especially in case of late‐onset EB. Given the increasing number of registered patients at our centre, the calculated figures might therefore still be an underestimation.
Risk of misclassification
Another strength of our Dutch‐EB‐Reg is the long period of systematic data collection and longitudinal follow‐up. During the investigated time period, significant efforts were made to adequately monitor the patient records and identify affected family members. Despite the high level of expertise of our centre, patients may have been misclassified. However, this number is expected to be low, as in more than ninety per cent of the patients the diagnosis was genetically confirmed (Fig. 2). In the group of ‘Unsolved’ patients (n = 22) DNA analysis could not provide a genetic diagnosis using the gold standard techniques at the time of visiting our centre, neither with Sanger sequencing nor with a panel of multiple EB‐related genes. This may be due to technical shortcomings, because the diagnosis of EB is incorrect, the presence of unusual mutations or mutations in genes not known at the time of diagnostics, or mutations in as yet unknown genes. Over the years developments have been made in the registration and (sub)classification of the different subtypes of EB. Although we have made attempts to correctly convert the diagnoses from earlier consensus classifications to the latest, these changes may have led to misclassifications and fluctuating incidences and prevalences over time. This may be particularly true for EBS, as the EB‐related skin fragility disorders were previously included in the EB spectrum as suprabasal EBS (Fig. 1, Table S1, Supporing Information). 1
Distribution of EB subtypes
The distribution of the subtypes of EB in the Dutch‐EB‐Reg (EBS 45.7%, JEB 18.8%, DEB 34.7% (DDEB 23.1% and RDEB 11.4%) and Kindler EB 0.9%) differ from those reported in other countries. 6 , 7 , 16 , 20 , 21 , 22 Noteworthy, the population of JEB‐patients in the Dutch‐EB‐Reg is relatively higher than reported in other countries. Worldwide variations in the population and level of immigration (e.g. ethnic background, consanguineous marriages, spectrum of mutations) may affect the epidemiology and distribution of the subtypes of EB per region. 20 , 23 , 24 During the investigated time period, the Netherlands received an increasing number of immigrants, especially from countries where consanguineous marriages are more common than in the native Dutch population (StatLine, CBS), which may have resulted in an increasing number of patients with recessive, and usually more severe, subtypes of EB.
Epidemiological trends
Over the years, the point‐prevalence of EB, particularly EBS and DEB, has progressively increased, representing an increasing number of registered EB‐patients at our centre. Interestingly, the point‐prevalence of JEB has reached a plateau since 2014. This may be attributed to the fact that the point‐prevalence of JEB mainly consists of the non‐lethal JEB‐subtypes, as the average life expectancy of patients with JEB‐severe and JEB‐PA in the Dutch‐EB‐Reg was 4.7 months and 6 days, respectively. Apparently, we have been able to register all other JEB‐patients in the Dutch‐EB‐Reg, in contrast to the relatively milder and more common EBS and DDEB. Over the years, the annual incidence rates showed a fluctuating pattern. The high incidence of EBS in 2011 is likely a chance finding, all patients were crosschecked.
EB and mortality
We found that 72.6% of the deceased EB‐patients died as a direct consequence of EB‐related complications. JEB‐patients experienced the highest mortality rate, with an average survival of 6 days for JEB‐pyloric atresia (n = 6), 4.7 months for JEB‐severe (n = 29) and 62.8 years for JEB‐intermediate (n = 12). The AAD of patients with JEB‐pyloric atresia and JEB‐severe in the Dutch‐EB‐Reg is low in comparison with other studies. 10 , 25 The deceased patients with RDEB‐severe had an average AAD of 18.5 years (n = 12), lower than the earlier reported 29.4 years (n = 4) by Kho et al. 10 However, due to the relatively small number of deceased RDEB‐patients in both the Dutch‐EB‐Reg (with high standard deviations) and other studies, and the young age of the alive RDEB‐patients in the Dutch cohort, the question is whether these data are comparable and representative for the whole RDEB population. Future studies have to show whether the life expectancy of RDEB‐severe patients is truly that low.
Foreign EB‐patients referred to our expertise centre
Since the referral of the first EB‐patients from Belgium in 1997, an increasing number of EB‐patients were referred to our centre by specialists from foreign countries for diagnostic testing and/or to receive medical care. This demonstrates the need and value of cross‐country collaboration of healthcare professionals in the diagnosis and care of EB‐patients and rare diseases in general, which might be impossible to organize for each disease in each country individually, and from which the patients can only benefit.
