

Abstract
We study the interaction and metalation reaction of a free base 5,10,15,20‐terakis(4‐cyanophenyl)porphyrin (2HTCNPP) with post‐deposited Zn atoms and the targeted reaction product Zn‐5,10,15,20‐terakis(4‐cyanophenyl)porphyrin (ZnTCNPP) on a Ag(111) surface. The investigations are performed with scanning tunneling microscopy at room temperature after Zn deposition and subsequent heating. The goal is to obtain further insights in the metalation reaction and the influence of the cyanogroups on this reaction. The interaction of 2HTCNPP with post‐deposited Zn leads to the formation of three different 2D ordered island types that coexist on the surface. All contain a new species with a bright appearance, which increases with the amount of post‐deposited Zn. We attribute this to metastable SAT (“sitting atop”) complexes formed by Zn and the macrocycle, that is, an intermediate in the metalation reaction to ZnTCNPP, which occurs upon heating to 500 K. Interestingly, the activation barrier for the successive reaction of the SAT complex to the metalated ZnTCNPP species can also be overcome by a voltage pulse applied to the STM tip.
Keywords: metalation, porphyrinoids, SAT complex, scanning tunneling microscopy, surface chemistry
Metalation with zinc: Post‐deposition of Zn onto 2HTCNPP leads to the formation of a SAT complex, which reacts to ZnTCNPP upon heating or application of a voltage pulse.

1. Introduction
The fabrication of functional nanoscale materials by a bottom‐up approach has become a major topic in science during the past years. This growing demand of miniaturization is present in areas such as catalysis, energy storage and conversion, electronic and photonic devices.[ 1 , 2 ] The adsorption of functional organic molecules on metal surfaces has proven to be a suitable method for the construction of devices at the nanoscale.[ 1 , 3 , 4 ] In this context, porphyrins are promising candidates since they offer interesting properties like a conformationally flexible macrocycle with a reactive coordination center and the possibility for functionalization at the periphery. Heme (Fe‐porphyrin) and chlorophyll (Mg‐porphyrin) are particularly important molecules in nature, which offer different functionalities according to their metal center. While heme is responsible for oxygen transport in mammals, chlorophyll is involved in the photosynthesis of plants. The porphyrin core is known to coordinate a variety of different metal atoms. [5] The most common oxidation state of the metal ions is 2+, e. g. for Cu and Zn, while Co, Ni and Fe can form 2+ and 3+. [6] This huge variety in functionality due to a different metal core offers great potential for a variety of applications.[ 7 , 8 , 9 , 10 ] In particular for functional nanomaterials the understanding of the metalation reaction of porphyrins on surfaces thereby is of utmost importance.
The metalation reaction on surfaces takes place with either pre‐ or post‐deposited metal atoms. [6] For Fe and Co, it occurs already at room temperature (RT) while moderate annealing is required for metals like Cu and Zn.[ 11 , 12 , 13 ] This observation indicates different activation energies for different metal atoms. Additionally, porphyrins are known to undergo a so‐called self‐metalation reaction on reactive single crystal surfaces like Cu, Fe and Ni, in which the macrocycle reacts directly with metal atoms present on the surface.[ 14 , 15 , 16 , 17 ] The mechanism of this very complex surface reaction has been topic of several previous studies, and the question arose whether it is a one‐step reaction or intermediate states exist like the “sitting atop” (SAT), which has been proposed for metalation in solution,[ 18 , 19 , 20 ] as well as at the solid‐vacuum interface.[ 21 , 22 , 23 ] In this adcomplex, the metal ion coordinates with the macrocycle while the pyrrolic hydrogen atoms are still bound to the nitrogen atoms. Shubina et al. investigated the metalation of 2HTPP with bare Zn atoms on a Ag(111) with XPS and compared it to DFT calculations of the gas‐phase reaction. [24] They found a similar complex with Zn weakly bound to the nitrogen atoms of the macrocycle and the two hydrogen atoms still attached to the two pyrrolic nitrogen atoms. The rate limiting step for metalation is the transfer of the first hydrogen to the metal atom. This step varies for different metal centers. For Zn, evidence for the formation of the adcomplex was found upon heating a coadsorbed Zn and 2HTPP layer on Ag(111) to 300 K, while metalation only occurred upon heating to 500 K. For coadsorbed Fe and 2HTPP on Ag(111), the adcomplex could be identified already at 100 K, as concluded from a shift of the iminic N 1s peak by 0.6 eV to higher binding energies compared to 2HTPP. [22] Metalation was completed already at 350 K, indicating a much lower activation energy as compared to Zn, in line with the calculations by Shubina et al. [24] Yamada et al. were able to image these three different states of the metalation reaction of 2HTPP with low‐temperature STM for Fe on Ag(111) at 78.5 K. The original 2HTPP, a 2HTPP precursor adcomplex “α” state (Fe atom on top), and a 2HTPP precursor adcomplex “γ” state (Fe atom inside) were identified by DFT calculations, STM topography and scanning tunneling spectroscopy (STS). [25] One interesting question in this context is, whether for a metal with a higher activation energy for metalation, such as Zn, the adcomplex can also be observed at room temperature, which is closer to operating conditions of real devices.
