

LRP1-targeted St-Cl–polyglutamate conjugates as multivalent neuroprotective/neurotrophic therapeutics for Alzheimer’s disease.
Abstract
Alzheimer’s disease (AD), the most prevalent form of dementia, remains incurable mainly due to our failings in the search for effective pharmacological strategies. Here, we describe the development of targeted multimodal polypeptide-based nanoconjugates as potential AD treatments. Treatment with polypeptide nanoconjugates bearing propargylamine moieties and bisdemethoxycurcumin or genistein afforded neuroprotection and displayed neurotrophic effects, as evidenced by an increase in dendritic density of pyramidal neurons in organotypic hippocampal culture. The additional conjugation of the Angiopep-2 targeting moiety enhanced nanoconjugate passage through the blood-brain barrier and modulated brain distribution with nanoconjugate accumulation in neurogenic areas, including the olfactory bulb. Nanoconjugate treatment effectively reduced neurotoxic β amyloid aggregate levels and rescued impairments to olfactory memory and object recognition in APP/PS1 transgenic AD model mice. Overall, this study provides a description of a targeted multimodal polyglutamate-based nanoconjugate with neuroprotective and neurotrophic potential for AD treatment.
INTRODUCTION
Alzheimer’s disease (AD) is an age-related, irreversible form of dementia characterized by the progressive deterioration of cognitive capacity (1). AD represents a growing concern in developed countries, with nearly 50 million people affected; moreover, the incidence of AD will inevitably increase due to the ongoing expansion of the aged population, thereby negatively affecting global health care systems (2). The accumulation of β amyloid (Aβ) peptide in toxic oligomers and amyloid plaques in the brain plays a crucial role in AD in a pathogenic cascade involving tau aggregation, synaptic dysfunction, neuronal death, and loss of cognitive capacity (3). It is now clear that AD is a multifactorial disease with several additional pathogenic mechanisms, including inflammation, oxidative damage, iron dysregulation, mitochondrial dysfunction, and altered cholesterol metabolism (4). Current AD treatments generally fail to modify the course of the disease; instead, most therapeutics merely delay the onset of symptoms. Disease-modifying therapies (DMTs) under investigation tend to be monomodal in nature, often designed to specifically tackle Aβ accumulation (5), and suffer from problems related to the presence of the blood-brain barrier (BBB), which inhibits the passage of many therapeutics to the affected regions of the brain. Overall, therapeutic strategies that can pass through the BBB and modulate multiple disease-associated pathways may provide an enhanced means to treat diseases such as AD (6). To this end, we have now designed, characterized, and biologically evaluated targeted multimodal polypeptide-based nanoconjugates for the treatment of AD.
We used polyglutamic acid (PGA), a water-soluble, biodegradable, and multivalent polypeptide with high loading capacity, and demonstrated clinical benefits (7, 8), as the nanocarrier for our multimodal treatment approach. Because of a highly controlled and versatile polymerization process and proven structure-activity relationships (9), linear PGA (as a homopolymer or in multiblocks) is a key component of polymer-drug conjugates and polymeric micelles (some in an advanced clinical stage) used in cancer treatment and tissue regeneration (8). PGA-based globular structures, such as our recently described sphere-like cross-linked self-assembled star-shaped PGAs (St-Cl), exhibit extended blood circulation times and, therefore, represent an ideal option to maximize passive targeting (9). In addition, St-Cl exhibit sizes of 80 to 100 nm, which lies within a size range (>5 and <200 nm) that supports transport across biological barriers (6), and possess a large surface for ligand conjugation and exposure.
Multimodal therapies for AD under clinical evaluation have used drug combinations that target different pathways, such as cromolyn and ibuprofen (ALZT-OP1, AZTherapies Inc., phase 3) (10) or dextromethorphan and quinidine (AVP-923/Nuedexta, Avanir Therapeutics, phase 4) (11) among notable others (12). Following this strategy, we modified St-Cl-PGAs by postpolymerization approaches through rationally designed linkers (12, 13) to bear a combination of neuroprotective propargylamine residues (Pr) and naturally occurring compounds that target different AD pathological pathways—either bisdemethoxycurcumin (BDMC), a polyphenolic curcuminoid derived from Curcuma longa (14), or genistein, an isoflavone obtained from soybeans among other plants (15).
The Pr moieties present in monoamine oxidase–B (MAO-B) inhibitors such as Selegiline (Plurimen, Orion Pharma), Rasagiline (Azilect, Teva Inc), M30, and, more recently, ASS234 and Contilisant (as preclinical examples) (16) act as neuroprotective components. The inhibition of MAO-B in glia affects the AD pathophysiological cascade (17) by promoting anti-inflammatory or antiapoptotic responses, stabilizing mitochondrial membrane potential, increasing levels of neurotrophic factors, and influencing amyloid precursor protein (APP) processing (18). In addition, propargylamine itself has demonstrated to provide neuroprotective and neurorestorative effects independently of MAO-B inhibition (19).
Curcuminoids are multimodal drugs that selectively bind to Aβ plaques to inhibit/disrupt fibril formation (20–23), clear phosphorylated tau protein (22), counteract acetylcholinesterase and β-secretase inhibitors (43), antagonize multiple steps of the inflammatory cascade (25), and exert potent antioxidant activity (26). Said effects attenuate cognitive deficits, neuroinflammation, and plaque pathology in AD models (21). Isoflavones, and genistein in particular, have been evaluated for the prevention of osteoporosis and cardiovascular diseases (27), as antioxidants, anti-inflammatories, and anticancer agents (28) and as a means to ameliorate postmenopausal syndrome (29). Genistein intake improves spatial learning and memory in AD animal models (30) and is now under clinical evaluation in patients with AD (NCT01982578, phase 2). Genistein functions, in part, by inducing the peroxisome proliferator–activated receptor γ–mediated increased release of apolipoprotein E by astrocytes to clear Aβ plaques from the brain (31, 32). Genistein also modulates tau pathology by blocking intracellular calcium levels (33), acts as a scavenger of reactive oxygen species, and prompts an anti-inflammatory response (34).
To enhance the ability of multimodal polypeptide-based nanoconjugates to cross the BBB, we modified our nanocarrier to also carry Angiopep-2 (ANG), a specific peptide ligand for the low-density lipoprotein receptor–related protein 1 (LRP1) that promotes transcytosis from blood to brain and thereby represents a target for delivery of substances into the central nervous system (CNS) (7, 35, 36). In AD, LRP1 expression in the brain endothelium mediates the rapid removal of Aβ from the brain via transport across the BBB, making LRP1 a therapeutic target (35). Although LRP1 becomes down-regulated in late-stage AD, patients with early-stage AD present with high expression levels of LRP1 in human brain endothelium, neurons, and activated astrocytes that surround Aβ plaques (37). Thus, we hypothesize that the expression of LRP1 in the CNS and its presence in Aβ plaques in both the APP/PS1 transgenic (TG) AD mouse model and the human AD brain makes ANG an optimal ligand choice for our early-stage neuroprotective approach (38).
Our results demonstrate that targeted multimodal polypeptide-based nanoconjugates effectively pass through BBB, diffuse through the brain, and become internalized by disease-relevant cell types within the CNS. By means of an exhaustive study with BDMC as conjugated drug, we achieved proof of concept for our CNS targeted nanoconjugates, a strategy that was further validated in vivo with our second selected drug, genistein. Furthermore, studies in the AβPPswe/PS1dE9 (APP/PS1) AD mouse model provide biochemical and behavioral evidence for enhanced therapeutic outcomes following treatment with our nanoconjugates when compared to treatment with unconjugated and untargeted drugs. Overall, we believe that our LRP1-targeted multimodal PGA-based nanoconjugates may represent a platform for the development of enhanced therapies for AD and other disorders of the CNS.
RESULTS
Targeted cross-linked star-polyglutamates cross the BBB and become internalized by cells of the CNS
Our previous results evaluating different forms of PGA suggested that larger architectures provided for enhanced blood residence time, a key feature that can influence targeting (9). St-Cl (Fig. 1) (diameter of 80 to 100 nm) exhibits a size (>5 and <200 nm) that promotes passage through biological barriers (6) with a large surface for ligand conjugation/exposure and a suitable elimination time (t1/2ß of 115 hours) (9). The three-dimensional structure is based on the self-assembly and subsequent click chemistry–mediated cross-linking of orthogonal star PGA chains containing either azide or alkyne (propargylamine) units. We modified the PGA backbone with an excess of Pr before self-assembly and cross-linking through amide bonds to afford 6% mol (of glutamic acid units) of free Pr, generating St-Cl-Pr. As neuroprotective effects have been attributed to Pr residues within existing neuroprotective drugs such as Rasagiline (18), we hypothesize that Pr does not necessarily need to be released from the structure to exert its effects. Furthermore, given the intrinsic biodegradability of PGA by lysosomal proteases, we chose the straightforward amide linker for Pr conjugation to PGA.
Fig. 1. Schematic representation of the polymeric structures used within this study.
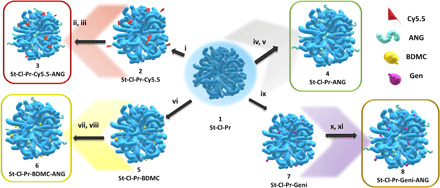
(i, vi, and ix) N,N′-diisopropylcarbodiimide/4-dimethylaminopyridine, N,N-dimethylformamide, 72 hours, room temperature. (ii, iv, vii, and x) 4-(4,6-Dimethoxy-1,3,5-triazin-2-yl)-4-methylmorpholinium chloride, cysteamine pyridyl dithiol, H2O, 24 hours, room temperature. (iii, v, viii, and xi) Hepes buffer (pH 7.4), 16 hours, room temperature.
We next labeled St-Cl-Pr with the near-infrared fluorescence dye Cy5.5 through an amide linker to allow in vivo tracking (St-Cl-Pr-Cy5.5, Fig. 1) and ANG via a bioresponsive disulfide linker (St-Cl-Pr-Cy5.5-ANG, Fig. 1) (8, 13). All conjugates displayed a similar size (around 40 nm in diameter) and z potential (−30 to −40 mV) according to dynamic light scattering measurements and corroborated by cryo–transmission electron microscopy (TEM) images (further details regarding conjugate characterization such as dye/peptide loading can be found in table S1 and fig. S1).
We evaluated whole body biodistribution in wild-type (WT) mice up to 24 hours after intravenous administration of St-Cl-Pr-Cy5.5 and St-Cl-Pr-Cy5.5-ANG to explore compound fate and the possible enhancement of brain accumulation by ANG (see fig. S2). While both St-Cl-Pr-Cy5.5 and St-Cl-Pr-Cy5.5-ANG exhibited renal excretion profiles, the inclusion of ANG prompted higher conjugate accumulation in organs such as the liver and kidney at later time points; however, we did not observe any toxicity, as evidenced by evaluating potential weight loss. St-Cl-Pr-Cy5.5-ANG offered more substantial brain accumulation at early time points when compared to nontargeted St-Cl-Pr-Cy5 [≈1.5% injected dose (ID)] (see figs. S2 and S3) and a clear brain distribution in different areas (fig. S3). To note, the presence of conjugated BDMC did not significantly alter the ability of St-Cl-Pr-Cy5.5-ANG to reach the brain (≈1.3% ID for St-Cl-Pr-BDMC-Cy5.5-ANG) (see fig. S2C).
