

Abstract
Prolactin (PRL) cooperates with other factors to orchestrate mammary development and lactation, and is epidemiologically linked to higher risk for breast cancer. However, how PRL collaborates with oncogenes to foster tumorigenesis and influence breast cancer phenotype is not well understood. To understand its interactions with canonical Wnt signals, which elevate mammary stem cell activity, we crossed heterozygous NRL-PRL mice with ApcMin/+ mice and treated pubertal females with a single dose of mutagen. PRL in the context of ApcMin/+ fueled a dramatic increase in tumor incidence in nulliparous mice, compared to ApcMin/+ alone. Although carcinomas in both NRL-PRL/ApcMin/+ and ApcMin/+ females acquired a mutation in the remaining wildtype Apc allele and expressed abundant β-catenin, PRL-promoted tumors displayed higher levels of Notch-driven target genes and Notch-dependent cancer stem cell activity, compared to β-catenin-driven activity in ApcMin/+ tumors. This PRL-induced shift to dominant Notch signals was evident in preneoplastic epithelial hyperplasias at 120 days of age. In NRL-PRL/ApcMin/+ females, rapidly proliferating hyperplasias, characterized by β-catenin at cell junctions and high NOTCH1 expression, contrasted with slower growing lesions with nuclear β-catenin in ApcMin/+ females. These studies demonstrate that PRL can powerfully modulate the incidence and phenotype of mammary tumors, shedding light on mechanisms whereby PRL elevates risk of breast cancer.
Keywords: breast cancer, prolactin, β-catenin, APC, Notch, Wnt
1. Introduction
Prolactin (PRL) cooperates with other hormones and growth factors to orchestrate normal mammary development and lactation [1]. Like many critical regulators of mammary function, PRL has been epidemiologically implicated in the risk for breast cancer. Large prospective nested case control studies of women in the U.S. and Europe have demonstrated that women with higher circulating levels of PRL have a heightened risk for breast cancer, although significant findings regarding menopausal status and breast cancer subtype differ somewhat between these reports [2, 3]. Breast cancer develops in the context of dysregulation of multiple pathways and mutations. How PRL collaborates with other players that control mammary growth and differentiation to foster tumorigenesis and influence the phenotype of breast cancers is not well understood.
Genetically modified mouse models of both PRL/ PRL receptor ablation and PRL overexpression have linked PRL to development of mammary tumors (for review, [4]). In the NRL-PRL mouse, transgenic mammary expression of PRL mimics the local PRL synthesis observed in women [5, 6]. Nulliparous NRL-PRL females in the FVB/N genetic background spontaneously develop aggressive ER+ adenocarcinomas after a long latency [7, 8], paralleling the human epidemiologic data. Analysis of PRL actions and crosstalk with ovarian steroids prior to visible lesions in this model revealed that PRL increases mammary progenitor/ stem cells, associated with modulation of transcriptional networks that drive epithelial differentiation [9]. Transcriptome analysis over the course of PRL-induced oncogenesis revealed heightened Notch activity in hyperplastic lesions and established tumors [8].
Wnts are a diverse family of secreted ligands, which initiate multiple signals essential for mammary development (reviewed in [10-12]). The best characterized downstream signaling cascade is the “canonical pathway”, which is mediated by nuclear β-catenin. Wnt signals increase β-catenin levels by reducing activity of a destruction complex, which includes the tumor suppressor Adenomatous polyposis coli (APC) and targets β-catenin for proteasomal degradation. The resulting elevated levels of β-catenin permit translocation to the nucleus where it associates with TCF transcription factors to regulate target genes. β-catenin is also an important component of adherens junctions, where it associates with cadherins and α-catenin to form polarized epithelium [13]. Although their actions in clinical breast cancer remain poorly understood, Wnts and the plethora of secreted, membrane and intracellular proteins that modulate their actions are widely dysregulated across breast cancer subtypes [14-18]. In mouse models, elevated canonical Wnt signals expand mammary stem cells, and result in complex squamous tumors, frequently containing ductal, acinar, and myoepithelial components (reviewed in [11, 12]).
Breast tissue in women may be exposed to both endocrine pituitary PRL and locally produced PRL throughout their lives [6, 19, 20]. In order to understand how PRL may influence responses to canonical Wnt signals in mammary tumorigenesis, we crossed heterozygous NRL-PRL mice with ApcMin/+ mice. The ApcMin point mutation creates a protein truncated at aa850 [21], which reduces degradation of β-catenin, thereby raising canonical Wnt signals. Although originally identified as a murine model for human familial adenomatous polyposis, the germline ApcMin allele also leads to mammary cancer in heterozygous females, particularly in response to exposure to chemical mutagens and ionizing radiation during puberty [22, 23]. Importantly, the germline ApcMin/+ mutation, unlike mammary promoter-driven β-catenin transgenes, does not select for mammary cell subtype. In combination with the locally secreted ligand of the NRL-PRL model, it permits unbiased evaluation of the net effects of the interactions of these factors on mammary pathology.
Our results show that PRL in the context of ApcMin/+ dramatically increased the incidence of mammary tumors, compared to ApcMin/+ alone. Carcinomas in both ApcMin/+ and NRL-PRL/ApcMin/+ females exhibited diverse histotypes, similar patterns of β-catenin and ERα expression, and acquisition of a mutation in the remaining wildtype (WT) Apc allele. However, tumors in NRL-PRL/ApcMin/+ females displayed higher cancer stem cell (CSC) activity, and self-renewal was dependent on Notch signals rather than canonical Wnt signals as observed in tumors in ApcMin/+ females. Examination of mammary glands from age-matched females prior to palpable tumors revealed that despite similar numbers of epithelial hyperplasias in NRL-PRL/ApcMin/+ females, these lesions were larger and proliferated more rapidly than hyperplasias in ApcMin/+ females, and expressed high levels of Notch receptors, in contrast to the canonical Wnt signals in ApcMin/+ females. These studies demonstrate that PRL cooperates with elevated β-catenin to powerfully modulate the incidence and signaling pathways of mammary tumors, shedding light on mechanisms whereby PRL contributes to the risk of breast cancer.
2. Materials and methods
2.1. Reagents
The following antibodies were used for immunohistochemical analyses: 5-bromo-2-deoxyuridine (BrdU, MAS-250) from Accurate Scientific (Westbury, NY); estrogen receptor α (ERα; SC-542) from Santa Cruz Biotechnology, Inc. (Santa Cruz, CA); β-catenin (610153) from BD Biosciences (San Jose, CA); Notch1 (Ab52627) from Abcam (Cambridge, MA). iCRT14 (SML-0203), γ-Secretase Inhibitor (S2188; CAS Number 200810-93-1) and BrdU (B5002) were obtained from Sigma-Aldrich (St. Louis, MO).
