

Abstract
Sarcomas are a heterogeneous group of malignancies with mesenchymal lineage differentiation. The discovery of neurotrophic tyrosine receptor kinase (NTRK) gene fusions as tissue-agnostic oncogenic drivers has led to new personalized therapies for a subset of patients with sarcoma in the form of tropomyosin receptor kinase (TRK) inhibitors. NTRK gene rearrangements and fusion transcripts can be detected with different molecular pathology techniques, while TRK protein expression can be demonstrated with immunohistochemistry. The rarity and diagnostic complexity of NTRK gene fusions raise a number of questions and challenges for clinicians. To address these challenges, the World Sarcoma Network convened two meetings of expert adult oncologists and pathologists and subsequently developed this article to provide practical guidance on the management of patients with sarcoma harboring NTRK gene fusions. We propose a diagnostic strategy that considers disease stage and histologic and molecular subtypes to facilitate routine testing for TRK expression and subsequent testing for NTRK gene fusions.
Keywords: entrectinib, larotrectinib, neurotrophic tyrosine receptor kinase, sarcoma, tropomyosin receptor kinase
INTRODUCTION
Sarcomas are a heterogeneous group of malignancies that exhibit mesenchymal lineage differentiation. They arise in either soft tissue (~80%) or bone (~20%) and comprise ~70 malignant subtypes (per World Health Organization classification), each with distinct underlying biology and clinical behavior.1 Sarcomas account for approximately 1% of all adult cancers and 20% of all pediatric solid tumors.2 Complete resection (with or without radiation and/or chemotherapy) forms the mainstay of curative management for most subtypes in the localized setting. For patients diagnosed with locally advanced or metastatic disease, or those with disease recurrence following surgery, treatment options include systemic therapy and potential local approaches such as radiation, isolated limb perfusion, surgery, and ablation techniques.3 The median overall survival of patients with advanced soft tissue sarcomas is approximately 20 months, with most patients deriving only transient benefit from palliative chemotherapy.4,5 Therefore, there is a clear unmet need for more effective systemic therapies for patients with advanced/metastatic sarcomas.
As our understanding of the molecular basis of tumorigenesis has improved with advances in diagnostic technology, precision oncology approaches to the treatment of sarcomas have emerged. A classic example of this is mutational profiling of KIT, PDGFRA, and other genes to predict sensitivity of gastrointestinal stromal tumors (GISTs) to imatinib and other KIT/PDGFRA tyrosine kinase inhibitors.6–11 More recently, the discovery of neurotrophic tyrosine receptor kinase (NTRK) gene fusions as pan-tumor oncogenic drivers has provided new precision medicine-based treatment options for a subset of patients with sarcoma.12 The rarity and diagnostic complexity of this particular biomarker raise a number of questions and challenges for clinicians.
To address these issues, the World Sarcoma Network, a think tank gathering national and international sarcoma groups for the past 10 years, convened two consensus meetings of expert adult oncologists and pathologists to discuss diagnostic challenges and propose a diagnostic strategy in this area. We subsequently developed this article to provide practical guidance on how to optimally integrate the NTRK gene fusion biomarker into the clinical management of patients with sarcoma.
NTRK GENE FUSIONS
The NTRK genes NTRK1 (chromosome 1q23.1), NTRK2 (chromosome 9q21.33), and NTRK3 (chromosome 15q25.3) are typically involved in normal neuronal development and encode the tropomyosin receptor kinase (TRK) proteins, traditionally known as TRKA, TRKB, and TRKC, respectively.12,13 Wild-type TRK proteins are activated through oligomerization mediated by the binding of neurotrophin ligands.14 Subsequent downstream signaling contributes to central nervous system (CNS) development and regulation of appetite, body weight, memory, mood, movement, pain, and proprioception.15–20
NTRK gene fusions have been identified in a diverse range of adult and pediatric tumor types.12 These fusions result from inter- or intra-chromosomal rearrangements leading to juxtaposition of the 3′ region of an NTRK gene (encoding the full kinase domain) with the 5′ region of a partner gene (encoding an oligomerization or other protein-association domain), ultimately producing a constitutively active TRK fusion protein.21 Fusions involving NTRK gene 5′ regions have also been reported, although the pathogenicity of these is unclear.22 In addition to fusions, NTRK gene point mutations and amplifications have been identified in a variety of different cancer types; however, roles for these aberrations in tumorigenesis have not been established.22
While rare in most common tumor types (e.g. lung and colorectal cancers), NTRK gene fusions are reported to be recurrent in a subset of rare tumor types (e.g. secretory carcinoma of the salivary gland, secretory carcinoma of the breast, congenital mesoblastic nephroma, pediatric melanoma, and infantile fibrosarcoma).21,23,24 One of the first discovered and most well characterized fusions, ETV6-NTRK3, resulting from a t(12;15)(p13;q25) translocation, is present in >90% of infantile fibrosarcomas.25,26 By contrast, NTRK fusions have been identified in other adult and pediatric sarcomas at a frequency of <1%.23,27,28 Recent studies investigating NTRK fusions among mesenchymal neoplasms have identified a number of emerging new soft tissue tumor entities displaying various phenotypes, which resemble lipofibromatosis, fibrosarcoma, and malignant peripheral nerve sheath tumors (Table 1). A significant number of these NTRK fusion-positive tumors show co-expression of S100 protein and CD34, while the rest have a nonspecific immunophenotype.27,29–32 The published literature on NTRK gene fusion frequency in sarcomas is limited and more data are needed.
Table 1.
Frequency of NTRK gene fusions identified in sarcomas
| Study | Testing method | Proportion of NTRK fusions identified | NTRK fusion-positive sarcoma subtypes | NTRK genes and fusion partners involved |
|---|---|---|---|---|
| Agaram et al.29 | FISH, RNA MPS | 71% (10/14) | Lipofibromatosis-like neural tumor | 1 TPR-NTRK1 1 TPM3-NTRK1 4 LMNA-NTRK1 |
| Bourgeois et al.68 | RT-PCR | 91% (10/11) | IFS | ETV6-NTRK3 |
| Bui et al.69 | Targeted DNA MPS | 0.7% (1/152) | Myopericytoma | NR |
| Chang et al.70 | Targeted RNA MPS | 33% (3/9) | IMT | ETV6-NTRK3 |
| Chmielecki et al.71 | Targeted RNA MPS | 1% (4/324) | IFS (n = 2), assorted soft tissue sarcoma (n = 1), hemangioma (n = 1), bone sarcoma (n = 1) | SQSTM1-NTRK1 (n = 1), other fusions partners NR |
| Church et al.72 | FISH | 96% (25/26) | IFS | NTRK3 |
| Croce et al.73 | Targeted RNA MPS | 54% (7/13) | Uterine and vaginal sarcomas resembling fibrosarcoma | 6 TPM3-NTRK1, 1 EML4-NTRK3 |
| Gatalica et al.51 | Targeted RNA MPS | 0.4% (2/478) | 1 STS (poorly differentiated sarcoma with possible myofibroblastic differentiation), 1 uterine sarcoma (intermediate to high-grade sarcoma of uterine origin, with myxoid stroma and no specific line of differentiation) | 1 TPM3-NTRK1, 1 SPECC1L-NTRK3 |
| Rosen et al.60 | Targeted RNA MPS | 1% (11/944) | Sarcoma NOS [9/770 (1%)], uterine sarcoma [2/174 (1%)] | NR |
| Shi et al.74 | Targeted DNA MPS | 0.5% (1/186) overall [4% (1/24) in quad-negative tumors] | GIST | ETV6-NTRK3 |
| Solomon et al.52 | Targeted DNA and/or RNA MPS |
0.7% (13/1915) | IFS (n = 2), lipofibromatosis-like neural tumor (n = 2), uterine sarcoma (n = 2), uterine high-grade pleomorphic sarcoma, high-grade spindle cell sarcoma, malignant spindle cell sarcoma, spindle cell sarcoma, angiosarcoma, S-100 positive malignant spindle cell neoplasm, low grade sarcoma (all n = 1) | LMNA-NTRK1 (n = 4), TPM3-NTRK1 (n = 3), ETV6-NTRK3 (n = 2), TPR-NTRK1, TPM4-NTRK3, EEF1A1-NTRK3, PEAR1-NTRK1 (all n = 1) |
| 18% (3/17) | IMT | ETV6-NTRK3 | ||
| Stransky et al.63 | TCGA RNA-seq dataset | 1% (1/103) | Sarcoma | TPM3-NTRK1 |
| Surrey et al.75 | Targeted RNA MPS | 4% (2/45) | Sarcomas (other) | 1 TFG-NTRK3, 1 RBPMS-NTRK3 |
| Suurmeijer et al.31 | FISH, targeted RNA MPS | 60% (15/25) | Malignant peripheral nerve sheath tumor-like | 8 LMNA-NTRK1, 3 TPM3-NTRK1, 1 SPECC1L-NTRK2, 1 TPR-NTRK1, 2 NTRK1 with unknown fusion partners |
| Yamamoto et al.76 | MPS (TBC), IHC | 5% (2/40) | IMT | ETV6-NTRK3 |
| Zhu et al.77 | Targeted RNA MPS | 3% (5/184) | Lipofibromatosis-like neural tumor (n = 2), IFS (n = 1), IMT (n = 1), sarcoma NOS (n = 1) | 2 ETV6-NTRK3 2 TPM3-NTRK1 1 LMNA-NTRK1 |
GIST, gastrointestinal stromal tumor; IFS, infantile fibrosarcoma; IHC, immunohistochemistry; IMT, inflammatory myofibroblastic tumor; LPS, liposarcoma; MPS, massive parallel sequencing; NOS, not otherwise specified; NR, not reported; RT-PCR, reverse transcription polymerase chain reaction; STS, soft tissue sarcoma; TCGA, The Cancer Genome Atlas.