Conclusion
This study shows that EB might be more common than assumed before and emphasizes the importance of thorough reporting systems and registries worldwide, which is invaluable for the design and execution of (upcoming) therapeutic trials. Besides, it provides an insight into the extensive need for (specialized) medical care of EB‐patients.
Ethical approval
This study contains routinely collected healthcare data.
Supporting information
Figure S1. Distribution of the genes involved in patients with an EB‐related skin fragility disorder in the Dutch Epidermolysis Bullosa Registry for the time period 1988–2018, n = 26.
Table S1. Epidemiological data and distribution of EB‐related skin fragility disorders in the Netherlands for the time period 1988–2018, n = 26.
Appendix S1. Epidemiological data on EB‐related skin fragility disorders from the Dutch EB Registry.
Acknowledgements
We thank our EB‐patients and their families for their trust and cooperation in our centre. This manuscript is a tribute to the late professor Marcel F. Jonkman, initiator of the EB Center in Groningen and the Dutch‐EB‐Reg. We are grateful to him for his tremendous contribution to the knowledge of EB and the care of EB‐patients in the Netherlands.
Conflict of interest
None.
Funding source
Stichting Vlinderkind (Dutch Butterfly Child foundation) has attributed to this study by financially supporting the care of patients with epidermolysis bullosa at our centre.
References
- 1. Fine JD, Bruckner‐Tuderman L, Eady RA et al. Inherited epidermolysis bullosa: updated recommendations on diagnosis and classification. J Am Acad Dermatol 2014; 70: 1103–1126. [DOI] [PubMed] [Google Scholar]
- 2. Has C, Bauer JW, Bodemer C et al. Consensus reclassification of inherited epidermolysis bullosa and other disorders with skin fragility. Br J Dermatol 2020; 183: 614–627. [DOI] [PubMed] [Google Scholar]
- 3. Duipmans JC, Jonkman MF. Interdisciplinary management of epidermolysis bullosa in the public setting: the Netherlands as a model of care. Dermatol Clin 2010; 28: 383–386. [DOI] [PubMed] [Google Scholar]
- 4. Has C, Bolling MC, Charlesworth AV et al. Clinical practice guidelines for laboratory diagnosis of epidermolysis bullosa. Br J Dermatol 2020; 182: 574–592. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Turcan I, Pasmooij AMG, van den Akker PC et al. Heterozygosity for a novel missense mutation in the ITGB4 gene associated with autosomal dominant epidermolysis bullosa. JAMA Dermatol 2016; 152: 558–562. [DOI] [PubMed] [Google Scholar]
- 6. Fine JD. Epidemiology of inherited epidermolysis bullosa based on incidence and prevalence estimates from the national epidermolysis bullosa registry. JAMA Dermatol 2016; 152: 1231–1238. [DOI] [PubMed] [Google Scholar]
- 7. Dănescu S, Has C, Senila S, Ungureanu L, Cosgarea R. Epidemiology of inherited epidermolysis bullosa in Romania and genotype‐phenotype correlations in patients with dystrophic epidermolysis bullosa. J Eur Acad Dermatol Venereol 2015; 29: 899–903. [DOI] [PubMed] [Google Scholar]
- 8. Hernandez‐Martín A, Aranegui B, Escámez MJ et al. Prevalence of dystrophic epidermolysis bullosa in Spain: a population‐based study using the 3‐source capture‐recapture method: evidence of a need for improvement in care. Actas Dermosigiliogr 2013; 29: 899–903. [DOI] [PubMed] [Google Scholar]
- 9. Yuen WY, Duipmans JC, Molenbuur B, Herpertz I, Mandema JM, Jonkman MF. Long‐term follow‐up of patients with Herlitz type junctional epidermolysis bullosa. Br J Dermatol 2012; 167: 374–382. [DOI] [PubMed] [Google Scholar]
- 10. Kho YC, Rhodes LM, Robertson SJ et al. Epidemiology of epidermolysis bullosa in the antipodes: the australasian epidermolysis bullosa registry with a focus on herlitz junctional epidermolysis bullosa. Arch Dermatol 2010; 146: 635–640. [DOI] [PubMed] [Google Scholar]