2.2. Experimental mice and treatment administration
NRL-PRL mice in the FVB/N strain background [24] and ApcMin/+ mice in the C57BL/6J strain were generated as described [22]. Primers used for genotyping are shown in Suppl. Table S1. Mice were bred, housed and handled in accordance with the Guide for Care and Use of Laboratory Animals in AAALAC-accredited facilities, and all procedures were approved by the University of Wisconsin-Madison Animal Care and Use Committee (G005201). Heterozygous NRL-PRL females were crossed with ApcMin/+ males to produce F1 females. Experimental mice were injected with single dose of the mutagen, ENU (50mg/kg body weight) at puberty (38-42d of age), which increases the incidence of mammary tumors in the ApcMin/+ model [22, 25, 26]. For some experiments, mice were injected with 200 mg/kg body weight BrdU 1h prior to euthanasia to label cells undergoing DNA synthesis. Mice were observed daily for overall health and biweekly for palpable mammary tumors. Mammary glands from 16-20 mice of each genotype were collected from an age-matched cohort at 120 days of age, and the remainder at end stage (defined as tumors of 1.5cm in diameter, or when animals exhibited loss of body condition or reached 500 days of age, whichever occurred first).
2.3. Sequencing of a mutational hotspot in the Apc gene
In order to identify somatic mutations in a region of the Apc gene previously shown to be a common site of nonsense mutations in mutant Apc-driven mammary tumors [26, 27], we sequenced both strands of this region by the Sanger method, as described previously [8]. The region encoding amino acids 1512-1579 was amplified by primers tagged with M1340For (forward strand) and M13R (reverse strand) (Suppl. Table S1) using Phusion Hot Start (New England Biolabs M0535S) and 5ng of DNA. The PCR product was purified using the QIAquick PCR Purification Kit (Qiagen 28104) and commercially sequenced (Genewiz, South Plainfield, NJ). The traces for the forward and reverse sequences were manually inspected using Sequence Scanner Software 2 v2.0 (ThermoFisher).
2.4. Tumorsphere assays
Single cell suspensions were prepared from end stage tumors, and the ability of these cells to form colonies under nonadherent conditions (primary tumorspheres) and renew this capacity (secondary tumorspheres) [28, 29] was assessed as described [30]. To examine the importance of canonical Wnt and Notch signals for self-renewal of this activity in vitro, freshly seeded cells were cultured with vehicle (0.1% DMSO), 100μM iCRT14, or 50μM γ-Secretase Inhibitor (GSI) during development of primary tumorspheres, and ability to form secondary tumorspheres in the absence of further treatment was quantified.
2.5. Quantitative real-time PCR
Total RNA was isolated from end stage tumors or isolated mammary epithelial cells [30], and specific transcripts were quantified by qRT-PCR as described [31] using the primers shown in Suppl. Table S1. Results were calculated using the comparative CT method and normalized to 18S RNA.
2.6. Histological and whole mount analyses
Mammary tissues were histologically examined, and apoptotic and BrdU-labeled cells in epithelial hyperplasias were quantified as described [24, 30]. Ductal branching was quantified as described [32]. For analyses of large hyperplasias, the left mammary chains from age-matched 120 day old females were whole-mounted and stained with carmine alum [33]. Volumes of individual lesions which were grossly visible under a dissecting microscope were calculated by: (the largest diameter x (the smallest diameter)2 x 0.4).
2.7. Statistical analyses
Statistical analyses were performed using Prism v.8.30 (GraphPad Software, Inc., San Diego, CA), or MSTAT, v.6.6.1 (N. Drinkwater, McArdle Laboratory for Cancer Research). Statistical tests are indicated in the figure legends. Differences were considered significant at p<0.05.
3. Results
3.1. PRL dramatically increases the incidence of mammary tumors in the context of ApcMin/+
The role of the Wnt pathway in breast cancer has been extensively studied, including examination of the susceptibility of ApcMin/+ females to mutagen-induced mammary carcinogenesis [22, 25, 26]. To determine how PRL may influence this process, we crossed heterozygous NRL-PRL FVB/N with ApcMin/+ C57BL/6J mice, generating the four genotypes in the F1 genetic background (Fig. 1A). At puberty, when the developing gland is more susceptible to carcinogenesis [34, 35], females were injected with single dose of the mutagen, ENU, and nulliparous females were monitored for development of mammary tumors. As expected, very few wildtype females developed mammary lesions (Fig. 1B). In contrast to the high penetrance in the FVB/N strain [24], locally elevated transgenic PRL in the absence of the Apc mutation promoted few tumors in this F1 background (11% incidence). The C57BL/J6 alleles reduce carcinogenesis driven by many mammary oncogenes (e.g., [36-38]). Similar to an earlier report in the same F1 strain background [25], 33% of ENU-treated ApcMin/+ females developed mammary tumors. However, the combination of NRL-PRL and ApcMin/+ increased the incidence of mammary tumors to 76%, indicating that PRL strongly potentiates ApcMin/+-induced tumorigenesis (P=0.008, logrank test).
Fig. 1. Prolactin potentiates tumorigenesis in the ApcMin/+ background.

(A) Experimental Design. Heterozygous NRL-PRL FVB/N females were crossed with ApcMin/+ C57BL/6Jmales, generating the four genotypes in the F1 background. Pubertal mice were injected with ENU as described in the Methods. A cohort of age-matched animals was examined at 120 days of age, and the remainder allowed to age until end stage. (B) The PRL transgene increased mammary tumor incidence in the ApcMin/+ background (ApcMin/+ vs. NRL-PRL/ApcMin/+, p=0.008, logrank test), but not tumor latency (Kaplan-Meier analysis, followed by Mantel-Cox test, ns). N= WT, 28; NRL-PRL, 28; ApcMin/+, 30; NRL-PRL/ApcMin/+, 40. (C) Tumors that developed in ApcMin/+ and NRL-PRL/ApcMin/+ females displayed a similar range of histotypes, and abundant ERα expression. Tumors in NRL-PRL/ApcMin/+ shown; ApcMin/+ tumors shown in Suppl. Fig. S1A, and NRL-PRL alone in Suppl. Fig. S1C. Left, hematoxylin and eosin (H&E) stained representative histotypes (i, ii, glandular; iii, iv, microacinar; v, vi, papillary; vii, viii, adenosquamous). Right, ERα expression by immunohistochemistry; insets as shown. (D) Tumors that developed in ApcMin/+ and NRL-PRL/ApcMin/+ females exhibited similarly heterogeneous cellular localization of β-catenin; insets as shown. Tumors in NRL-PRL/ApcMin/+ shown; ApcMin/+ tumors in Suppl. Fig. S1B, and NRL-PRL alone in Suppl. Fig. S1D. (i, ii, glandular; iii, iv, microacinar; v, vi, papillary; vii, viii, adenosquamous). White arrows indicate β-catenin at cell junctions, black arrowheads point to nuclear β-catenin. Black asterisks denote cells with little detectable β-catenin, at background. Scale bars, 100 μm. Original magnifications, H&E, x 100; ERα, β-catenin x 200.
