

Abstract
P-glycoprotein (Pgp) is a member of the ABC transporter superfamily with high physiological importance. Pgp nucleotide binding domains (NBDs) drive the transport cycle through ATP binding and hydrolysis. We use molecular dynamics simulations to investigate the ATP hydrolysis-induced conformational changes of NBDs. Five systems, including all possible ATP/ADP combinations in the NBDs and the APO system are simulated. ATP/ADP exchange induces NBDs conformational changes mostly within the conserved signature motif, resulting in relative orientational changes of the NBDs. Nucleotide removal leads to additional orientational changes in the NBDs, allowing their dissociation. Furthermore, we capture putative hydrolysis-competent configurations where the conserved glutamate in the Walker-B motif acts as a catalytic base capturing a water molecule likely initiating ATP hydrolysis.
Keywords: ABC transporters, P-glycoprotein, ATP hydrolysis, conformational changes, molecular dynamics
Introduction
ATP-binding cassette (ABC) transporters are a superfamily of integral membrane proteins that harness the energy of ATP to actively transport diverse substrates across the cellular membrane [1]. This superfamily exists in all three kingdoms of life and includes both exporters and importers [2, 3, 4]. ABC importers provide essential nutrients and cofactors, whereas ABC exporters expel diverse xenobiotics, toxins, lipids, hormones, peptides, and proteases [3]. The lack of substrate selectivity in ABC exporters can be attributed to the poor sequence similarities in their membrane-spanning subunits [4].
The simplest model of transport in ABC transporters suggests alternating between two different conformations, namely, an inward-facing (IF) and an outward-facing (OF) conformational state, representing states in which the substrate binding site is accessible from the intracellular and extracellular sides, respectively [1, 5]. The structure of ABC transporters consists of two transmembrane domains (TMDs) that provide the necessary binding site and passageway for the transported substrate, and two cytoplasmic nucleotide-binding domains (NBDs) [6]. Two ATP molecules bind at the interface of the two NBDs through multiple electrostatic interactions and induce the formation of a closed NBD dimer [7]. Many studies have shown the importance of the ATPase activity in ABC transporters, suggesting that NBD dimerization, ATP hydrolysis, and substrate transport are intimately linked [7, 8, 9, 10, 11, 12, 13].
The NBDs are highly conserved in ABC transporters [14] and contain a number of functional loops and motifs directly involved in and important for ATP binding and hydrolysis [15, 16, 17]. In particular, the Walker-A motif (also known as P-loop), the Walker-B motif, the ABC signature motif, A-loop (an aromatic residue that interacts with the adenine base of the bound nucleotide), a sequence containing a conserved glutamine (Q-loop), X-loop, H-loop, and D-loop of the NBDs are involved in binding to the nucleotide, capturing the catalytic water, mediating ATP hydrolysis, and subsequently powering the transport cycle [1, 16, 18, 19].
ABCA1, or P-glycoprotein (Pgp), is a member of the ABC transporter superfamily with significant physiological and clinical importance [20, 21]. Pgp hyperactivity can confer multidrug resistance (MDR) to cancer cells by pumping anti-cancer drugs to the extracellular side of the membrane, thereby preventing them from reaching therapeutic concentrations in the target cells [22, 20]. Several investigations have described possible mechanisms for substrate-stimulated ATPase activity of Pgp, suggesting two different coupling models associating ATP hydrolysis with substrate translocation. The first model suggests that ATP hydrolysis takes place after substrate translocation is completet, in order to convert Pgp from a collapsed post-translocation state to an open IF state [23, 24, 25]. The second model proposes a two-stage ATP-hydrolysis process, which powers isomerization of Pgp from an IF to an OF-like conformation [26]. Despite the differences, both models agree that ATP binding/hydrolysis and the subsequent conformational changes in the NBDs play a vital role in substrate translocation and the Pgp transport cycle [16, 27]. Nevertheless, mechanistic details of ATP hydrolysis and its induced NBD conformational changes at an atomic level remain elusive.
Molecular Dynamics (MD) simulation is a powerful technique to investigate structural dynamics of membrane-embedded proteins at high spatial and temporal resolutions [11, 28, 29, 30]. Previous MD studies on ABC transporters focusing on the impact of binding and hydrolysis of the nucleotide to the NBDs have addressed various functional and mechanistic features in this superfamily. For example, MD simulations on the NBD dimer of HlyB, an ABC exporter, revealed multiple conformational changes in the contact area between the two NBDs after ATP hydrolysis, indicating that the ATPase activity and NBD conformational changes are linked [31]. In another study, MD simulations of different nucleotide- (ATP or ADP)-bound configurations of Sav1866, another ABC exporter, in pre- and post-hydrolysis states, showed that the NBD conformational changes are triggered as a consequence of ATP hydrolysis. It has also been shown that major ATP-induced rearrangements are not restricted to only the NBDs, but can extend to the TMDs as well [32]. Simulations of ATP and ADP/Pi (inorganic phosphate) bound to the NBDs in MalK suggested that the closed form of the NBD dimer can only be maintained with two bound ATP molecules, and that the dimer opening is a direct effect of ATP hydrolysis rather than the dissociation of the hydrolysis products [33]. Along with classical MD, quantum mechanical (QM) calculations have been employed to study the ATP hydrolysis mechanism and details of the nucleophilic attack by the catalytic water in ABC transporters. A hybrid computational study combining quantum and molecular mechanics (QM/MM) investigated the underlying mechanism of ATP hydrolysis in BtuCD-F, an ABC importer, revealing that ATP hydrolysis takes place when a water molecule is close to the ATP γ-phosphate at the right distance and angle, initiating a nucleophilic attack at the ATP γ-phosphate [34].
In the present study, the NBDs local as well as global conformational changes due to changes in the nucleotide state are investigated using unbiased MD simulations of Pgp in multiple nucleotide-bound states. Our focus here is neither substrate transport nor the details of ATP hydrolysis mechanism in Pgp. Rather by placing the system in different bound states, we will study how the NBDs react to such chnages in terms of protein conformational changes, both locally within the nucleotide binding site/region, and globally as it pertains to the coupling of the NBDs to TMDs. A total of five different systems, including all possible combinations of ATP and ADP (ATP/ATP, ATP/ADP, ADP/ATP, and ADP/ADP) bound to the two NBDs along with the APO (nucleotide-free) systems are constructed in their membrane-bound forms and simulated. Key NBD functional motifs that are involved in and mediate nucleotide binding and ATP hydrolysis are explored and analyzed. Our results suggest that NBDs’ local conformational changes take place mostly within the signature motif followed by abolishing of nucleotide (ATP and ADP) interactions with other motifs and loops in the nucleotide binding site, whereas global conformational changes trigger NBDs dissociation through changes in the relative orientation of the NBDs. Additionally, the conserved glutamate present in the Walker-B motif (E556/E1201 in NBD1/NBD2), is observed to occasionally trap a putative catalytic water molecule, thereby, generating a hydrolysis-competent state optimal for ATP hydrolysis. Through the course of the simulations, multiple such hydrolysis-competent states were captured with the putative catalytic water present in either NBD, suggesting that in Pgp both NBDs are likelly capable of ATP hydrolysis. In general, our simulation results suggest that both global and local conformational changes in the NBDs are initiated as a result of ATP hydrolysis and nucleotide dissociation.
Materials and Methods
System preparation
The starting system consists of an ATP/Mg2+ bound Pgp embedded in a 1-palmitoyl-2-oleoylphosphatidylcholine (POPC)/cholesterol (CL) membrane (1:1 molar ratio, 289 k atoms). The initial configuration was built from a cryo-EM structure epresenting a pre-hydrolysis state of human Pgp (PDB ID: 6C0V [35]). This experimental structure was stabilized by mutations that prevent ATP hydrolysis, where the two glutamates at the ATP binding sites were replaced by two glutamines (E556Q/E1201Q mutations) [35]. These two mutations were reversed (Q to E) during the construction of the simulation systems. There is also a missing loop in the initial cryo-EM structure (residues 90–108), which was modeled, using MOE [36]. The protonation state of each titratable residue was determined using PROPKA [37, 38]. Due to the importance of NBDs hydration in ATP hydrolysis, these parts were solvated using Dowser [39]. Initial membrane configuration, as well as water solvation and 150 mM NaCl were generated using CHARMM-GUI [40, 41]. The generated system was initially equilibrated using the default CHARMM-GUI’s protocol. Specifically, membrane was minimized initially for 10,000 steps and pre-equilibrated for 2.5 ns while restraining the protein backbones and lipid head groups in order to relax lipid tails and protein side-chains. This step was followed by a 1-ns unrestrained simulation. The final structure was equilibrated for 100 ns and used as the starting configuration for constructing the five final systems (Figure 1A). The equilibrated structure was then subjected to multiple ATP to ADP transformations by removing the γ-phosphates in order to generate all possible combinations of ATP or ADP bound to the NBDs in Pgp (Figure 1A). Since lipid-protein interactions were not a point of attention in our study, all the systems were generated from the same equilibrated state with the same initial lipid configuration. In order to improve statistics and sampling, each system was simulated in three replicates independently with initial atom velocities randomly assigned from the Maxwell–Boltzmann distribution for the desired temperature [42].
FIGURE 1.