TARGETED THERAPY FOR TRK FUSION CANCERS
NTRK gene fusions (but not other NTRK alterations) appear to be primary oncogenic drivers in the tumors that harbor them. The encoded fusion proteins feature constitutive tyrosine kinase activity that may be targeted clinically with a number of agents that are either approved or in development.12,33
Larotrectinib
Larotrectinib is a first-in-class, ATP-competitive, small-molecule inhibitor of TRK. It is highly potent, with IC50 values in the range of 6.5–10.6 nM, and highly selective for TRKA, B, and C, with binding affinities over 100-fold greater than for a panel of other kinases.24,34,35 Larotrectinib is approved by the US Food and Drug Administration (FDA) and European Medicines Agency (EMA) for use in adult and pediatric patients with solid tumors harboring an NTRK gene fusion who have disease that is locally advanced or metastatic, or where surgery is likely to result in severe morbidity, and who have no satisfactory treatment options.36,37 Patients with a known resistance mutation are not indicated for larotrectinib treatment.
Larotrectinib has demonstrated robust efficacy in a combined analysis of three phase I/II trials in adults and children with TRK fusion cancers, irrespective of age or tumor type.38 In an integrated dataset of 159 patients, investigator-assessed objective response rate (ORR) was 79% [95% confidence interval (CI) 72% to 85%] and median duration of response was 35.2 months (median follow-up 12.9 months). The median time to response was 1.8 months.39 Objective responses and durable disease control were also observed in the subsets of patients with primary CNS tumors or non-CNS solid tumors with brain metastases.40,41 Larotrectinib-related adverse events of grade 3–4 occurred in 13% of patients, and dose reductions and treatment discontinuations due to treatment-related adverse events occurred in 8% and 2% of patients, respectively.39 Long-term follow-up is ongoing. The favorable safety profile of larotrectinib, together with robust clinical efficacy, translated into rapid, sustained, and clinically meaningful improvements in quality of life in the majority of patients.42
Among the 17 different tumor types represented in the larotrectinib dataset, the most common (47%) were sarcomas.39 Of 71 patients with a sarcoma, two (3%) had an osteosarcoma and a dedifferentiated chondrosarcoma, four (6%) had a GIST, 29 (41%) had infantile fibrosarcoma, and 36 (51%) had other soft tissue tumors, including adult fibrosarcoma, inflammatory myofibroblastic tumor, infantile myofibromatosis, lipofibromatosis, malignant peripheral nerve sheath tumor, myopericytoma, spindle cell sarcoma, high-grade endometrial stromal tumor, and synovial sarcoma.43 The histologic subtypes of these patients were captured as reported by the investigators; however, due to the very rare nature of the subtypes reported and the varied nomenclature used in sarcoma pathology, a central pathology review and efficacy analysis by sarcoma subtype is warranted. Furthermore, data on other driver alterations in these patients would be informative.
The ORR with larotrectinib in adult and pediatric patients with sarcoma harboring an NTRK fusion was 74% (95% CI 52% to 90%) and 94% (95% CI 82% to 99%), respectively. Objective responses were observed in patients with soft tissue sarcomas, GISTs, and infantile fibrosarcoma. Of two patients with a bone sarcoma, one had a partial response and one had stable disease. At a median follow-up of 15.6, 13.0, and 14.1 months, median duration of response, progression-free survival, and overall survival were not estimable (NE; range 1.6+ to 44.2+), 28.3 (95% CI 16.8-NE), and 44.4 (95% CI 44.4-NE) months, respectively (Table 2). Grade 3–4 adverse events related to larotrectinib were reported in 13% of patients.43
Table 2.
Efficacy of TRK inhibitors in patients with sarcoma harboring NTRK gene fusionsa
| Larotrectinib n = 7143 |
Entrectinib n = 1347 |
|
|---|---|---|
| Objective response rate, % | 87 (95% CI 77–94) | 46 (95% CI 19–75) |
| Median duration of response, months | NE (range 1.6+ to 44.2+) | 10.3 (95% CI 4.6–15.0) |
| Median progression-free survival, months | 28.3 (95% CI 16.8-NE) | 11.0 (95% CI 6.5–15.7) |
| Median overall survival, months | 44.4 (95% CI 44.4-NE) | 16.8 (95% CI 10.6–20.9) |
CI, confidence interval; MPS, massive parallel sequencing; NE, not estimable; NTRK, neurotrophic tyrosine receptor kinase receptor; PCR, polymerase chain reaction; TRK, tropomyosin receptor kinase.
In the larotrectinib clinical trials, NTRK gene fusions were detected by local MPS, according to the procedures and analytic pipelines established by each laboratory, or by FISH. All tests were carried out in a Clinical Laboratory Improvement Amendments-certified (or equivalent) laboratory. In the entrectinib clinical trials, NTRK gene fusions were detected by central RNA MPS (Trailblaze Pharos, Ignyta, San Diego, CA, USA) or local molecular testing (FISH, quantitative PCR, or DNA or RNA MPS).
Entrectinib
Entrectinib is a multi-targeted, pan-TRK, ROS1, and ALK inhibitor. It has low to sub-nanomolar enzymatic activity against TRKA, TRKB, TRKC, ROS1, and ALK (IC50 values of 1.7, 0.1, 0.1, 0.2, and 1.6 nM, respectively).44 Entrectinib is FDA-approved for use in adult and pediatric patients ≥12 years of age with solid tumors harboring an NTRK gene fusion who have disease that is metastatic or where surgery is likely to result in severe morbidity, and who have progressed following treatment or have no satisfactory alternative therapy. Patients with a known resistance mutation are not indicated for entrectinib treatment. Entrectinib is also FDA-approved for patients with metastatic non-small cell lung cancer (NSCLC) harboring a ROS1 gene fusion.45
Entrectinib demonstrated tumor-agnostic efficacy in an integrated analysis of 54 patients with TRK fusion cancers in one of three phase I/II trials. Independently assessed ORR was 57% (95% CI 43% to 71%) and median duration of response was 10.4 months (median follow-up 12.9 months). Clinically meaningful and durable intracranial responses were seen in patients with brain metastases. Adverse events with entrectinib were mainly grade 1 or 2 and the proportion of patients with dose reductions and treatment discontinuations due to a treatment-related adverse event was 27% and 4%, respectively.46
Among 13 patients with sarcoma in the overall entrectinib clinical trial dataset, six subtypes were identified: cervix adenosarcoma, dedifferentiated chondrosarcoma, endometrial stromal sarcoma, follicular dendritic cell sarcoma, GIST, and malignant peripheral nerve sheath tumor. Of note, there were no patients with infantile fibrosarcoma enrolled in these trials. The ORR for the sarcoma subset was 46%. Median duration of response, progression-free survival, and overall survival were 10.3 (95% CI 4.6–15.0), 11.0 (95% CI 6.5–15.7), and 16.8 (95% CI 10.6–20.9) months, respectively (Table 2).47
METHODS OF NTRK GENE FUSION TESTING
NTRK gene rearrangements and fusion transcripts can be detected with different molecular pathology techniques such as FISH, reverse transcription polymerase chain reaction (RT-PCR), and massive parallel sequencing (MPS), while TRK protein expression can be demonstrated by immunohistochemistry (IHC) (Table 3).48,49
Table 3.