To better understand the brain distribution of St-Cl-Pr-Cy5.5-ANG in an AD scenario, we performed a brain biodistribution study in the APP/PS1 double TG AD mouse model after intravenous injection of the St-Cl-Pr-Cy5.5-ANG through the tail vein. We observed the diffusion of St-Cl-Pr-Cy5.5-ANG throughout the brain vasculature into the brain parenchyma, suggesting an efficient passage through the BBB (Fig. 2A). Histological analysis revealed the presence of the compound in different brain areas, including the cerebral cortex, striatum, corpus callosum, hippocampus, ventricular-subventricular zone, rostral migratory stream, and olfactory bulb (Fig. 2, B to D). In general, we observed St-Cl-Pr-Cy5.5-ANG in the vascular walls, with a prominent signal in endothelial cells and astrocyte endfeet processes enwrapping blood vessels. We also encountered St-Cl-Pr-Cy5.5-ANG in the cytoplasm of astrocytes, neurons, and microglia within the brain parenchyma. Within the cerebral cortex, we encountered abundant levels of St-Cl-Pr-Cy5.5-ANG distributed in blood vessel walls and surrounding glial fibrillary acidic protein (GFAP)+ astrocytes participating in the BBB. We also found punctuate foci of St-Cl-Pr-Cy5.5-ANG in the cytoplasm of other parenchymal GFAP+ astrocytes, neuronal nuclear protein (NeuN)+ neurons, and ionized calcium-binding adapter molecule 1 (Iba1)+ microglia (see Fig. 2B). We also discovered the presence of St-Cl-Pr-Cy5.5-ANG in astrocytes, neurons, and microglia in the granule cell layer of the dentate gyrus of the hippocampus and olfactory bulb key neurogenic areas that can prompt neuroregeneration (Fig. 2, C and D) (39). Quantitative analysis enabled us to investigate differences in the distribution of Cy5.5 fluorescent intensity between different brain regions and time points. As expected, fluorescence presented a decreasing trend between short (30 min) and long (4 hours) times after intravenous administration of St-Cl-Pr-Cy5.5-ANG (non-significant). We found that 30 min after administration, Cy5.5 signal was significantly more abundant in layers II/III of the somatosensory cortex than in the hippocampus [P = 0.026; two-way analysis of variance (ANOVA) followed by Šidák’s post hoc test], but we did not find significant differences between the analyzed regions at longer times (Fig. 2E). These results confirm that St-Cl-Pr-Cy5.5-ANG fluorescence remains stable in important brain regions for AD progression for at least 4 hours after injection. Subsequent investigations of the subcellular localization of St-Cl-Pr-Cy5.5-ANG observed the clear colocalization of St-Cl-Pr-Cy5.5-ANG with lysosomes [lysosomal-associated membrane protein 1 (Lamp1), green] in cells of the cerebral cortex and olfactory bulb. These data provide evidence for lysosomotropic drug delivery after conjugate endocytosis (Fig. 2F) (40).
Fig. 2. Neural cell uptake of St-Cl-Pr-Cy5.5-ANG after passage through the BBB.
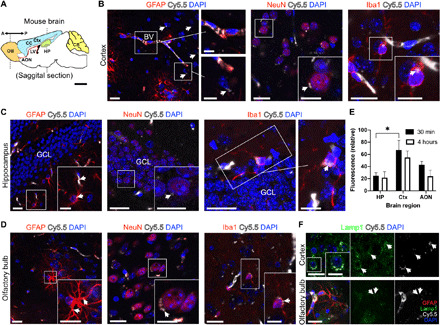
(A) Schematic of an adult mouse brain representing the two antero-posterior coronal levels studied for the presence of St-Cl-Pr-Cy5.5-ANG. AON, anterior olfactory nucleus; Ctx, cerebral cortex; CB, cerebellum; CC, corpus callosum; HP, hippocampus; LV, lateral ventricle; OB, olfactory bulb. (B) The presence of St-Cl-Pr-Cy5.5-ANG in the cerebral cortex, showing abundant accumulation in the blood vessel (BV) walls and surrounding GFAP+ astrocytes (left), and in punctate regions (arrows) in the cytoplasm of NeuN+ neurons (middle) and Iba1+ microglia (right). (C and D) Distribution of St-Cl-Pr-Cy5.5-ANG (arrows) in GFAP+ astrocytes (left), NeuN+ neurons (middle), and Iba1+ microglia (right) in the granule cell layer (GCL) of the dentate gyrus of the hippocampus (C) and the olfactory bulb (D). (E) Quantification of Cy5.5 fluorescent intensity between different brain regions at short (30 min) and long (4 hours) time points after intravenous administration of St-Cl-Pr-Cy5.5-ANG. All data shown are relative fluorescence intensity values corrected for background autofluorescence of the same brain regions in vehicle-injected mice (means ± SEM; n = 4 mice per time point). Two-way ANOVA followed by Šidák’s post hoc test, *P < 0.05. (F) St-Cl-Pr-Cy5.5-ANG (arrows) localizes to lysosomes (Lamp1, green) in cells of the cerebral cortex (top) and olfactory bulb (bottom). Scale bars, 20 μm in panoramic images and 10 μm in insets.
Characterized and in vitro–tested multimodal polypeptide-based nanoconjugates disrupt fibril formation
After confirming the ability of St-Cl-Pr-Cy5.5-ANG to effectively pass through the BBB and become internalized by multiple cell types of various brain regions, we next sought to conjugate St-Cl-Pr with BDMC to create a previously undescribed therapeutic strategy for AD treatment. As previously mentioned, curcuminoids bear enormous potential as multimodal drugs for the treatment of AD; however, the low water solubility, poor oral absorption in both humans and animals, and low systemic bioavailability of curcuminoids such as BDMC have hampered their clinical development. We chose BDMC due to its higher stability under physiological conditions (pH 7.4) compared to other curcuminoids (20, 41). Our synthetic strategy followed a bottom-up approach using St-Cl with the mentioned excess of Pr units followed by the conjugation of BDMC through a biodegradable ester linker (achieving around 14% weight BDMC loading) (13). St-Cl-Pr-BDMC exhibited a hydrodynamic radius of 40 nm and a z potential of around −38 mV (see the Supplementary Materials for further details regarding characterization).
Before proceeding to the evaluation of in vivo efficacy, we first assessed the in vitro ability of our untargeted St-Cl-Pr-BMDC conjugate to maintain/improve the therapeutic effect of the BDMC. Previous studies have established a curcuminoid concentration range of 0.1 to 1 μM as sufficient to induce a therapeutic benefit by diminishing oxidative stress. Moreover, the median inhibitory concentration value for curcuminoids for Aβ aggregation and lipid peroxidation also lies within the concentration range of 0.1 to 1 μM (20, 21).
The evaluation of St-Cl-Pr-BDMC and St-Cl-Pr-BDMC-ANG administration to the SH-SY5Y neuroblastoma model cell line (Fig. 3A) as well as to primary neurons (Fig. 3B) revealed a lack of substantial toxicity up to the concentrations tested (see experimental details in the Supplementary Materials). BDMC release from our nanocarriers relies on a dual mechanism based on both the pH sensitivity of the drug linker and the carrier degradation by enzymes in vivo. PGA is well known to be degraded both in vitro and in vivo by cysteine proteases found in the lysosomes, with cathepsin B playing a central role (42). Moreover, we have already demonstrated the degradation of star-shaped PGA by incubation with cathepsin B (43). Thus, we studied the effect of the linker’s pH sensitivity by following the kinetics of BDMC release from St-Cl-Pr-BDMC under hydrolytic conditions. Therefore, we incubated the samples of the nontargeted St-Cl-Pr-BDMC at 37°C and at different pH values including 5.0 (lysosome), 6.5 (endosome), and 7.4 (blood) for up to 96 hours. We observed a sustained and controlled BDMC release profile, as analyzed by high-performance liquid chromatography (HPLC), with approximately 20% of conjugated BDMC released within the first 48 hours at pH 5.0, thereby demonstrating the capacity for lysosomotropic drug delivery (Fig. 3C); however, pH values of 6.5 and 7.4 displayed a slower release profile (Fig. 3C), an indication of physiological stability. While these experiments provided information regarding the contribution of pH sensitivity to drug release, we discovered a plateau in our method of extraction and quantification of BDMC, deriving from a threshold concentration of free drug that drives the precipitation of the sample due to hydrophobic forces. Therefore, this influence can lead to miscalculations in the cumulative drug release. However, in the in vivo settings, we envisage complete release of BDMC from the nanoconjugates due to this dual mechanism.
Fig. 3. In vitro cell viability, drug release, and fibril formation inhibition capacity of St-Cl-Pr-BDMC.
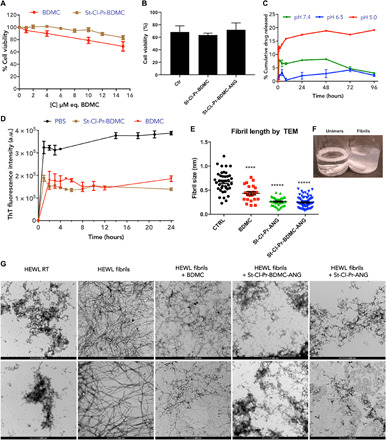
(A) Cell viability of BDMC and St-Cl-Pr-BDMC in the SH-SY5Y cell line as measured by MTS cell viability assay. n > 3, means ± SEM. (B) Cell viability of St-Cl-Pr-BDMC and St-Cl-Pr-BDMC-ANG in primary neural culture as measured by PI-labeled nuclei. n > 3, means ± SEM. (C) Drug release profiles at different pH values (5.0, 6.5, and 7.4) for St-Cl-Pr-BDMC. Time course experiments were carried out in triplicate. n > 3, means ± SEM. (D) ThT fluorescence intensity changes over time in HEWL samples incubated with BDMC and St-Cl-Pr-BDMC at 10 μM BDMC-equivalents n > 3, means ± SEM. a.u., arbitrary units. (E) Quantification of fibril length obtained by analyzing TEM micrographs using ImageJ. Photo credit: Aroa Duro-Castaño, CIPF. (F) Image depicts an example of unimers and fibrils used in the experiments. (G) Images of HEWL unimers and HEWL fibrils upon heating at 60°C and vigorous stirring for 24 hours (pH 2.0) (20). TEM image visualization of the effect of free BDMC, St-Cl-Pr-ANG and St-Cl-Pr-BDMC-ANG on HEWL fibril formation or disruption. Scale bars, 500 nm, in all cases, apart from HEWL fibrils (second-down), 200 nm. Photo credit: I. Conejos-Sánchez (CIPF).