Despite the pronounced effect of PRL on tumor incidence, mammary tumors that developed in both ApcMin/+ and NRL-PRL/ApcMin/+ females displayed a similar range of tumor histotypes [39], which differed only by restriction of the papillary histotype to NRL-PRL/ApcMin/+ tumors (Fig. 1C, Fig. S1A, Fig. S2). All tumors in both genotypes robustly expressed nuclear ERα. In light of the critical role for β-catenin in the ApcMin/+ model, we examined its expression and location by immunohistochemistry. All tumors in both ApcMin/+ and NRL-PRL/ApcMin/+ females exhibited strong β-catenin expression, which was heterogeneously located at cell junctions, and in the cytoplasm and nucleus (Fig. 1D, Fig. S1B). In contrast, the few tumors that formed in NRL-PRL females accumulated little β-catenin (Fig. S1C, D).
Mutagen-induced mammary tumors in ApcMin/+ females are characterized by acquisition of somatic mutations in the remaining WT Apc allele, which are especially common in a “hotspot” spanning codons 1512-1579 [26, 27] (Fig. S3A). We therefore sequenced this region of the Apc gene in a subset of tumors with representation across histotypes. As expected, all mammary tumors from ApcMin/+ females (9/9) showed a “second hit” in this region. Thirteen of 17 tumors in NRL-PRL/ApcMin/+ females also exhibited a mutation in this region, which was independent of histotype (Fig. S3B). The frequency of this additional Apc mutation was not significantly different between genotypes (P=0.263), demonstrating that transgenic PRL does not significantly alter this feature of ApcMin/+-facilitated mammary tumorigenesis. Together, these data show that the established tumors that develop in both genotypes are morphologically similar, and exhibit robust β-catenin expression.
3.2. Notch signals dominate in tumors in NRL-PRL/ApcMin/+ females, in contrast to canonical Wnt signals in ApcMin/+females
In order to examine other characteristics of these tumors which may be influenced by PRL, we assessed CSC activity using the established tumorsphere assay [28, 29]. Tumors from NRL-PRL/ApcMin/+ females exhibited about 40% higher activity as primary tumorspheres (Fig. 2A). We previously demonstrated that both Wnt and Notch signals contributed to self-renewal of CSC activity in PRL-induced tumors in the FVB/N genetic background [30]. In order to determine the importance of these pathways in ApcMin/+ and NRL-PRL/ApcMin/+ tumors in the F1 genetic background, we exposed tumor cells to inhibitors during formation of primary tumorspheres, and then examined the ability to form secondary tumorspheres in the absence of any inhibitors. Cells were treated with vehicle, iCRT14, which potently inhibits β-catenin responsive transcription, or a GSI, which blocks Notch signaling by preventing its cleavage at the cell surface. As shown in Fig. 2B, iCRT14 exposure decreased secondary sphere formation in cells from ApcMin/+ tumors (~55%), whereas GSI had no effect. In contrast, GSI reduced secondary spheres from NRL-PRL/ApcMin/+ tumors by almost 70%, while iCRT14 elicited little response. These results demonstrate that CSC self-renewal in tumors in the ApcMin/+ and NRL-PRL/ApcMin/+ models is controlled by strikingly different signaling pathways.
Fig. 2. Tumors that develop in NRL-PRL/ ApcMin/+ females exhibit GSI-dependent cancer stem cell activity and elevated transcripts for Notch target genes.

(A) The frequency of CSC activity was assessed by the ability of tumor cells to form primary tumorspheres as described in the Methods (tumorspheres/ 10,000 cells). Mean ± S.E.M., N=3. (*, p<0.05; unpaired Student’s t test). (B) Ability to renew CSC activity was assessed by the ability of cells treated with DMSO, GSI, or iCRT14 (iCRT) during formation of primary tumorspheres to form secondary tumorspheres (tumorspheres/ 10,000 cells). Mean ± S.E.M., N=3. (*, p<0.05; **, p<0.01; one way ANOVA followed by Tukey Multiple Comparison post tests). (C) Relative levels of transcripts for Notch (i) and canonical Wnt (ii) target genes were determined by qRT-PCR. Mean ± S.E.M., N=3. (*, p<0.05; **, p<0.01, unpaired Student’s t test).
To examine the relative activity of canonical Wnt and Notch pathways in the total heterogeneous tumor cell population, we examined levels of transcripts for well-established target genes of both signaling cascades. These results mirrored the dependence of CSC self-renewal: NRL-PRL/ApcMin/+ tumors displayed higher levels of Notch regulated mRNAs (Hes1, Hey1), in contrast to higher levels for targets of canonical Wnt signals (Axin2, Tcf4) in tumors from ApcMin/+ females (Fig. 2C).
3.3. Mammary glands in NRL-PRL/ApcMin/+ females exhibit larger epithelial hyperplasias
In order to understand the mechanisms underlying the PRL-increased tumor incidence and altered pathway activation in the context of the ApcMin/+ mutation, we examined mammary glands of ENU-treated nulliparous females at 120 d of age, prior to palpable tumors. Examination of whole mount preparations from all four genotypes revealed substantial differences. Only a few small lesions were visible in the glands from wildtype or NRL-PRL mice (Fig. 3Ai, ii), as expected from the low tumor incidence in these genotypes. In contrast, multiple large epithelial hyperplasias were readily detectable in both ApcMin/+ and NRL-PRL/ApcMin/+ glands (Fig. 3Ai, ii). Although the number of these lesions was similar between these two genotypes (Fig. 3Aii), they were significantly larger in NRL-PRL/ApcMin/+ glands (Fig. 3Aiii). A closer examination of the ductal trees showed that NRL-PRL/ApcMin/+ glands exhibited significantly more branching compared to WT and ApcMin/+ glands (Fig. S4A, B). Microscopic examination confirmed the greater incidence of abnormalities in ApcMin/+ and NRL-PRL/ApcMin/+ animals (Fig. S4C, D, Fig. 4A). Interestingly, NRL-PRL females, which developed few tumors and few large hyperplasias in the F1 genetic background, also displayed detectable, although infrequent, abnormalities (irregular ductal epithelium, small hyperplasias) (Fig. S4C, D), which are prominent features of the NRL-PRL model in the FVB/N strain background in the absence of mutagen [24].
Fig. 3. PRL increases the size, but not the number of epithelial hyperplasias in the ApcMin/+ genetic background.