Methods of NTRK gene fusion testing
| FISH | IHC | RT-PCR | MPS | |
|---|---|---|---|---|
| Advantages | Available in many clinical laboratories | Widely available | Widely available | Allows simultaneous detection of fusions between NTRK1—3 and any number of known or novel fusion partner genes |
| Rapid turnaround time | Rapid turnaround time | Rapid turnaround time | ||
| Relatively inexpensive | Inexpensive | nexpensive | ||
| Allows identification of specific cell types harboring the NTRK fusion | ||||
| Disadvantages | Requires specific expertise | Detection of wild-type protein expression, especially in tumors with neural and myogenic differentiation | Requires knowledge of fusion partner gene sequence | Not routinely conducted in all clinical laboratories |
| False negativity rate of ≤30% | False negativity rate of ~10%; this rate may be higher in tumors harboring NTRK3 fusions | Challenging to test for fusions involving multiple NTRK and fusion partner genes in parallel | Relatively long turnaround time | |
| Does not distinguish between in-frame and out-of-frame fusion events | Relatively expensive | |||
| DNA MPS may miss NTRK2 and NTRK3 fusions due to large introns RNA MPS requires high quality RNA | ||||
| Variable detection rates of different panels | ||||
| May identify non-oncogenic NTRK rearrangements |
MPS, massive parallel sequencing; NTRK, neurotrophic tyrosine receptor kinase receptor.
Fluorescence in situ hybridization
FISH employs fluorescently labeled DNA probes that anneal to specific regions within or flanking a gene(s) of interest. To detect a particular NTRK gene fusion, colocalization of FISH probes to each gene component of the fusion can be demonstrated.27 In practice, however, it is more feasible to use break-apart FISH probes that flank each of the three NTRK genes and demonstrate rearrangement without identifying the fusion partner gene.27,34 The efficient break-apart approach avoids the need to develop an unrealistically large number of FISH probe sets for uncommon fusions and also detects novel fusions with as yet uncharacterized fusion partners. FISH is available in many clinical laboratories, has a short turnaround time, and is relatively inexpensive; however, specific expertise is required to interpret test results, particularly in paraffin sections where nuclear slicing can result in artifacts. A false-negative rate of up to 30% has been reported with FISH in pediatric sarcomas.50 Furthermore, FISH does not distinguish between in-frame and out-of-frame fusion events.
Immunohistochemistry
IHC typically employs an antibody that binds to antigens common to the C-terminal domain of all three TRK proteins (pan-TRK IHC) to detect elevated TRK protein expression (Table 4). This method relies on the fact that most normal cells express low levels of TRK while tumor cells harboring an NTRK gene fusion typically display elevated TRK protein levels. Nevertheless, TRKA, TRKB, and TRKC expression can be observed by IHC in some normal cells: neurons, myenteric plexus, endothelial cells, and podocytes. In adult tissue, expression is restricted to smooth muscle, testes, and neuronal components. These components can be used as internal (endothelial cells, myenteric plexus) or external (podocytes in renal tissue) positive IHC controls. IHC represents a useful indirect readout for NTRK gene rearrangements. However, variable rates of sensitivity (75%−88%) and specificity (81%−96%) have been reported,51,52 which may be explained by the use of different antibodies, different IHC detection protocols, and poor or excessive tissue fixation. Any of these variables can impact IHC staining and intensity. The overall positive and negative predictive values in one IHC study have been reported to be 11.2% and 99.8%, respectively (using pan-TRK antibody clone EPR17341).51 Moreover, sensitivity has been shown to vary according to the NTRK gene involved, with lower sensitivity reported for NTRK3 (55%−79%) compared with NTRK1 (88%−96%) and NTRK2 (89%−100%).51,52 A TRK IHC signal should be considered as positive when staining of ≥1% of tumor cells is observed.53 Furthermore, the subcellular pattern of pan-TRK IHC staining may indicate the nature of the underlying gene fusion, with nuclear staining suggestive of NTRK3 fusions and moderate to strong, diffuse cytoplasmic staining suggestive of NTRK1/NTRK2 fusions.49,53,54
Table 4.
Commercially available pan-TRK antibodies
| Clone | Company | Label | Studies |
|---|---|---|---|
| EPR17341 | Abcam | RUO | Hechtman et al.78 Leica Bond 3 (ER2 retrieval, dilution 6 μg/ml) Solomon et al.52 Leica Bond 3 (ER2 retrieval, dilution 6 μg/ml) Gatalica et al.51 Ventana Benchmark or Dako Autostainer Rudzinski et al.79 Ventana Benchmark (CC1 retrieval, 1:500 dilution) Hung et al.80 Manual (1/300 dilution) Xu et al.81 Leica Bond 3 |
| EPR17341 (prediluted) | Ventana | CE/IVD | Feng et al.82 Ventana assay on dedicated platform |
| EPR18413 | Abcam | RUO | Not reported |
| C17F1 | Cell Signaling Technology | RUO | Murphy et al.83 Dako Autostainer (dilution 1:25, cocktail with anti-ALK and anti-ROS1 antibodies) |
| A7H6R | Cell Signaling Technology | RUO | Not reported |
All antibodies are rabbit monoclonal and bind to the TRK C-terminal domain.
IVD, in vitro diagnostic; TRK, tropomyosin receptor kinase; RUO, research use only.
One benefit of IHC over molecular analyses is that it provides evidence of the expression of the protein target of TRK inhibitors. Of six patients with primary resistance to larotrectinib among the initial 55 patients treated in clinical trials, three had tumor material available for central analysis and in all three cases, pan-TRK IHC did not demonstrate elevated TRK protein expression, indicating that the rearrangements detected by molecular testing did not yield chimeric proteins with an intact TRK C-terminus in these cases. One additional patient harbored an NTRK3 kinase domain mutation that conferred resistance.38
IHC is widely available in clinical laboratories, allows a rapid turnaround time, and is far less expensive than FISH. Although more data on sensitivity and specificity are needed, pan-TRK IHC is considered to have a false-negative rate of ~10%.52 In a study of seven patients with soft tissue spindle cell tumors and NTRK3 rearrangement detected by both IHC and FISH, rearrangements were only confirmed by RNA sequencing in three cases.30 Therefore, sarcomas with a high probability of harboring an NTRK fusion but with negative pan-TRK IHC staining should be considered for confirmatory testing with a genomic method (FISH or MPS). IHC may prove to be a valuable screening tool to highlight NTRK rearrangements, with the exception of CNS and neuroendocrine tumors where IHC is not a reliable screening tool because of endogenous elevated TRK expression. Furthermore, many sarcomas with myogenic or neural differentiation may display focal TRK expression.53 Thus, in these tumors, only diffuse staining should be considered positive. Strong TRK IHC staining can also be found in cases with NTRK gene amplification, supporting the requirement to confirm the presence of NTRK rearrangements with an orthogonal molecular method (Figure 1).30
Figure 1. Examples of positive TRK IHC staining.
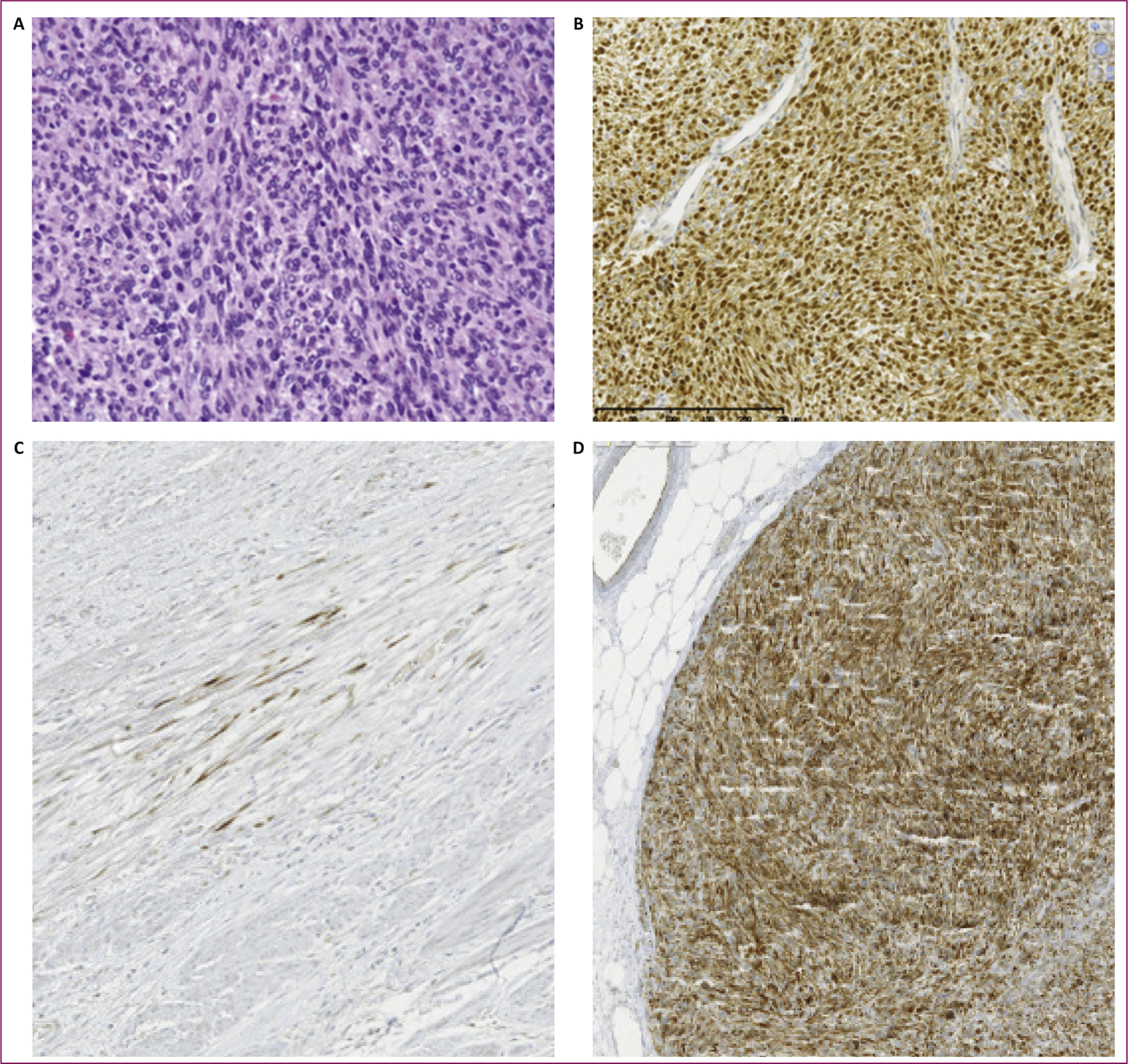
(A) ETV6-NTRK3 fusion infantile fibrosarcoma stained by hematoxylin and eosin and (B) pan-TRK IHC with A7H6R clone (Cell Signaling Technology) and ultraView detection. (C) Focal staining in a leiomyosarcoma without NTRK gene alterations. (D) Intense staining in a leiomyosarcoma with NTRK1 copy number gain. IHC, immunohistochemistry; NTRK, neurotrophic tyrosine receptor kinase receptor; TRK, tropomyosin receptor kinase.