We next used hen egg white lysozyme (HEWL) and Aβ1–42 peptide as model systems for amyloid formation to provide proof of concept for the in vitro interaction of St-Cl-Pr-BDMC with fibrils (44, 45). HEWL fibrillation is one of the best known and accepted models to study protein aggregation and amyloid cytotoxicity (44, 46). Moreover, this model has been extensively applied to study the interaction among curcuminoids and amyloid fibrils and to investigate the potential of curcuminoids as therapy against amyloid fibrillation (47). Therefore, we first compared the activity of 10 μM drug-equivalents St-Cl-Pr-BDMC and 10 μM free BDMC as inhibitors of fibril formation through the thioflavin T (ThT) fluorescence assay that monitors HEWL fibril dynamics over time (48). We coincubated HEWL during fibril formation [phosphate-buffered saline (PBS) used as negative control], finding that both free BDMC and St-Cl-Pr-BDMC inhibited fibril formation to a similar extent (Fig. 3D) and with comparable results to those obtained in literature (47). Notably, an increase in the concentration to 50 μM drug-equivalents failed to improve our findings (fig. S4). Coincubation of free BDMC during HEWL fibril formation prevented fibril clumping, as clearly observed in the TEM images (Fig. 3, D to F). To verify that the introduction of ANG do not diminish the fibril disruption capacity of the conjugate, we also coincubate with the LRP1-targeted version of St-Cl-Pr-BDMC and St-Cl-Pr-BDMC-ANG (Fig. 1, full characterization in fig. S1 and table S1). This conjugate also led to notably shorter fibrils and the presence of granular nonfibrillar amorphous protein aggregates similar to those observed in the control HEWL sample (not induced to form fibrils). Of particular note, an “empty” nanoconjugate (St-Cl-Pr-ANG) also displayed inherent disruptor/inhibitory activity itself, which may occur from the intercalation of the negatively charged PGA polymer within the fibril structure, as has been described for linear PGA and other polyanions/polycations (49). Again, we encountered shorter fibrils after incubating preformed HEWL fibrils with St-Cl-Pr-ANG (Fig. 3, D and F, and fig. S5A). We confirmed these observations by 1H nuclear magnetic resonance (NMR) (fig. S5B) by analyzing the fibril disrupting activity of St-Cl-Pr-ANG, St-Cl-Pr-BDMC-ANG, and free BDMC by means of monitoring the fraction of solubilized protein upon coincubation. In all cases, treated fibrils resulted in a higher peak intensity, which corresponds to increased protein solubilization (inhibition of fibril formation) in the trend St-Cl-Pr-BDMC-ANG > BDMC > St-Cl-Pr-ANG (fig. S5B). Although St-Cl-Pr-ANG prompted the lowest disparity, we confirmed the significant disruption or inhibition of fibril formation, in agreement with the ThT and TEM studies (Fig. 3 and fig. S4).
To note, the capability of fibril disruption for free BDMC, St-Cl-Pr-ANG, and St-Cl-Pr-BDMC-ANG was corroborated with an Aβ1–42 peptide fibrillation assay, another well-accepted model to study protein aggregation and amyloid cytotoxicity (45). Drug coincubation during Aβ1–42 peptide fibril formation, again, notably prevented fibril clumping as clearly seen in the TEM images, with a slight trend for St-Cl-Pr-ANG > St-Cl-Pr-BDMC-ANG > BDMC (fig. S6).
St-Cl-Pr-BDMC inhibits Aβ-induced neurotoxicity and induces a neurotrophic effect in hippocampal organotypic cultures
Before moving to in vivo studies, we compared the effects of St-Cl-Pr-BDMC and free BDMC exposure in organotypic cultures from the entorhinal cortex hippocampus (50), as these brain regions accumulate the high-density extracellular deposits of Aβ peptide in patients with AD that are partially responsible for the progressive memory loss and cognitive impairment (1, 20). As the BBB component is absent in this culture setup, we used the untargeted St-Cl-Pr-BDMC for these experiments.
We pretreated organotypic cultures with St-Cl-Pr-BDMC and free BDMC before Aβ1–42-triggered injury to evaluate neuroprotection and used propidium iodide (PI) staining to quantify the density of degenerated cells in a given region. In our case, the region of interest is the CA1 region of the hippocampus (cornus ammonis 1), where several studies have found neurodegeneration induced by Aβ peptides (51). Before these experiments, we investigated the adequate uptake and distribution of the nanoconjugates throughout the organotypic slice upon addition to culture media (fig. S7) followed by the viability of the organotypic cultures after a 48-hour treatment with St-Cl-Pr-BDMC in the absence of Aβ peptides to optimize the dose range. In general, St-Cl-Pr-BDMC failed to induce significant changes in PI-positive nuclei density in the stratum pyramidale of CA1, up to 0.5 μM drug-equivalents of St-Cl-Pr-BDMC, where we observed an almost threefold increase in cytotoxicity compared to untreated controls (see Fig. 4A). Encouragingly, the pretreatment of organotypic cultures with St-Cl-Pr-BDMC and free BDMC before the induction of neurotoxicity by Aβ1–42 significantly decreased the density of PI-labeled nuclei when compared with cultures treated with the Aβ1–42 peptide (20) only (P = 0.006 versus P = 0.034 for St-Cl-Pr-BDMC and BDMC, respectively). We failed to observe an effect with the control nanoconjugate (St-Cl-Pr) (Fig. 4B and fig. S8). Overall, this ex vivo study provided evidence that St-Cl-Pr-BDMC can prevent neurotoxicity induced by the Aβ1–42 peptide.
Fig. 4. Analysis of St-Cl-Pr-BDMC capability to inhibit Aβ-induced neurotoxicity and to induce a neurotrophic effect in hippocampal organotypic cultures.
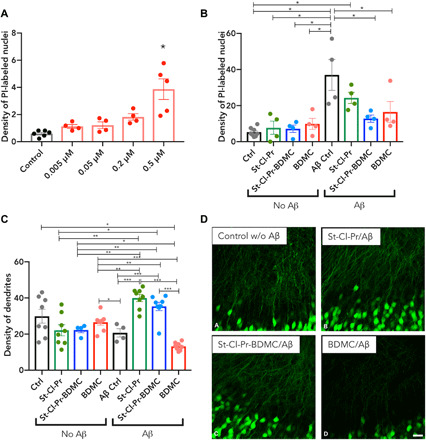
(A) Changes in density (nuclei/1000 μm2) of PI-stained nuclei in the pyramidal layer of the CA1 region of hippocampal organotypic cultures comparing control cultures treated with vehicle and cultures treated with different concentrations of St-Cl-Pr-BDMC (0.005, 0.05, 0.2, and 0.5 μM drug-equivalents). Asterisks indicate statistically significant differences after ANOVA analyses followed Bonferroni’s post hoc tests. n > 3, means ± SEM (20). (B) Changes in the density (nuclei/1000 μm2) of PI-stained nuclei in the pyramidal layer of the CA1 region of hippocampal organotypic cultures when comparing control cultures treated with vehicle and cultures treated with the polymer St-Cl-Pr, St-Cl-Pr-BDMC, or free BDMC (0.05 μM). The distinct groups were subsequently treated with vehicle (no Aβ) or Aβ1–42 peptide (Aβ). Asterisks indicate statistically significant differences after ANOVA analyses followed Bonferroni’s post hoc tests. n > 3, means ± SEM (20). (C) Graph representing the changes in dendrite density of CA1 pyramidal neurons measured in the stratum radiatum (optical density, arbitrary units) of hippocampal organotypic cultures when compared with control cultures treated with St-Cl-Pr, St-Cl-Pr-BDMC, or free BDMC (0.05 μM). The distinct groups were subsequently treated with vehicle (no Aβ) or Aβ1–42 peptide (Aβ). Asterisks in bars indicate statistically significant differences between groups after ANOVA analyses followed by Tukey or Games-Howell post hoc tests. (D) Confocal microscopic analysis of dendrite density of pyramidal neurons in the stratum radiatum of CA1 of hippocampal organotypic cultures. All microphotograph images are derived from single confocal planes. Scale bar, 25 μm.
To confirm the safety of our nanoconjugates, we also measured any abnormal increase of astrocytes (astrogliosis) and microglia (microgliosis), which reflects the induction of inflammation, through the immunohistochemical detection of GFAP and the histochemical detection of Tomato lectin. Notably, treatment of organotypic cultures with the free BDMC or St-Cl-Pr-BDMC failed to induce significant increases in GFAP+ reactive astrocytes or microglial cell density in the absence or presence of the Aβ1–42 peptide (fig. S9), which ensured an adequate therapeutic window for further studies.
To also evaluate the neurotrophic effect of the nanoconjugates, we used organotypic cultures from slices containing the entorhinal cortex and the hippocampus of Thy1–yellow fluorescence protein (YFP) TG mice, which display fluorescent excitatory neurons in the CA1 region of the hippocampus, to quantify dendritic density (20). We indirectly measured modifications to dendritic arborization via the quantification of YFP fluorescence intensity in the stratum radiatum of CA1. Treatment of organotypic cultures with St-Cl-Pr, St-Cl-Pr-BDMC, or free BDMC in the absence of Aβ1–42 peptide–induced neurotoxicity failed not only to produce deleterious effects on dendritic spine density in the basal dendrites of CA1 pyramidal neurons but also to elicit neurotrophic effects (Fig. 4, C and D, and figs. S10 to S12). Unexpectedly, we failed to observe a significant reduction in dendritic density in organotypic cultures following treatment with Aβ1–42 peptide (Fig. 4, C and D, and fig. S10), which should promote deleterious effects due to its toxicity. However, while we failed to observe any neurotrophic effects when analyzing Aβ1–42 peptide–treated cultures pretreated with free BDMC, pretreatment with St-Cl-Pr and St-Cl-Pr-BDMC before Aβ insult resulted in a robust and significant increase in dendritic density (P > 0.001 in both cases compared to those receiving only Aβ1–42 and P > 0.001 compared to free BDMC treatment) (Fig. 4, C and D). These results may indicate that St-Cl-Pr and St-Cl-Pr-BDMC exert neurotrophic activities when primed by Aβ1–42.
Given the lack of effect of free BDMC and the neurotrophic effect of St-Cl-Pr, we suggest that the Pr moieties within the nanocarrier [in the knowledge that MAO-B inhibitors increase the levels of neurotrophic factors (18)] may promote this neuroprotective effect. Furthermore, PGA, as a polyanionic carrier, may trigger a “scavenger effect,” as shown by TEM and 1H NMR (Fig. 3 and fig. S5). The sequestration of the Aβ1–42 peptide by the PGA component of St-Cl-Pr before triggering any apoptotic signaling should be further explored to understand this behavior fully; however, we did not observe significant numbers of degenerated dendrites (aberrant dendrites presenting frequent swellings and disruption of the fluorescence) in organotypic cultures under any conditions (figs. S10 and S12).
In vivo evaluation in a AβPPswe/PS1dE9 Alzheimer’s murine model demonstrates the ability of St-Cl-Pr-ANG to efficiently carry drug cargoes across the BBB
The promising results obtained in the organotypic cultures encouraged us to evaluate the ability of our developed conjugates to ameliorate amyloid deposition and improve memory and cognition in the double TG AβPPswe/PS1dE9 (APP/PS1) mouse AD model. Amyloid plaques deposits are the main hallmark of AD, and the APP/PS1 mouse displays progressive amyloidosis and begins to develop plaques by 6 months with abundant deposits in the hippocampus and cortex by 9 months (52). APP/PS1 mice also suffer from progressive olfactory dysfunction by 8 months, which correlated with cell degeneration due to high levels of Aβ in the olfactory epithelium and bulb (53). Although often unnoticed, olfactory deficits are considered early clinical presentations of neurodegeneration with high prevalence in several forms of dementia, reaching up to 100% in AD (54). The APP/PS1 mouse also develops synaptic loss by the early age of 4 months and suffers from behavioral and cognitive changes at 8 months that worsen at 12 months (55). We used 8-month-old APP/PS1 mice that display early symptoms of AD to achieve proof of concept of early treatment therapy with our nanoconjugates. To note, significant BBB-enhanced permeability has been only detected in APP/PS1 animals older than 12 months (56, 57).
To validate the versatility and effectiveness of targeted multimodal polypeptide-based nanoconjugates to bypass the BBB, we also assessed the conjugation/delivery of genistein as an alternative drug to BDMC with a different mechanism of action. Genistein, a well-known natural isoflavone from soybeans, exerts antioxidant and neuroprotective effects when orally administered to the APP/PS1 mouse model (32). We conjugated genistein through a biodegradable ester linker as for BDMC, and fully characterized the resultant nanoconjugate (St-Cl-Pr-Geni-ANG) (Fig. 1 and table S1).