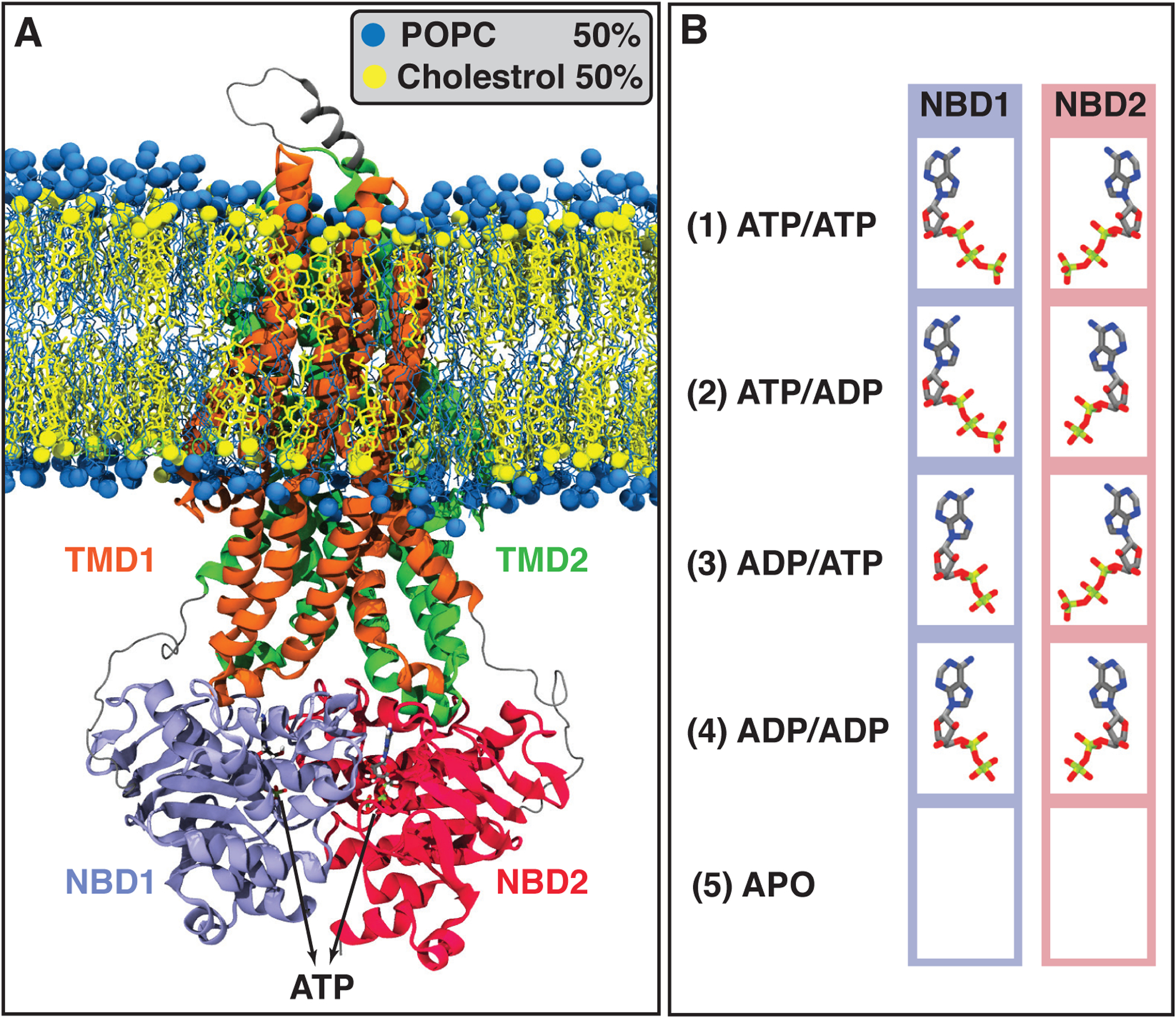
Initial systems configuration including all possible combinations of ATP or ADP bound to the NBDs, as well as the APO (nucleotide-free) system. (A) Pgp ATP-bound structure embedded in a POPC/Cholesterol lipid bilayer (molar ratio 1:1) after 100 ns of MD equilibration. POPC and cholesterol lipids are colored in blue and yellow, respectively. Pgp’s different domains, TMD1, TMD2, NBD1, and NBD2, are colored in orange, green, purple, and red, respectively. (B) The bound states simulated in this study, including all possible combinations of binding nucleotides (ATPs and ADPs), and the APO system which does not include any nucleotide. Each bound state was simulated in three independent simulations.
A total of five systems representing five different nucleotide-bound states of Pgp were simulated: (1) ATP/ATP with ATP bound to both NBD1 and NBD2, (2) ATP/ADP with ATP and ADP bound to NBD1 and NBD2, respectively, (3) ADP/ATP with ADP and ATP bound to NBD1 and NBD2, respectively, (4) ADP/ADP with ADP bound to both NBD1 and NBD2, and, (5) APO system with no nucleotide bound (Figure 1B). Each system was simulated in three replicates each for 200 ns, collectively providing 600 ns simulation time for each of the five bound states with a total of fifteen systems and a total simulation time of 3 μs.
MD simulation protocol
All MD simulations in this study were performed using NAMD2 [43, 44]. The fully atomistic CHARMM36 force field [45, 46] was used to calculate the forces in all the simulations. TIP3 model [47] was employed for water molecules. A 12-Å cutoff˙ was used for short-range, non-bonded interactions, with a switching distance starting at 10 Å. Long-range electrostatic interactions were calculated using the particle mesh Ewald (PME) method [48] with a grid density of 1 Å−3, and a PME interpolation order of 6. All bonds involving hydrogen atoms were kept rigid using the SHAKE algorithm [49]. Temperature was set to 310 K by using Langevin thermostat with a damping coeffcient of 1.0 ps−1. Pressure was maintained at 1 atm by a Nosé-Hoover Langevin piston barostat (period: 50 fs, decay: 25 fs) [50, 51]. All simulations were run in a flexible cell allowing the dimensions of the periodic cell to change independently while keeping the cell aspect ratio in the x-y plane constant. The simulation timestep was set to 2 fs. Lennard-Jones and PME forces were updated every timestep. Atomic coordinates were saved every 10 ps, providing a sufficiently detailed sampling of the trajectories.
Analysis
All MD trajectories were visualized with VMD [52] and analyzed using in-house scripts. Plots were generated using GNUplot [53]. In order to characterize the local conformational changes induced by ATP hydrolysis at the binding site, root mean square fluctuations (RMSF), as well as pairwise distances between different regions of the NBDs interacting with the nucleotides were calculated for all simulated systems. Data were collected from the last 100 ns of each simulation and averaged over all three replicates for each system.
The nucleotide binding region was defined as all amino acids within 3 Å of the nucleotides. The residues within this radius were divided into five main regions, namely, Walker-A motif part (I) (S429/S1072, G430/G1073, and C431/C1074 in NBD1/NBD2), Walker-A motif part (II) (G432/G1075, K433/K1076, S434/S1077, and T435/T1078 in NBD1/NBD2), part of the signature motif (Q530/Q1175, S532/S1177, G533/G1178, and G534/G1179 located in NBD1/NBD2 and interacting with the bound nucleotide in the opposing NBD), A-loop (Y401/Y1044 in NBD1/NBD2), and part of the Q-loop (Q475/Q1118 in NBD1/NBD2). It should be noted that the signature motif in each NBD interacts closely with the bound nucleotide in the opposing NBD and is therefore considered to be a critical part of the nucleotide binding site. As described, residues interacting with the nucleotides in the Walker-A motif are divided into two parts since the first part (Residues 429/1072 to 431/1074) interacts mostly with the ATP γ-phosphate, while the second part (Residues 432/1075 to 435/1078) interacts with the α- and β-phosphates.
The center of mass (COM) coordinate of all the atoms within each of the five regions is calculated and used as a single point to represent the respective region. The pairwise distances between each of these five regions are measured and employed to construct a 10 dimensional phase space. Given the high dimensionality of the generated phase space, we used time-lagged independent component analysis (TICA) with a lag time of 1 ns to reduce the dimensionality of the constructed phase space by mapping the data onto a 2D space [54], allowing comparison of the results from different simulations more easily.
NBDs’ global conformational changes are analyzed in a phase space consisting of orientational collective variables. In this calculation, the backbone Cα atoms within each NBD are selected and their relative orientational changes are monitored with respect to the initial structure through the course of simulations using quaternions (four dimensional vectors representing the orientation of an object in 3D) available in the collective variables (COLVARS) modules [55, 56] of NAMD and VMD. The quaternions are then converted to Euler angles, representing each NBD’s orientational changes. The differences between the first, second, and third Euler angles of the two NBDs, which are donted here as ϕ, θ, and ψ angles, corresponding to relative roll, pitch, and yaw motions of the NBDs, respectively, represent NBDs’ relative orientational changes with respect to the x, y, and z axes, respectively (Figure S1). Out of these, we used the first two Euler angles (ϕ and θ) to represent the global conformational changes of the NBDs. The third Euler angle, ψ, did not show any significant changes in any of the simulated systems and, therefore, was not used in the constructed phase space. All the angles were calculated in reference to the ATP bound (ATP/ATP) structure with this system placed at the origin of the phase space. The angles were calculated for each system (ATP/ATP, ATP/ADP, ADP/ATP, ADP/ADP, and APO) and averaged over the last 100 ns of each three simulations to obtain a single point in the phase space.
Results and Discussion
The simulation trajectories have been analyzed with regard to four major aspects: (1) local conformational changes in the nucleotide binding pocket due to ATP hydrolysis; (2) global conformational changes in the NBDs initiated by ATP hydrolysis, which are analyzed by monitoring NBDs relative orientations; (3) formation of putative hydrolysis-competent states of Pgp and detecting the catalytic base responsible for trapping a putative catalytic water; and, (4) observing multiple hydrolysis-competent states with putative catalytic water present in either NBDs.