Another important topic in the context of metalation is the question, whether the activation energy can be influenced by different ligands attached to the macrocycle. Recently, Lepper et al. were able to show that the self‐metalation rate of 2HTPP on Cu(111) can be modified by cyano‐groups attached to the phenyl ligands. [26] They observed the metalation rate to decrease significantly with increasing number of cyanophenyl ligands. In these studies, no information of reaction intermediates such as the adcomplex is available.
Herein, we report a comprehensive study of the interaction and reaction of a free base 5,10,15,20‐terakis(4‐cyanophenyl)porphyrin (2HTCNPP) with deposited Zn atoms on a Ag(111) surface. As a reference, we also investigated the targeted reaction product, that is, Zn‐5,10,15,20‐terakis(4‐cyanophenyl)porphyrin (ZnTCNPP). Both molecules and their interaction with Zn are studied with a scanning tunneling microscope at RT with the goal to get further insights in the metalation reaction, and the influence of the cyano groups on this reaction.
2. Results and Discussion
In order to study possible chemical reactions of 2HTCNPP with post‐deposited Zn on Ag(111), we first separately studied the adsorption of 2HTCNPP and the potential reaction product ZnTCNPP at RT; the corresponding STM images are shown in Figure 1. At low coverages, individual 2HTCNPP molecules are exclusively found on the step edges. On the terraces, only streaky features are observed, which are attributed to a 2D gas of fast diffusing molecules (see Figure S1 in the Supporting Information). Upon increasing the surface coverage, a long‐range‐ordered 2D supramolecular structure is found. [27] The individual molecules are clearly resolved with submolecular resolution, as is evident from Figure 1 (left panel). A slight depression marks the center of the bright macrocycle, and the four cyanophenyl groups at periphery of the molecule can be identified. The molecules adsorb in the so‐called “saddle shape” conformation and assemble in a square arrangement with lattice vectors of a=1.49±0.04 nm and b=1.45±0.04 nm, with an enclosed angle of α=87°±5°, which is in good agreement with the reported literature values. [27] Notably, the individual molecules are rotated relative to unit cell vectors a/b of the superstructure by 11°/22°, but are aligned along the substrate high symmetry directions (indicated by white arrows in the Figure 1).
Figure 1.
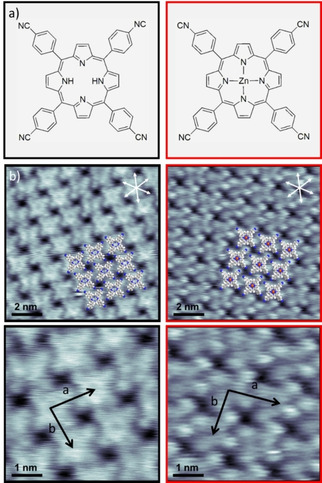
a) Chemical structure of 2HTCNPP (black) and ZnTCNPP (red). b) Constant‐current STM images of an island of 2HTCNPP (black, U=−850 mV, I=19 pA) and ZnTCNPP (red, U=−190 mV, I=30 pA) on Ag(111) at RT and higher resolution of the respective unit cells. Overlaid molecular models indicate the structure of the ordered 2HTCNPP and ZnTCNPP layers; the central Zn atom in ZnTCNPP is depicted in red.
The corresponding metalloporphyrin, ZnTCNPP, shows an overall very similar behavior. At low coverages (not shown) a 2D gas of fast diffusing molecules is identified from streaky features in the STM images, with some molecules adsorbed to step edges at RT. An increase in molecular coverage leads to the formation of long‐range ordered 2D supramolecular islands; see Figure 1 (right panel). The appearance within the islands is slightly different to that of 2HTCNPP. It is dominated by four bright protrusions due to the cyanophenyl groups at the periphery of the molecule, with the macrocycle being less bright and no obvious depression in its center. The central Zn atom is not visible at the applied tunneling conditions, since the 4s and 3d orbitals are completely occupied. Within the margins of error, the unit cell vectors a=1.51±0.04 nm and b=1.49±0.04 nm and the enclosed angle of α=85°±5° are the same as for 2HTCNPP. This behavior is expected due to the stabilization via attractive intermolecular interactions (hydrogen bonds between cyano groups and neighboring H atoms of phenyl groups), which are not affected by the Zn atom in the center of the molecule.