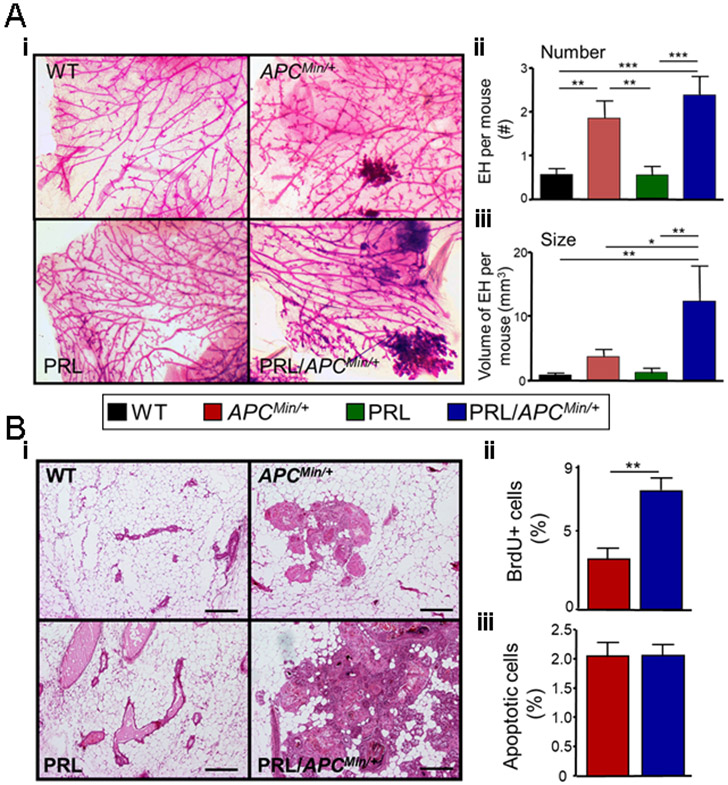
Pubertal females of the four genotypes were treated with ENU, and at 120 days of age, mammary glands were examined (Fig. 1A). (A) Mammary glands from ApcMin/+ and NRL-PRL/ApcMin/+ females exhibited large hyperplastic lesions, compared to WT or NRL-PRL females. (i) Representative micrographs of whole mounted mammary glands stained with carmine alum (genotypes as shown). (ii) ApcMin/+ and NRL-PRL/ApcMin/+ glands contained similar numbers of large lesions. [Number of large epithelial hyperplasias (EH) in the left chain of mammary glands 2-5 of individual females]. (iii) Hyperplasias in NRL-PRL/ApcMin/+ glands were larger than those in ApcMin/+ glands. (Total volume of large EH per mouse, see Methods). (ii, iii) Mean ± S.E.M., N=16-20 mice. Statistical differences among the different genotypes was determined by the Kruskal-Wallis test followed by the Dunn’s Multiple Comparison post test. (*, p<0.05; **, p<0.01; ***, p<0.005). (B) Hyperplastic lesions in NRL-PRL/ApcMin/+ glands proliferated more rapidly than those in ApcMin/+ females. (i) Hematoxylin/ eosin stained glands. Scale bars, 500 μm. Rates of proliferation (ii) and apoptosis (iii) in epithelial cells in large hyperplasias in NRL-PRL/ApcMin/+ and ApcMin/+ glands were determined as described in the Methods. Mean ± S.E.M.; N=6 mice. (**, p<0.01, unpaired Student’s t test.). Rates of apoptosis were not significantly different.
Fig. 4. Large hyperplastic lesions in mammary glands from ENU-treated ApcMin/+ and NRL-PRL/ApcMin/+ females exhibit different localization of β-catenin and expression of Notch receptors, which are associated with different levels of target genes regulating proliferation and cell fate.

(A) Left, representative large hyperplasias from genotypes shown, stained with H&E. Arrows indicate squamous changes. Right, representative immunohistochemistry for β-catenin, showing markedly different localization between these genotypes. Insets as shown. Scale bars, 100μm. Original magnifications, x200. (B) Relative levels of transcripts in mammary epithelial cell preparations were determined by qRT-PCR. (i) indicators of Wnt and Notch signals. (ii) cell cycle regulators. (iii) Sox genes. Mean ± S.E.M., N=3-4. (*, p<0.05; **, p<0.01, unpaired Student’s t test). (C) Relative levels of transcripts for Notch receptors in mammary epithelial cell preparations were determined by qRT-PCR. Mean ± S.E.M., N=3-4. (*, p<0.05; unpaired Student’s t test). (D) Representative immunohistochemistry for NOTCH1, showing strikingly different levels of expression between these genotypes. Insets as shown. Scale bars, 100μm. Original magnifications, x200.
Large epithelial hyperplasias were only observed in the mammary glands of NRL-PRL/ApcMin/+ and ApcMin/+ mice, so we further analyzed cell turnover in these genotypes. Consistent with the larger size in NRL-PRL/ApcMin/+ compared to ApcMin/+ mammary glands (Fig. 3Ai, ii), the rate of proliferation was higher in these lesions in NRL-PRL/ApcMin/+ glands (Fig. 3Bi, ii). However, no difference in apoptosis was observed (Fig. 3Biii). Together, these data suggest that PRL promotes lesion progression in part by increasing proliferation.
3.4. Preneoplastic lesions in NRL-PRL/ApcMin/+ females exhibit elevated Notch signals
In order to understand the role of β-catenin in the large epithelial hyperplasias present in the 120 day old females, we examined its expression/ location by immunohistochemistry. As shown in Fig. 4A, β-catenin was located primarily in the nucleus of cells in these structures in ApcMin/+ mice, whereas in NRL-PRL/ApcMin/+ females, it was strikingly enriched at cell junctions. In contrast, β-catenin did not accumulate in morphologically normal ductal epithelium in either genotype (Fig. S5). Mammary epithelial cell preparations from ApcMin/+ females contained significantly higher levels of Axin2 mRNA, consistent with nuclear β-catenin (Fig. 4Bi). Interestingly, ApcMin/+ cells also contained higher levels of transcripts for Nrarp, a negative regulator of Notch signals [40]. NRL-PRL/ApcMin/+ mammary epithelial cells contained lower levels of mRNA for the cell cycle inhibitor Cdkn2c, which predicts expansion of luminal progenitors [41] and is repressed by Notch signals [42], and higher levels of Ccne1 transcripts, which are increased by Notch [43], consistent with the observed increased proliferation (Fig. 4Bii). Levels of Ccnd1 mRNA did not differ between these genotypes (data not shown). SOX transcription factors influence cell fate in multiple tissues, and interact with both β-catenin and Notch signals [18, 44]. As shown in Fig. 4Biii, mammary epithelial cells from ApcMin/+ females expressed much higher levels of Sox2 mRNA relative to cells from NRL-PRL/ApcMin/+ females, whereas Sox9 mRNA was more modestly affected.
To further examine Notch signaling in 120 day old females, we determined relative transcript levels for the receptors, NOTCH 1-4. Notch1 and Notch2 mRNAs, but not Notch3 or Notch4, were significantly higher in mammary epithelial preparations from NRL-PRL/ApcMin/+ compared to ApcMin/+ females (Fig. 4C), and NOTCH1 protein was readily detected in hyperplasias in NRL-PRL/ApcMin/+, but not ApcMtn/+ mice (Fig. 4D). In contrast, epithelial cells in morphologically normal ducts exhibited little detectable NOTCH1 expression in either genotype (Fig. S6). Together, these data demonstrate that elevated PRL exerts dramatic effects on hyperplastic mammary epithelia in the context of ApcMin/+, increasing proliferation, and shifting the dominant signaling pathway from canonical Wnt signals toward Notch signals.