Hydrolysis induced local conformational changes
The bound nucleotides in our simulations are stabilized due to their tight interactions with the nucleotide binding site motifs and loops. In particular, the adenine base of the nucleotide interacts with A-loop, whereas the phosphates form contacts with Walker-A and signature motifs (Figure 2A), as also reported in previous structural and MD studies on ABC transporters [16, 19, 33, 57]. RMSF values for the nucleotide binding site in NBD1 (Figure 2A), calculated using the starting structure as the reference and averaged over all three replicates, show stable nucleotide binding to the Walker-A motif in the case of ATP/ATP, ATP/ADP, ADP/ATP, and ADP/ADP systems (Figure 2B). In contrast, the stability of this region diminishes significantly in the APO system, indicating the importance of the nucleotide binding in stabilizing the Walker-A motif through several non-bonded interactions between this motif’s side-chains and the nucleotide phosphates, which was also reported in previous mutagenesis and protein modeling studies on ABC transporters [18]. ATP to ADP transformation does not seem to destabilize the Walker-A motif since this motif mostly interacts with the α- and β-phosphates of the bound nucleotide. This observation is consistent with a previous study on HlyB [58] emphasizing the importance of the NBD’s conserved motifs in ABC transporters for ATP binding, the ATPase activity, and substrate translocation. However, RMSF values corresponding to the signature motif residues increase significantly after ATP to ADP transformation (comparing the ADP/ATP and ADP/ADP systems to ATP/ATP and ATP/ADP systems, respectively), reflecting the importance of the ATP γ-phosphate in stabilizing the signature motif. Q1175 (part of the signature motif) was observed to be the most sensitive binding residue to ATP hydrolysis in the ADP/ATP, ADP/ADP, and APO systems. RMSF values for this residue can reach 3.5 Å for the APO and 1.5 Å for both the ADP/ADP and ADP/ATP systems. Signature motif RMSF values obtained for the ATP/ADP system are different in comparison to the ATP/ATP system, indicating that ATP hydrolysis in NBD2 and the consequent conformational changes in both NBDs may be due to cooperativity between the two NBDs. This cooperativity between the NBDs in Pgp was also reported in vanadate trapping studies on NBDs catalytic site and ATPase activity [59, 60]. Furthermore, based on the nucleotide binding site RMSFs calculated for NBD1 in all simulated systems, Y401 (A-loop) does not display a clear pattern of conformational changes, indicating that A-loop is not directly involved in ATP hydrolysis, likely due to its lack of direct interactions between with γ-phosphate, consistent with previous mutagenesis and modeling studies on ABC transporters [18]. Finally, Q1175 RMSF only changes in the case of the APO system, confirming that this residue does not have a substantial interaction with the ATP γ-phosphate. Therefore, only the complete dissociation of the nucleotide can disturb Q1175 and increase its RMSF, whereas ATP to ADP transformation shows no direct effect on this residue.
FIGURE 2.
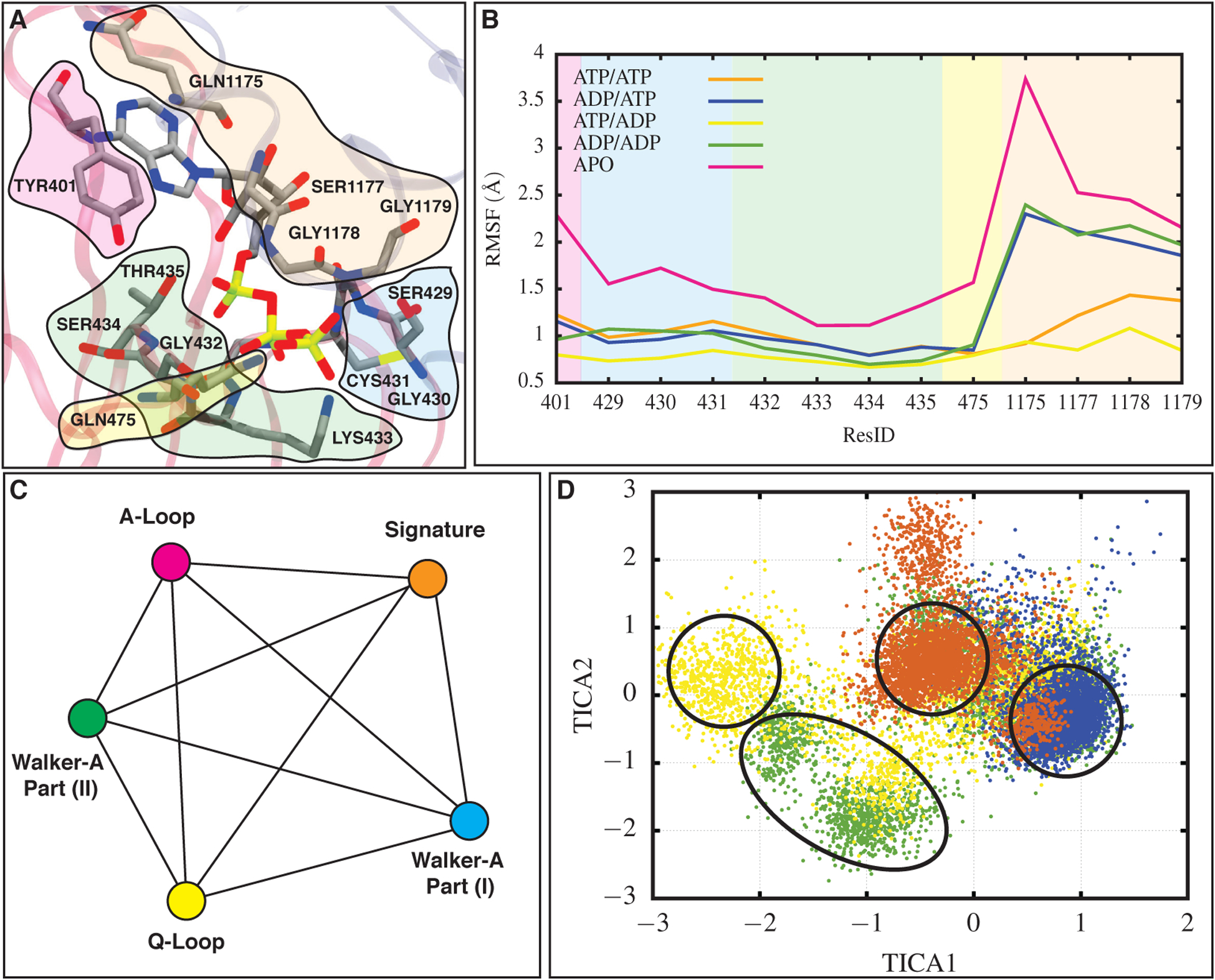
Nucleotide binding site conformational changes in NBD1 for all simulated systems induced by ATP hydrolysis. (A) Nucleotide binding site in NBD1 for one of the ATP/ATP systems. Residues belonging to the A-loop, Walker-A motif part (I), Walker-A motif part (II), the signature motif, and the Q-loop are colored in magenta, blue, green, orange, and yellow, respectively. (B) RMSF values for the residues in the five regions highlighted in panel (A), calculated in different bound states of Pgp and averaged over the last 100 ns of all three replicas for each system. Background colors correspond to different regions of the nucleotide binding site and are consistent with colors in (A). As reflected in its high RMSF, the interactions between the signature motif and ATP are affected the most after ATP hydrolysis. (C) A network used to calculate the pairwise distances between different nucleotide binding regions. Each line represents a distance and each node corresponds to a region within the binding site. Colors here are consistent with (A). (D) The reduced dimension distance phase space constructed for NBD1. Colors representing each bound state are consistent with lines in (B). Highest population of ATP/ATP, ATP/ADP, ADP/ATP, and ADP/ADP systems occupy different regions in the phase space indicating a different configuration of the nucleotide binding site in each bound state (see Figure S4 for 10 calculated pairwise distances in NBD1 used to construct the phase space).
The constructed distance phase space for the nucleotide binding site (Figure 2C and D) in NBD1 contains four main clusters, each representing a different bound state. We excluded the APO system from the phase space due to its substantial variation from the other systems which would excessively stretch the phase space, making the comparison between ATP- and ADP-bound systems difficult. The highest-population density of clusters containing the four nucleotide-bound systems fall into four separate islands with small overlap (Figure 2D). This separation in the phase space speaks to differences in nucleotide-NBD1 binding and interactions in these four different bound states. ATP to ADP transformation in NBD1 induces a new configuration in comparison to the ATP-bound structure, due to the γ-phosphate removal. The ADP/ADP cluster occupies a region with the highest population quite far from the ATP/ATP and ADP/ATP clusters, indicating a substantial difference in the nucleotide binding pocket in NBD1 due to two ATP hydrolysis reactions in the NBDs (Figure 2D). The ADP/ADP system shows some overlap with the ADP/ATP system as well since both systems contain ADP in their NBD1 and may sample the same NBD1 configuration through the course of the simulations. The ATP/ADP system is scattered in different areas of the phase space with a significant population two units away in TICA1 direction with respect to the ATP/ATP cluster (Figure 2D), which can be due to different interactions between the signature motif and A-loop with the nucleotide in NBD1 (Figure S2). Similar TICA2 values observed, on the other hand, may correspond to similar interactions between the Walker-A motif and Q-loop with the nucleotide (Figure S2). This suggests that ATP hydrolysis in NBD2 can induce conformational changes in the nucleotide binding pocket in NBD1, since the ATP/ADP system shows a different population in comparison to the ATP/ATP system. This observation provides additional evidence for potential cooperativity between the NBDs in Pgp. This behavior may be caused by abolishment of the non-bonded interactions between the signature motif and ATP γ-phosphate, since resdiues in this motif shows the highest RMSF values after each ATP to ADP transformation in comparison to other motifs and loops interacting with ATP.