Reverse transcription polymerase chain reaction
RT-PCR uses primers flanking the breakpoint region in the transcript encoded by the fused genes, with the 3′ primer annealing to an NTRK gene and the more 5′ primer annealing to the relevant fusion partner gene. When present in the tumor, the targeted portion of the fusion transcript will be amplified yielding a positive RT-PCR result. RT-PCR is a widely established technique and is rapid and inexpensive. Multiplex RT-PCR can be carried out using primer sets specific to a number of known NTRK fusion genes in a single assay. However, this method does not detect NTRK gene fusions with unknown partner genes; thus, a negative RT-PCR result cannot exclude the presence of a fusion. Therefore, RT-PCR is only recommended in settings where a specific type of fusion is expected following histologic-based triage, for instance, ETV6-NTRK3 in infantile fibrosarcoma, or as a complementary method for gene rearrangements detected by FISH.
Massive parallel sequencing
MPS allows for the simultaneous detection of fusions between NTRK1—3 and any number of fusion partner genes, depending upon the particular assay used. Targeted MPS with a panel of primers that hybridize to select regions in predefined genes is the preferred method. DNA-based MPS assays are not the best approach for identifying all NTRK fusions, especially those involving the NTRK2 and NTRK3 genes because of their large introns. Targeted RNA MPS allows for more systematic detection of NTRK fusion transcripts. Adequately designed targeted RNA MPS panels allow for the detection of novel NTRK gene fusion partners, and there are a number of commercially available assays that cover all three NTRK genes. However, it should be noted that different commercial panels have shown some remarkable differences in detection rate.55 MPS is not routinely conducted in all clinical laboratories, has a relatively long turnaround time, and is quite expensive, particularly if only a limited number of tests are required. However, various groups are continuing to develop RNA MPS platforms that can detect in parallel the multitude of fusion genes observed in sarcomas.
In addition to functional NTRK fusion transcripts, RNA-based MPS assays may identify non-oncogenic aberrant NTRK rearrangements (incidental genomic alterations) that do not yield constitutively active fusion proteins. Clinicians should be aware of this possibility and understand how to interpret complex MPS data reports. If expression of the fusion protein is in doubt, IHC may be a useful confirmatory tool. Similarly, NTRK point mutations occur more often than gene fusions and may also be identified by MPS assays; however, these mutations are not considered predictive of treatment response.56
TESTING FOR NTRK GENE FUSIONS IN SARCOMAS
Given the robust efficacy and favorable safety profiles of TRK inhibitors demonstrated in patients with TRK fusion sarcomas, testing for NTRK gene fusions should be incorporated into the clinical management of patients with sarcoma, with prioritization in specific stages and subtypes, as discussed below. The rarity of these oncogenic drivers presents a number of challenges, including the cost of testing, limited resources, limited tumor tissue, and the complexities of integrating a new molecular test into the current diagnostic workup. However, the overall benefit of molecular testing in the diagnosis and clinical management of patients with sarcoma has been demonstrated in large multicenter studies, similar to what has been shown for lung cancer.57,58 While sequence-based testing methods (RNA MPS or RT-PCR) are recommended for the detection of productive NTRK gene rearrangements, IHC with a validated antibody against TRK proteins (most easily with a pan-TRK antibody) may be used as a fast and less expensive pre-screening tool. Furthermore, selecting histotypes negative for pathognomonic genetic alterations (other translocations, kinase mutations, MDM2/CDK4 amplification) could allow exclusion of ~45% of all sarcomas from NTRK gene fusion testing, given the mutual exclusivity of such driver alterations.59,60
NTRK fusion testing may be prioritized in disease settings where TRK inhibitor therapy is most relevant, also considering that some of the recently reported NTRK-rearranged entities tend to behave indolently. The majority of sarcoma patients are diagnosed while still localized and these tumors are amenable to curative surgical resection without the need for systemic therapy. Therefore, NTRK fusion testing in primary, resectable sarcomas may not be necessary (except when used for definitive diagnosis, such as in the case of putative infantile fibrosarcomas). Nevertheless, for patients at high risk of relapse, NTRK gene fusion testing might provide clinically actionable information for later in the disease course. Testing for NTRK gene fusions should be carried out in patients with locally advanced, unresectable tumors or in those with metastatic disease failing conventional therapies.
Sarcomas with a high NTRK fusion frequency (priority 1)
Given the potential cost and resource limitations of universal testing, we propose a three-tiered diagnostic algorithm for the prioritization of NTRK gene fusion testing according to the likelihood of finding a fusion (Figure 2). The highest priority for NTRK fusion testing is given to the histologic subtypes that commonly or non-infrequently harbor NTRK gene fusions, such as infantile fibrosarcomas61 and ALK and ROS1 fusion-negative inflammatory myofibroblastic tumors.62 These entities should be tested upfront for NTRK fusions in all situations53 and the test should ideally be ordered by the pathologist following central pathologic diagnosis. In fact, NTRK fusion testing is often conducted as part of the diagnostic process for suspected infantile fibrosarcomas. For histologic subtypes with a high pre-test probability of harboring an NTRK fusion, we recommend the use of FISH, IHC, or MPS. A negative FISH result should be confirmed by MPS. For a positive FISH result, confirmation that the fusion is in-frame by MPS or RT-PCR should be considered in parallel to treatment. MPS confirmation of a negative IHC result is recommended for cases with typical histology. For cases with positive IHC results, treatment may be considered concurrently with confirmatory MPS.
Figure 2. Recommended algorithm for NTRK gene fusion testing in sarcomas.