Discussion
Despite the epidemiologic association of PRL with increased risk for development of breast cancer [2, 3, 45, 46], its contributions to oncogenic processes and the resulting phenotype of breast cancers are not well understood. Here we utilized defined mouse models to examine the influence of this hormone on tumorigenesis driven by canonical Wnt signals, which expand mammary stem cell populations and drive oncogenesis in multiple experimental models. The murine genetic background employed here permitted relatively few mammary tumors in response to either the NRL-PRL transgene or ApcMin/+ alone, revealing a powerful synergistic increase in tumor incidence when both genetic modifications were present. Although tumors in both NRL-PRL/ApcMin/+ and ApcMin/+ females expressed abundant β-catenin at heterogeneous locations, tumors that developed in the presence of elevated PRL displayed higher levels of Notch downstream targets and Notch-dependent CSC activity, in contrast to the β-catenin-driven activity observed in ApcMin/+ tumors. Tumors in NRL-PRL/ApcMin/+ females were preceded by rapidly proliferating epithelial hyperplasias, characterized by β-catenin located at cell junctions and high levels of Notch receptors, compared to more slowly growing hyperplasias with nuclear β-catenin that developed in the absence of elevated PRL.
Wnt and Notch are important developmental pathways which are frequently altered in many cancers [13, 16-18, 47, 48]. Canonical Wnt signals and SOX2 contribute to maintenance of pluripotency/ stem cell activity of many tissues, including the mammary gland [11, 17, 18, 49-52]. In contrast, Notch signals dominate in more differentiated progenitor subpopulations in many cell lineages [50, 51, 53]. Although expressed in several mammary epithelial cell subpopulations, Notch receptors, especially NOTCH1, have been linked most closely to an ER- luminal progenitor population [50, 54, 55]. SOX9 plays a role in determining luminal/ basal cell fate and marks ER- luminal progenitor cells [56, 57]. Accumulating evidence suggests that these cells are important cells of origin for luminal as well as BRCA1 basal breast cancers (reviewed in [50, 58]). Further, elevated NOTCH1 has been implicated in the progression of ductal carcinoma in situ [59]. Although Wnt and Notch signals are inversely related in the hierarchies of multiple tissues in development as well as tumorigenesis [60-63], these pathways can cooperate in cancer, and growing evidence implicates dysregulation of both pathways in treatment resistance of established breast cancers (reviewed in [18, 53, 58, 64, 65]).
Our studies show that the PRL-induced shift from dominant Wnt to Notch signals in the presence of elevated β-catenin is evident in preneoplastic mammary lesions. As expected, epithelial hyperplasias in ApcMin/+ females exhibited strong nuclear β-catenin localization and transcriptional activity, associated with high Sox2 transcripts. Consistent with the inverse relationship observed in multiple other tissues (reviewed in [18, 53, 58, 64, 66]), Notch signals were inhibited in the presence of these strong canonical Wnt signals, indicated by elevated Nrarp mRNA, a negative regulator of Notch transcriptional activity [40]. PRL did not significantly alter the acquisition of somatic mutations in the region of the Apc gene previously associated with mammary tumors [26, 27], and consistently, large epithelial hyperplasias in NRL-PRL/ApcMin/+ mice also accumulated β-catenin. However, in NRL-PRL/ApcMin/+ females, β-catenin was enriched at cell junctions, rather than the nucleus, and was associated with lower canonical Wnt signals and Sox2 mRNA and high NOTCH1 expression. The membrane Notch receptors in these hyperplasias may mediate both canonical and noncanonical Notch activity. The latter may alter β-catenin trafficking and contribute to its enriched membrane localization in these lesions [67, 68].
Our data indicate that PRL can robustly alter signaling pathways in mammary lesions early in pathogenesis in the presence of the oncogenic driver, elevated β-catenin, with dramatic consequences for the incidence and CSC activity of the resulting tumors. The augmented Notch signals and rapid proliferation in epithelial hyperplasias of NRL-PRL/ApcMin/+ females are consistent with observed PRL actions in the absence of other oncogenes in the FVB/N genetic background. There, the NRL-PRL transgene increases a luminal progenitor population by modulating transcriptional programs driving maturation [9], and tumorigenesis is associated with elevated Notch signals [8]. Moreover, PRL impacts cell cycle regulators and drives proliferation of mammary epithelial cells in multiple experimental mouse models, including NRL-PRL, and in breast cancer cell lines in vitro [4, 69, 70]. Together, these observations suggest that in the current study, PRL facilitates the transition of β-catenin/SOX2 expanded stem cell populations fueled by mutations in Apc into rapidly proliferating Notch-driven lesions, which efficiently progress to adenocarcinomas.
This study extends our understanding of the mechanisms whereby PRL can cooperate with other oncogenes to promote mammary tumorigenesis, illuminating the epidemiologic link between PRL exposure and increased cancer risk. PRL has been reported to promote mammary tumorigenesis in conjunction with multiple oncogenes, with implications for several breast cancer subtypes [7, 71], suggesting context dependent actions. The importance of the cell populations in early lesions has been shown to be an important determinant of tumor phenotype [72]. Moreover, single cell sequencing has revealed a spectrum of cell differentiation states within experimental tumors, underscoring the complexity of CSC activity [73]. Understanding how endogenous factors which regulate the mammary epithelial hierarchy, including hormones such as prolactin, collaborate with oncogenic processes to promote dysregulated breast cancers is essential for formulation of preventative and therapeutic strategies, and identification of patients who will benefit.
Supplementary Material
Highlights:
Prolactin dramatically increased mammary tumor incidence in the context of ApcMin/+
Prolactin/ApcMin/+ tumors exhibited Notch-rather than Wnt-dependent CSC activity
Prolactin induced Notch dominance and rapid proliferation in early lesions
Acknowledgements
The authors are grateful to Dr. Norman Drinkwater (UW McArdle Laboratories) for assistance with statistical analyses, and Drs. Richard Halberg, Caroline Alexander, Fern Murdoch, Hua Wang, Jennifer Brockman and Lindsay Hinck for insightful discussions. We appreciate the assistance of Sarah Nikolai (Laubmeier), Lindsey Moderski and Kevin Joyce with histological analyses.
Funding Sources
This work was supported by the National Institutes of Health [R01CA157675 and R01CA179556 (LAS), T32ES007015 (MPS) and P30CA014520 (University of Wisconsin Carbone Cancer Center)]. Its contents are solely the responsibility of the authors and do not necessarily represent the official views of the NIEHS, NIH.
4. Abbreviations:
- APC
Adenomatous polyposis coli
- BrdU
5-bromo-2-deoxyuridine
- CSC
cancer stem cell
- ENU
N-ethyl-N-nitrosourea
- ER
estrogen receptor
- GSI
gamma secretase inhibitor
- PRL
prolactin
- TCF
T-cell factor/lymphoid enhancer factor
- WT
wildtype
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Conflict of Interest
The authors declare no potential conflicts of interest.
Declaration of interests
The authors declare that they have no known competing financial interests or personal relationships that could have appeared to influence the work reported in this paper.