Our results suggest a similar nucleotide binding pattern in NBD2. Similar to NBD1, the signature motif fluctuates the most after ATP conversion to ADP in NBD2 based on the nucleotide binding site RMSF (Figure S3), as well as COM distance calculations between different regions within the nucleotide binding site and the nucleotides (Figure S2). However, some differences in nucleotides binding of NBD1 and NBD2 are observed that are worth mentioning. RMSF values of the nucleotide binding site in NBD2 change significantly in the APO system with the largest deviation taking place in the Walker-A motif (Figure S3). Furthermore, pairwise distance calculations between different regions of the nucleotide binding site, as well as the signature and Walker-A motifs root-mean-square deviation (RMSD) show more fluctuations in NBD2 for the APO system, indicating a more flexible binding site after dissociation of the nucleotides in comparison to NBD1 (Figure S4, S5, and S6). Differences in hydrolysis-induced conformational changes in NBD1 and NBD2 are not surprising, in light of a previous infrared spectroscopy study also suggesting that the NBDs have an asymmetric structure and possibly different functions in the Pgp catalytic cycle [61].
Hydrolysis-induced global conformational changes
We monitored global conformational changes of the NBDs, induced by the change in the nucleotide state, by measuring their relative orientations as quantified by the ϕ and θ angles (see Methods), representing the roll and pitch motion of the NBDs, respectively. ATP conversion to ADP in one of the NBDs (either in NBD1 or NBD2, corresponding to the ADP/ATP and ATP/ADP systems, respectively) changes the θ angle by ~1.5° (Figure 3A, left panel). Hydrolysis of two ATPs (the ADP/ADP system) opens up the θ angle by more than 3°, almost twice as in the case of one ATP hydrolysis. Finally, transitioning from the ATP/ATP to the APO system is accompanied by a change of 6° in θ opening. These results indicate that each ATP hydrolysis is responsible for almost 1.5° opening of θ and removing the nucleotides (APO structure) can add up an additional 3–3.5° change in θ (Figure 3A, left panel). These changes in θ may consequently facilitate the dissociation of the NBDs (Figure S7), bringing Pgp to a post-hydrolysis state [11]. The relatively large orientational changes, as well as larger distances observed between the Walker-A and signature motifs in the APO structure compared to the ATP/ADP-bound systems (Figure S4, S5, and S8) suggest that NBDs dissociation may be initiated as a direct effect of nucleotide dissociation and not just ATP hydrolysis (Figure S7), in line with previously reported studies investigating conformational changes during the catalytic cycle of Pgp and ABC exporter MRP1 [62, 63].
FIGURE 3.
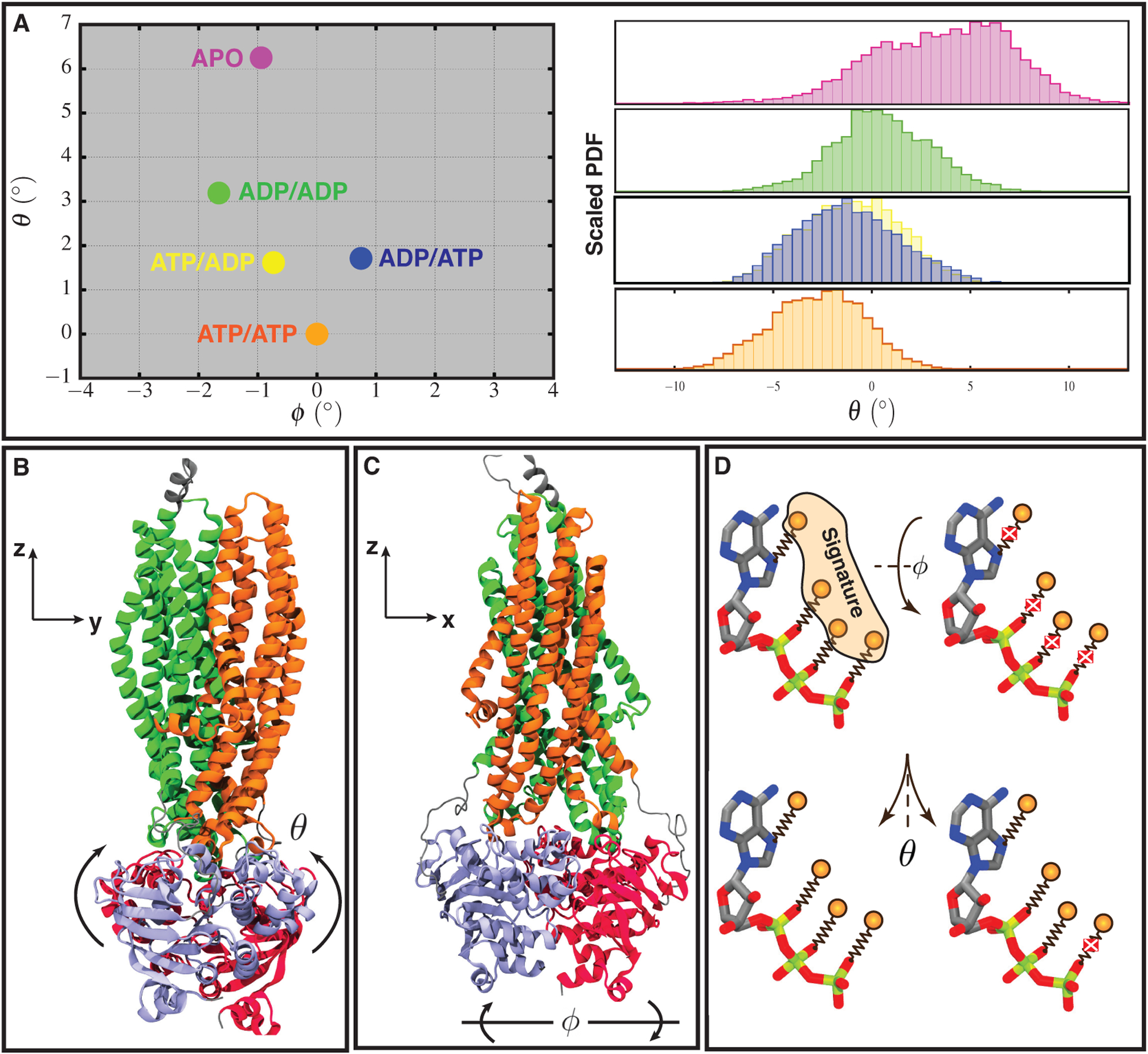
Global conformational changes in NBDs induced by ATP hydrolysis. (A) Left: the constructed phase space illustrating global conformation changes in NBDs with ATP/ATP, ATP/ADP, ADP/ATP, ADP/ADP, and APO systems shown in orange, yellow, blue, green, and magenta, respectively. Each angle is averaged over the last 100 ns of all three replicas. Right: histogram distributions of θ angles obtained from the last 100 ns of all three simulation replicas (see Figure S8 for ϕ distributions). The y axis is the scaled probability distribution function (PDF), with the highest probability within each distribution set to one. (B, C) Euler angles used to construct the phase space and their corresponding axes. Coloring in (B) and (C) are consistent with each other and they represent different domains of Pgp with TMD1, TMD2, NBD1, and NBD2 shown in orange, green, purple, and red, respectively. (D) Schematic representation of ATP binding to the signature motif and binding/unbinding direction. Springs represent non-bonded interactions between ATP oxygen atoms and side-chain of residues located in the signature motif, which are represented with orange spheres. Each cross represents dissociation of a non-bonded interaction. Changes in NBDs ϕ angle tend to break many non-bonded interactions between nucleotide and the signature motif at the same time, whereas NBDs opening in θ direction dissociates each interaction at a time making the NBDs dissociation more favorable in this direction.
Comparison of the θ angle histogram distributions (Figure 3A, right panel) of different bound states illustrates that in the ATP/ATP system the θ distribution is narrower than the other four systems. During progressive ATP hydrolysis as we move toward the APO system, the θ histograms become wider due to the decrease in the NBDs stability. This illustrates the role of ATPs in stabilizing the NBD dimerization and neutralizing the repulsion between the signature and Walker-A motifs due to their electrostatic positive charges, as also described in previous studies [18]. Histogram distributions of the ATP/ADP and ADP/ATP systems show a perfect overlap in the θ values, suggesting that each ATP hydrolysis plays a vital role in triggering the NBDs’ global conformational changes, as was also previously shown [62], and that ATP hydrolysis in each NBD has an equal contribution to θ opening.
We observed changes in the ϕ angle as well, however, θ exhibits a more consistent and significant change. The change in ϕ was observed to be less than 2° for the ADP/ADP system with respect to the ATP/ATP system. We identified a less than 1° change in ϕ after one ATP hydrolysis in NBDs, which takes place in opposite directions in the ATP/ADP and ADP/ATP systems (Figure 3A, left panel). Based on these results, it seems that the dissociation of the nucleotides (going from ADP/ADP to APO) closes back the ϕ angle by 1°, bringing the NBDs orientation closer to the initial ATP/ATP structure. This change may result from disruption of the ADPs-NBDs interactions and the higher flexibility of the motifs at the nucleotide binding sites in the APO system. Therefore, θ shows a monotonic trend in the constructed phase space, whereas ϕ changes in a non-monotonic fashion (Figure S8). The small and non-monotonic changes in the ϕ angle in comparison to θ are likely due to the tight binding of the nucleotides to the signature motif, making the θ direction/path more favorable on the constructed θ-ϕ phase space (Figure 3A, B, C, and S1). Furthermore, ATP to ADP transformation and subsequent nucleotide dissociation each abolishes part of the nucleotide-signature motif non-bonded interactions, resulting in a sequential opening of NBDs in the θ direction (Figure 3A and D).