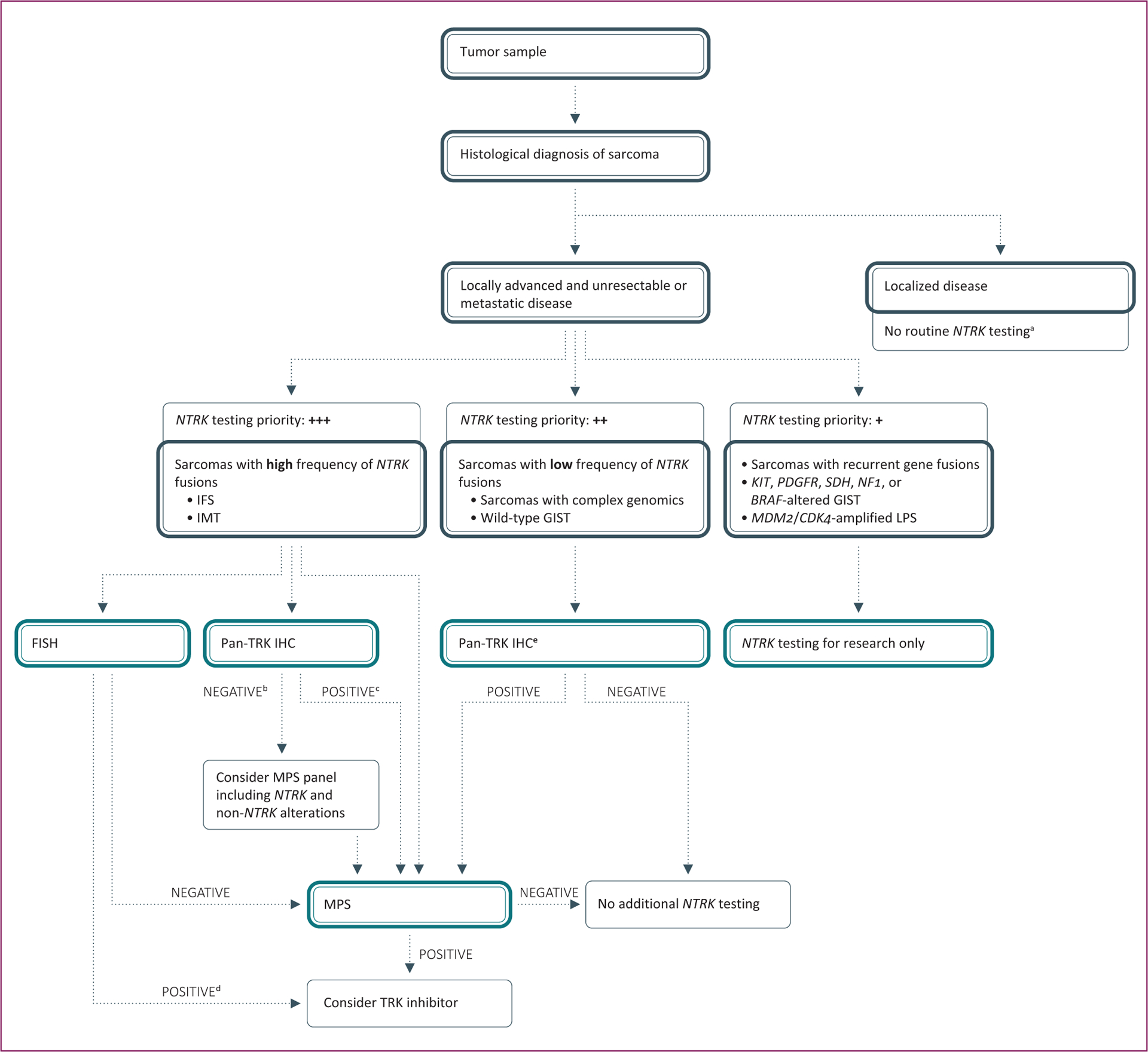
GIST, gastrointestinal stromal tumor; IFS, infantile fibrosarcoma; IHC, immunohistochemistry; IMT, inflammatory myofibroblastic tumor; LPS, liposarcoma; MPS, massive parallel sequencing; NTRK, neurotrophic tyrosine receptor kinase; RT-PCR, reverse transcription polymerase chain reaction; TRK, tropomyosin receptor kinase.
a For patients at high risk of relapse, NTRK gene fusion testing might provide clinically actionable information for later in the disease course.
b If histology is typical then confirmation by MPS is recommended.
c Treatment may be considered concurrently with confirmatory MPS.
d Consider parallel validation by MPS or RT-PCR to confirm that fusion is in-frame.
e Avoid IHC screening in cases with myogenic and neural differentiation due to the high rate of false positivity.
Sarcomas with a low NTRK fusion frequency (priority 2)
NTRK gene fusions are thought essentially to be mutually exclusive to other primary oncogenic drivers.59 In a study of patients with various tumor types, 31% of NTRK fusion-negative cases harbored activating MAPK pathway alterations compared with only 1.5% (n = 1) of NTRK fusion-positive cases.60 In another study, among 103 sarcomas tested for recurrent kinase fusions in one study, one sample had an NTRK1 gene fusion but no other concurrent fusions.63 Therefore, for sarcoma subtypes where NTRK gene fusions are rare, NTRK fusion screening should only be routinely done in cases already known to lack canonical oncogene alterations, such as wild-type GISTs and sarcomas with complex genomics. Sarcomas with recurrent gene fusions, GISTs with KIT, PDGFR, SDH, NF1, or BRAF alterations, and liposarcomas with MDM2 or CDK4 amplification may be excluded from routine NTRK fusion testing. Of this subset, however, tumors that do not show specific lineage differentiation (i.e. positive only for vimentin) may be enriched in molecular alterations including NTRK fusions.
Sarcomas with canonical oncogene alterations (priority 3)
Very infrequent situations of NTRK gene fusions co-occurring with other driver alterations in untreated tumors have been reported; however, the NTRK fusion appears to exert oncogenic dominance in these rare cases.59,60 Therefore, NTRK fusion testing in tumors with canonical pathognomonic alterations may be valuable in a research context in order to provide data on frequency and clinical significance of co-occurring NTRK gene fusions. While NTRK gene fusions have been identified in a range of sarcoma subtypes (Table 1), comprehensive data on NTRK fusion frequency in different sarcoma subtypes are lacking. Therefore, the majority of soft tissue and bone sarcomas should continue to be studied until there are sufficient data to guide future diagnostic approaches. Comprehensive data about NTRK gene fusion frequency in different sarcoma subtypes and correlation with morphological features would better inform the optimal approach to NTRK gene fusion screening in sarcomas and should be collected. In this regard, a prospective registry and retrospective collection of TRK fusion sarcoma cases would be valuable and a study is planned in Spain and France where all soft tissue sarcomas will be prospectively screened with pan-TRK IHC, with positive cases then confirmed by MPS.
CLINICAL MANAGEMENT OF TRK FUSION SARCOMAS
The labeled indications for larotrectinib (FDA and EMA) and entrectinib (FDA) include patients for whom surgery is likely to result in severe morbidity or who have no satisfactory alternative therapy. Therefore, the advantages and disadvantages of TRK inhibitors compared with other available therapies should be discussed by the patient and the treating physician.
In addition to the overall efficacy of TRK inhibitors described earlier (Table 2), efficacy of neoadjuvant larotrectinib therapy has also been demonstrated in situations where surgery would otherwise result in life-changing operations (e.g. amputation). Five children with locally advanced TRK fusion sarcomas (three with infantile fibrosarcomas and two with other soft tissue sarcomas) achieved a partial response to neoadjuvant larotrectinib and underwent resection after a median of six treatment cycles. Resections were R0 (negative resection margins with no tumor at the inked resection margin) in three patients, R1 (microscopic residual tumor at the resection margin) in one patient and R2 (incomplete resection with macroscopic residual tumor) in one patient. Three patients achieved complete or near-complete pathological responses and at last follow-up remained disease-free 7–15 months after surgery.64 While these data are encouraging, the question of if and when to discontinue TRK inhibitor therapy following a complete response still remains.
For patients with metastatic disease requiring systemic therapy, treatment with larotrectinib or entrectinib is approved after failure of standard therapies and may be valuable after standard first-line treatment given the rapid, durable responses and tolerability observed. In clinical trials of larotrectinib and entrectinib, responses were typically observed at the time of the first protocol-mandated tumor assessment, and pseudo-progression is uncommon with these therapies; therefore, it may be possible to quickly evaluate treatment response. However, it should be noted that no data exist for larotrectinib or entrectinib compared or combined with standard systemic cytotoxic therapies. Furthermore, the long-term safety profile of TRK inhibitors remains unknown and requires further study.
NTRK gene fusions have been shown to persist in tumors over time,60 suggesting that they remain the dominant oncogenic driver over the course of different treatments. This provides the rationale for a sequential TRK inhibitor treatment approach in patients with TRK fusion cancer, similar to current practice in oncogene-addicted (e.g. EGFR, ALK) NSCLC. The next-generation TRK inhibitors selitrectinib and repotrectinib have shown encouraging activity in patients who had progressed on larotrectinib or entrectinib due to acquired resistance mutations in the TRK kinase domain, including patients with sarcoma.65–67
SUMMARY
The emergence of NTRK gene fusions as clinically actionable biomarkers marks a new era in precision oncology, with the tumor-agnostic approvals of larotrectinib and entrectinib representing milestones in drug development. TRK inhibitors provide new personalized treatment options with the potential to extend survival and improve quality of life in some patients with sarcoma harboring NTRK gene fusions. Integrating NTRK fusion testing into the current diagnostic workup of patients with sarcoma is particularly challenging due to the rarity of this biomarker. Here, we propose a diagnostic strategy to address this that considers disease stage and histologic and molecular subtypes to facilitate routine testing for TRK expression and subsequent testing for NTRK gene fusions.