- 11. Castori M, Floriddia G, De Luca N et al. Herlitz junctional epidermolysis bullosa: laminin‐5 mutational profile and carrier frequency in the Italian population. Br J Dermatol 2008; 158: 38–44. [DOI] [PubMed] [Google Scholar]
- 12. Tadini G, Gualandri L, Colombi M et al. The Italian registry of hereditary epidermolysis bullosa. [in Italian] G Ital Dermatol Venereol 2005; 140: 359–372. [Google Scholar]
- 13. Fine JD, Johnson LB, Suchindran C, Moshell A, Gedde‐Dahl T. The epidemiology of inherited EB: findings within American, Canadian, and European study populations. In Fine JD, Bauer EA, McGuire J, Moshell A eds. Epidermolysis Bullosa: Clinical, Epidemiologic, and Laboratory Advances, and the Findings of the National Epidermolysis Bullosa Registry, Johns Hopkins University Press, Baltimore, MD, 1999: 101–113. [Google Scholar]
- 14. Horn HM, Priestley GC, Eady RA, Tidman MJ. The prevalence of epidermolysis bullosa in Scotland. Br J Dermatol 1997; 136: 560–564. [PubMed] [Google Scholar]
- 15. Abahussein AA, Al‐zayir AA, Mostafa WZ, Okoro AN. Epidermolysis bullosa in the eastern province of Saudi Arabia. Int J Dermatol 1993; 32: 579–581. [DOI] [PubMed] [Google Scholar]
- 16. McKenna KE, Walsh MY, Bingham EA. Epidermolysis bullosa in Northern Ireland. Br J Dermatol 1992; 127: 318–321. [DOI] [PubMed] [Google Scholar]
- 17. Pavicić Z, Kmet‐Vizintin P, Kansky A, Dobrić I. Occurrence of hereditary bullous epidermolyses in Croatia. Pediatr Dermatol 1990; 7: 108–110. [DOI] [PubMed] [Google Scholar]
- 18. Inaba Y, Kitamura K, Ogawa H, Manabe M, Sasai Y. A study on the estimation of prevalence of epidermolysis bullosa in Japan. [in Japanese] Nihon Hifuka Gakkai Zasshi 1989; 99: 1021–1026. [PubMed] [Google Scholar]
- 19. Kero M. Occurrence of epidermolysis bullosa in Finland. Acta Derm Venereol 1984; 64: 57–62. [PubMed] [Google Scholar]
- 20. Abu Sa'd J, Indelman M, Pfendner E et al. Molecular epidemiology of hereditary epidermolysis bullosa in a Middle Eastern population. J Invest Dermatol 2006; 126: 777–781. [DOI] [PubMed] [Google Scholar]
- 21. Feinstein JA, Jambal P, Peoples K et al. Assessment of the timing of milestone clinical events in patients with epidermolysis bullosa from North America. J Am Acad Dermatol 2019; 155: 196–203. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Vahlquist A, Tasanen K. Epidermolysis bullosa care in Scandinavia. Dermatol Clin 2010; 28: 425–427. [DOI] [PubMed] [Google Scholar]
- 23. Murata T, Masunaga T, Ishiko A, Shimizu H, Nishikawa T. Differences in recurrent COL7A1 mutations in dystrophic epidermolysis bullosa: ethnic‐specific and worldwide recurrent mutations. Arch Dermatol Res 2004; 295: 442–447. [DOI] [PubMed] [Google Scholar]
- 24. Nakano A, Lestringant GG, Paperna T et al. Junctional epidermolysis bullosa in the Middle East: clinical and genetic studies in a series of consanguineous families. J Am Acad Dermatol 2002; 46: 510–516. [DOI] [PubMed] [Google Scholar]
- 25. Dank JP, Kim S, Parisi MA et al. Outcome after surgical repair of junctional epidermolysis bullosa‐pyloric atresia syndrome: a report of 3 cases and review of the literature. Arch Dermatol 1999; 135: 1243–1247. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Figure S1. Distribution of the genes involved in patients with an EB‐related skin fragility disorder in the Dutch Epidermolysis Bullosa Registry for the time period 1988–2018, n = 26.
Table S1. Epidemiological data and distribution of EB‐related skin fragility disorders in the Netherlands for the time period 1988–2018, n = 26.
Appendix S1. Epidemiological data on EB‐related skin fragility disorders from the Dutch EB Registry.