Mg2+ coordination and capturing hydrolysis-competent states in both NBDs
The ATP γ-phosphate in our simulations is tightly coordinated with a Mg2+, also present in the initial cryo-EM structure. The Mg2+ position in the ATP-bound systems (ATP/ATP in both NBDs, ATP/ADP in NBD1, and ADP/ATP in NBD2) is highly conserved and remained stable in the course of the simulations, where the ion is coordinated with the Q-loop (Q475/Q1118 in NBD1/NBD2), a SER residue from the Walker-A motif (S434/S1077 in NBD1/NBD2), β- and γ-phosphates from ATP, and two water molecules (Figure 4A). In the ADP-bound systems (ADP/ADP in both NBDs, ATP/ADP in NBD2, and ADP/ATP in NBD1), we observe that Mg2+ is coordinated with another water molecule due to the lack of the γ-phosphate interactions (Figure 4B). The two water molecules coordinating Mg2+ in the ATP-bound structures remain bound in ADP-bound systems as well. This indicates that neither of these two water molecules is the catalytic water participating in ATP hydrolysis since they remain present even after the ATP to ADP conversion, as was also observed in a previous QM/MM study to investigate the molecular mechanism of ATP hydrolysis in ABC importer BtuCD-F [34]. The electrostatic interactions between Mg2+ and these two coordinated water molecules may prevent them from initiating a nucleophilic attack on the ATP γ-phosphate and consequently ATP hydrolysis. However, we observed in our ATP-bound simulations another water molecule approaching the nucleotide binding site, with a substantial occupancy, presenting a strong candidate for initiating the ATP hydrolysis. This water is present and coordinated with the catalytic glutamate and γ-phosphate of ATP, and may represent a putative catalytic water (Figure 4A).
FIGURE 4.
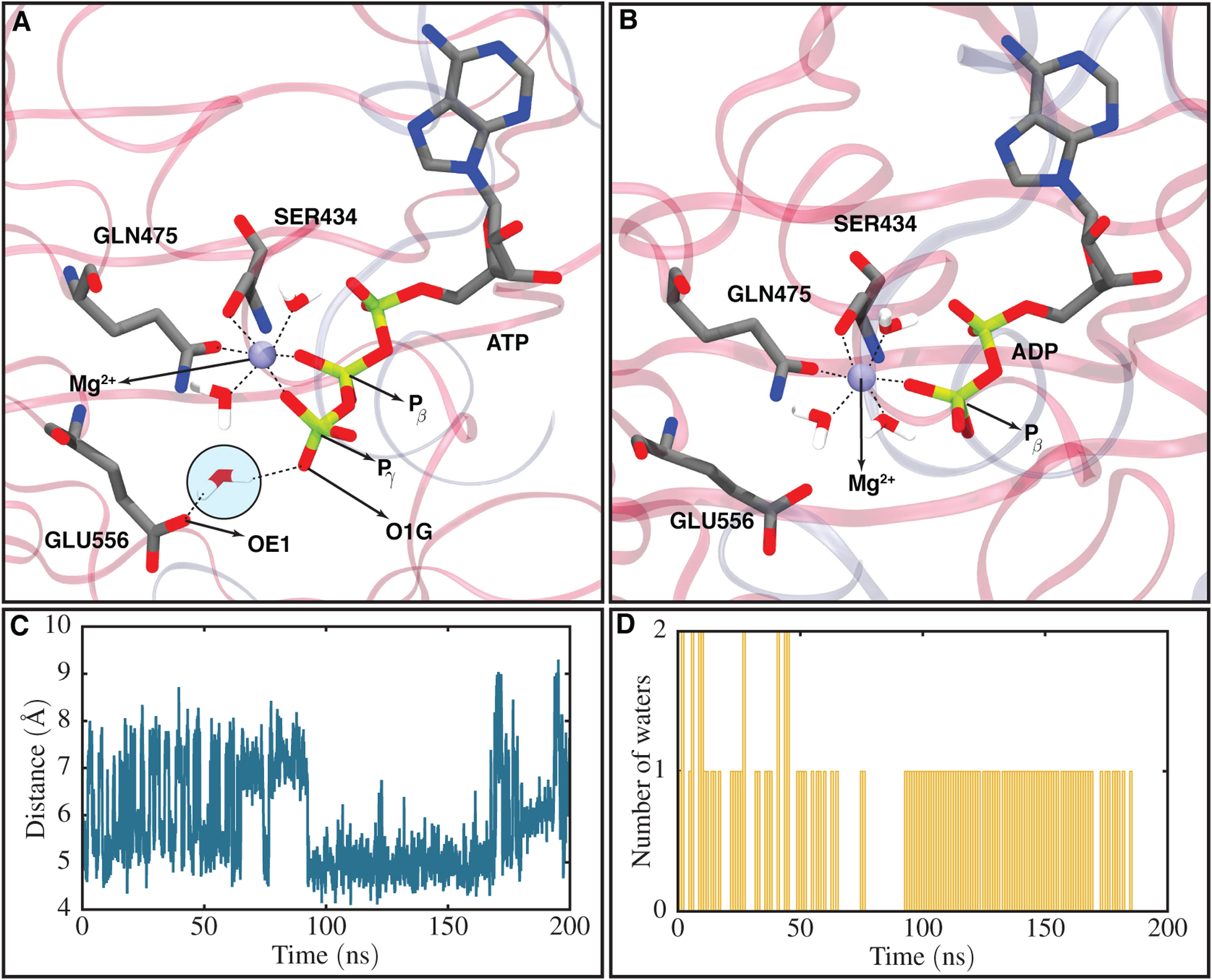
Mg2+ coordinations in ATP- and ADP-bound structures and a hydrolysis competent state captured for one of the ATP/ATP systems. (A) Mg2+ six coordinations with the β- and γ-phosphate of ATP, two water molecules, Q-loop, and SER434 (part of the Walker-A motif), as well as a captured putative catalytic water coordinating with one of the catalytic glutamate carboxy oxygens (OE1) and one of the ATP γ-phosphate oxygens (O1G). The candidate catalytic water is highlighted in a circle. (B) Mg2+ six coordinations in an ADP-bound system including interactions with β-phosphate of ADP, three water molecules, the Q-loop, and SER434. (C) Distance between the catalytic glutamate carboxy oxygen (OE1) and ATP γ-phosphate phosphorus (Pγ) atoms with respect to time in one of the ATP/ATP systems. The catalytic glutamate is found to be the catalytic residue capturing the putative catalytic water in Pgp. (D) Number of water molecules trapped in a region within 3 Å of both the catalytic glutamate and the γ-phosphate with respect to time for the same system presented in (C). The putative catalytic water can only be captured when the catalytic glutamate is within a close distance of the γ-phosphate (4.5 Å in this case).
We observed in our simulations that the putative catalytic water can be captured only if the catalytic glutamate is present at a close distance to the γ-phosphate. Distance calculation between one of the oxygens located in the catalytic glutamate side-chain carboxy group (named OE1 in the CHARMM force-field) and the γ-phosphate phosphorus (Pγ) atom in NBD1 for one of the ATP/ATP systems during the course of 200 ns of simulation shows that the catalytic glutamate has two energetically favorable states, one 4.5 Å and another 8.0 Å away from the phosphorus atom (Figure 4C). The simulations reveal that H-loop (H587 in NBD1) coordination with the catalytic glutamate plays a key role in stabilizing the catalytic glutamate in each state (Figure S9). The putative catalytic water can be trapped when the catalytic glutamate is close to the γ-phosphate (4.5 Å distance). In order to better describe the dependence of the putative catalytic water on specific arrangement of side chains in the binding site, we calculated the number of water molecules in NBD1 within 3 Å of the catalytic glutamate side-chain and γ-phosphate atoms for the same ATP/ATP system used for the above distance calculation during 200 ns of equilibrium simulation (Figure 4D). The results show that the distance between the catalytic glutamate and γ-phosphate and the number of tightly bound water molecules are highly correlated. Between 0 and 70 ns of the simulation, the distance between the catalytic glutamate and γ-phosphate fluctuates from 4.5 to 8 Å, making the catalytic glutamate and γ-phosphate unable to stably trap any water molecule. The water count in this time period fluctuates between 0, 1, and 2, with the water molecules remaining transient and only staying in the pocket for a very short time. This time period is followed by an approximately 20 ns of the glutamate being at 8 Å distance from the γ-phosphate (Figure 4C), causing no water trapping in the pocket. After this, we observe a time period between 95 and 165 ns, where the glutamate is present at a close distance to γ-phosphate (4.5 Å). In this time period a stable water in the pocket tightly binds to the two oxygen atoms from the glutamate and the γ-phosphate. This water in our simulation has the characteristics of a potential catalytic water, due to the high occupancy time (more than 70 ns) and being present at a close distance with respect to the γ-phosphate that is necessary for a nucleophilic attack. This suggestion is consistent with previous computational and experimental studies using QM/MM and mutagenesis of the catalytic glutamate, respectively [34, 64]. The proximity of this water to the glutamate carboxy group can polarize the water for the nucleophilic attack. We believe this state of Pgp captured in our simulations can be characterized as a hydrolysis-competent state due to the existence of a putative catalytic water that may enable ATP hydrolysis by the NBDs. Furthermore, the simulations suggest that the glutamate can be characterized as the catalytic base in Pgp, playing an important role in capturing the catalytic water, with mutation in this loop found to abolish Pgp ATPase activity, as observed in previous Pgp structural studies [35].
In the course of 600 ns simulations (three replicates each simulated for 200 ns) for each system, the putative catalytic water was captured in multiple occasions. These include: (1) the ATP/ATP system between 95 and 170 ns of the first replica in NBD1, (2) the ATP/ATP system in the second replica between 175 and 185 ns in NBD2, (3) the ATP/ADP system in the second replica within a time period between 55 and 125 ns in NBD1, and finally, (4) the ADP/ATP system in the third replica between 110 and 175 ns in NBD2 (Figure 5A, B, C, and D, respectively). The nature of all the captured hydrolysis-competent states are similar, meaning that the catalytic glutamate (E556/E1201 in NBD1/NBD2) is at a close distance with respect to ATP γ-phosphate (4.5 to 5.5 Å between the glutamate carboxy oxygen and the γ-phosphate phosphorus atom), subsequently capturing the putative catalytic water (Figure 5 left and middle panels). Therefore, based on our results, the putative catalytic water can be captured in both ATP-bound NBD1 or NBD2 (Figure 5 right panels), implying that both NBDs in Pgp are capable of ATP binding and hydrolysis, as was also suggested in previous studies [59].