After post‐deposition of Zn atoms onto the 2HTCNPP islands at room temperature a drastic change is observed. The interaction with post‐deposited Zn leads to the formation of three different new island types, A, B and C, which coexist on the surface, as is evident from Figure 2b (for further images see Figure S2 in the SI). Notably, in all three the molecularly resolved structure of some molecules changes towards a bright appearance, with a central double protrusion (depending on scan direction and tunneling conditions sometimes only a single broad protrusion is seen). The rest of molecules still exhibit the features characteristic of 2HTCNPP. The bright appearance of this new species is remarkably similar in the different island types. The number of bright species increases approximately proportional with the amount of deposited Zn (this is demonstrated by subsequent Zn deposition, which is shown in SI Figure S3). The bright appearance clearly differs from that of ZnTCNPP, which is dominated by the four protrusions from the cyanophenyl groups; see Figure 1 (right). This observation rules out a metalation reaction at this stage. Furthermore, the bright protrusions are not stationary and occasionally hop to a different molecule between STM frames (see Figure S4 in the SI). This behavior suggests a loosely bound, mobile Zn adatom. Indeed, in earlier DFT calculations Shubina et al. proposed the formation of a so called “SAT” (sitting atop) complex for 2HTPP plus Zn, in which the Zn atom coordinates the macrocycle without the release of hydrogen. [24] This complex, which can also be denoted as 2H‐SAT complex (as both pyrrolic hydrogens are still in place), is the first intermediate in the metalation reaction of 2HTPP to ZnTPP. In order to progress in the reaction pathway to form ZnTPP an activation barrier of 32.6 kcal mol−1 is needed to overcome. [24] This correlates with experiments performed by Kretschmann et al., who showed that an annealing step to 550 K is required for the metalation of 2HTPP with Zn. [13] We propose a similar reaction pathway also for deposition of Zn onto a 2HTCNPP layer on Ag(111). Within this interpretation, we assign the bright protrusions seen in the three new structures in Figure 2 to metastable SAT complexes formed by Zn and the macrocycle, prior to the metalation reaction to ZnTCNPP and the release of hydrogen.
Figure 2.

a) High resolution STM image of 2HTCNPP (U=−850 mV, I=19 pA). b) The addition of Zn forms the three structures seen in the right overview (U=−1660 mV, I=30 pA). c) High‐resolution STM image of structure A (U=−810 mV, I=34 pA). d) High resolution STM image of structure B, where each molecule has on average six neighbors (U=−1660 mV, I=30 pA). e,f) High resolution STM image of structure C with regular missing molecules within the structure (U=−1660 mV, I=30 pA). g,h) High resolution STM image of structure C (different contrast; measured in Campinas/Brazil) with circular features within the holes of the structure (U=−1488 mV, I=13 pA). Overlaid molecular models indicate the structure of the different layers. The central Zn adatom in the SAT complex is depicted in yellow, and the coordinating Ag adatoms in green.
The first and dominant island type, A, is shown in Figure 2c, which shows a square ordered 2D island (a=1.52±0.04 nm, b=1.48±0.04 nm, α=85°±5°). Notably, both the bright SAT complexes and the remaining 2HTCNPP molecules are rotated against the lattice vectors of the island structures (best seen for molecules with double protrusion appearance), but are aligned along the high directions axis of the Ag(111) substrate. This arrangement is similar to that seen for pure 2HTCNPP in Figure 2a and Figure 1. It indicates that island‐type A results from the adsorption of Zn onto 2HTCNPP in the square islands without a rearrangement of the porphyrin molecule.
The second island type, B, (Figure 2d) has a densely packed, but disordered structure, with each molecule surrounded by six other molecules. This disorder makes it impossible to give a unit cell. The overall interaction theme within the structure is that three CN‐groups of neighboring molecules point towards each other. The double protrusions are predominantly orientated parallel to Ag(111) high symmetry directions.
The third island type, C, is a regular porous 2D structure built by pores, which are surrounded by eight molecules each (Figure 2e). Alternatively, one can describe this structure as an arrangement of two alternating types of rows: in the first, each site along the row is occupied and in the second only every second site occupied. The structure has quite a number of defects by occasional filling of the pores by a molecule and a break of the alternating row symmetry. Figure 2g shows the same structure in a better resolution measured with a different STM (see Experimental Section). Under these conditions, circular bright features are observed within the pores. We attribute these features to fast diffusing Zn metal atoms, which diffuse between the different cyano groups of the molecules. The alternative interpretation as molecules sitting slightly above the pore in a very mobile state, which do not fit in the pores, appears unlikely, since the apparent height is lower than that of neighboring molecules. The molecules are oriented again along the high symmetry direction of the substrate but now also along the rows. The overall lattice is again close to quadratic (a=1.66±0.08 nm, b=1.50±0.07 nm, α=88°±5°). However, in contrast to 2HTCNPP, ZnTCNPP and structure A, the lattice vectors are now aligned along the substrate high symmetry directions and are not rotated by 11°/22°. Notably, the different lattice sites are occupied by either a non‐metalated 2HTCNPP, a SAT complex or an empty pore, which makes a non‐homogeneous structure.