References
- [1].Oakes SR, Rogers RL, Naylor MJ, Ormandy CJ, Prolactin regulation of mammary gland development, J. Mammary Gland Biol. Neoplasia, 13 (2008) 13–28. [DOI] [PubMed] [Google Scholar]
- [2].Tworoger SS, Eliassen AH, Zhang X, Qian J, Sluss PM, Rosner BA, Hankinson SE, A 20-year prospective study of plasma prolactin as a risk marker of breast cancer development, Cancer Res., 73 (2013) 4810–4819. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [3].Tikk K, Sookthai D, Johnson T, Rinaldi S, Romieu I, Tjonneland A, Olsen A, Overvad K, Clavel-Chapelon F, Baglietto L, Boeing H, Trichopoulou A, Lagiou P, Trichopoulos D, Palli D, Pala V, Tumino R, Rosso S, Panico S, Agudo A, Menendez V, Sanchez MJ, Amiano P, Huerta Castano JM, Ardanaz E, Bueno-de-Mesquita HB, Monninkhof E, Onland-Moret C, Andersson A, Sund M, Weiderpass E, Khaw KT, Key TJ, Travis RC, Gunter MJ, Riboli E, Dossus L, Kaaks R, Circulating prolactin and breast cancer risk among pre- and postmenopausal women in the EPIC cohort, Ann. Oncol, 25 (2014) 1422–1428. [DOI] [PubMed] [Google Scholar]
- [4].Arendt LM, Schuler LA, Transgenic models to study actions of prolactin in mammary neoplasia, J. Mammary Gland Biol. Neoplasia, 13 (2008) 29–40. [DOI] [PubMed] [Google Scholar]
- [5].McHale K, Tomaszewski JE, Puthiyaveettil R, Livolsi VA, Clevenger CV, Altered expression of prolactin receptor-associated signaling proteins in human breast carcinoma, Mod. Pathol, 21 (2008) 565–571. [DOI] [PubMed] [Google Scholar]
- [6].Marano RJ, Ben-Jonathan N, Minireview: Extrapituitary prolactin: an update on the distribution, regulation, and functions, Mol. Endocrinol, 28 (2014) 622–633. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [7].O'Leary KA, Shea MP, Schuler LA, Modeling prolactin actions in breast cancer in vivo: insights from the NRL-PRL mouse, Adv. Exp. Med. Biol, 846 (2015) 201–220. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [8].Campbell KM, O'Leary KA, Rugowski DE, Mulligan WA, Barnell EK, Skidmore ZL, Krysiak K, Griffith M, Schuler LA, Griffith OL, A spontaneous aggressive ERα+ mammary tumor model is driven by Kras activation, Cell Rep., 28 (2019) 1526–1537. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [9].O'Leary KA, Shea MP, Salituro S, Blohm CE, Schuler LA, Prolactin Alters the mammary epithelial hierarchy, increasing progenitors and facilitating ovarian steroid action, Stem Cell Rep., 9 (2017) 1167–1179. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [10].van Amerongen R, Nusse R, Towards an integrated view of Wnt signaling in development, Development, 136 (2009) 3205–3214. [DOI] [PubMed] [Google Scholar]
- [11].Incassati A, Chandramouli A, Eelkema R, Cowin P, Key signaling nodes in mammary gland development and cancer: beta-catenin, Breast Cancer Res., 12 (2010) 213. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [12].Alexander CM, Goel S, Fakhraldeen SA, Kim S, Wnt signaling in mammary glands: plastic cell fates and combinatorial signaling, Cold Spring Harb. Perspect. Biol, 4 (2012) a008037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [13].Clevers H, Nusse R, Wnt/beta-catenin signaling and disease, Cell, 149 (2012) 1192–1205. [DOI] [PubMed] [Google Scholar]
- [14].Geyer FC, Lacroix-Triki M, Savage K, Arnedos M, Lambros MB, MacKay A, Natrajan R, Reis-Filho JS, beta-Catenin pathway activation in breast cancer is associated with triple-negative phenotype but not with CTNNB1 mutation, Mod. Pathol 24 (2011) 209–231. [DOI] [PubMed] [Google Scholar]
- [15].Koval A, Katanaev VL, Dramatic dysbalancing of the Wnt pathway in breast cancers, Sci. Rep, 8 (2018) 7329. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [16].Sanchez-Vega F, Mina M, Armenia J, Chatila WK, Luna A, La KC, Dimitriadoy S, Liu DL, Kantheti HS, Saghafinia S, Chakravarty D, Daian F, Gao Q, Bailey MH, Liang WW, Foltz SM, Shmulevich I, Ding L, Heins Z, Ochoa A, Gross B, Gao J, Zhang H, Kundra R, Kandoth C, Bahceci I, Dervishi L, Dogrusoz U, Zhou W, Shen H, Laird PW, Way GP, Greene CS, Liang H, Xiao Y, Wang C, Iavarone A, Berger AH, Bivona TG, Lazar AJ, Hammer GD, Giordano T, Kwong LN, McArthur G, Huang C, Tward AD, Frederick MJ, McCormick F, Meyerson M, Cancer N Genome Atlas Research, Van Allen EM, Cherniack AD, Ciriello G, Sander C, Schultz N, Oncogenic signaling pathways in the cancer genome atlas, Cell, 173 (2018) 321–337 e310. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [17].van Schie EH, van Amerongen R, Aberrant WNT/CTNNB1 Signaling as a therapeutic target in human breast cancer: weighing the evidence, Front. Cell Dev. Biol, 8 (2020) 25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [18].Oliphant MUJ, Kong D, Zhou H, Lewis MT, Ford HL, Two sides of the same coin: the role of developmental pathways and pluripotency factors in normal mammary stem cells and breast cancer metastasis, J. Mammary Gland Biol. Neoplasia, 25 (2020) 85–102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [19].Phillipps HR, Yip SH, Grattan DR, Patterns of prolactin secretion, Mol. Cell Endocrinol, 502 (2020) 110679. [DOI] [PubMed] [Google Scholar]
- [20].Bernard V, Young J, Binart N, Prolactin - a pleiotropic factor in health and disease, Nat. Rev. Endocrinol, 15 (2019) 356–365. [DOI] [PubMed] [Google Scholar]
- [21].Su LK, Kinzler KW, Vogelstein B, Preisinger AC, Moser AR, Luongo C, Gould KA, Dove WF, Multiple intestinal neoplasia caused by a mutation in the murine homolog of the APC gene, Science, 256 (1992) 668–670. [DOI] [PubMed] [Google Scholar]
- [22].Moser AR, Mattes EM, Dove WF, Lindstrom MJ, Haag JD, Gould MN, Apc-min, a mutation in the murine Apc gene, predisposes to mammary carcinomas and focal alveolar hyperplasias, Proc. Natl. Acad. Sci.,USA, 90 (1993) 8977–8981. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [23].Imaoka T, Okamoto M, Nishimura M, Nishimura Y, Ootawara M, Kakinuma S, Tokairin Y, Shimada Y, Mammary tumorigenesis in ApcMin/+ mice is enhanced by X irradiation with a characteristic age dependence, Radiat. Res, 165 (2006) 165–173. [DOI] [PubMed] [Google Scholar]
- [24].Rose-Hellekant TA, Arendt LM, Schroeder MD, Gilchrist K, Sandgren EP, Schuler LA, Prolactin induces ERα-positive and ERα-negative mammary cancer in transgenic mice, Oncogene, 22 (2003) 4664–4674. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [25].Moser AR, Hegge LF, Cardiff RD, Genetic background affects susceptibility to mammary hyperplasias and carcinomas in Apc(min)/+ mice, Cancer Res., 61 (2001) 3480–3485. [PubMed] [Google Scholar]