FIGURE 5.
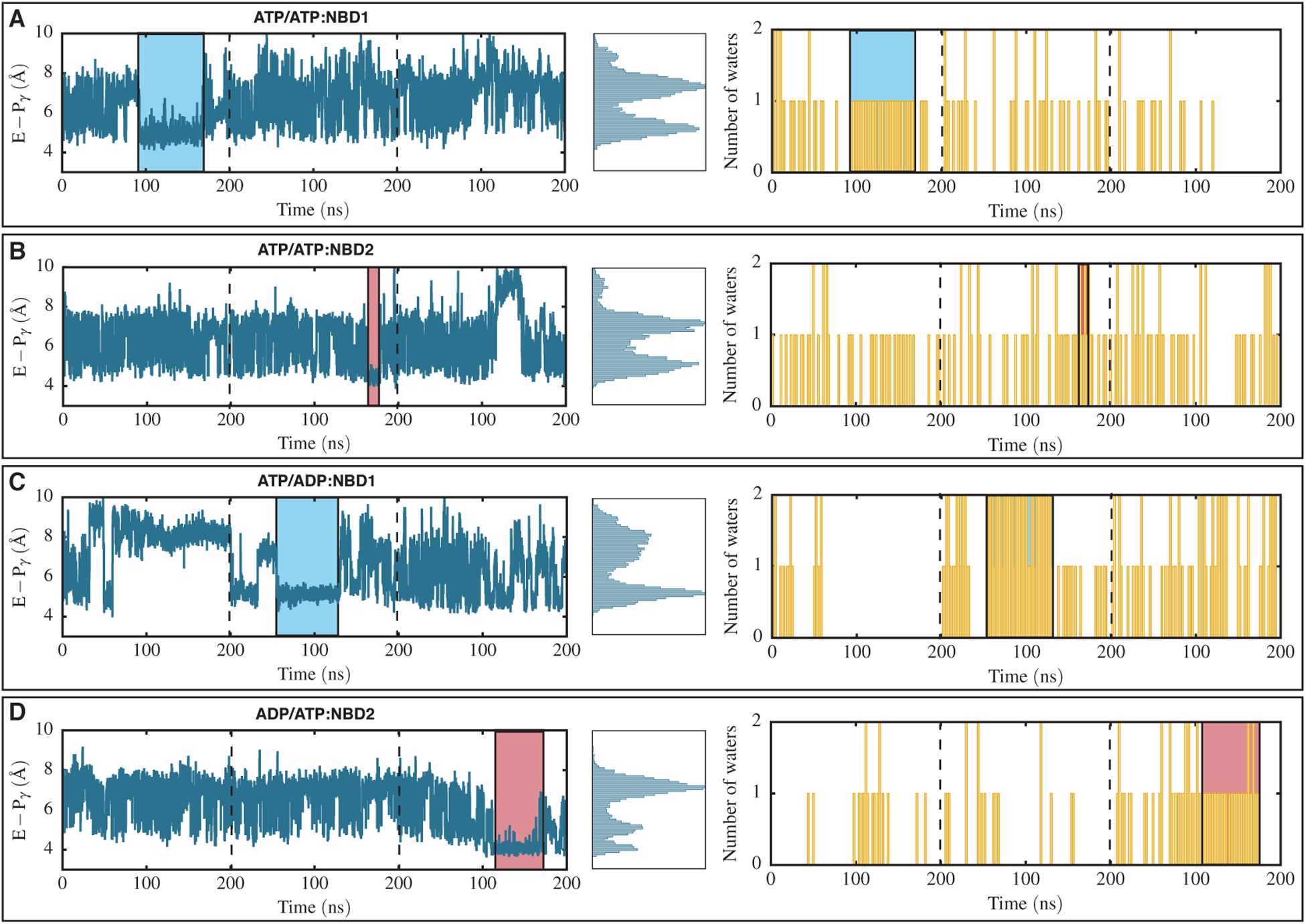
Hydrolysis-competent states captured in any of the simulated bound states. (A, B) NBD1 and NBD2 of the ATP/ATP system, respectively. (C, D) NBD1 of the ATP/ADP system and NBD2 of the ADP/ATP system, respectively. Left panels correspond to distances between the catalytic glutamate carboxy oxygen and the γ-phosphate phosphorus atoms with respect to time for all three simulation replicas separated by dashed lines. Middle panels represent the histogram distributions of distances calculated in the left panels. Right panels show the number of water molecules captured in a region within 3 Å of the catalytic glutamate and the γ-phosphate with respect to time for the same systems presented on the left. Blue and red boxes correspond to a captured putative catalytic water in NBD1 and NBD2, respectively. The putative catalytic water can be captured in both NBDs indicating that in Pgp both NBDs are catalytic active.
Even though the catalytic glutamate is capable of capturing the putative catalytic water in both the NBDs, the occupancy time of the catalytic water in the binding pocket vary when comparing NBD1 and NBD2. The occupancy time of the putative catalytic water in NBD1 and in the ATP/ATP and ATP/ADP systems are 75 and 70 ns, respectively (Figure 5 left panels), whereas, this time in NBD2 and in the ATP/ATP and ADP/ATP systems were measured to be 10 and 65 ns, respectively (Figure 5 left panels). During a total sampling of 1200 ns for the ATP/ATP and ATP/ADP systems (each simulated for 600 ns), the total occupancy times of the putative catalytic water in NBD1 and NBD2 are 145 ns and 75 ns, respectively (overall 220 ns). The occupancy time corresponds to 12 % and 6 % probability of trapping the putative catalytic water in NBD1 and NBD2, respectively, and 18 % probability of finding this water in either NBDs, which matches with the similar probability reported in a QM/MM study investigating molecular mechanism of ATP hydrolysis in the ABC transporter BtuCD-F [34]. One might speculate that NBD1 is more likely to hydrolyze the bound ATP, and that NBDs may in fact alternate in the catalysis, as has been suggested in earlier studies [59]. We note, however, that this observation may be biased by the limited simulation time nd much longer simulations are needed to make firmer conclusions.
Concluding Remarks
We have performed MD simulations on Pgp in different nucleotide-bound states of the two NBDs, including all possible ATP/ADP binding combinations and the APO system, to study the coupling between ATP hydrolysis and local and global conformational changes in NBDs. Each system was replicated three times and run for 200 ns. Within the timescale of the simulations, we find that local conformational changes in NBDs induced by the change in the nucleotide state are initiated mostly from the signature motif. Residues in this motif show the highest RMSF values in comparison to other loops and motifs at the nucleotide binding site after ATP hydrolysis in either NBDs. While the dissociation of the hydrolyzed nucleotide shows differential effects on the two NBDs, they both become more flexible in response, resulting in their facilitated dissociation.
During ATP hydrolysis and nucleotide dissociation, NBDs also undergo global conformational changes, which are characterized in our study using two orientational collective variables, Euler angles θ and ϕ, representing relative pitch and roll motions of the NBDs, respectively. Our results suggest that may be a more favorable direction for the global conformational changes in NBDs. The ATP to ADP transformation and the nucleotide dissociation steps each abolishes part of the nucleotide-signature non-bonded interactions, resulting in a sequential opening of the NBDs along the θ direction.
In the ATP-bound sites, we observe that the conserved glutamate of the Walker-B motif (E556/E1201 in NBD1/NBD2) may function as a catalytic base in Pgp, trapping a putative catalytic water in an optimal position for the nucleophilic attack on the ATP γ-phosphate. Multiple hydrolysis-competent configurations are captured in both NBDs during the course of 3 μs of MD simulations. The occupancy time of the putative catalytic waters in these states varies between 10 to 75 ns. These results further suggest that both NBDs in Pgp are catalytic, although the putative catalytic water in NBD1 shows a slightly longer occupancy time.
Altogether, the local and global conformational changes of NBDs induced by ATP conversion to ADP suggest that in Pgp, ATP hydrolysis can trigger a series of orientational changes in the NBDs, which start mainly form the signature motif of each NBD (Figure 6), and that the nucleotide dissociation step (formation of the APO state) further promotes NBDs dissociation. As a part of this catalytic cycle, ATP hydrolysis and the nucleotide dissociation generate different binding states of the transporter, accompanied with various degrees of local, as well as global conformational changes in the NBDs (Figure 6).
FIGURE 6.