As binding motif of this structure, we tentatively propose a cyano‐metal‐cyano coordination. The cyano groups of three molecules are pointing towards each other, which implies a metal coordination center with a +3 oxidation state, assuming that only one metal atom is coordinated (Figure 2e and h). For Zn, a +3 oxidation state is highly unlikely, due to its electronic structure. Alternatively, we propose that Ag adatoms, which are present on the surface, serve as coordination centers. Comparable Au‐coordination structures formed by cyano‐functionalized triarylamines have indeed been observed on a Au(111) surface, [28] where similar to our observations, pores are surrounded by molecules forming long‐range‐ordered structures. These molecules have a threefold symmetry which results a hexagonal structure, while in our case the fourfold symmetry of the porphyrin core yields a square structure.
The absence of this type of coordination prior to Zn evaporation onto 2HTCNPP indicates that the presence of Zn is needed for the formation of the peculiar structure of island type C. From the fact this structure is not observed upon Zn deposition onto ZnTCNPP (see below), we conclude that Zn must play an active role for the formation of this structure. We tentatively propose that the geometric/electronic structure of the SAT complex is modified, as compared to 2HTCNPP and also ZnTCNPP, such that the formation of the coordination network becomes energetically favorable. In addition, increasing the deposition of Zn could also lead to an increase of the amount of Ag adatoms due to the filling of vacancies/defects in the underlying Ag surface layer by Zn. Especially upon heating Ag is known to dissolve Zn up to 40 atomic percent at 550 K. [29]
Analyzing the differences between the three island types, it appears that types A and B represent two subsequent steps towards type C upon deposition of Zn onto 2HTCNPP. In the first step Zn forms a SAT complex with 2HTCNPP, but in type A the long‐range structure remains unchanged. In a next step the molecule tends to arrange towards type C, that is, a quadratic lattice with holes, where the unit cell vectors are aligned along the substrate high symmetry direction. As driving force, we propose the formation of the Ag‐coordinated network, with three molecules coordinated to one Ag adatom. Type B has a disordered intermediate structure during this reorganization, in which three neighboring molecules are already coordinated, but the differently oriented quadratic lattice is not yet formed. The coexistence of structures A, B and C indicates that the energetic differences are small. Otherwise, exclusively type C islands would be expected.
As mentioned above, we do not observe the formation of the pore structure upon deposition of Zn onto a pre‐adsorbed ZnTCNPP layer directly after deposition, and also not after annealing to temperatures up to 500 K. Considering the fact that the peripheries of 2HTCNPP and ZnTCNPP are identical, this observation further indicates that Zn‐adatoms are not coordinating the pore network, further supporting the idea of CN−Ag−CN bonds.
Interestingly, after Zn deposition onto ZnTCNPP a different highly ordered pattern is very frequently observed. This phenomenon is also present for 2HTCNPP and 2HTCNPP+Zn (see SI, Figure S5), but is particularly pronounced for ZnTCNPP (Figure 3a–c). For ZnTCNPP+Zn, we observe an outstandingly pronounced long range order, which extends across substrate step edges. The long‐range order is characterized by extended parallel rows of molecules with identical orientation, which form a square lattice (see Figure 3a). Occasionally, the periodicity perpendicular to the rows is broken by single rows of molecules with different azimuthal orientation (see Figure 3a and enlarged image in Figure 3b), which represents a “line defect” in the long range ordered structure. The distance between the defects can vary (with e. g. 3 to 8 regular rows in between – see Figure 3a), or can also be periodic (with e. g. 3 regular rows in between – see Figure 3c). This break of symmetry is not attributed to a domain boundary, but rather to a line defect since the island orientation remains the same after the row of different orientation. The two islands left and right of the line defect row are shifted by half a molecule. Interestingly, this line defect (that is, the single row with molecules of different orientation), always starts at a kink of a Ag(111) step edge and extends over step edges like a carpet. In other words, the island grows in a regular fashion along straight step edges. Starting at kinks, one row of molecules is tilted against the molecules in the regular islands; the molecules in this row are aligned along one of the high symmetry axis of the Ag(111) substrate. Looking at the images in Figure 3a and 3c, we observe that the molecular order of the regular rows and also the defect rows extend over many step edges. For the defect rows, this requires (or indicates) a kink at each step edge, which means that the kinks have the same regularity as the defect rows. Considering the fact that one expects no correlation of the kinks between neighboring step edges, one has to conclude that the porphyrin layer is able to modify the morphology of the step edges such that the kinks are aligned along the molecular defect rows. The fact that this type of molecular order is most pronounced after Zn deposition on ZnTCNPP (where no Zn is needed for the formation the SAT complex), could indicate that the deposited Zn preferentially adsorbs the step edges. At the step edge, Zn has a high mobility and thus can be arranged such that the molecular order of regular rows and defect rows is maintained over the step edges.
Figure 3.

a–c) High resolution STM images (Erlangen) of ZnTCNPP+Zn (Ua=−1000 mV, I=30 pA; Uc=−455 mV, I=30 pA; Uc=−441 mV, I=30 pA). d) Schematic drawing of the correlation between step edge shape and orientation of the rows (red arrow indicates differently orientated row).