- [26].Keller RR, Gestl SA, Lu AQ, Hoke A, Feith DJ, Gunther EJ, Carcinogen-specific mutations in preferred Ras-Raf pathway oncogenes directed by strand bias, Carcinogenesis, 37 (2016) 810–816. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [27].Kuraguchi M, Ohene-Baah NY, Sonkin D, Bronson RT, Kucherlapati R, Genetic mechanisms in Apc-mediated mammary tumorigenesis, PLoS. Genet, 5 (2009) e1000367. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [28].Guo W, Keckesova Z, Donaher JL, Shibue T, Tischler V, Reinhardt F, Itzkovitz S, Noske A, Zurrer-Hardi U, Bell G, Tam WL, Mani SA, van Oudenaarden A, Weinberg RA, Slug and Sox9 cooperatively determine the mammary stem cell state, Cell, 148 (2012) 1015–1028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [29].Shaw FL, Harrison H, Spence K, Ablett MP, Simoes BM, Farnie G, Clarke RB, A detailed mammosphere assay protocol for the quantification of breast stem cell activity, J. Mammary Gland Biol. Neoplasia, 17 (2012) 111–117. [DOI] [PubMed] [Google Scholar]
- [30].Shea MP, O'Leary KA, Fakhraldeen SA, Goffin V, Friedl A, Wisinski KB, Alexander CM, Schuler LA, Anti-estrogen therapy increases plasticity and cancer stemness of prolactin-induced ERα+ mammary carcinomas, Cancer Res., (2018) 1672–1684. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [31].Arendt LM, Rugowski DE, Grafwallner-Huseth TL, Garcia-Barchino MJ, Rui H, Schuler LA, Prolactin-induced mouse mammary carcinomas model estrogen resistant luminal breast cancer, Breast Cancer Res., 13 (2011) R11–25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [32].Stanko JP, Fenton SE, Quantifying branching density in rat mammary gland whole-mounts using the sholl analysis method, JoVE, (2017) doi: 10.3791/55789. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [33].O'Leary KA, Jallow F, Rugowski DE, Sullivan R, Sinkevicius KW, Greene GL, Schuler LA, Prolactin activates ERα in the absence of ligand in female mammary development and carcinogenesis in vivo, Endocrinology, 154 (2013) 4483–4492. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [34].Russo J, Yang X, Hu YF, Bove BA, Huang Y, Silva ID, Tahin Q, Wu Y, Higgy N, Zekri A, Russo IH, Biological and molecular basis of human breast cancer, Front. Biosci, 3 (1998) D944–D960. [DOI] [PubMed] [Google Scholar]
- [35].Land CE, Tokunaga M, Koyama K, Soda M, Preston DL, Nishimori I, Tokuoka S, Incidence of female breast cancer among atomic bomb survivors, Hiroshima and Nagasaki, 1950-1990, Radiat. Res, 160 (2003) 707–717. [DOI] [PubMed] [Google Scholar]
- [36].Kuperwasser C, Hurlbut GD, Kittrell FS, Dickinson ES, Laucirica R, Medina D, Naber SP, Jerry DJ, Development of spontaneous mammary tumors in BALB/c p53 heterozygous mice. A model for Li-Fraumeni syndrome, Am. J. Pathol, 157 (2000) 2151–2159. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [37].Rose-Hellekant TA, Schroeder MD, Brockman JL, Zhdankin O, Bolstad R, Chen KS, Gould MN, Schuler LA, Sandgren EP, Estrogen receptor positive mammary tumorigenesis in TGFa transgenic mice progresses with progesterone receptor loss, Oncogene, 26 (2007) 5238–5246. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [38].Blackburn AC, Hill LZ, Roberts AL, Wang J, Aud D, Jung J, Nikolcheva T, Allard J, Peltz G, Otis CN, Cao QJ, Ricketts RS, Naber SP, Mollenhauer J, Poustka A, Malamud D, Jerry DJ, Genetic mapping in mice identifies DMBT1 as a candidate modifier of mammary tumors and breast cancer risk, Am. J. Pathol, 170 (2007) 2030–2041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [39].Cardiff RD, Anver MR, Gusterson BA, Hennighausen L, Jensen RA, Merino MJ, Rehm S, Russo J, Tavassoli FA, Wakefield LM, Ward JM, Green JE, The mammary pathology of genetically engineered mice: the consensus report and recommendations from the Annapolis meeting, Oncogene, 19 (2000) 968–988. [DOI] [PubMed] [Google Scholar]
- [40].Lamar E, Deblandre G, Wettstein D, Gawantka V, Pollet N, Niehrs C, Kintner C, Nrarp is a novel intracellular component of the Notch signaling pathway, Genes Dev., 15 (2001) 1885–1899. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [41].Pei XH, Bai F, Smith MD, Usary J, Fan C, Pai SY, Ho IC, Perou CM, Xiong Y, CDK inhibitor p18(INK4c) is a downstream target of GATA3 and restrains mammary luminal progenitor cell proliferation and tumorigenesis, Cancer Cell, 15 (2009) 389–401. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [42].Shin DM, Shaffer DJ, Wang H, Roopenian DC, Morse HC 3rd, NOTCH is part of the transcriptional network regulating cell growth and survival in mouse plasmacytomas, Cancer Res., 68 (2008) 9202–9211. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [43].Zender S, Nickeleit I, Wuestefeld T, Sorensen I, Dauch D, Bozko P, El-Khatib M, Geffers R, Bektas H, Manns MP, Gossler A, Wilkens L, Plentz R, Zender L, Malek NP, A critical role for notch signaling in the formation of cholangiocellular carcinomas, Cancer Cell, 23 (2013) 784–795. [DOI] [PubMed] [Google Scholar]
- [44].Sarkar A, Hochedlinger K, The sox family of transcription factors: versatile regulators of stem and progenitor cell fate, Cell Stem Cell, 12 (2013) 15–30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [45].Gabrielson M, Ubhayasekera K, Ek B, Andersson Franko M, Eriksson M, Czene K, Bergquist J, Hall P, Inclusion of plasma prolactin levels in current risk prediction models of premenopausal and postmenopausal breast cancer, JNCI Cancer Spectr., 2 (2018) pky055. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [46].Shemanko CS, Prolactin receptor in breast cancer: marker for metastatic risk, J. Mol. Endocrinol, 57 (2016) R153–R165. [DOI] [PubMed] [Google Scholar]
- [47].Ranganathan P, Weaver KL, Capobianco AJ, Notch signalling in solid tumours: a little bit of everything but not all the time, Nat. Rev. Cancer, 11 (2011) 338–351. [DOI] [PubMed] [Google Scholar]