Analysis of nanoconjugate efficiency used several treatment groups in two mouse strains from the same genetic background; untreated WT mice, untreated TG mice, TG mice intravenously injected with St-Cl-Pr-ANG, St-Cl-Pr-BDMC-ANG, free BDMC, or St-Cl-Pr-Geni-ANG; and TG mice administered with free genistein via oral gavage (see Materials and Methods for experimental details) (31). We failed to observe any signs of toxicity (animal weight loss) in any animal during the entire experiment (fig. S13). Toxicity was also analyzed by determining plasma levels of lactate dehydrogenase (LDH) (fig. S14A), creatinine (fig. S14B), and serum-based liver function markers [aspartate aminotransferase (AST) and alanine aminotransferase (ALT)] (fig. S14, C and D). We did not find any significant differences between the different groups, indicating the safe profile of the tested nanoconjugates. The different treatments significantly inhibited the increased plasma levels of LDH, creatinine, and AST-to-ALT activity ratio observed in TG mice, with values returning to those observed in WT animals (fig. S14). Of note, studies have described a crucial role for the liver in AD-associated metabolic dysfunction and an association between elevated AST-to-ALT activity ratio and AD (58). Upon treatments, the AST-to-ALT ratio encountered was reduced even up to the basal WT level; therefore, this could support the neuroprotective role of the tested conjugates (fig. S14D).
We also evaluated the levels of Aβ1–40 and Aβ1–42 toxic peptides in the brain (Fig. 5, A and B, respectively). We found a statistically significant reduction in Aβ1–40 levels for both St-Cl-Pr-BDMC-ANG and St-Cl-Pr-ANG (P ≤ 0.05 and 0.01, respectively, compared with untreated TG). As discussed previously (Fig. 3 and fig. S5), the apparent therapeutic effect of St-Cl-Pr-ANG may derive from electrostatic interactions. Upon analysis of Aβ1–42 levels, we found a decreasing trend in Aβ1–42 concentrations in total brains (Fig. 5B) after all treatments with the cross-linked carrier, BDMC (free or conjugated), and genistein (free or conjugated). However, we only observed statistical significance for free genistein treatment (P ≤ 0.05) compared to untreated TG mice (Fig. 5B).
Fig. 5. In vivo activity of LRP1-targeted conjugates bearing BDMC or genistein in AβPP/PS1 Alzheimer’s murine model.
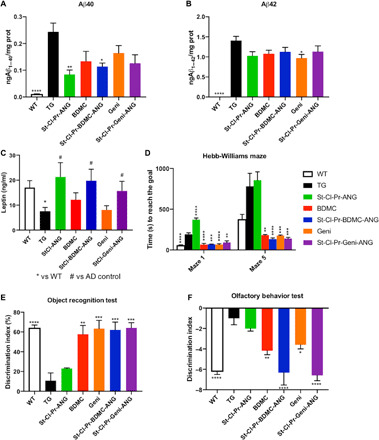
(A and B) Aβ40 or Aβ42 levels determined in whole brain homogenates by enzyme-linked immunosorbent assay (ELISA). (C) Plasma leptin levels determined by the Quantikine ELISA Leptin Immunoassay Kit. (D) Hippocampal learning evaluated using the Hebb-Williams maze. (E) Recognition memory studied using the novel object recognition test. (F) Olfactory deficits assessed by the odor habituation test. Data are expressed as means ± SD. Significance is shown as *P ≤ 0.05, **P ≤ 0.01, ***P ≤ 0.001, and ****P < 0.0001 versus TG, n = 5 to 10 animals per group.
The adipokine leptin may also be considered an interesting plasma marker in AD incidence as reported in Lieb et al. (59). Leptin can facilitate long-term potentiation and synaptic plasticity in the hippocampus, promote β amyloid clearance, and improve memory function in animal models of aging and AD as well as in human patients. In humans, higher leptin levels were associated with a lower risk of incident dementia and AD (59). As shown in Fig. 5C, we found a statistically significant reduction of plasma leptin levels in TG animals when compare to WT (P ≤ 0.01). While free drugs were not able to significantly alter plasma leptin concentration, all treatments containing St-Cl-Pr-ANG (with and without conjugated drug) significantly enhanced leptin levels compared to TG control group (P ≤ 0.05), suggesting that this preventive effect could be directly correlated with our polypeptidic carrier. Subsequent behavioral evaluation used an analysis of hippocampal learning using Hebb-Williams mazes (Fig. 5D), recognition memory through novel object recognition (Fig. 5E), and olfactory memory via odor discrimination test (Fig. 5F).
We evaluated spatial learning using two training mazes and two different problem mazes (1 and 5), with maze 5 being significantly more difficult than maze 1 (P < 0.001). Untreated TG animals performing maze 1 required significantly more time to reach their goal compared to WT animals (P < 0.0001) while this difference was not as clear in maze 5, where both WT and untreated TG mice performed significantly worse than in maze 1 (Fig. 5D). Encouragingly, TG mice treated with free BDMC, free genistein, St-Cl-Pr-BDMC-ANG, and St-Cl-Pr-Gen-ANG needed a significantly shorter time to complete both mazes when compared to untreated TG mice (P < 0.001), although the nanocarrier St-Cl-Pr-ANG failed to induce any improvements in maze completion times in TG mice (Fig. 5D). There existed no significant differences between the mice treated with free BDMC, free genistein, St-Cl-Pr-BDMC-ANG, and St-Cl-Pr-Gen-ANG, who all displayed results similar to WT untreated animals.
Novel object recognition was expressed using the discrimination index (%), a measure that discriminates between new and familiar objects (see the Supplementary Materials). Untreated TG mice (10.7 ± 15.6%) suffered from a significant impairment in recognition memory compared to untreated WT mice (64.3 ± 5.6%) (Fig. 5E); however, treatment of TG mice with free BDMC, free genistein, St-Cl-Pr-BDMC-ANG, and St-Cl-Pr-Geni-ANG, but not St-Cl-Pr-ANG, prompted a return to values similar to the untreated WT mice (57.8 ± 17.8% for BDMC, 63.4 ± 18.6% for genistein, 62.0 ± 19.5% for St-Cl-Pr-BDMC-ANG, 64.0 ± 12.2% for St-Cl-Pr-Geni-ANG, and 23.0 ± 1.0% for St-Cl-Pr-ANG) (Fig. 5E).
While often overlooked, olfactory loss represents one of the first clinical events during AD progression in patients and occurs in 8-month-old APP/PS1 mice (60). An evaluation of odor habituation (see the Supplementary Materials) revealed significant olfactory deficits in untreated TG mice when compared to untreated WT mice (P < 0.0001) (Fig. 5F). While treatment of TG mice with St-Cl-Pr-ANG failed to significantly influence olfactory parameters when compared to untreated TG mice, free BDMC (P < 0.01), free genistein (P < 0.05), and St-Cl-Pr-BDMC-ANG and St-Cl-Pr-Geni-ANG (P < 0.0001 in both cases) significantly restored olfactory memory as demonstrated by a reduced discrimination index. Encouragingly, only St-Cl-Pr-BDMC-ANG and St-Cl-Pr-Geni-ANG treatment induced values similar to those encountered in the untreated WT mice (Fig. 5F).
DISCUSSION
Even given the ongoing exploration for DMTs for AD, patient outcomes remain poor, partly due to the multifactorial nature of the disease and the presence of the BBB as an obstacle to therapeutic agents. Any improvements in cognition with current drugs under study remain insufficient and are usually accompanied by severe side effects (5). In this respect, curcuminoids are well-tolerated compounds that perform as multimodal drugs, acting at the majority of AD pathological pathways (61), by impeding Aβ fibril clumping and promoting their disruption (21), acting as nonsteroidal anti-inflammatory drugs that modify microglial activity (61), anti-oxidants (26), AChE inhibitors and β-secretase inhibitors (24), reducing phosphorylated tau protein burden (22), and improving neuronal density and BBB stability (63). However, the low systemic bioavailability of curcuminoids (mainly due to low water solubility and low stability) negatively affects clinical outcomes.
In an attempt to address the abovementioned problems, a variety of curcumin nanoformulations from adjuvants to micelles, dendrimers, nanoparticles, liposomes, or nanocrystals have been exploited for the treatment of neurodegenerative diseases (23). Some have demonstrated potential in terms of increased curcumin bioavailability and transport across the BBB. For instance, curcumin gold nanoparticles can inhibit amyloid fibrillation and dissolve amyloid fibrils (64). Poly(n-butylcyanoacrylate) nanoparticles were reported to enhance the transport of curcumin to the brain tissue and extend the therapeutic time (65). Curcumin nanoliposomes have demonstrated to bind Aβ plaques in postmortem AD brains (66), as well as selectively bind and inhibit the aggregation of Aβ in vitro (67). An encapsulated curcumin formulation (Nanocurc) based on micellar aggregates of cross-linked and random copolymers of N-isopropylacrylamide, with N-vinyl-2-pyrrolidone and poly(ethyleneglycol)monoacrylate (PEG-A), has shown increased curcumin concentration as well as antioxidant activity in AD mouse brains (68). In a more recent study and relevant to our results, boronic acid and curcumin loaded targeted micelles based on PEG-polylysine showed an improvement in memory and Aβ burden in vivo after intravenous weekly administration for over 3 months, due to a synergy between the active agents (62). Nevertheless, most of them rely on the use of biopersistent or long-term degrading nanomaterials, which represents a limitation for the treatment of chronic conditions such as AD. In this line of research, we have used a biodegradable polypeptide carrier (PGA) as an alternative means of improve drug pharmacokinetics and pharmacodynamics, to facilitate multimodal treatment approaches, and to enhance passage through the BBB. Polypeptide-based nanocarriers demonstrate huge potential in overcoming barriers to brain delivery with a number of preclinical examples using ligand-installed approaches summarized in the recent literature (36). PGA carriers have proven biodegradability, biocompatibility, adequate bodily secretion, and affordable industry scalability, while a U.S. Food and Drug Administration orphan-drug designation for glioblastoma for linear PGA aided bench-to-bedside translation (8). Our rationally designed star-shaped cross-linked PGA-based carriers (St-Cl) (9) allow for greater circulation time and higher brain accumulation after functionalization with the ANG targeting agent, an advanced ligand that targets LRP1 that has been evaluated in clinical trials (69). We also included neuroprotective and neuro-restorative propargylamine (Pr) groups as part of the multivalent design (5).
The conjugation of ANG to a nanocarrier can allow efficient BBB crossing via noninvasive transcytosis through interaction with LRP1 on the surface of the endothelial cells of the BBB (70). ANG affords higher transcytosis and brain parenchymal accumulation than transferrin, lactoferrin, (71) and the transport of large cargos (70, 72). For instance, ANG-conjugated paclitaxel (ANG1005) is now in phase 3 clinical trials for brain tumors (69). In AD, the use of endothelial LRP1 as a brain shuttle to cross the BBB has not been reported widely as this strategy has proven challenging due to the ongoing controversy regarding LRP1 expression in AD. Studies support a cell type–dependent dysregulation of LRP1 expression in AD that changes during disease progression. Animal models of aging and AD show a cell type–dependent increase (73) or decrease of LRP1 expression (74). Although in advanced AD progression LRP1 expression significantly drops, contributing to Aβ retention and cognition impairment, studies on early AD stages show high expression of LRP1 in brain endothelium (75) as well as in neurons and activated astrocytes surrounding senile plaques (37). Thus, in our aim to develop neuroprotective therapies for AD, the expression of LRP1 in the BBB/brain in early-stage AD and its presence in amyloid plaques provide a basis for the selection of ANG as an adequate targeting strategy (38).