Notably, the observed structure is quite similar to a highly ordered striped [2+1] structure of ZnTPP on Ag(100) observed by Rangan et al. [30] In this structure, one double row of 2 identically oriented molecules is separated by 1 row of molecules, which are azimuthally rotated relative to the molecules in the double rows. This specific structure also extends over step edges and is assigned to a metastable ordered phase which can be transformed to a quadratic phase of equally oriented molecules by heating to 500 K. Furthermore, a similar effect with alternating rows was seen for the supramolecular ordering of a chlorophenyl porphyrin on Ag(111). [31]
For the ZnTCNPP+Zn system studied here, the rotation of the molecules in the line defect row breaks the dipole‐dipole interactions of the cyano groups between neighboring molecules (see Figure 3d). This lost interaction is compensated by forming a N−H bond between a CN‐group and a H atom of the neighboring pyrrole group of the macrocycle. This shows how much the molecules prefer adsorption sites at the step edges and that the islands start to grow from there.
In a next step, the 2HTCNPP+Zn layer (island types A, B and C; Figure 2) was annealed to check whether the metastable SAT complexes can be metalated, as was the case for 2HTPP plus Zn.[ 13 , 24 ] In Figure 4, we compare high resolution STM images of directly deposited ZnTCNPP (a) and of 2HTCNPP+Zn after annealing to 500 K (b). The molecular appearances in both images are virtually identical, with the four bright protrusions resembling the cyanophenyl groups, and the Zn atom in the center of the macrocycle invisible. Also, the unit cell of the 500 K‐annealed 2HTCNPP+Zn layer (a=1.54±0.04 nm, b=1.46±0.04 nm, α=85°±5°) matches that of ZnTCNPP (a=1.51±0.04 nm, b=1.49±0.04 nm, α=85°±5°), within the margin of error. This similarity is taken as strong evidence that metalation is indeed possible by annealing and further corroborates our interpretation of the SAT complex as a metastable intermediate in the metalation reaction. The uniform appearance of the STM image after heating to 500 K indicates that the bright protrusions which were observed for all three coexisting island types after Zn deposition at RT all transform into the same quadratic structure (with some disordered regions) after metalation, which does not show any sign of a Ag coordinated network. Notably, after the metalation, the molecular rows, and thus the unit cell vectors of all porphyrins are rotated relative to the high symmetry directions of the substrate.
Figure 4.

a) High resolution STM images (Erlangen) of ZnTCNPP (Ua=−190 mV, I=30 pA), b) High resolution STM images (Erlangen) of 2HTCNPP+Zn+500 K (Ub=−1000 mV, I=22 pA), c) High resolution STM images (Erlangen) of 2HTCNPP+Zn+pulse(+2.73 V) (Uc=−1660 mV, I=30 pA).
Most interestingly, temperature is not the only possible option to overcome the energy barrier to induce a conformational change or metalation reaction. During the course of our experiments, we observed a peculiar behavior of the SAT complex species after a tip pulse. After applying a positive voltage pulse of 1.5 to 4 V (referred to the sample), the molecular appearance of a small area within the island changed. Higher voltages could not be studied, since the contrast of the overall image changed or the tip drifted, and negative voltage pulses did not induce such a change. Since the appearance of the molecules around the switched species remains unchanged, we attribute our observation to a voltage‐induced change of the molecule and not a change in overall contrast (e. g. due to a tip change). To get further insights into this phenomenon, we measured “STM movies” of the same spot: In Figure 5, we show ten successive frames, each measured within 10 sec, which resembles a total of 100 s monitored during the movie (the full movie is provided as Movie M1 in the SI). In the middle of frame 3 a pulse (2.73 V) was applied, and the change of the structure was observed afterwards. This behavior was observed for the bright protrusions in all three island types (see Figure S6 and also the corresponding Movie M2 in the SI). Remarkably, the structural change did not occur instantaneous, but happened over time by switching of 4 to 7 molecules in between frames. Looking at the molecular appearance and comparing it to 2HTCNPP and ZnTCNPP, the four cyanophenyl groups are clearly observable and the bright protrusion of the SAT complex vanishes. While a bias‐induced transfer of the coordinated metal atom to the tip has been observed, e. g., for lead‐phthalocyanine molecules, [32] we rule out such a process here for two reasons: (i) it is hard to imagine that subsequent demetalation of neighboring molecules would occur on the timescale of seconds, and (ii) metalation can be also induced thermally (see above).
Figure 5.

Consecutive STM frames of an 2HTCNPP island (structure A) covered with Zn (Uc=−1660 mV, I=30 pA). During Frame 3, a+2.73 V tip pulse was executed, which leads to a change in molecular conformation and supramolecular ordering (compare frames 9 and 10). Complementary data for structures B and C are shown in Figure S6 in the SI.
