
Fig. 2. Formulation excipients, insulin aggregation, and stabilization techniques.
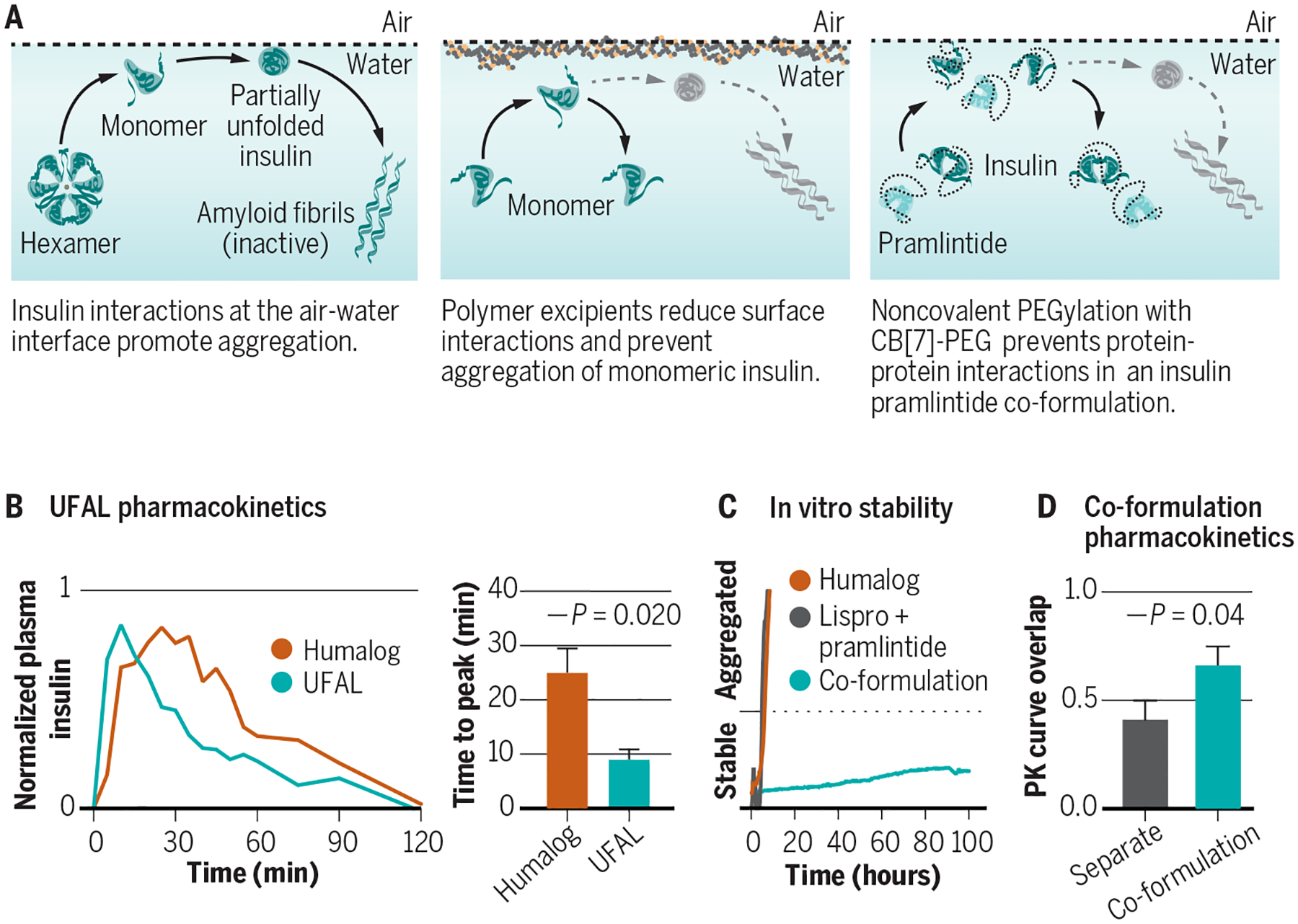
(A) Aggregation of biopharmaceuticals such as insulin typically occurs as a result of protein-protein interactions at a hydrophobic interface (e.g., the air-water or vial-water interfaces) that nucleate aggregation events. (B) Excipients, such as tonicity modifiers, antimicrobial preservatives, and stabilizing agents can affect insulin association state and are carefully chosen to balance stability in the vial and absorption upon subcutaneous administration. (C) Ultrafast acting insulins aim to shift the equilibrium of insulin association states from the insulin hexamer towards the insulin monomer to promote more rapid absorption and commensurate onset of action, as well as to reduce the duration of action. New excipient platforms look at displacing insulin from the air-water and vial-water interfaces using amphiphilic copolymers to prevent protein-protein aggregation. (D) Judicious design of polyacrylamide-based copolymer excipients can generate an ultrafast absorbing lispro (UFAL) formulation comprising mostly monomeric insulin that is significantly more stable even than current commercial fast-acting insulin formulations (e.g., Humalog). (E) Pharmacokinetic exposure curve and time to peak exposure for this UFAL formulation in diabetic pigs indicates a 2.8-fold decrease in the time-to-peak when compared to Humalog. (F) Simultaneous non-covalent PEGylation of insulin and pramlintide using cucurbit[7]uril-poly(ethylene glycol) (CB[7]-PEG) provides a protective “wrapper” on each protein that allows for stable co-formulation of the two historically incompatible therapeutics at pH=7. (G) Supramolecular PEGylation with CB[7]-PEG enables insulin-pramlintide co-formulations to be more stable to stressed aging than even commercial Humalog. (H) Pharmacokinetic curves for a CB[7]-PEG stabilized insulin-pramlintide co-formulation in diabetic pigs demonstrating increased overlap of insulin and pramlintide action compared to the current clinical approach of separate administrations. Adapted from (24, 38).