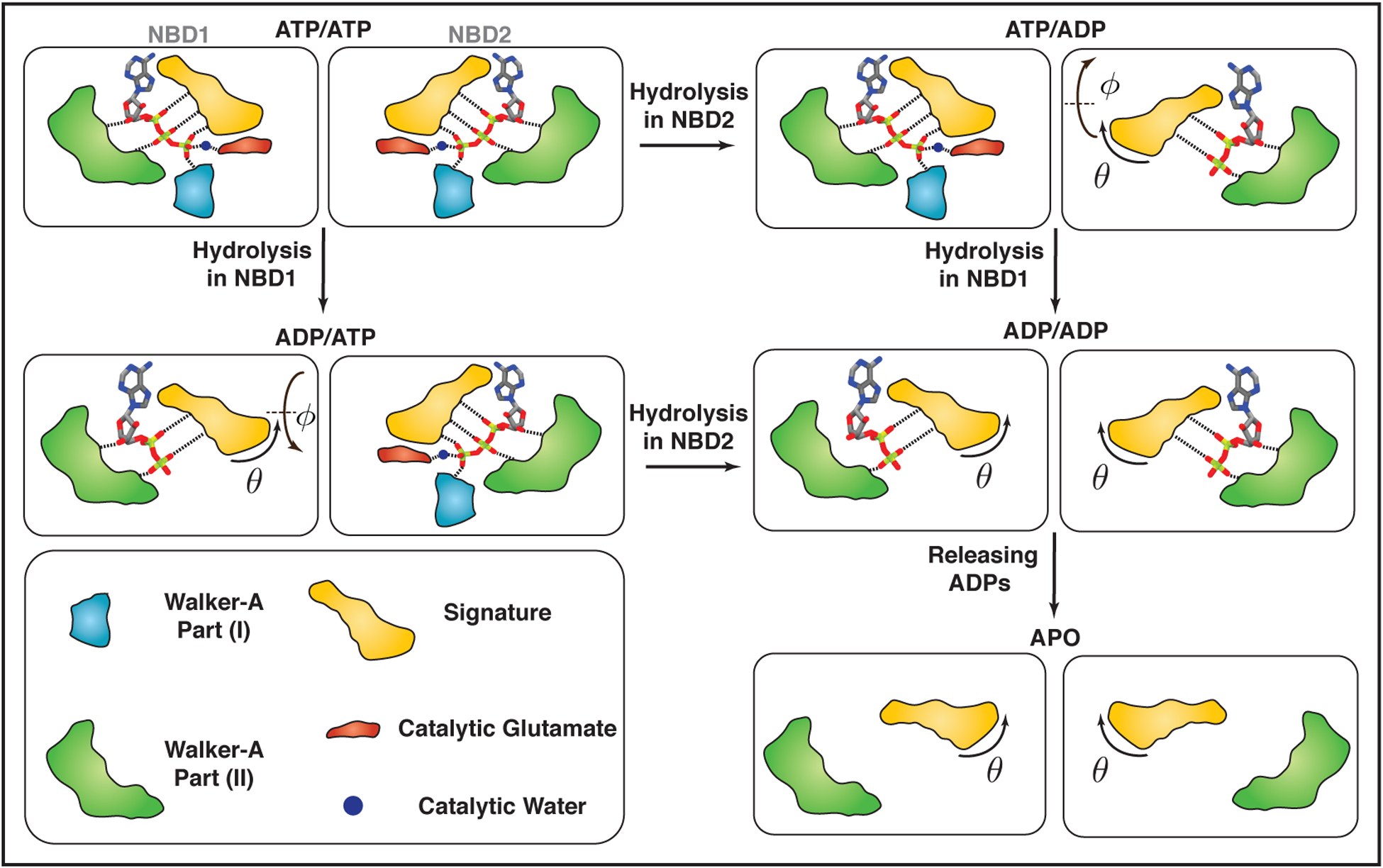
Schematic representation of coupling between NBDs conformational changes and ATPase activity in Pgp. Different regions of NBDs interacting with the nucleotides are colored differently with Walker-A motif part (I), Walker-A motif part (II), the signature motif, the catalytic glutamate, and putative catalytic water shown in light blue, green, orange, red and dark blue, respectively. Each dashed line represents a tight non-bonded interaction. ADP-bound NBDs lack γ-phosphate and therefore motifs interacting with the γ-phosphate are removed from those NBDs. Local conformational changes mostly affect the signature motif. Global conformational changes mainly take place in the θ direction in a monotonic fashion, meaning, both ATP hydrolysis and dissociation of the nucleotides open up θ by a certain value and play a role in NBDs global conformational changes. The change in θ mostly comes from the signature motif and its rotation. Unlike θ, ϕ changes in a non-monotonic fashion upon ATP hydrolysis. Furthermore, both NBDs are catalytic active, i.e., capture of catalytic waters and subsequent ATP hydrolysis are feasible in both NBDs.
Supplementary Material
Acknowledgments
The authors acknowledge support by the National Institutes of Health under award numbers P41-GM104601 (to E.T.) and R01-GM123455 (to E.T.). We also acknowledge computing resources provided by Blue Waters at National Center for Supercomputing Applications (NCSA), Extreme Science and Engineering Discovery Environment (XSEDE grant MCA06N060 to E.T.), and Microsoft Azure (grant TG-MCB200137 to E.T.).
Footnotes
Supporting information and trajectories
More information can be found in the online version of the article: LINK. All the MD trajectories are deposited in line with FAIR data principles and can be found here.
references
- 1.Dawson RJ, Locher KP. Structure of a bacterial multidrug ABC transporter. Nature 2006;443(7108):180–185. [DOI] [PubMed] [Google Scholar]
- 2.Oldham ML, Davidson AL, Chen J. Structural insights into ABC transporter mechanism. Curr Opin Struct Biol 2008;18(6):726–733. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Jones PM, George AM. Mechanism of the ABC transporter ATPase domains: catalytic models and the biochemical and biophysical record. Crit Rev Biochem Mol Biol 2013;48(1):39–50. [DOI] [PubMed] [Google Scholar]
- 4.Locher KP, Lee AT, Rees DC. The E. coli BtuCD structure: A framework for ABC transporter architecture and mechanism. Science 2002;296(5570):1091–1098. [DOI] [PubMed] [Google Scholar]
- 5.Hohl M, Briand C, Grütter MG, Seeger MA. Crystal structure of a heterodimeric ABC transporter in its inward-facing conformation. Nat Struct Mol Biol 2012;19(4):395–402. [DOI] [PubMed] [Google Scholar]
- 6.Hollenstein K, Dawson RJ, Locher KP. Structure and mechanism of ABC transporter proteins. Curr Opin Struct Biol 2007;17(4):412–418. [DOI] [PubMed] [Google Scholar]
- 7.Sauna ZE, Ambudkar SV. About a switch: how P-glycoprotein (ABCB1) harnesses the energy of ATP binding and hydrolysis to do mechanical work. Mol Cancer Ther 2007;6(1):13–23. [DOI] [PubMed] [Google Scholar]
- 8.Sauna ZE, Ambudkar SV. Evidence for a requirement for ATP hydrolysis at two distinct steps during a single turnover of the catalytic cycle of human P-glycoprotein. Proc Natl Acad Sci USA 2000;97(6):2515–2520. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Al-Shawi MK, Polar MK, Omote H, Figler RA. Transition state analysis of the coupling of drug transport to ATP hydrolysis by P-glycoprotein. J Biol Chem 2003;278(52):52629–52640. [DOI] [PubMed] [Google Scholar]
- 10.Senior AE, Al-Shawi MK, Urbatsch IL. ATP hydrolysis by multidrug-resistance protein from Chinese hamster ovary cells. J Bioenerg Biomembr 1995;27(1):31–36. [DOI] [PubMed] [Google Scholar]
- 11.Moradi M, Tajkhorshid E. Mechanistic picture for conformational transition of a membrane transporter at atomic resolution. Proc Natl Acad Sci USA 2013;110(47):18916–18921. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Kerr KM, Sauna ZE, Ambudkar SV. Correlation between steady-state ATP hydrolysis and vanadate-induced ADP trapping in Human P-glycoprotein. Evidence for ADP release as the rate-limiting step in the catalytic cycle and its modulation by substrates. J Biol Chem 2001;276(12):8657–8664. [DOI] [PubMed] [Google Scholar]
- 13.Wise JG. Catalytic transitions in the human MDR1 P-glycoprotein drug binding sites. Biochemistry 2012;51(25):5125–5141. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Dean M, Hamon Y, Chimini G. The human ATP-binding cassette (ABC) transporter superfamily. J Lipid Res 2001;42(7):1007–1017. [PubMed] [Google Scholar]
- 15.Hollenstein K, Frei DC, Locher KP. Structure of an ABC transporter in complex with its binding protein. Nature 2007;446(7132):213–216. [DOI] [PubMed] [Google Scholar]
- 16.Szöllősi D, Rose-Sperling D, Hellmich UA, Stockner T. Comparison of mechanistic transport cycle models of ABC exporters. Biochim Biophys Acta 2018;1860(4):818–832. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Ledwitch KV, Gibbs ME, Barnes RW, Roberts AG. Cooperativity between verapamil and ATP bound to the efflux transporter P-glycoprotein. Biochem Pharmacol 2016;118:96–108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Ambudkar SV, Kim IW, Xia D, Sauna ZE. The A-loop, a novel conserved aromatic acid subdomain upstream of the Walker A motif in ABC transporters, is critical for ATP binding. FEBS Lett 2006;580(4):1049–1055. [DOI] [PubMed] [Google Scholar]
- 19.Jones PM, George AM. The ABC transporter structure and mechanism: perspectives on recent research. Cell Mol Life Sci 2004;61:682–699. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Gottesman MM, Pastan I, Ambudkar SV. P-glycoprotein and multidrug resistance. Curr Opin Genet Dev 1996;6(5):610–617. [DOI] [PubMed] [Google Scholar]
- 21.Lee CA, Cook JA, Reyner EL, Smith DA. P-glycoprotein related drug interactions: clinical importance and a consideration of disease states. Expert Opin Drug Metab Toxicol 2010;6(5):603–619. [DOI] [PubMed] [Google Scholar]
- 22.Ambudkar SV, Kimchi-Sarfaty C, Sauna ZE, Gottesman MM. P-glycoprotein: from genomics to mechanism. Oncogene 2003;22:7468–7485. [DOI] [PubMed] [Google Scholar]
- 23.Alam A, Kowal J, Broude E, Roninson I, Locher KP. Structural insight into substrate and inhibitor discrimination by human P-glycoprotein. Science 2019;363(6428):753–756. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Higgins CF, Linton KJ. The ATP switch model for ABC transporters. Nat Struct Mol Biol 2004;11(10):918–926. [DOI] [PubMed] [Google Scholar]
- 25.Janas E, Hofacker M, Chen M, Gompf S, van der Does C, Tampé R. The ATP hydrolysis cycle of the nucleotide-binding domain of the mitochondrial ATP-binding cassette transporter Mdl1p. J Biol Chem 2003;278(29):26862–26869. [DOI] [PubMed] [Google Scholar]
- 26.Verhalen B, Dastvan R, Thangapandian S, Peskova Y, Koteiche HA, Nakamoto RK, Tajkhorshid E, Mchaourab HS. Energy transduction and alternating access of the mammalian ABC transporter P-glycoprotein. Nature 2017;543(7647):738–741. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Zoghbi ME, Mok L, Swartz DJ, Singh A, Fendley GA, Urbatsch IL, Altenberg GA. Substrate-induced conformational changes in the nucleotide-binding domains of lipid bilayer–associated P-glycoprotein during ATP hydrolysis. J Biol Chem 2017;292(50):20412–20424. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Muller MP, Jiang T, Sun C, Lihan M, Pant S, Mahinthichaichan P, Trifan A, Tajkhorshid E. Characterization of Lipid-Protein Interactions and Lipid-mediated Modulation of Membrane Protein Function Through Molecular Simulations. Chem Rev 2019;119:6086–6161. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Jiang T, Wen PC, Trebesch N, Zhao Z, Pant S, Kapoor K, Shekhar M, Tajkhorshid E. Computational Dissection of Membrane Transport at a Microscopic Level. Trends Biochem Sci 2019;. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Ferreira RJ, Ferreira MJU, dos Santos DJVA. Insights on P-glycoprotein’s efflux mechanism obtained by molecular dynamics simulations. J Chem Theory Comput 2012;8:1853–1864. [DOI] [PubMed] [Google Scholar]