Routine genome-wide MPS in sarcomas may not currently be cost-effective due to the small number of additional genomic alterations to be tested. However, IHC provides a valuable pre-screening tool and focused MPS panels, such as those that detect NTRK gene fusions and other key gene fusions in parallel, are particularly relevant for sarcomas. Further research is necessary to fully establish the sensitivity and specificity of pan-TRK IHC. Furthermore, multinational comparative studies are encouraged to increase the reproducibility of MPS assays. Finally, prospective studies will be essential to determine the frequency of NTRK gene fusions in different sarcoma subtypes and correlation with morphological, biological, and clinical features in order to better inform the optimal approach to NTRK gene fusion screening.
ACKNOWLEDGEMENTS
Under the authors’ conceptual guidance, medical writing support was provided by Michael Sheldon, PhD and editorial support was provided by Annabel Ola, MSc, both of Scion, London, UK, according to Good Publication Practice guidelines and supported by an independent grant from Bayer HealthCare Pharmaceuticals, Inc.
FUNDING
GDD was supported in part by the Ludwig Center at Harvard; the Dr Miriam and Sheldon G. Adelson Medical Research Foundation; and the Pan Mass Challenge (no grant numbers). IMS is supported by the National Institutes of Health/National Cancer Institute (grant number K08 CA241085). JYB was supported by NetSARC (INCA & DGOS) and RREPS (INCA & DGOS), RESOS (INCA & DGOS), EURACAN (EC 739521), LYRICAN (INCA-DGOS-INSERM 12563), LabEx DEvweCAN (ANR-10-LABX-0061), Institut Convergence PLASCAN (17-CONV-0002), RHU4 DEPGYN (ANR-18-RHUS-0009).
DISCLOSURE
KB has received consultant fees from Bayer and Merck, and research support from Bayer, Merck, and Eli Lilly. GDD discloses scientific consultancy with sponsored research to Dana-Farber from Bayer, Pfizer, Novartis, Roche/Genentech, Epizyme, LOXO Oncology, AbbVie, GSK, Janssen, Pharma-Mar, ZioPharm, Daiichi Sankyo, Adaptimmune, and Mirati; scientific consultancy for GSK, EMD Serono, Sanofi, ICON plc, WCG/Arsenal Capital, Polaris Pharmaceuticals, MJ Hennessy/OncLive, C4 Therapeutics, Synlogic, and MED-SCAPE; consultant/SAB member with minor equity holding for G1 Therapeutics, Caris Life Sciences, Champions Biotechnology, Bessor Pharma, Erasca Pharmaceuticals, RELAY Therapeutics, and Caprion/HistoGeneX; board of directors member and scientific advisory board consultant with minor equity for Blueprint Medicines, Merrimack Pharmaceuticals (ended Oct 2019), and Translate BIO; royalties from Novartis to Dana-Farber for use of patent of imatinib in GIST; non-financial interests with McCann Health, Alexandria Real Estate Equities, and AACR Science Policy and Government Affairs Committee (Chair). JD discloses advisory/consultancy roles for Amgen, Novartis, Lilly, GSK, Pierre Fabre, and Eisai; institutional research funding from Novartis, Lilly, GSK, Roche/Genentech, BMS, AstraZeneca, and BeiGene. PH discloses consultancy fees from Roche and Pfizer, and research support from Novartis. HJ has a co-appointment at Orion Pharma, has received fees from Neutron Therapeutics, and owns stocks of Orion Pharma and Sartar Therapeutics. YKK discloses consultancy for Taiho, Ono, Merck, Daehwa, BMS, Astellas, Zymeworks, ALX ONCOLOGY, Amgen, Novartis, MacroGenics, and Surface Oncology. SP discloses grant support from Blueprint Medicines and Hutchison MediPharma; consultancy for Daiichi Sankyo, Epizyme, Dova, Decimera, Bayer, and Immune Design. FPL discloses advisory/consultancy roles for Bayer and Roche, and institutional research grants from Bayer. PR has received honoraria for lectures and advisory boards from Novartis, MSD, Roche, BMS, Sanofi, Merck, Amgen, Pfizer, Pierre Fabre, and Blueprint Medicines, outside the scope of this report. WDT discloses advisory/consultancy roles for Lilly, EMD Serono, Eisai, Janssen, Immune Design, Daiichi Sankyo, Blueprint Medicines, LOXO Oncology, GSK, Agios Pharmaceuticals, NanoCarrier, Deciphera, Certis Oncology Solutions, and Atropos Therapeutics; patent for CDK4 inhibitor companion diagnostic (14/854 329) pending to MSKCC/SKI; stock ownership in Certis Oncology Solutions and Atropos Therapeutics; participation in FDA ODAC meeting for pexidartinib. DT is the CEO of a non-profit company, Omico, which undertakes precision oncology activities across Australia. He has received honoraria or consultant fees from Roche, Pfizer, Bayer, Astra Zeneca, Merck, and the Maine Cancer Genome Initiative. He has received research support from Roche, Pfizer, Bayer, AstraZeneca, Amgen, Eisai, Illumina, and Sun Pharma. JZ discloses honoraria from Pfizer, Merck Serono, Specialized Therapeutics, Targovax, Halozyme, Gilead Sciences, and Bayer; advisory/consultancy roles for Pfizer, Merck Serono, Targovax, MSD, Sirtex Medical, Halozyme, Lipotek, Novella, and Khloris Biosciences; institutional research funding from Bayer, Merck Serono, Roche, BMS, Pfizer, AstraZeneca, Specialized Therapeutics, Baxalta/Shire, Lilly, Boehringer-Ingelheim, and MSD; travel/accommodation/expenses from Merck Serono, AstraZeneca, MSD, Deciphera, and Sirtex; shareholder/stockholder/stock options in GW Pharmaceuticals, Aimmune, Vertex, Bluebird Bio, Alnylam, BioMarin, Sage Therapeutics, Dova Pharmaceuticals, Therapeutics MD, Juno Therapeutics, Kite Pharma, Kiadis Pharma, CSL Ltd, Cochlear, Amarin, Freq Therapeutics, Global Blood Therapeutics, Gilead, uniQure, Sangamo, Acceleron, Zogenix, Myovant Science, and Khloris Biosciences; board of directors for Praxis Australia; non-remunerated activities for Australian Clinical Trials Alliance (Chair), National Oncology Alliance (co-Chair), and All.Can Australia (co-Chair). All other authors have declared no conflicts of interest.