There is no consensus on required ligand density (i.e., peptide quantification) to promote sufficient BBB entry (70). Preclinical data regarding the ID per gram (% ID/g) achieved in brain tissue are often missed or misleading and use many different measurement techniques; however, numerous studies have reported ID/g values up to <7% (76), with even very low percentages boosting activity by several orders of magnitude (69). Analysis in WT mice and in an APP/PS1 AD mouse model provided evidence that our LRP1-targeted nanocarrier with the chosen ANG % penetrates the BBB and diffuses through the brain where it becomes internalized by various CNS cell types, likely through an endocytic pathway as we observed lysosomal colocalization. Our targeted nanocarrier displayed abundant localization in the brain vasculature, with substantial levels of fluorescence signal observed in blood vessel walls in the APP/PS1 mouse model. This vasculature distribution, which included brain regions such as the cortex and neurogenic areas such as the hippocampal dentate gyrus or the olfactory bulb, suggests a possible use of St-Cl-Pr-ANG as a drug depot, providing a sustained drug supply from the endothelium to CNS cells. All the cell types analyzed contained internalized carrier in their cytoplasm, including neurons, astrocytes, and microglia. The nanocarrier frequently colocalized with Lamp1, supporting the notion that the polymer is preferentially uptake by endocytic specialized cell types in AD, such as astrocytes and microglia, rather than neurons (77). Although proof of concept of BBB entry was achieved in this study with the selected ANG %, our design could benefit in the future of the improvement in BBB crossing by fine-tuning of the ligand density in our nanocarrier as reported in recent models (78). Multivalency provided by the use of our nanocarrier could be exploited to achieve binding only to cell targets with certain number of receptors within a specified range (range selectivity) (78), thus increasing selectivity and minimizing off-target effects.
We observed inherent fibril disruption activity of St-Cl-Pr/ St-Cl-Pr-ANG in vitro and in vivo, where our carrier notably decreased Aβ1–40 levels in the APP/PS1 mouse model. In addition, in vivo treatment with the nanoconjugates, returned the expression of key plasma AD markers, such as the AST-to-ALT ratio (58) or leptin (59), which displayed significantly alterations in the TG APP/PS1 mice, to levels observed in WT mice. This clearly supports the neuroprotective role of the tested conjugates. Also, St-Cl-Pr-BDMC-ANG provided neuroprotection upon Aβ peptide insult in hippocampal organotypic cultures and robust enhancement of dendritic density of pyramidal neurons not achieved by the free form of the drug. The multimodality of St-Cl-Pr-BDMC-ANG obtained promising results in cognition in vivo, ameliorating symptoms of the early onset of AD, such as olfactory dysfunction, lost on recognition memory and hippocampal learning in 8-month-old APP/PS1 AD mice. Notably, St-Cl-Pr-BDMC-ANG–treated APP/PS1 mice fully recovered olfactory capacity when compared to treatment with the free drugs or St-Cl-Pr-ANG. One possible explanation for this result is the abundant fluorescent signal of the labeled version of the conjugate detected in the olfactory bulb, ventricular-subventricular zone, and rostral migratory stream (Fig. 2), all of which are involved in olfactory memory and plasticity through olfactory bulb neuronal turnover (39, 53, 54). This result together with the good safety profile could be an indicator of successful early therapy. Our strategy’s versatility was validated with a second drug, genistein, whose respective conjugate exhibited similar significant results in vivo.
Overall, our findings support St-Cl-Pr-ANG as an excellent platform for the construction of multivalent neuroprotective/neurotrophic therapeutic approaches for early-stage AD, as well as additional combinations of active agents for the treatment of other neurological disorders. The differential brain biodistribution with prominent presence in neurogenic regions suggests additional applications for this targeted polypeptidic carrier as a good candidate to enhance the performance of a multivalent therapeutic approach, not only by facilitating the diffusion of the bioactives to the different cells of the CNS but also by acting as a depot, securing the sustained release of the drug(s) during a prolonged time period and, consequently, enhancing drug(s) bioavailability in the brain.
MATERIALS AND METHODS
Materials
All reagent grade chemicals were obtained from Sigma-Aldrich/MERCK (Dorset, UK) and used without further purification unless otherwise indicated. All solvents were of analytical grade and were dried and freshly distilled. Deuterated chloroform-d1, dimethyl sulfoxide-d6, and D2O were purchased from Deutero GmbH (Kastellaun, Germany). Cy5.5 (6S-IDCC) was obtained from Mivenion GmbH (Berlin, Germany). ANG-cysteine Ac-TFFYGGSRGKRNNFKTEEYC was obtained from Selleck Chemicals LLC (Houston, Texas, USA). BDMC was obtained from TCI chemicals (Oxford, UK). Optimem-1 was obtained from Gibco (Dublin, Ireland), and Aβ1–42 was purchased from Tocris Bioscience (Bristol, UK). Aβ1–16 (6E10) monoclonal antibody was purchased from Covance (Harrogate, UK). Preparative size exclusion chromatography was performed using Sephadex G-25 superfine from GE Healthcare (Chicago, Illinois, USA) and PD MiniTrap G-10 columns containing 2.1 ml of Sephadex G-10. Dialysis was performed in a Millipore ultrafiltration device fitted with a 3 or 5 kDa molecular weight cutoff regenerated cellulose membrane (Vivaspin, Sartorius, Goettingen, Germany) (20).
Synthetic procedures and characterization techniques
The synthetic procedures and main characterization techniques are described in full in the Supplementary Materials.
Drug release kinetics
Drug release kinetics of St-Cl-Pr-BDMC were performed at three different pH values (5.0, 6.5, and 7.4) as previously reported (20). Briefly, St-Cl-Pr-BDMC was dissolved at 4 mg/ml in PBS buffer at different pH levels and subsequently aliquoted in 100-μl samples. Samples were incubated at 37°C and freeze-dried at different time points, resuspended in 100 μl of a double distilled water/acetonitrile (50/50), and injected into HPLC (eluent A was double distilled water and eluent B was acetonitrile). Samples were analyzed using the following gradient: from 40% B to 80% B over 20 min using LiChrospher 100 RP 18, 5.0 μm (dimension: length × ID = 125 × 4.0 mm) from Sigma-Aldrich. BDMC retention time was 5.98 min. Experiments were carried out in triplicate. The percentage of drug release was established by performing a calibration curve with BDMC dissolved in double distilled water/acetonitrile (50/50) and injected under the same conditions as for the release samples (20).
HEWL lysozyme fibril formation assays (20)
ThT fluorescence assay
HEWL fibrils were formed using HEWL (2 mg/ml) in acidic buffer 12 mM HCl containing 140 nM NaCl and 2.7 mM KCl (pH 2). Samples were stirred at 60°C for up to 24 hours for fibril formation. The kinetics of fibril formation was monitored from time zero using the ThT-based titration method. Typically, a 100 μM solution of ThT was prepared by dissolving ThT powder in PBS (pH 7.4). Aliquots of 20 μl of HEWL solution were collected at various intervals and mixed with 100 μl of ThT solution in a dark 96-well plate. Samples were left to stabilize for 5 min, and fluorescence was measured at 590 nm in the plate reader Victor Wallace. For the evaluation of compounds in the prevention of fibril formation, fibrils were incubated with 10 or 50 μM BDMC equivalents of the compounds [or free BDMC dissolved in ethanol (maximum ethanol percentage 1% in the final volume)] dissolved in a minimum volume of PBS buffer (pH 7.4). In control samples, the same amount of PBS buffer without compound was added (20).
Transmission electron microscopy
Images were obtained from an FEI Tecnai G2 Spirit transmission electron microscope (FEI Europe, Eindhoven, the Netherlands) using a Morada digital camera (Olympus Soft Image Solutions GmbH, Münster, Germany). For sample preparation, samples were applied directly onto carbon film on 200 mesh square grids in copper. Any excess of the sample was carefully removed, and the grids were immediately stained with one drop of 0.1% phosphotungstic acid for 30 s. Fibril length was measured using ImageJ software, using areas on the grid with a low concentration of fibrils where starting and end terms were clearly defined. Values were analyzed by one-way ANOVA. Statistical significance was set at P ≤ 0.0001. All results are expressed as means ± SEM.
Organotypic hippocampal slice cultures
Transverse hippocampal organotypic slice cultures were obtained from the brain of P7 mouse pups [B6.Cg-Tg(Thy1-YFP)HJrs/J] using a McIIwain Tissue Chopper (350 μM) (20, 79). Briefly, brains were placed into Petri dishes filled with cold (4°C) sterile dissecting medium (1% glucose, 0.2% penicillin/streptomycin, and 0.5% l-glutamine in Gey’s balanced salt sodium). The overlying pia were gently removed, and coronal cuts were used to remove portions of the rostral and caudal poles, leaving the frontoparietal region intact. The right and left cortices were cut simultaneously in the coronal plane at a thickness of 350 μM using a McIlwain Tissue Chopper. Slices obtained were transferred into dissecting medium and separated gently by agitation. Slices containing the hippocampus and entorhinal cortex were placed on moistened translucent membranes of tissue culture inserts (0.4 μM, Millicell-CM, Millipore, Bedford, MA, USA) and immersed in 1 ml of Serum-OPTIMEM culture medium [25% heat-inactivated horse serum, 25% Hank’s balanced salt solution, and 50% Optimem-1 supplemented with glucose (10 μl/ml)]. Three slices were cultured in the same insert, and six inserts were placed together in six-well plates. To ensure that slices from control and treated groups were cultured under identical conditions, three inserts from each plate were designated as the control group and the other three treated groups. Cultures were stored in a humid atmosphere at 37°C in 5% CO2 for 16 days (Heracell 150i, Thermo Fisher Scientific), and the medium was changed three times per week by replacing 0.5 ml of the total 1 ml volume (20).
Organotypic culture viability assays
Compounds under analysis were dissolved in double distilled water and added to the culture media at different concentrations at day 16 of organotypic culture. Compounds were incubated within the cultures for 48 hours. Tissue cultures were then stained with PI (10 μM) for 10 min, washed with PBS (three times), and then fixed with 4% paraformaldehyde (PFA). Tissue samples were again washed with PBS (three times), left to dry, and mounted using Dako glycergel media containing DAPI (4′,6-diamidino-2-phenylindole). Cell damage was assessed by fluorescent image analysis of PI uptake. Cultures were observed with an upright confocal microscope (Leica TCS STED confocal microscope). Images were captured and analyzed using Fiji Image Software (80). After the capture of images, the density of PI nuclei was analyzed in the pyramidal layer of CA1. For each experimental group, means ± SEM were determined, and the resulting values were analyzed by one-way ANOVA with the number of slices as the “n.” Significant effects were further analyzed by Bonferroni post hoc test, using the IBM SPSS statistics software (version 22) (20).
Aβ1–42 neurotoxicity induction in organotypic culture
For the establishment of Aβ1–42 toxicity, 1 mg of Aβ1–42 peptide was dissolved in double distilled water to prepare a stock solution of Aβ1–42 (0.5 mM) that was stored at −20°C. Before use, Aβ1–42 was aggregated at 37°C for 72 hours. To establish the induction of neurotoxicity, on day 16 of the organotypic culture, Aβ1–42 peptide was added by replacing half of the media, obtaining a final concentration of 1 μM. In the case of pretreated cultures (controls with and without Aβ1–42), the St-Cl-Pr-BDMC dissolved in double distilled water was added at different concentrations (0.05 and 0.2 μM) on days 14 (prevention) and 16 (maintenance) (20).