One can envisage two possible voltage‐induced reactions pathways of the SAT complex. The first would be the back reaction to 2HTCNPP under the release of the Zn atom, and the second is the formation of ZnTCNPP under the release of hydrogen. Comparing the appearance of the switched molecules with 2HTCNPP, we do not observe the bright macrocycle characteristic for the free base species. Moreover, there is no evidence of released Zn atoms or fast diffusing species, or Zn diffusion of SAT complexes to neighboring 2HTCNPP. We thus conclude that the SAT complex reacts towards ZnTCNPP. The newly formed species arrange into a regular patterns of ∼20 molecules (e. g. 4 times 5) with a quadratic lattice (a=1.54±0.04 nm, b=1.46±0.04 nm, α=86°±5°), like that of ZnTCNPP or 2HTCNPP. The question now arises, why the switching occurs not only at the time of the pulse but continues afterwards on the time scale of seconds. One possibility is that the hydrogen released in the course of the reaction of the SAT complex to ZnTCNPP adsorbs as atomic hydrogen on the surface, thereby modifying the local electronic properties such that the activation barrier for the metalation reaction of neighboring SAT complexes is lowered. The fact the metalation happens only with positive bias is difficult to address without calculation. As one possible explanation, we suggest that upon applying a positive voltage pulse, one populates some specific molecular states in the LUMO which induce deprotonation and as a consequence metalation. In the metalated porphyrin probably the central Zn atom interacts with the surface, and thus a reversible switching by applying negative voltage was also not observed. Wang et al. have studied the reversibility metalation/demetalation of the Sn atom in SnPc. [33] They have found that the injection of electrons in the LUMO+1 gives rise to a negatively charged and thus transiently reduced molecule. Upon leaving the molecule, the electron may deposit energy to vibrational degrees and thus initiate the molecular switching. However, for molecules with a stronger contact to metal surface rapid transfer of the attached electron may suppress switching, as is observed here.
3. Conclusions
The goal of this study was to investigate the metalation reaction of the tetracyanophenyl‐functionalized free base porphyrin 2HTCNPP with post‐deposited Zn atoms to the metalloporphyrin ZnTCNPP on a Ag(111) surface, and to identify possible reaction intermediates by scanning tunneling microscopy at RT. The study was performed using two different experimental setups, one in Erlangen/Germany and one in Campinas/Brazil, with both studies yielding identical results. After Zn deposition onto a preadsorbed 2HTCNPP layer at RT, we observe the formation of three different 2D ordered island types that coexist on the surface. For all three of them, we find a new species with a bright appearance, the amount of which increases with the amount of post‐deposited Zn. We attribute this species to metastable SAT (“sitting atop”) complexes. In this SAT complex, the metal ion coordinates with the macrocycle while the pyrrolic hydrogen atoms are still bound to the nitrogen atoms. This metastable SAT complex has been previously observed by Shubina et al. in the corresponding metalation reaction of the non‐cyano‐functionalized 2HTPP to ZnTPP, with a characteristic signature in XPS. [24] Upon heating to 500 K, the activation barrier for the subsequent reaction of the intermediate SAT complex to the metalated porphyrin is overcome, yielding ZnTCNPP and gaseous dihydrogen. Interestingly, the activation barrier for the successive reaction of the SAT complex to the metalated ZnTCNPP species can also be overcome by a voltage pulse applied to the STM tip.
One additional interesting observation is the mentioned formation of three characteristic adlayer structures after the deposition of Zn at RT. While two of the structures (A and B) can easily by interpreted as modifications of the initial structure of 2HTCNPP induced by the post‐deposited Zn atoms, yielding the SAT complex, the third structure (C) has a very characteristic appearance with regular unoccupied sites (holes). We propose this structure to be a coordination network, where three cyanophenyl groups coordinate to Ag adatoms from the substrate. Notably, the formation of this structure seems to require adsorbed SAT complexes, since it is not observed upon post‐deposition of Zn onto a ZnTCNPP. When performing this experiment, however, we find the formation of an extremely long‐range ordered quadratic phase, which extends over step‐edges. This phase contains line defects, which are related to kinks in the steps. The fact that the defect lines extend over the step edges is taken as indication that the porphyrin layer is able to modify the morphology of the step edges such that the kinks are aligned along the molecular defect rows.
Experimental Section
The study was performed using two different experimental setups, one in Erlangen/Germany and one in Campinas/Brazil, with both studies yielding identical results.
The experiments and sample preparations in Erlangen were performed in a two‐chamber ultrahigh vacuum (UHV) system at a background pressure in the low 10−10 mbar regime. The variable temperature scanning tunneling microscope (STM) is an RHK UHV VT STM 300 with RHK SPM 1000 electronics. All STM images were acquired at room temperature (RT) in constant current mode with a Pt/Ir tip and the bias was applied to the sample. The STM images were processed with WSxM software [34] and moderate filtering (Gaussian smoothing, background subtraction) was applied for noise reduction. The preparation of the clean Ag(111) surface was done by repeated cycles of Ar+ sputtering (600 eV) and annealing to 850 K. The 2HTCNPP molecules were deposited onto the metal substrates held at RT, by thermal sublimation from a home‐built Knudsen cell (nominal temperature: 430 °C).