REFERENCES
- 1.Board WCoTE. Soft Tissue and Bone Tumours: WHO Classification of Tumours. 5th ed. France: Iarc; 2020. [Google Scholar]
- 2.Siegel RL, Miller KD, Jemal A. Cancer statistics, 2020. CA Cancer J Clin. 2020;70:7–30. [DOI] [PubMed] [Google Scholar]
- 3.Dangoor A, Seddon B, Gerrand C, et al. UK guidelines for the management of soft tissue sarcomas. Clin Sarcoma Res. 2016;6:20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Harris SJ, Maruzzo M, Thway K, et al. Metastatic soft tissue sarcoma, an analysis of systemic therapy and impact on survival. J Clin Oncol. 2015;33:10545. [Google Scholar]
- 5.Maki RG, Wathen JK, Patel SR, et al. Randomized phase II study of gemcitabine and docetaxel compared with gemcitabine alone in patients with metastatic soft tissue sarcomas: results of sarcoma alliance for research through collaboration study 002 [corrected]. J Clin Oncol. 2007;25:2755–2763. [DOI] [PubMed] [Google Scholar]
- 6.Corless CL, Fletcher JA, Heinrich MC. Biology of gastrointestinal stromal tumors. J Clin Oncol. 2004;22:3813–3825. [DOI] [PubMed] [Google Scholar]
- 7.Wardelmann E, Pauls K, Merkelbach-Bruse S, et al. Gastrointestinal stromal tumors carrying PDGFRalpha mutations occur preferentially in the stomach and exhibit an epithelioid or mixed phenotype. Verh Dtsch Ges Pathol. 2004;88:174–183. [PubMed] [Google Scholar]
- 8.Corless CL, Schroeder A, Griffith D, et al. PDGFRA mutations in gastrointestinal stromal tumors: frequency, spectrum and in vitro sensitivity to imatinib. J Clin Oncol. 2005;23:5357–5364. [DOI] [PubMed] [Google Scholar]
- 9.Duensing A, Medeiros F, McConarty B, et al. Mechanisms of oncogenic KIT signal transduction in primary gastrointestinal stromal tumors (GISTs). Oncogene. 2004;23:3999–4006. [DOI] [PubMed] [Google Scholar]
- 10.Gastrointestinal Stromal Tumor Meta-Analysis Group. Comparison of two doses of imatinib for the treatment of unresectable or metastatic gastrointestinal stromal tumors: a meta-analysis of 1,640 patients. J Clin Oncol. 2010;28:1247–1253. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Heinrich MC, Maki RG, Corless CL, et al. Primary and secondary kinase genotypes correlate with the biological and clinical activity of sunitinib in imatinib-resistant gastrointestinal stromal tumor. J Clin Oncol. 2008;26:5352–5359. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Cocco E, Scaltriti M, Drilon A. NTRK fusion-positive cancers and TRK inhibitor therapy. Nat Rev Clin Oncol. 2018;15:731–747. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Amatu A, Sartore-Bianchi A, Bencardino K, et al. Tropomyosin receptor kinase (TRK) biology and the role of NTRK gene fusions in cancer. Ann Oncol. 2019;30:viii5–viii15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Cunningham ME, Greene LA. A function-structure model for NGF-activated TRK. EMBO J. 1998;17:7282–7293. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Greco A, Villa R, Fusetti L, et al. The Gly571Arg mutation, associated with the autonomic and sensory disorder congenital insensitivity to pain with anhidrosis, causes the inactivation of the NTRK1/nerve growth factor receptor. J Cell Physiol. 2000;182:127–133. [DOI] [PubMed] [Google Scholar]
- 16.Indo Y, Tsuruta M, Hayashida Y, et al. Mutations in the TRKA/NGF receptor gene in patients with congenital insensitivity to pain with anhidrosis. Nat Genet. 1996;13:485–488. [DOI] [PubMed] [Google Scholar]
- 17.Klein R, Smeyne RJ, Wurst W, et al. Targeted disruption of the trkB neurotrophin receptor gene results in nervous system lesions and neonatal death. Cell. 1993;75:113–122. [PubMed] [Google Scholar]
- 18.Xu B, Goulding EH, Zang K, et al. Brain-derived neurotrophic factor regulates energy balance downstream of melanocortin-4 receptor. Nat Neurosci. 2003;6:736–742. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Yeo GSH, Connie Hung C-C, Rochford J, et al. A de novo mutation affecting human TrkB associated with severe obesity and developmental delay. Nat Neurosci. 2004;7:1187–1189. [DOI] [PubMed] [Google Scholar]
- 20.Cordon-Cardo C, Tapley P, Jing SQ, et al. The trk tyrosine protein kinase mediates the mitogenic properties of nerve growth factor and neurotrophin-3. Cell. 1991;66:173–183. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Vaishnavi A, Le AT, Doebele RC. TRKing down an old oncogene in a new era of targeted therapy. Cancer Discov. 2015;5:25–34. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Okamura R, Boichard A, Kato S, et al. Analysis of NTRK alterations in pan-cancer adult and pediatric malignancies: implications for NTRK-targeted therapeutics. JCO Precis Oncol. 2018;2018. 10.1200/PO.18.00183. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Amatu A, Sartore-Bianchi A, Siena S. NTRK gene fusions as novel targets of cancer therapy across multiple tumour types. ESMO Open. 2016;1:e000023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Kummar S, Lassen UN. TRK inhibition: a new tumor-agnostic treatment strategy. Target Oncol. 2018;13:545–556. [DOI] [PubMed] [Google Scholar]
- 25.Knezevich SR, McFadden DE, Tao W, et al. A novel ETV6-NTRK3 gene fusion in congenital fibrosarcoma. Nat Genet. 1998;18:184–187. [DOI] [PubMed] [Google Scholar]
- 26.Davis JL, Lockwood CM, Albert CM, et al. Infantile NTRK-associated mesenchymal tumors. Pediatr Dev Pathol. 2018;21:68–78. [DOI] [PubMed] [Google Scholar]
- 27.Chiang S, Cotzia P, Hyman DM, et al. NTRK fusions define a novel uterine sarcoma subtype with features of fibrosarcoma. Am J Surg Pathol. 2018;42:791–798. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Haller F, Knopf J, Ackermann A, et al. Paediatric and adult soft tissue sarcomas with NTRK1 gene fusions: a subset of spindle cell sarcomas unified by a prominent myopericytic/haemangiopericytic pattern. J Pathol. 2016;238:700–710. [DOI] [PubMed] [Google Scholar]
- 29.Agaram NP, Zhang L, Sung YS, et al. Recurrent NTRK1 gene fusions define a novel subset of locally aggressive lipofibromatosis-like neural tumors. Am J Surg Pathol. 2016;40:1407–1416. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Suurmeijer AJ, Dickson BC, Swanson D, et al. The histologic spectrum of soft tissue spindle cell tumors with NTRK3 gene rearrangements. Genes Chromosomes Cancer. 2019;58:739–746. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Suurmeijer AJH, Dickson BC, Swanson D, et al. A novel group of spindle cell tumors defined by S100 and CD34 co-expression shows recurrent fusions involving RAF1, BRAF, and NTRK1/2 genes. Genes Chromosomes Cancer. 2018;57:611–621. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Suurmeijer AJH, Kao YC, Antonescu CR. New advances in the molecular classification of pediatric mesenchymal tumors. Genes Chromosomes Cancer. 2019;58:100–110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Drilon A TRK inhibitors in TRK fusion-positive cancers. Ann Oncol. 2019;30:viii23–viii30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Doebele RC, Davis LE, Vaishnavi A, et al. An oncogenic NTRK fusion in a patient with soft-tissue sarcoma with response to the tropomyosin-related kinase inhibitor LOXO-101. Cancer Discov. 2015;5:1049–1057. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Ghilardi JR, Freeman KT, Jimenez-Andrade JM, et al. Administration of a tropomyosin receptor kinase inhibitor attenuates sarcoma-induced nerve sprouting, neuroma formation and bone cancer pain. Mol Pain. 2010;6:87. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Vitrakvi. Summary of product characterisitics. Available at https://www.ema.europa.eu/en/documents/product-information/vitrakvi-epar-product-information_en.pdf. Accessed September 19, 2020.
- 37.Vitrakvi prescribing information. Available at https://www.accessdata.fda.gov/drugsatfda_docs/label/2018/211710s000lbl.pdf. Accessed September 19, 2020.
- 38.Drilon A, Laetsch TW, Kummar S, et al. Efficacy of larotrectinib in TRK fusion-positive cancers in adults and children. N Engl J Med. 2018;378: 731–739. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Hong DS, DuBois SG, Kummar S, et al. Larotrectinib in patients with TRK fusion-positive solid tumours: a pooled analysis of three phase 1/2 clinical trials. Lancet Oncol. 2020;21:531–540. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Drilon A, DuBois SG, Farago AF, et al. Activity of larotrectinib in TRK fusion cancer patients with brain metastases or primary central nervous system tumors. J Clin Oncol. 2019;37:2006. [Google Scholar]
- 41.Doz F, Geoerger B, DuBois SG, et al. RARE-45. Activity of larotrectinib in TRK fusion cancer patients with primary central nervous system tumors. Neuro-Oncology. 2019;21:vi231. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Kummar S, Mascarenhas L, Geoerger B, et al. Patient-reported outcomes from two global multicenter clinical trials of children and adults with tropomyosin receptor kinase (TRK) fusion cancer receiving larotrectinib. J Clin Oncol. 2019;37:6602. [Google Scholar]
- 43.Demetri G, Albert CM, Tan DSW, et al. Larotrectinib efficacy and safety in patients with TRK fusion sarcomas. Connective tissue oncology society (CTOS). Final program 2019, p40. Available at https://www.ctos.org/Portals/0/PDF/2019%20CTOS%20Final%20Program.pdf. Accessed September 19, 2020. [Google Scholar]
- 44.Anderson D, Ciomei M, Banfi P, et al. 310 inhibition of Trk-driven tumors by the pan-Trk inhibitor RXDX-101. Eur J Cancer. 2014;50:101. [Google Scholar]
- 45.Rozlytrek prescribing information. Available at https://www.accessdata.fda.gov/drugsatfda_docs/label/2019/212725s000lbl.pdf. Accessed September 19, 2020.
