
FIGURE 2.
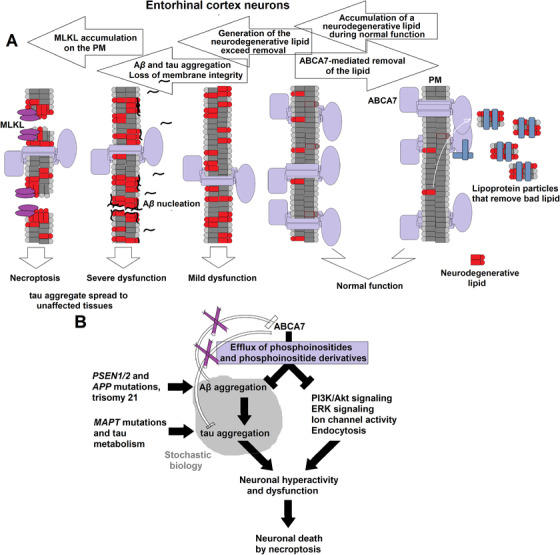
The altered lipidostasis hypothesis of AD. A, A proposed sequence of events leading from an imbalance in the generation and disposal of a neurodegenerative lipid to death of the specifically vulnerable neurons of the entorhinal cortex. Neurons of the entorhinal cortex accumulate a neurodegenerative lipid during normal functioning, and ABCA7 removes this lipid from the cell by exporting it into the extracellular milieu. When the rate of ABCA7‐mediated removal is lower than the rate of generation, concentration of the neurodegenerative lipid in neurons increases. Cell membranes enriched for the lipid catalyze Aβ aggregation. Soluble Aβ aggregates cause wide‐spread dysfunction, induce tau aggregation, and damage cell membranes. The neurodegenerative lipid further directly acts on cell membranes and disrupts normal functions. Activation of mixed lineage kinase domain‐like (MLKL) protein leads to death of the vulnerable neurons thorough necroptosis, while aggregated tau diffuses to healthy neurons to initiate a secondary pathogenic process. B, The proposed ABCA7 functions that forestall AD pathogenesis. ABCA7 suppresses Aβ aggregation and regulates levels of phosphatidylinositol and its derivatives. Purple X's indicate that ABCA7 does not act directly on tau and that Aβ aggregates do not affect ABCA7. Mutations inPSEN1/2andAPPorAPPduplication in trisomy 21 increase Aβ concentration and reduce the need for cell membranes to catalyze Aβ aggregation, thus obviating ABCA7 neuroprotection.MAPTmutations and changes in tau metabolism likewise cause tau dysfunction that then leads to development of tauopathies