In Campinas, the experiments were performed in a two interconnected UHV chambers (sample preparation and STM) running with a base pressure in the low 10−10 mbar range. The variable temperature STM is a SPECS Aarhus 150 STM operated with a SPECS SPC 260 controller. The STM measurements were performed in a constant current mode with a W tip cleaned in situ by Ar+ sputtering. STM bias voltages are applied to the sample. The images were analyzed using the WSxM and Gwyddion softwares.[ 34 , 35 ] The Ag(111) crystal was prepared by sequential Ar+ sputtering cycles at 600 V @ 5 μA cm−2 for 30 min followed by annealing at ∼790 K for 10 min. The 2HTCNPP molecules were sublimated using a home‐made Knudsen cell with a quartz crucible (nominal temperature: 350 °C) while the Ag(111) sample was held at room temperature (RT). Zn atoms were post‐deposited on 2HTCNPP/Ag(111) from ultrapure Zn shots (Alfa Aesar, 99.999 %) inserted in a well degassed Mo crucible in a FOCUS‐Omicron EFM3 e‐beam evaporator.
Conflict of interest
The authors declare no conflict of interest.
Supporting information
As a service to our authors and readers, this journal provides supporting information supplied by the authors. Such materials are peer reviewed and may be re‐organized for online delivery, but are not copy‐edited or typeset. Technical support issues arising from supporting information (other than missing files) should be addressed to the authors.
Supplementary
Supplementary
Supplementary
Acknowledgements
The authors gratefully acknowledge funding by the German Research Foundation (DFG) through Research Unit FOR 1878 (funCOS), the Collaborative Research Center SFB 953 (project number 182849149) at the Friedrich‐Alexander‐Universität Erlangen‐Nürnberg and São Paulo Research Foundation (FAPESP) through project numbers 2007/54829‐5 and 2007/08244‐5. M.L., J.K. and H.M are grateful for travel grants from DAAD. R.C.C.F. and A.S. are grateful for travel grants from CAPES (Brazil) under the Brazilian‐German collaborative project PROBRAL (1925/2016). Open access funding enabled and organized by Projekt DEAL.
J. Kuliga, R. C. de Campos Ferreirra, R. Adhikari, S. Massicot, M. Lepper, H. Hölzel, N. Jux, H. Marbach, A. de Siervo, H.-P. Steinrück, ChemPhysChem 2021, 22, 396.
Contributor Information
Prof. Dr. Abner de Siervo, Email: asiervo@ifi.unicamp.br.
Prof. Dr. Hans‐Peter Steinrück, Email: hans-peter.steinrueck@fau.de.
References
- 1. Barth J. V., Costantini G., Kern K., Nature 2005, 437, 671–679. [DOI] [PubMed] [Google Scholar]
- 2. Sosa-Vargas L., Kim E., Attias A.-J., Mater. Horiz. 2017, 4, 570–583. [Google Scholar]
- 3. Jung T., Schlittler R., Gimzewski J., Nature 1997, 386, 696. [Google Scholar]
- 4. De Feyter S., De Schryver F. C., Chem. Soc. Rev. 2003, 32, 139–150. [DOI] [PubMed] [Google Scholar]
- 5. Marbach H., Acc. Chem. Res. 2015, 48, 2649–2658. [DOI] [PubMed] [Google Scholar]
- 6. Gottfried J. M., Surf. Sci. Rep. 2015, 70, 259–379. [Google Scholar]
- 7. Wang L., Li H., Deng J., Cao D., Curr. Org. Chem. 2013, 17, 3078–3091. [Google Scholar]
- 8. Gutiérrez-Cerón C., Páez M. A., Zagal J. H., J. Solid State Electrochem. 2016, 20, 3199–3208. [Google Scholar]
- 9. Walter M. G., Rudine A. B., Wamser C. C., J. Porphyrins Phthalocyanines 2010, 14, 759–792. [Google Scholar]
- 10. Jurow M., Schuckman A. E., Batteas J. D., Drain C. M., Coord. Chem. Rev. 2010, 254, 2297–2310. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Gottfried J. M., Flechtner K., Kretschmann A., Lukasczyk T., Steinrück H.-P., J. Am. Chem. Soc. 2006, 128, 5644–5645. [DOI] [PubMed] [Google Scholar]
- 12. Buchner F., Schwald V., Comanici K., Steinruck H. P., Marbach H., ChemPhysChem 2007, 8, 241–243. [DOI] [PubMed] [Google Scholar]