- 46.Doebele RC, Drilon A, Paz-Ares L, et al. Entrectinib in patients with advanced or metastatic NTRK fusion-positive solid tumours: integrated analysis of three phase 1–2 trials. Lancet Oncol. 2020;21:271–282. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Liu SV, Paz-Ares L, Hu J, et al. Entrectinib in NTRK fusion-positive sarcoma: integrated analysis of patients enrolled in STARTRK-2, STARTRK-1 AND ALKA-372–001. Connective tissue oncology society (CTOS). Final program 2019, p41. Available at https://www.ctos.org/Portals/0/PDF/2019%20CTOS%20Final%20Program.pdf. Accessed September 19, 2020. [Google Scholar]
- 48.Wong D, Yip S, Sorensen PH. Methods for identifying patients with tropomyosin receptor kinase (TRK) fusion cancer. Pathol Oncol Res. 2020;26:1385–1399. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Penault-Llorca F, Rudzinski ER, Sepulveda AR. Testing algorithm for identification of patients with TRK fusion cancer. J Clin Pathol. 2019;72: 460–467. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Davis JL, Lockwood CM, Stohr B, et al. Expanding the spectrum of pediatric NTRK-rearranged mesenchymal tumors. Am J Surg Pathol. 2018;43:435–445. [DOI] [PubMed] [Google Scholar]
- 51.Gatalica Z, Xiu J, Swensen J, Vranic S. Molecular characterization of cancers with NTRK gene fusions. Mod Pathol. 2018;32:147–153. [DOI] [PubMed] [Google Scholar]
- 52.Solomon JP, Linkov I, Rosado A, et al. NTRK fusion detection across multiple assays and 33,997 cases: diagnostic implications and pitfalls. Mod Pathol. 2020;33:38–46. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Solomon JP, Benayed R, Hechtman JF, Ladanyi M. Identifying patients with NTRK fusion cancer. Ann Oncol. 2019;30:viii16–viii22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Marchio C, Scaltriti M, Ladanyi M, et al. ESMO recommendations on the standard methods to detect NTRK fusions in daily practice and clinical research. Ann Oncol. 2019;30:1417–1427. [DOI] [PubMed] [Google Scholar]
- 55.Pfarr N, Kirchner M, Lehmann U, et al. Testing NTRK testing: wet-lab and in silico comparison of RNA-based targeted sequencing assays. Genes Chromosomes Cancer. 2020;59:178–188. [DOI] [PubMed] [Google Scholar]
- 56.Hong DS, Bauer TM, Lee JJ, et al. Larotrectinib in adult patients with solid tumours: a multi-centre, open-label, phase I dose-escalation study. Ann Oncol. 2019;30:325–331. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Gounder MM, Ali SM, Robinson V, et al. Impact of next-generation sequencing (NGS) on diagnostic and therapeutic options in soft-tissue and bone sarcoma. J Clin Oncol. 2017;35:11001. [Google Scholar]
- 58.Italiano A, Di Mauro I, Rapp J, et al. Clinical effect of molecular methods in sarcoma diagnosis (GENSARC): a prospective, multicentre, observational study. Lancet Oncol. 2016;17:532–538. [DOI] [PubMed] [Google Scholar]
- 59.Jiao X, Lokker A, Snider J, et al. Co-occurrence of NTRK fusions with other genomic biomarkers in cancer patients. Ann Oncol. 2019;30:v29–v30. [Google Scholar]
- 60.Rosen EY, Goldman DA, Hechtman JF, et al. TRK fusions are enriched in cancers with uncommon histologies and the absence of canonical driver mutations. Clin Cancer Res. 2020;26:1624–1632. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Pavlick D, Schrock AB, Malicki D, et al. Identification of NTRK fusions in pediatric mesenchymal tumors. Pediatr Blood Cancer. 2017;64. 10.1002/pbc.26433. [DOI] [PubMed] [Google Scholar]
- 62.Alassiri AH, Ali RH, Shen Y, et al. ETV6-NTRK3 is expressed in a subset of ALK-negative inflammatory myofibroblastic tumors. Am J Surg Pathol. 2016;40:1051–1061. [DOI] [PubMed] [Google Scholar]
- 63.Stransky N, Cerami E, Schalm S, et al. The landscape of kinase fusions in cancer. Nat Commun. 2014;5:4846. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.DuBois SG, Laetsch TW, Federman N, et al. The use of neoadjuvant larotrectinib in the management of children with locally advanced TRK fusion sarcomas. Cancer. 2018;124:4241–4247. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Hyman D, Kummar S, Farago A, et al. Phase I and expanded access experience of LOXO-195 (BAY 2731954), a selective next-generation TRK inhibitor (TRKi). In: Proceedings of the 110th Annual Meeting of the American Association for Cancer Research. Atlanta, GA: AACA; 2019. Abstract CT127. [Google Scholar]
- 66.Drilon A, Ou S-HI, Cho BC, et al. Repotrectinib (TPX-0005) is a next-generation ROS1/TRK/ALK inhibitor that potently inhibits ROS1/TRK/ALK solvent-front mutations. Cancer Discov. 2018;8:1227–1236. [DOI] [PubMed] [Google Scholar]
- 67.Drilon A, Zhai D, Deng W, et al. Repotrectinib, a next generation TRK inhibitor, overcomes TRK resistance mutations including solvent front, gatekeeper and compound mutations. Presented at the AACR Annual Meeting 2019. March 29-April 3, 2019; Atlanta, GA. Abstract# 4000 2019. [Google Scholar]
- 68.Bourgeois JM, Knezevich SR, Mathers JA, Sorensen PH. Molecular detection of the ETV6-NTRK3 gene fusion differentiates congenital fibrosarcoma from other childhood spindle cell tumors. Am J Surg Pathol. 2000;24:937–946. [DOI] [PubMed] [Google Scholar]
- 69.Bui NQ, Przybyl J, Trabucco SE, et al. A clinico-genomic analysis of soft tissue sarcoma patients reveals CDKN2A deletion as a biomarker for poor prognosis. Clin Sarcoma Res. 2019;9:12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Chang JC, Zhang L, Drilon AE, et al. Expanding the molecular characterization of thoracic inflammatory myofibroblastic tumors beyond ALK gene rearrangements. J Thorac Oncol. 2019;14:825–834. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Chmielecki J, Bailey M, He J, et al. Genomic profiling of a large set of diverse pediatric cancers identifies known and novel mutations across tumor spectra. Cancer Res. 2017;77:509–519. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Church AJ, Calicchio ML, Nardi V, et al. Recurrent EML4-NTRK3 fusions in infantile fibrosarcoma and congenital mesoblastic nephroma suggest a revised testing strategy. Mod Pathol. 2018;31:463–473. [DOI] [PubMed] [Google Scholar]
- 73.Croce S, Hostein I, Longacre TA, et al. Uterine and vaginal sarcomas resembling fibrosarcoma: a clinicopathological and molecular analysis of 13 cases showing common NTRK-rearrangements and the description of a COL1A1-PDGFB fusion novel to uterine neoplasms. Mod Pathol. 2019;32:1008–1022. [DOI] [PubMed] [Google Scholar]
- 74.Shi E, Chmielecki J, Tang CM, et al. FGFR1 and NTRK3 actionable alterations in “Wild-Type” gastrointestinal stromal tumors. J Transl Med. 2016;14:339. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Surrey LF, MacFarland SP, Chang F, et al. Clinical utility of custom-designed NGS panel testing in pediatric tumors. Genome Med. 2019;11:32. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Yamamoto H, Nozaki Y, Kohashi K, et al. Diagnostic utility of pan-Trk immunohistochemistry for inflammatory myofibroblastic tumours. Histopathology. 2020;76:774–778. [DOI] [PubMed] [Google Scholar]
- 77.Zhu G, Benayed R, Ho C, et al. Diagnosis of known sarcoma fusions and novel fusion partners by targeted RNA sequencing with identification of a recurrent ACTB-FOSB fusion in pseudomyogenic hemangioendo-thelioma. Mod Pathol. 2019;32:609–620. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Hechtman JF, Benayed R, Hyman DM, et al. Pan-Trk immunohistochemistry is an efficient and reliable screen for the detection of NTRK fusions. Am J Surg Pathol. 2017;41:1547–1551. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Rudzinski ER, Lockwood CM, Stohr BA, et al. Pan-Trk immunohistochemistry identifies NTRK rearrangements in pediatric mesenchymal tumors. Am J Surg Pathol. 2018;42:927–935. [DOI] [PubMed] [Google Scholar]
- 80.Hung YP, Jo VY, Hornick JL. Immunohistochemistry with a pan-TRK antibody distinguishes secretory carcinoma of the salivary gland from acinic cell carcinoma. Histopathology. 2019;75: 54–62. [DOI] [PubMed] [Google Scholar]
- 81.Xu B, Haroon Al Rasheed MR, Antonescu CR, et al. Pan-Trk immunohistochemistry is a sensitive and specific ancillary tool for diagnosing secretory carcinoma of the salivary gland and detecting ETV6–NTRK3 fusion. Histopathology. 2020;76:375–382. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Feng J, Ebata K, Hansen F, et al. 80PTRK wild-type and fusion protein expression in solid tumors: characterization by immunohistochemistry and in situ hybridization. Ann Oncol. 2018;29(suppl_6). [Google Scholar]
- 83.Murphy DA, Ely HA, Shoemaker R, et al. Detecting gene rearrangements in patient populations through a 2-step diagnostic test comprised of rapid IHC enrichment followed by sensitive next-generation sequencing. Appl Immunohistochem Mol Morphol. 2017;25:513–523. [DOI] [PMC free article] [PubMed] [Google Scholar]