Quantification of dendritic density of Thy1-YFP fluorescent excitatory neurons in organotypic culture
Modifications in dendritic arborization were measured indirectly by quantifying the intensity of Thy1-YFP fluorescence in the stratum radiatum of CA1. Following the same procedure as for neurotoxicity studies described before, treated and untreated organotypic sections were incubated with a 4% PFA solution in phosphate buffer [0.1 M (pH 7.4)] for 30 min. After washing with PBS, stacks of confocal images from the stratum radiatum were obtained with an upright confocal microscope (Leica TSE). We selected single confocal planes in which the complete dendritic trees in stratum radiatum could be identified. Four square regions (5625 μm2) from these images were randomly selected and analyzed using Fiji Image Software. We calculated mean optical densities for each square, and the means ± SEM were determined and the resulting values were analyzed by one-way ANOVA with the number of slices as the n. Significant effects were further analyzed by Tukey or Games-Howell post hoc tests, using IBM SPSS statistics software (version 22).
In vivo animal model
The AβPPswe/PS1dE9 (APP/PS1) mouse model of AD mice (52) is from the C57BL/6J strain genetic background. Mice were obtained from the Jackson laboratory (USA) (http://jaxmice.jax.org/strain/004462.html) and raised in the animal facility of the Faculty of Medicine, Universitat de València (Spain). Animals were housed with ad libitum access to water and food at constant temperature and humidity with a 12-hour light/dark cycle. All animals used in this work were 8-month-old females that were ovariectomized (81) to discount the effect of endogenous estrogens.
Mouse groups were distributed as follows: WT, WT mice with the same genetic background as the AβPP/PS1 mice; TG, AβPP/PS1 mice; TG animals treated with empty targeted polymer (St-Cl-Pr-ANG) solution in sterile PBS at the corresponding polymer concentration of that of St-Cl-Pr-BDMC-ANG, intravenously injected in the lateral tail vein every 2 days for 2 weeks; TG animals treated with St-Cl-Pr-BDMC-ANG solution in sterile PBS at 5 mg/kg eq-BDMC, intravenously injected in the lateral tail vein every 2 days for 2 weeks; TG treated with St-Cl-Pr-Gen-ANG solution in sterile PBS, intravenously injected at 0.022 mg/kg drug equivalents in the lateral tail vein every 2 days for 2 weeks; TG treated with free BDMC in sterile PBS solution with 2.5 (w/v) Tween 20, intravenously injected at 5 mg/kg injected through the lateral tail vein every 2 days for 2 weeks; TG treated with genistein dissolved in water for oral administration at 0.022 mg/kg per day every day for a week.
Dosage was related to mouse body weight before every injection or oral administration. All methods were carried out in accordance with the approved guidelines, and the different experimental designs were approved by the ethics committee of the Universitat de València (codes: A1348072223268, A1361200660910, A1389269795198, and A1402567995618), following the animal care guidelines of the European Commission 2010/63/UE.
Behavioral analysis
Behavioral protocols can be found in the Supplementary Materials.
Determination of Aβ concentration
Aβ concentration in total brain was measured by an Aβ1–40 and an Aβ1–42 enzyme-linked immunosorbent assay (ELISA) kits (both Invitrogen, Camarillo, CA, USA). Sample preparation, processing, and detection were performed according to the manufacturer’s instructions. Statistical analysis used a one-way ANOVA with one variable (treatment) followed by a Tukey multiple comparison post hoc test. Statistical significance was set at P ≤ 0.05. All results are expressed as means ± SEM.
Acknowledgments
We would like to acknowledge CNB-CiB (CSIC) cryo-EM facility, Neuropharmatest for experimental support and helpful discussion on the organotypic culture model and S. P. Atkinson for help in manuscript development. We acknowledge the experimental support of D. Charbonnier, S. Vila, and S. Đorđević from the Polymer Therapeutics Lab at CIPF; P. García-Tárraga at the Laboratory of Comparative Neurobiology of the University of Valencia; M. J. Sanz at the University of Valencia for the Leptin assay; V. Moreno-Manzano from the Neuronal and Tissue Regeneration Laboratory at CIPF for providing with the primary neurons; M. Palomino-Schätzlein from the NMR Service at CIPF; and A. Hernández from the Confocal Microscopy service at CIPF and Alicia Martínez from the Cytomics Service at CIPF. Funding: This work was supported by the European Research Council (grant ERC-CoG-2014-648831 “MyNano” to M.J.V.); the Spanish Ministry of Science, Innovation, and Universities (SAF2016-80427-R to M.J.V., PID2019-108806RB-I00 to M.J.V., PCI2018-093062 to V.H.-P., and PCIN-2017-117 to C.B.); the Valencian Council for Innovation, Universities, Science and Digital Society (PROMETEO/2019/075 to J.M.G.-V. and PROMETEO/2019/097 to J.V.); the EU Joint Programming Initiative “A Healthy Diet for a Healthy Life” (JPI HDHL INTIMIC-085 to C.B.); Donation of the “Asociación de Jubilados La Pobla Llarga” (Spain); and MINECO/FEDER, UE; European Regional Development Fund (ERDF) included in the ERDF operational program of Comunidad Valenciana 2014-2020. Author contributions: A.D.-C. and M.J.V. developed the concept and design of the project. Design, synthesis, and physic-chemical characterization of the nanoconjugates were performed by A.D.-C. Biodistribution studies in WT mice were performed by A.D.-C. AD brain biodistribution was performed by V.H.-P., J.M.G.-V., and A.A. Drug-release profiles and in vitro safety studies were performed by A.D.-C. Fibrillation studies were performed by A.D.-C. and I.C.-S. Organotypic hippocampal cultures were performed by Neuropharmatest. The in vivo model and molecular analyses were performed by C.B., A.A., C.M.-B., and M.I. The behavioral tests were conducted by M.C.B.-G., M.R.-A., and J.M. Analysis and interpretation of the data included the contribution of all authors. M.J.V. coordinated the whole study. The manuscript was written by A.D.-C., I.C.-S., and M.J.V. with contributions of V.H.-P. and C.B. and was revised by A.A., V.H.-P., J.M.G.-V., J.V., and M.J.V. All authors have read and have given approval for the final version of the manuscript. Competing interests: A.D.-C. and M.J.V. are inventors on an EU and U.S. granted patent related to this work filed by CIPF (no. WO2017025298A1 filed 7 August 2015 and published 16 February 2017). The authors declare that they have no other competing interests. Data and materials availability: All data needed to evaluate the conclusions in the paper are present in the paper and/or the Supplementary Materials. Additional data related to this paper may be requested from the authors.
SUPPLEMENTARY MATERIAL
Supplementary material for this article is available at http://advances.sciencemag.org/cgi/content/full/7/13/eabf9180/DC1
REFERENCES AND NOTES
- 1.Bateman R. J., Xiong C., Benzinger T. L., Fagan A. M., Goate A., Fox N. C., Marcus D. S., Cairns N. J., Xie X., Blazey T. M., Holtzman D. M., Santacruz A., Buckles V., Oliver A., Moulder K., Aisen P. S., Ghetti B., Klunk W. E., McDade E., Martins R. N., Masters C. L., Mayeux R., Ringman J. M., Rossor M. N., Schofield P. R., Sperling R. A., Salloway S., Morris J. C.; Dominantly Inherited Alzheimer Network , Clinical and biomarker changes in dominantly inherited Alzheimer’s disease. N. Engl. J. Med. 367, 795–804 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Alzheimer’s Statistics (2021); www.alzheimers.net/resources/alzheimers-statistics/) [accessed 13 January 2021]
- 3.Burns A., Iliffe S., Alzheimer’s disease. BMJ 338, b158 (2009). [DOI] [PubMed] [Google Scholar]
- 4.A. Onur Keskin, N. Durmaz, G. Uncu, E. Erzurumluoglu, Z. Yıldırım, N. Tuncer, D. Özbabalık Adapınar, Geriatric Medicine and Gerontology (IntechOpen, 2019). [Google Scholar]
- 5.Oset-Gasque M. J., Marco-Contelles J., Alzheimer’s disease, the “one-Molecule, One-Target” paradigm, and the multitarget directed ligand approach. ACS Chem. Nerosci. 9, 401–403 (2018). [DOI] [PubMed] [Google Scholar]
- 6.Wechsler M. E., Vela Ramirez J. E., Peppas N. A., 110th Anniversary: Nanoparticle mediated drug delivery for the treatment of Alzheimer’s disease: Crossing the blood-brain barrier. Ind. Eng. Chem. Res. 58, 15079–15087 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Rodriguez-Otormin F., Duro-Castano A., Conejos-Sánchez I., Vicent M. J., Envisioning the future of polymer therapeutics for brain disorders. Wiley Interdiscip. Rev. Nanomed. Nanobiotechnol. 11, e1532 (2019). [DOI] [PubMed] [Google Scholar]
- 8.Melnyk T., Đorđević S., Conejos-Sánchez I., Vicent M. J., Therapeutic potential of polypeptide-based conjugates: Rational design and analytical tools that can boost clinical translation. Adv. Drug Deliv. Rev. 160, 136–169 (2020). [DOI] [PubMed] [Google Scholar]
- 9.Duro-Castano A., Nebot V. J., Niño-Pariente A., Armiñán A., Arroyo-Crespo J. J., Paul A., Feiner-Gracia N., Albertazzi L., Vicent M. J., Capturing “Extraordinary” soft-assembled charge-like polypeptides as a strategy for nanocarrier design. Adv. Mater. 29, 1702888 (2017). [DOI] [PubMed] [Google Scholar]
- 10.AZTherapies - Harnessing the Power of Neuro-Immunology – Pipeline (2021); https://aztherapies.com/pipeline/ [accessed 13 January 2021].
- 11.AVP-923 | ALZFORUM (2021); www.alzforum.org/therapeutics/avp-923; Therapeutics Search | ALZFORUM; www.alzforum.org/therapeutics/search?fda_statuses=&target_types=&therapy_types%5B%5D=36536&conditions%5B%5D=145&keywords-entry=&keywords= [accessed 13 January 2021].
- 12.Barz M., Duro-Castano A., Vicent M. J., A versatile post-polymerization modification method for polyglutamic acid: Synthesis of orthogonal reactive polyglutamates and their use in “click chemistry”. Polym. Chem. 4, 2989 (2013). [Google Scholar]
- 13.Córdoba-David G., Duro-Castano A., Castelo-Branco R. C., González-Guerrero C., Cannata P., Sanz A. B., Vicent M. J., Ortiz A., Ramos A. M., Effective nephroprotection against acute kidney injury with a star-shaped polyglutamate-curcuminoid conjugate. Sci. Rep. 10, 2056 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Schaffer M., Schaffer P. M., Zidan J., Sela G. B., Curcuma as a functional food in the control of cancer and inflammation. Curr. Opin. Clin. Nutr. Metab. Care 14, 588–597 (2011). [DOI] [PubMed] [Google Scholar]
- 15.Jung S., Murphy P. A., Sala I., Isoflavone profiles of soymilk as affected by high-pressure treatments of soymilk and soybeans. Food Chem. 111, 592–598 (2008). [Google Scholar]
- 16.do Carmo Carreiras M., Ismaili L., Marco-Contelles J., Propargylamine-derived multi-target directed ligands for Alzheimer’s disease therapy. Bioorg. Med. Chem. Lett. 30, 126880 (2020). [DOI] [PubMed] [Google Scholar]
- 17.Romero A., Marco-Contelles J., Ramos E., Highlights of ASS234: A novel and promising therapeutic agent for Alzheimer’s disease therapy. Neural Regen. Res. 15, 30–35 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Amit T., Bar-Am O., Mechlovich D., Kupershmidt L., Youdim M. B. H., Weinreb O., The novel multitarget iron chelating and propargylamine drug M30 affects APP regulation and processing activities in Alzheimer’s disease models. Neuropharmacology 123, 359–367 (2017). [DOI] [PubMed] [Google Scholar]
- 19.Bar-Am O., Weinreb O., Amit T., Youdim M. B. H., Regulation of Bcl-2 family proteins, neurotrophic factors, and APP processing in the neurorescue activity of propargylamine. FASEB J. 19, 1899–1901 (2005). [DOI] [PubMed] [Google Scholar]
- 20.A. Duro-Castano, Well-defined polyglutamates as carriers for the treatment of neurodegenerative diseases, thesis, (University of Valencia-CIPF, Valencia, SP, 2015); https://roderic.uv.es/handle/10550/47191.