- 13. Kretschmann A., Walz M. M., Flechtner K., Steinruck H. P., Gottfried J. M., Chem. Commun. (Camb.) 2007, 568–570. [DOI] [PubMed] [Google Scholar]
- 14. González-Moreno R., Sánchez-Sánchez C., Trelka M., Otero R., Cossaro A., Verdini A., Floreano L., Ruiz-Bermejo M., García-Lekue A., Martín-Gago J. Á., J. Phys. Chem. C 2011, 115, 6849–6854. [Google Scholar]
- 15. Doyle C. M., Krasnikov S. A., Sergeeva N. N., Preobrajenski A. B., Vinogradov N. A., Sergeeva Y. N., Senge M. O., Cafolla A. A., Chem. Commun. 2011, 47, 12134–12136. [DOI] [PubMed] [Google Scholar]
- 16. Diller K., Klappenberger F., Marschall M., Hermann K., Nefedov A., Wöll C., Barth J., J. Chem. Phys. 2012, 136, 014705. [DOI] [PubMed] [Google Scholar]
- 17. Goldoni A., Pignedoli C. A., Di Santo G., Castellarin-Cudia C., Magnano E., Bondino F., Verdini A., Passerone D., ACS Nano 2012, 6, 10800–10807. [DOI] [PubMed] [Google Scholar]
- 18. Fleischer E. B., Wang J. H., J. Am. Chem. Soc. 1960, 82, 3498–3502. [Google Scholar]
- 19. Hsiao Y.-W., Ryde U., Inorg. Chim. Acta 2006, 359, 1081–1092. [Google Scholar]
- 20. De Luca G., Romeo A., Scolaro L. M., Ricciardi G., Rosa A., Inorg. Chem. 2009, 48, 8493–8507. [DOI] [PubMed] [Google Scholar]
- 21. Bischoff F., Seufert K., Auwärter W., Seitsonen A. P., Heim D., Barth J. V., J. Phys. Chem. C 2018, 122, 5083–5092. [Google Scholar]
- 22. Röckert M., Franke M., Tariq Q., Steinrück H.-P., Lytken O., Chem. Phys. Lett. 2015, 635, 60–62. [Google Scholar]
- 23. Mielke J., Hanke F., Peters M. V., Hecht S., Persson M., Grill L., J. Am. Chem. Soc. 2015, 137, 1844–1849. [DOI] [PubMed] [Google Scholar]
- 24. Shubina T. E., Marbach H., Flechtner K., Kretschmann A., Jux N., Buchner F., Steinrück H.-P., Clark T., Gottfried J. M., J. Am. Chem. Soc. 2007, 129, 9476–9483. [DOI] [PubMed] [Google Scholar]
- 25. Inami E., Yamaguchi M., Nemoto R., Yorimitsu H., Krüger P., Yamada T. K., J. Phys. Chem. C 2020, 124, 3621–3631. [Google Scholar]
- 26. Lepper M., Köbl J., Zhang L., Meusel M., Holzel H., Lungerich D., Jux N., de Siervo A., Meyer B., Steinruck H. P., Marbach H., Angew. Chem. Int. Ed. 2018, 57, 10074–10079; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2018, 130, 10230–10236. [Google Scholar]
- 27. Lepper M., Schmitt T., Gurrath M., Raschmann M., Zhang L., Stark M., Hölzel H., Jux N., Meyer B., Schneider M. A., Steinrück H.-P., Marbach H., J. Phys. Chem. C 2017, 121, 26361–26371. [Google Scholar]
- 28. Gottardi S., Müller K., Moreno-López J. C., Yildirim H., Meinhardt U., Kivala M., Kara A., Stöhr M., Adv. Mater. Interfaces 2014, 1, 1300025. [Google Scholar]
- 29. Hansen M., Anderko K., in ‘Constitution of binary alloys’, McGraw-Hill, 1965. [Google Scholar]
- 30. Rangan S., Kim P., Ruggieri C., Whitelam S., Bartynski R. A., Phys. Rev. B 2019, 100. [Google Scholar]
- 31. de Campos Ferreira R. C., Perez Paz A., Mowbray D. J., Roulet J.-Y., Landers R., de Siervo A., J. Phys. Chem. C 2020. [Google Scholar]
- 32. Sperl A., Kröger J., Berndt R., J. Am. Chem. Soc. 2011, 133, 11007–11009. [DOI] [PubMed] [Google Scholar]
- 33. Wang Y., Kröger J., Berndt R., Hofer W. A., J. Am. Chem. Soc. 2009, 131, 3639–3643. [DOI] [PubMed] [Google Scholar]
- 34. Horcas I., Fernández R., Gomez-Rodriguez J., Colchero J., Gómez-Herrero J., Baro A., Rev. Sci. Instrum. 2007, 78, 013705. [DOI] [PubMed] [Google Scholar]
- 35. Nečas D., Klapetek P., Open Phys. 2012, 10, 181–188. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
As a service to our authors and readers, this journal provides supporting information supplied by the authors. Such materials are peer reviewed and may be re‐organized for online delivery, but are not copy‐edited or typeset. Technical support issues arising from supporting information (other than missing files) should be addressed to the authors.
Supplementary
Supplementary
Supplementary