- [48].Reyna MA, Haan D, Paczkowska M, Verbeke LPC, Vazquez M, Kahraman A, Pulido-Tamayo S, Barenboim J, Wadi L, Dhingra P, Shrestha R, Getz G, Lawrence MS, Pedersen JS, Rubin MA, Wheeler DA, Brunak S, Izarzugaza JMG, Khurana E, Marchal K, von Mering C, Sahinalp SC, Valencia A, P. Drivers, G. Functional Interpretation Working, Reimand J, Stuart JM, Raphael BJ, P. Consortium, Pathway and network analysis of more than 2500 whole cancer genomes, Nat. Commun, 11 (2020) 729. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [49].Wang Y, Dong J, Li D, Lai L, Siwko S, Li Y, Liu M, Lgr4 regulates mammary gland development and stem cell activity through the pluripotency transcription factor Sox2, Stem Cells, 31 (2013) 1921–1931. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [50].Fu NY, Nolan E, Lindeman GJ, Visvader JE, Stem cells and the differentiation hierarchy in mammary gland development, Physiol. Rev, 100 (2020) 489–523. [DOI] [PubMed] [Google Scholar]
- [51].Gehart H, Clevers H, Tales from the crypt: new insights into intestinal stem cells, Nat. Rev. Gastroenterol. Hepatol, 16 (2019) 19–34. [DOI] [PubMed] [Google Scholar]
- [52].Liu K, Lin B, Zhao M, Yang X, Chen M, Gao A, Liu F, Que J, Lan X, The multiple roles for Sox2 in stem cell maintenance and tumorigenesis, Cell Signal., 25 (2013) 1264–1271. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [53].Liu J, Sato C, Cerletti M, Wagers A, Notch signaling in the regulation of stem cell self-renewal and differentiation, Curr. Top. Dev. Biol, 92 (2010) 367–409. [DOI] [PubMed] [Google Scholar]
- [54].Rodilla V, Dasti A, Huyghe M, Lafkas D, Laurent C, Reyal F, Fre S, Luminal progenitors restrict their lineage potential during mammary gland development, PLoS Biol., 13 (2015) e1002069. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [55].Bouras T, Pal B, Vaillant F, Harburg G, Asselin-Labat ML, Oakes SR, Lindeman GJ, Visvader JE, Notch signaling regulates mammary stem cell function and luminal cell-fate commitment, Cell Stem Cell, 3 (2008) 429–441. [DOI] [PubMed] [Google Scholar]
- [56].Wang C, Christin JR, Oktay MH, Guo W, Lineage-biased stem cells maintain estrogen-receptor-positive and -negative mouse mammary luminal lineages, Cell Rep., 18 (2017) 2825–2835. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [57].Domenici G, Aurrekoetxea-Rodriguez I, Simoes BM, Rabano M, Lee SY, Millan JS, Comaills V, Oliemuller E, Lopez-Ruiz JA, Zabalza I, Howard BA, Kypta RM, Vivanco MD, A Sox2-Sox9 signalling axis maintains human breast luminal progenitor and breast cancer stem cells, Oncogene, 38 (2019) 3151–3169. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [58].Tharmapalan P, Mahendralingam M, Berman HK, Khokha R, Mammary stem cells and progenitors: targeting the roots of breast cancer for prevention, EMBO J., 38 (2019) e100852. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [59].Yuan X, Zhang M, Wu H, Xu H, Han N, Chu Q, Yu S, Chen Y, Wu K, Expression of Notch1 correlates with breast cancer progression and prognosis, PloS one, 10 (2015) e0131689. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [60].Shahi P, Seethammagari MR, Valdez JM, Xin L, Spencer DM, Wnt and Notch pathways have interrelated opposing roles on prostate progenitor cell proliferation and differentiation, Stem Cells, 29 (2011) 678–688. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [61].Meier-Abt F, Milani E, Roloff T, Brinkhaus H, Duss S, Meyer DS, Klebba I, Balwierz PJ, van Nimwegen E, Bentires-Alj M, Parity induces differentiation and reduces Wnt/Notch signaling ratio and proliferation potential of basal stem/progenitor cells isolated from mouse mammary epithelium, Breast Cancer Res., 15 (2013) R36. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [62].Collu GM, Hidalgo-Sastre A, Brennan K, Wnt-Notch signalling crosstalk in development and disease, Cell. Mol. Life Sci, 71 (2014) 3553–3567. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [63].Kaemmerer E, Jeon MK, Berndt A, Liedtke C, Gassler N, Targeting Wnt Signaling via Notch in Intestinal Carcinogenesis, Cancers, 11 (2019) 555. doi: 10.3390/cancers11040555. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [64].Braune EB, Seshire A, Lendahl U, Notch and Wnt dysregulation and its relevance for breast cancer and tumor initiation, Biomedicines, 6 (2018) 101. doi: 10.3390/biomedicines6040101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [65].Majumder S, Crabtree JS, Golde TE, Minter LM, Osborne BA, Miele L, Targeting Notch in oncology: the path forward, Nat. Rev. Drug Discov, (2020) doi: 10.1038/s41573-020-00091-3. [DOI] [PubMed] [Google Scholar]
- [66].Mollen EWJ, lent J, Tjan-Heijnen VCG, Boersma LJ, Miele L, Smidt ML, Vooijs M, Moving breast cancer therapy up a Notch, Front. Oncol, 8 (2018) 518. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [67].Andersen P, Uosaki H, Shenje LT, Kwon C, Non-canonical Notch signaling: emerging role and mechanism, Trends Cell Biol, 22 (2012) 257–265. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [68].Valenta T, Hausmann G, Basler K, The many faces and functions of beta-catenin, EMBO J., 31 (2012) 2714–2736. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [69].Schroeder MD, Symowicz J, Schuler LA, Prolactin modulates cell cycle regulators in mammary tumor epithelial cells, Mol. Endocrinol, 16 (2002) 45–57. [DOI] [PubMed] [Google Scholar]
- [70].Clevenger CV, Furth PA, Hankinson SE, Schuler LA, Role of prolactin in mammary carcinoma, Endocr. Rev, 24 (2003) 1–27. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [71].Johnston AN, Bu W, Hein S, Garcia S, Camacho L, Xue L, Qin L, Nagi C, Hilsenbeck SG, Kapali J, Podsypanina K, Nangia J, Li Y, Hyperprolactinemia-inducing antipsychotics increase breast cancer risk by activating JAK-STAT5 in precancerous lesions, Breast Cancer Res., 20 (2018) 42. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [72].Bu W, Liu Z, Jiang W, Nagi C, Huang S, Edwards DP, Jo E, Mo Q, Creighton CJ, Hilsenbeck SG, Leavitt AD, Lewis MT, Wong STC, Li Y, Mammary precancerous stem and non-stem cells evolve into cancers of distinct subtypes, Cancer Res., 79 (2019) 61–71. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [73].Yeo SK, Zhu X, Okamoto T, Hao M, Wang C, Lu P, Lu LJ, Guan JL, Single-cell RNA-sequencing reveals distinct patterns of cell state heterogeneity in mouse models of breast cancer, eLife, 9 (2020) doi: 10.7554/eLife.58810. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.