- 21.Yang F., Lim G. P., Begum A. N., Ubeda O. J., Simmons M. R., Ambegaokar S. S., Chen P., Kayed R., Glabe C. G., Frautschy S. A., Cole G. M., Curcumin inhibits formation of amyloid β oligomers and fibrils, binds plaques, and reduces amyloid in vivo. J. Biol. Chem. 280, 5892–5901 (2005). [DOI] [PubMed] [Google Scholar]
- 22.Shytle R. D., Tan J., Bickford P. C., Rezai-Zadeh K., Hou L., Zeng J., Sanberg P. R., Sanberg C. D., Alberte R. S., Fink R. C., Roschek B. Jr., Optimized turmeric extract reduces β-Amyloid and phosphorylated Tau protein burden in Alzheimer’s transgenic mice. Curr. Alzheimer Res. 9, 500–506 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Mandal M., Jaiswal P., Mishra A., Role of curcumin and its nanoformulations in neurotherapeutics: A comprehensive review. J. Biochem. Mol. Toxicol. 34, e22478 (2020). [DOI] [PubMed] [Google Scholar]
- 24.Akinyemi A. J., Okonkwo P. K., Faboya O. A., Onikanni S. A., Fadaka A., Olayide I., Akinyemi E. O., Oboh G., Curcumin improves episodic memory in cadmium induced memory impairment through inhibition of acetylcholinesterase and adenosine deaminase activities in a rat model. Metab. Brain Dis. 32, 87–95 (2017). [DOI] [PubMed] [Google Scholar]
- 25.Weber W. M., Hunsaker L. A., Gonzales A. M., Heynekamp J. J., Orlando R. A., Deck L. M., Vander Jagt D. L., TPA-induced up-regulation of activator protein-1 can be inhibited or enhanced by analogs of the natural product curcumin. Biochem. Pharmacol. 72, 928–940 (2006). [DOI] [PubMed] [Google Scholar]
- 26.Chan M. M., Huang H. I., Fenton M. R., Fong D., In vivo inhibition of nitric oxide synthase gene expression by curcumin, a cancer preventive natural product with anti-inflammatory properties. Biochem. Pharmacol. 55, 1955–1962 (1998). [DOI] [PubMed] [Google Scholar]
- 27.Sathyapalan T., Aye M., Rigby A. S., Thatcher N. J., Dargham S. R., Kilpatrick E. S., Atkin S. L., Soy isoflavones improve cardiovascular disease risk markers in women during the early menopause. Nutr. Metab. Cardiovasc. Dis. 28, 691–697 (2018). [DOI] [PubMed] [Google Scholar]
- 28.Hillman G. G., Singh-Gupta V., Al-Bashir A. K., Yunker C. K., Joiner M. C., Sarkar F. H., Abrams J., Mark Haacke E., Monitoring sunitinib-induced vascular effects to optimize radiotherapy combined with soy isoflavones in murine xenograft tumor. Transl. Oncol. 4, 110–121 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Liu Z. M., Ho S. C., Chen Y. M., Tang N., Woo J., Effect of whole soy and purified isoflavone daidzein on renal function-a 6-month randomized controlled trial in equol-producing postmenopausal women with prehypertension. Clin. Biochem. 47, 1250–1256 (2014). [DOI] [PubMed] [Google Scholar]
- 30.Sarkaki A., Amani R., Badavi M., Moghaddam A. Z., Aligholi H., Safahani M., Haghighizadeh M. H., Pre-treatment effect of different doses of soy isoflavones on spatial learning and memory in an ovariectomized animal model of Alzheimer’s disease. Pak. J. Biol. Sci. 11, 1114–1119 (2008). [DOI] [PubMed] [Google Scholar]
- 31.Valles S. L., Dolz-Gaiton P., Gambini J., Borras C., LLoret A., Pallardo F. V., Viña J., Estradiol or genistein prevent Alzheimer’s disease-associated inflammation correlating with an increase PPARγ expression in cultured astrocytes. Brain Res. 1312, 138–144 (2010). [DOI] [PubMed] [Google Scholar]
- 32.Bonet-Costa V., Herranz-Pérez V., Blanco-Gandía M., Mas-Bargues C., Inglés M., Garcia-Tarraga P., Rodriguez-Arias M., Miñarro J., Borras C., Garcia-Verdugo J. M., Viña J., Clearing amyloid-β through PPARγ/ApoE Activation by Genistein is a Treatment of Experimental Alzheimer’s Disease. J. Alzheimers Dis. 51, 701–711 (2016). [DOI] [PubMed] [Google Scholar]
- 33.Ye S., Wang T. T., Cai B., Wang Y., Li J., Zhan J. X., Shen G. M., Genistein protects hippocampal neurons against injury by regulating calcium/calmodulin dependent protein kinase IV protein levels in Alzheimer’s disease model rats. Neural Regen. Res. 12, 1479–1484 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Uddin M. S., Kabir M. T., Emerging signal regulating potential of genistein against Alzheimer’s disease: A promising molecule of interest. Front. Cell Dev. Biol. 7, 197 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Storck S. E., Pietrzik C. U., Endothelial LRP1 – A potential target for the treatment of Alzheimer’s Disease: Theme: Drug discovery, development and delivery in Alzheimer’s disease. Pharm. Res. 34, 2637–2651 (2017). [DOI] [PubMed] [Google Scholar]
- 36.Duro-Castano A., Leite D. M., Forth J., Deng Y., Matias D., Jesus C. N., Battaglia G., Designing peptide nanoparticles for efficient brain delivery. Adv. Drug Deliv. Rev. 160, 52–77 (2020). [DOI] [PubMed] [Google Scholar]
- 37.Arélin K., Kinoshita A., Whelan C. M., Irizarry M. C., Rebeck G. W., Strickland D. K., Hyman B. T., LRP and senile plaques in Alzheimer’s disease: Colocalization with apolipoprotein E and with activated astrocytes. Mol. Brain Res. 104, 38–46 (2002). [DOI] [PubMed] [Google Scholar]
- 38.Kanekiyo T., Bu G., The low-density lipoprotein receptor-related protein 1 and amyloid-β clearance in Alzheimer’s disease. Front. Aging Neurosci. 6, 93 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Sawada M., Sawamoto K., Mechanisms of neurogenesis in the normal and injured adult brain. Keio J. Med. 62, 13–28 (2013). [DOI] [PubMed] [Google Scholar]
- 40.Duncan R., The dawning era of polymer therapeutics. Nat. Rev. Drug Discov. 2, 347–360 (2003). [DOI] [PubMed] [Google Scholar]
- 41.Price L. C., Buescher R. W., Kinetics of alkaline degradation of the food pigments curcumin and curcuminoids. J. Food Sci. 62, 267–269 (1997). [Google Scholar]
- 42.Shaffer S. A., Baker-Lee C., Kennedy J., Lai M. S., de Vries P., Buhler K., Singer J. W., In vitro and in vivo metabolism of paclitaxel poliglumex: Identification of metabolites and active proteases. Cancer Chemother. Pharmacol. 59, 537–548 (2007). [DOI] [PubMed] [Google Scholar]
- 43.Duro-Castano A., England R. M., Razola D., Romero E., Oteo-Vives M., Morcillo M. A., Vicent M. J., Well-defined star-shaped polyglutamates with improved pharmacokinetic profiles as excellent candidates for biomedical applications. Mol. Pharm. 10, 3639–3649 (2015). [DOI] [PubMed] [Google Scholar]
- 44.Arnaudov L. N., De Vries R., Thermally induced fibrillar aggregation of hen egg white lysozyme. Biophys. J. 88, 515–526 (2005). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Yanagisawa D., Taguchi H., Yamamoto A., Shirai N., Hirao K., Tooyama I., Curcuminoid binds to amyloid-β1-42 oligomer and fibril. J. Alzheimers Dis. 24, 33–42 (2011). [DOI] [PubMed] [Google Scholar]
- 46.Meratan A., Ghasemi A., Nemat-Gorgani M., Membrane integrity and amyloid cytotoxicity: A model study involving mitochondria and lysozyme fibrillation products. J. Mol. Biol. 409, 826–838 (2011). [DOI] [PubMed] [Google Scholar]
- 47.Mohammadi F., Mahmudian A., Moeenia M., Hassani L., Inhibition of amyloid fibrillation of hen egg-white lysozyme by the natural and synthetic curcuminoids. RSC Adv. 6, 23148–23160 (2016). [Google Scholar]
- 48.Khurana R., Coleman C., Ionescu-Zanetti C., Carter S. A., Krishna V., Grover R. K., Roy R., Singh S., Mechanism of thioflavin T binding to amyloid fibrils. J. Struct. Biol. 151, 229–238 (2005). [DOI] [PubMed] [Google Scholar]
- 49.Conejos-Sánchez I., Cardoso I., Oteo-Vives M., Romero-Sanz E., Paul A., Sauri A. R., Morcillo M. A., Saraiva M. J., Vicent M. J., Polymer-doxycycline conjugates as fibril disrupters: An approach towards the treatment of a rare amyloidotic disease. J. Control. Release 198, 80–90 (2015). [DOI] [PubMed] [Google Scholar]
- 50.Frozza R. L., Horn A. P., Hoppe J. B., Simão F., Gerhardt D., Comiran R. A., Salbego C. G., A comparative study of β-Amyloid peptides Aβ1-42 and Aβ25-35 toxicity in organotypic hippocampal slice cultures. Neurochem. Res. 34, 295–303 (2009). [DOI] [PubMed] [Google Scholar]
- 51.Eslamizade M. J., Madjd Z., Rasoolijazi H., Saffarzadeh F., Pirhajati V., Aligholi H., Janahmadi M., Mehdizadeh M., Impaired memory and evidence of histopathology in CA1 pyramidal neurons through injection of Aβ1-42 peptides into the frontal cortices of rat. Basic Clin. Neurosci. 7, 31–41 (2016). [PMC free article] [PubMed] [Google Scholar]
- 52.Jankowsky J. L., Slunt H. H., Gonzales V., Savonenko A. V., Wen J. C., Jenkins N. A., Copeland N. G., Younkin L. H., Lester H. A., Younkin S. G., Borchelt D. R., Persistent amyloidosis following suppression of Aβ production in a transgenic model of Alzheimer disease. PLOS Med. 2, e355 (2005). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Yao Z. G., Jing H. Y., Wang D. M., Lv B. B., Li J. M., Liu F. F., Fan H., Sun X. C., Qin Y. J., Zhao M. Q., Valproic acid ameliorates olfactory dysfunction in APP/PS1 transgenic mice of Alzheimer’s disease: Ameliorations from the olfactory epithelium to the olfactory bulb. Pharmacol. Biochem. Behav. 144, 53–59 (2016). [DOI] [PubMed] [Google Scholar]
- 54.Zou Y. M., Lu D., Liu L. P., Zhang H. H., Zhou Y. Y., Olfactory dysfunction in Alzheimer’s disease. Neuropsychiatr. Dis. Treat. 12, 869–875 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Jansen D., Zerbi V., Janssen C. I. F., Dederen P. J. W. C., Mutsaers M. P. C., Hafkemeijer A., Janssen A. L., Nobelen C. L. M., Veltien A., Asten J. J., Heerschap A., Kiliaan A. J., A longitudinal study of cognition, proton MR spectroscopy and synaptic and neuronal pathology in aging wild-type and AβPPswe-PS1dE9 mice. PLOS ONE 8, e63643 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Wang Y., Liu J., Zhang Z., Wang X., Zhang C., Structure and permeability changes of the blood-brain barrier in APP/PS1 mice: An Alzheimer’s disease animal model. Neurochem. J. 5, 220–222 (2011). [Google Scholar]