- 31.Damas JM, Oliveira ASF, Baptista AM, Soares CM. Structural consequences of ATP hydrolysis on the ABC transporter NBD dimer: molecular dynamics studies of HlyB. Prot Sci 2011;20(7):1220–1230. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Oliveira AS, Baptista AM, Soares CM. Conformational changes induced by ATP-hydrolysis in an ABC transporter: A molecular dynamics study of the SAV1866 exporter. Proteins: Struct, Func, Bioinf 2011;79(6):1977–1990. [DOI] [PubMed] [Google Scholar]
- 33.Wen PC, Tajkhorshid E. Dimer opening of the nucleotide binding domains of ABC transporters after ATP hydrolysis. Biophys J 2008;95(11):5100–5110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Prieß M, Göddeke H, Groenhof G, Schäfer LV. Molecular mechanism of ATP hydrolysis in an ABC transporter. ACS Cent Sci 2018;4(10):1334–1343. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Kim Y, Chen J. Molecular structure of human P-glycoprotein in the ATP-bound, outward-facing conformation. Science 2018;359(6378):915–919. [DOI] [PubMed] [Google Scholar]
- 36.Vilar S, Cozza G, Moro S. Medicinal chemistry and the molecular operating environment (MOE): application of QSAR and molecular docking to drug discovery. Curr Topics Med Chem 2008;8(18):1555–1572. [DOI] [PubMed] [Google Scholar]
- 37.Søndergaard CR, Olsson MHM, Rostkowski M, Jensen JH. Improved treatment for ligands and coupling effects in empirical calculation and rationalization of pKa values. J Chem Theory Comput 2011;7:2284–2295. [DOI] [PubMed] [Google Scholar]
- 38.Olsson MH, Søndergaard CR, Rostkowski M, Jensen JH. PROPKA3: consistent treatment of internal and surface residues in empirical pKa predictions. J Chem Theory Comput 2011;7:525–537. [DOI] [PubMed] [Google Scholar]
- 39.Gumbart J, Trabuco LG, Schreiner E, Villa E, Schulten K. Regulation of the protein-conducting channel by a bound ribosome. Structure 2009;17(11):1453–1464. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Jo S, Kim T, Iyer VG, Im W. CHARMM-GUI: a web-based graphical user interface for CHARMM. J Comput Chem 2008;29:1859–1865. [DOI] [PubMed] [Google Scholar]
- 41.Lee J, Cheng X, Swails JM, Yeom MS, Eastman PK, Lemkul JA, Wei S, Buckner J, Jeong JC, Qi Y, Jo S, Pande VS, Case DA, Brooks CL III, MacKerell AD Jr, Klauda JB, Im W. CHARMM-GUI input generator for NAMD, GROMACS, AMBER, OpenMM, and CHARMM/OpenMM simulations using the CHARMM36 additive force field. J Chem Theory Comput 2016;12:405–413. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Buchete NV, Hummer G. Peptide folding kinetics from replica exchange molecular dynamics. Phys Rev E 2008;77(3):030902. [DOI] [PubMed] [Google Scholar]
- 43.Phillips JC, Braun R, Wang W, Gumbart J, Tajkhorshid E, Villa E, Chipot C, Skeel RD, Kale L, Schulten K. Scalable molecular dynamics with NAMD. J Comput Chem 2005;26:1781–1802. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Phillips JC, Hardy DJ, Maia JDC, Stone JE, Ribeiro JV, Bernardi RC, Buch R, Fiorin G, Hénin J, Jiang W, McGreevy R, Melo MCR, Radak B, Skeel RD, Singharoy A, Wang Y, Roux B, Aksimentiev A, Luthey-Schulten Z, Kalé LV, Schulten K, Chipot C, Tajkhorshid E. Scalable molecular dynamics on CPU and GPU architectures with NAMD. J Chem Phys 2020;153:044130. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Hart K, Foloppe N, Baker CM, Denning EJ, Nilsson L, Mackerell AD Jr. Optimization of the CHARMM additive force field for DNA: improved treatment of the BI/BII conformational equilibrium. J Chem Theory Comput 2012;8:348–362. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Klauda JB, Venable RM, Freites JA, O’Connor JW, Tobias DJ, Mondragon-Ramirez C, Vorobyov I, MacKerell AD Jr, Pastor RW. Update of the CHARMM all-atom additive force field for lipids: validation on six lipid types. J Phys Chem B 2010;114(23):7830–7843. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Jorgensen WL, Chandrasekhar J, Maudura JD, Impey RW, Klein ML. Comparison of Simple Potential Functions for Simulating Liquid Water. J Chem Phys 1983;79:926–935. [Google Scholar]
- 48.Darden T, York D, Pedersen L. Particle mesh Ewald: an N·log(N) method for Ewald sums in large systems. J Chem Phys 1993;98:10089–10092. [Google Scholar]
- 49.Ryckaert JP, Ciccotti G, Berendsen HJC. Numerical integration of the Cartesian equations of motion of a system with constraints: molecular dynamics of n-Alkanes. J Comp Phys 1977;23:327–341. [Google Scholar]
- 50.Martyna GJ, Tobias DJ, Klein ML. Constant pressure molecular dynamics algorithms. J Chem Phys 1994;101:4177–4189. [Google Scholar]
- 51.Feller SE, Zhang Y, Pastor RW. Constant pressure molecular dynamics simulation: The Langevin piston method. J Chem Phys 1995;103:4613–4621. [Google Scholar]
- 52.Humphrey W, Dalke A, Schulten K. VMD: visual molecular dynamics. J Mol Graphics 1996;14:33–38. [DOI] [PubMed] [Google Scholar]
- 53.Williams T, Kelley C, et al. , Gnuplot 4.4: an interactive plotting program; 2010.
- 54.Scherer MK, Trendelkamp-Schroer B, Paul F, Pérez-Hernández G, Hoffmann M, Plattner N, Wehmeyer C, Prinz JH, Noé F. PyEMMA 2: a software package for estimation, validation, and analysis of Markov models. J Chem Theory Comput 2015;11:5525–5542. [DOI] [PubMed] [Google Scholar]
- 55.Fiorin G, Klein ML, Hénin J. Using collective variables to drive molecular dynamics simulations. Mol Phys 2013;111(22–23):3345–3362. [Google Scholar]
- 56.Coutsias EA, Seok C, Dill KA. Using quaternions to calculate RMSD. J Chem Phys 2004;25(15):1849–1857. [DOI] [PubMed] [Google Scholar]
- 57.Smith PC, Karpowich N, Millen L, Moody JE, Rosen J, Thomas PJ, Hunt JF. ATP binding to the motor domain from an ABC transporter drives formation of a nucleotide sandwich dimer. Mol Cell 2002;10(1):139–149. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Zaitseva J, Jenewein S, Jumpertz T, Holland IB, Schmitt L. H662 is the linchpin of ATP hydrolysis in the nucleotide-binding domain of the ABC transporter HlyB. EMBO J 2005;24(11):1901–1910. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Urbatsch IL, Sankaran B, Bhagat S, Senior AE. Both P-glycoprotein nucleotide-binding sites are catalytically active. J Biol Chem 1995;270(45):26956–26961. [DOI] [PubMed] [Google Scholar]
- 60.Senior AE, Bhagat S. P-glycoprotein shows strong catalytic cooperativity between the two nucleotide sites. Biochemistry 1998;37(3):831–836. [DOI] [PubMed] [Google Scholar]
- 61.Vigano C, Julien M, Carrier I, Gros P, Ruysschaert JM. Structural and functional asymmetry of the nucleotide-binding domains of P-glycoprotein investigated by attenuated total reflection Fourier transform infrared spectroscopy. J Biol Chem 2002;277(7):5008–5016. [DOI] [PubMed] [Google Scholar]
- 62.Sauna ZE, Ambudkar SV. Characterization of the catalytic cycle of ATP hydrolysis by human P-glycoprotein the two ATP hydrolysis events in a single catalytic cycle are kinetically similar but affect different functional outcomes. J Biol Chem 2001;276(15):11653–11661. [DOI] [PubMed] [Google Scholar]
- 63.Wang L, Johnson ZL, Wasserman MR, Levring J, Chen J, Liu S. Characterization of the kinetic cycle of an ABC transporter by single-molecule and cryo-EM analyses. eLife 2020;9:e56451. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Sauna ZE, Müller M, Peng XH, Ambudkar SV. Importance of the conserved Walker B glutamate residues, 556 and 1201, for the completion of the catalytic cycle of ATP hydrolysis by human P-glycoprotein (ABCB1). Biochemistry 2002;41(47):13989–14000. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.