- 57.Kumar V., Lee J. D., Coulson E. J., Woodruff T. M., A validated quantitative method for the assessment of neuroprotective barrier impairment in neurodegenerative disease models. J. Neurochem. (2020). [DOI] [PubMed] [Google Scholar]
- 58.Nho K., Kueider-Paisley A., Ahmad S., Mahmoudian-Dehkordi S., Arnold M., Risacher S. L., Louie G., Blach C., Baillie R., Han X., Kastenmüller G., Trojanowski J. Q., Shaw L. M., Weiner M. W., Doraiswamy P. M., van Duijn C., Saykin A. J., Kaddurah-Daouk R.; Alzheimer’s Disease Neuroimaging Initiative and the Alzheimer Disease Metabolomics Consortium , Association of altered liver enzymes with Alzheimer disease diagnosis, cognition, neuroimaging measures, and cerebrospinal fluid biomarkers. JAMA Netw. Open 2, e197978 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Lieb W., Beiser A. S., Vasan R. S., Tan Z. S., Au R., Harris T. B., Roubenoff R., Auerbach S., DeCarli C., Wolf P. A., Seshadri S., Association of plasma leptin levels with incident Alzheimer disease and MRI measures of brain aging. JAMA Netw. Open 302, 2565–2572 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Wesson D. W., Levy E., Nixon R. A., Wilson D. A., Olfactory dysfunction correlates with amyloid-β burden in an Alzheimer’s disease mouse model. J. Neurosci. 30, 505–514 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Voulgaropoulou S. D., van Amelsvoort T. A. M. J., Prickaerts J., Vingerhoets C., The effect of curcumin on cognition in Alzheimer’s disease and healthy aging: A systematic review of pre-clinical and clinical studies. Brain Res. 1725, 146476 (2019). [DOI] [PubMed] [Google Scholar]
- 62.Lu Y., Guo Z., Zhang Y., Li C., Zhang Y., Guo Q., Chen Q., Chen X., He X., Liu L., Ruan C., Sun T., Ji B., Lu W., Jiang C., Microenvironment remodeling micelles for Alzheimer’s disease therapy by early modulation of activated microglia. Adv. Sci. 6, 1801586 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Potter K. A., Jorfi M., Householder K. T., Foster E. J., Weder C., Capadona J. R., Curcumin-releasing mechanically adaptive intracortical implants improve the proximal neuronal density and blood-brain barrier stability. Acta Biomater. 10, 2209–2222 (2014). [DOI] [PubMed] [Google Scholar]
- 64.Palmal S., Maity A. R., Singh B. K., Basu S., Jana N. R., Jana N. R., Inhibition of amyloid fibril growth and dissolution of amyloid fibrils by curcumin-gold nanoparticles. Chemistry 20, 6184–6191 (2014). [DOI] [PubMed] [Google Scholar]
- 65.Sun M., Gao Y., Guo C., Cao F., Song Z., Xi Y., Yu A., Li A., Zhai G., Enhancement of transport of curcumin to brain in mice by poly(n-butylcyanoacrylate) nanoparticle. J. Nanoparticle Res. 12, 3111–3122 (2010). [Google Scholar]
- 66.Mourtas S., Lazar A. N., Markoutsa E., Duyckaerts C., Anti-misiaris S. G., Multifunctional nanoliposomes with curcumin-lipid derivative and brain targeting functionality with potential applications for Alzheimer disease. Eur. J. Med. Chem. 80, 175–183 (2014). [DOI] [PubMed] [Google Scholar]
- 67.Lazar A. N., Mourtas S., Youssef I., Parizot C., Dauphin A., Dela-tour B., Antimisiaris S. G., Duyckaerts C., Curcumin-conjugated nanoliposomes with high affinity for Aβ deposits: Possible applications to Alzheimer disease. Nanomedicine 9, 712–721 (2013). [DOI] [PubMed] [Google Scholar]
- 68.Ray B., Bisht S., Maitra A., Maitra A., Lahiri D. K., Neuroprotective and neurorescue effects of a novel polymeric nanoparticle formulation of curcumin (NanoCurc™) in the neuronal cell culture and animal model: Implications for Alzheimer’s disease. J. Alzheimers Dis. 23, 61–77 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Guidotti G., Brambilla L., Rossi D., Peptides in clinical development for the treatment of brain tumors. Curr. Opin. Pharmacol. 47, 102–109 (2019). [DOI] [PubMed] [Google Scholar]
- 70.Tian X., Leite D. M., Scarpa E., Nyberg S., Fullstone G., Forth J., Matias D. L., Apriceno A., Poma A., Duro-Castano A., Vuyyuru M., Harker-Kirschneck L., Šarić A., Zhang Z., Xiang P., Fang B., Tian Y., Luo L., Rizzello L., Battaglia G., On the shuttling across the blood-brain barrier via tubule formation: Mechanism and cargo avidity bias. Sci. Adv. 6, eabc4397 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Niu X., Chen J., Gao J., Nanocarriers as a powerful vehicle to overcome blood-brain barrier in treating neurodegenerative diseases: Focus on recent advances. Asian J. Pharm. Sci. 14, 480–496 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Zhao Z., Sagare A. P., Ma Q., Halliday M. R., Kong P., Kisler K., Winkler E. A., Ramanathan A., Kanekiyo T., Bu G., Owens N. C., Rege S. V., Si G., Ahuja A., Zhu D., Miller C. A., Schneider J. A., Maeda M., Maeda T., Sugawara T., Ichida J. K., Zlokovic B. V., Central role for PICALM in amyloid-β blood-brain barrier transcytosis and clearance. Nat. Neurosci. 18, 978–987 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Erickson M. A., Niehoff M. L., Farr S. A., Morley J. E., Dillman L. A., Lynch K. M., Banks W. A., Peripheral administration of antisense oligonucleotides targeting the amyloid-β protein precursor reverses AβPP and LRP-1 overexpression in the aged SAMP8 mouse brain. J. Alzheimers Dis. 28, 951–960 (2012). [DOI] [PubMed] [Google Scholar]
- 74.Shibata M., Yamada S., Ram Kumar S., Calero M., Bading J., Frangione B., Holtzman D. M., Miller C. A., Strickland D. K., Ghiso J., Zlokovic B. V., Clearance of Alzheimer’s amyloid-β1-40 peptide from brain by LDL receptor-related protein-1 at the blood-brain barrier. J. Clin. Invest. 106, 1489–1499 (2000). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Demeule M., Currie J. C., Bertrand Y., Ché C., Nguyen T., Régina A., Gabathuler R., Castaigne J. P., Béliveau R., Involvement of the low-density lipoprotein receptor-related protein in the transcytosis of the brain delivery vector Angiopep-2. J. Neurochem. 106, 1534–1544 (2008). [DOI] [PubMed] [Google Scholar]
- 76.Min H. S., Kim H. J., Naito M., Ogura S., Toh K., Hayashi K., Kim B. S., Fukushima S., Anraku Y., Miyata K., Kataoka K., Systemic brain delivery of antisense oligonucleotides across the blood–brain barrier with a glucose-coated polymeric nanocarrier. Angew. Chemie Int. Ed. Engl. 59, 8173–8180 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Jones R. S., Minogue A. M., Connor T. J., Lynch M. A., Amyloid-β-induced astrocytic phagocytosis is mediated by CD36, CD47 and RAGE. J. Neuroimmune Pharmacol. 8, 301–311 (2013). [DOI] [PubMed] [Google Scholar]
- 78.Liu M., Apriceno A., Sipin M., Scarpa E., Rodriguez-Arco L., Poma A., Marchello G., Battaglia G., Angioletti-Uberti S., Combinatorial entropy behaviour leads to range selective binding in ligand-receptor interactions. Nat. Commun. 11, 4836 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Stoppini L., Buchs P.-A., Muller D., A simple method for organotypic cultures of nervous tissue. J. Neurosci. Methods 37, 173–182 (1991). [DOI] [PubMed] [Google Scholar]
- 80.Schindelin J., Arganda-Carreras I., Frise E., Kaynig V., Longair M., Pietzsch T., Preibisch S., Rueden C., Saalfeld S., Schmid B., Tinevez J. Y., White D. J., Hartenstein V., Eliceiri K., Tomancak P., Cardona A., Fiji: An open-source platform for biological-image analysis. Nat. Methods 9, 676–682 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.López-Grueso R., Gambini J., Abdelaziz K. M., Monleón D., Díaz A., El Alami M., Bonet-Costa V., Borrás C., Viña J., Early, but not late onset estrogen replacement therapy prevents oxidative stress and metabolic alterations caused by Ovariectomy. Antioxid. Redox Signal. 20, 236–246 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Conejos-Sánchez I., Duro-Castano A., Birke A., Barzb M., Vicenta M. J., A controlled and versatile NCA polymerization method for the synthesis of polypeptides. Polym. Chem. 4, 3182 (2013). [Google Scholar]
- 83.Stine W. B., Jungbauer L., Yu C., LaDu M. J., Preparing synthetic Aβ in different aggregation states. Methods Mol. Biol. 670, 13–32 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Kim Y. J., Kim S. Y., Sung D. K., Chang Y. S., Park W. S., Neuroprotective effects of L-carnitine against oxygen-glucose deprivation in rat primary cortical neurons. Korean J. Pediatr. 55, 238–248 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Ennaceur A., Delacour J., A new one-trial test for neurobiological studies of memory in rats. 1: Behavioral data. Behav. Brain Res. 31, 47–59 (1988). [DOI] [PubMed] [Google Scholar]
- 86.Prickaerts, Van Staveren W. C. G., Ik A., Ittersum M. M.-Van, Niewöhner U., Van der Staay F. J., Blokland A., De Vente J., Effects of two selective phosphodiesterase type 5 inhibitors, sildenafil and vardenafil, on object recognition memory and hippocampal cyclic GMP levels in the rat. Neuroscience 113, 351–361 (2002). [DOI] [PubMed] [Google Scholar]
- 87.Galsworthy M. J., Paya-Cano J. L., Liu L., Monleón S., Gregoryan G., Fernandes C., Schalkwyk L. C., Plomin R., Assessing reliability, heritability and general cognitive ability in a battery of cognitive tasks for laboratory mice. Behav. Genet. 35, 675–692 (2005). [DOI] [PubMed] [Google Scholar]
- 88.Rabinovitch M. S., Rosvold H. E., A closed-field intelligence test for rats. Can. J. Psychol. 5, 122–128 (1951). [DOI] [PubMed] [Google Scholar]
- 89.Sundberg H., Døving K., Novikov S., Ursin H., A method for studying responses and habituation to odors in rats. Behav. Neural Biol. 34, 113–119 (1982). [DOI] [PubMed] [Google Scholar]
- 90.Gao X., Cui Y., Levenson R. M., Chung L. W. K., Nie S., In vivo cancer targeting and imaging with semiconductor quantum dots. Nat. Biotechnol. 22, 969–976 (2004). [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary material for this article is available at http://advances.sciencemag.org/cgi/content/full/7/13/eabf9180/DC1