
Figure 1.
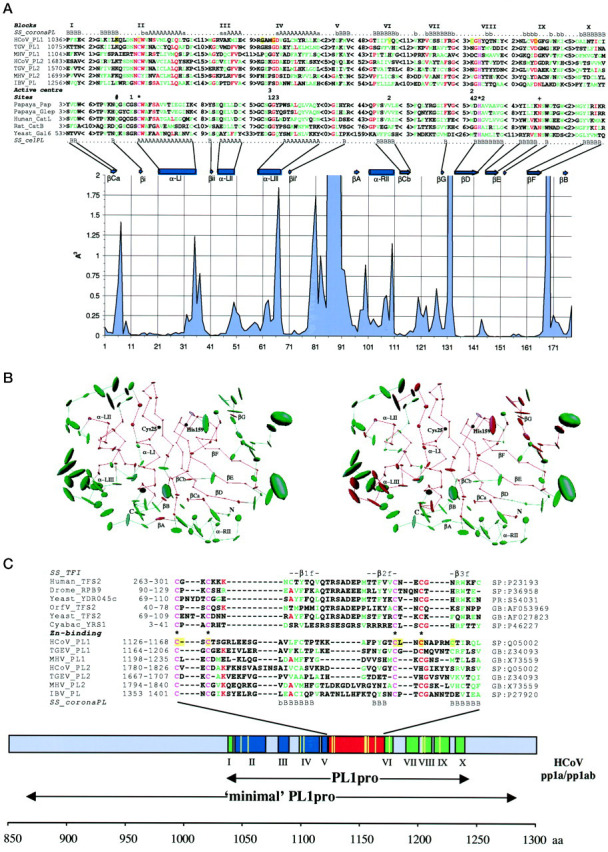
Fig. 1. Coronaviral and cellular PLpros: structural similarities and unique features.A, secondary structure-based sequence alignment of coronaviral and cellular PLpros. The primary structures of HCoV PL1pro and its coronaviral relatives (for accession numbers see C) were aligned using ClustalW program (43) in a stepwise manner and manually corrected with the ClustalX program (44) and the MACAW workbench (45). The main portion of this alignment is presented as 10 ungapped blocks. Only blocks II and III were statistically significant (p < 10−20 and p = 1.5−13, respectively), and blocks IV and VII, excluding MHV PL1pro, were conditionally significant, using a searching space between blocks III and VIII and between blocks IV and VIII, respectively. The validity of block VIII was previously confirmed by site-directed mutagenesis of conserved His for MHV and HCoV PL1pros and IBV PLpro (37, 41, 42). The secondary structures predicted by the PhD program are shown at the top (SS_coronaPL; A anda represent α-helix, and B and brepresent β-strand, predictions in capital letters have a reliability >5 and predictions inlowercase letters have a reliability of 5 and less (49)). The validity of this prediction was confirmed when similar secondary structure profiles were also returned for (i) the same alignment using the DSC program (50) and (ii) two automatically generated alignments containing either PL1pros or PL2pros encoded by HCoV and TGEV (not shown). The secondary structure profile of coronaviral PLpros was aligned with secondary structure elements conserved in the tertiary structure of 11 cellular PLpros (SS_celPL) (Protein Data Bank accession numbers: 1ppn, Papaya_Pap, papain (77);1gec, Papaya_Glep, glycyl endopeptidase (78); 1ppo, caricain (79);1yac, chymopapain (80); 1mem, cathepsin K (81); 1cjl, Human_CatL, cathepsin L (82); 1cte, Rat_Catb, cathepsin B (83); 2aim, trypanosoma cruzain (84); 2act, actinidin (85); 1gcb, yeast Gal6/bleomycin hydrolase (86)). The secondary structure alignment guided a sequence alignment of coronaviral and cellular proteases. A register of the alignment within each block was (arbitrarily) selected to maximize interfamily sequence similarity, although two or more poorly discriminated alignments were produced for all blocks except blocks II and VIII. When the three-dimensional structures of coronaviral PLpros become available, this alignment may need to be locally adjusted. For cellular PLpros, only a representative set of five sequences is shown. Coloring of the alignment of 12 sequences indicates the following:pink, invariant residues; red, residues conserved in >50% of the sequences; green, group of similar residues. The alignments of coronaviral and cellular PLpros highlight the active site residues of cellular proteases (66, 78). *, principal catalytic; +, “accessory” catalytic; 1, 2,3, and 4, substrate-binding pocket subsites S1, S2, S3, and S4, respectively; #, oxyanion hole-forming residue.Beneath the alignments, a plot displaying the positional structural variability (55) of cellular PLpros is shown.Above the plot, the positions of conserved secondary structure elements of cellular PLpros (66) as well as four conserved hydrogen-forming elements consisting of one residue (not marked) in the primary structure are displayed. Vertical axis, space variability at a position of the alignment; horizontal axis, numeration in the structural alignment containing only aligned residues. B, core structural residues of the cellular PLpros and residues conserved in cellular and coronaviral PLpros. Using the CORE package (55), a structural alignment of 11 cellular PLpros was converted into an average PL structure. It is characterized by the mean position of each C-α atom common in the family. The size of the ellipsoid around each of these atoms is proportional to the volume of atom variance. The two identical average PL structures, consisting of 178 atoms, are displayed in the “standard” papain orientation (66) featuring left-hand and right-hand domains as well as the interdomain active site cleft with the two catalytic residues of papain, Cys25 and His159. Conserved secondary structure elements of cellular PLpros are also marked. These structures are colored ingreen and red as follows. The left structure, the half of C-α atoms plus two atoms having the lowest space variance (91 atoms) are colored in red (core), and the remaining atoms are in green (noncore). The right structure, 109 atoms, whose residues were aligned with coronaviral PL residues in Fig. 1A, are shown in red (interfamily conserved residues), and the remaining atoms are in green. Note that the cellular PL core residues and the interfamily conserved residues are mainly from the same pool. C, A unique Zn2+finger connects the two domains of the PL fold of coronaviral PLpros. A region of the coronaviral PLpros between blocks V and VI was aligned as specified in Fig. 1A. Using the secondary structures predicted for the PLpros (SS_coronaPL) (50) and derived from the NMR structure (69) of the TFIIS Zn2+ ribbon (SS_TFI), an alignment of Zn2+ fingers of coronaviral PLpros and TFIIS was generated. The positions of these sequences in the corresponding proteins are given on the left, and accession numbers in the sequence data bases are shown on the right.Coloring of the alignment is as detailed for A. Residues involved in Zn2+ binding in TFIIS (69) are marked. A bar depicts the region of HCoV pp1a/pp1ab characterized in this study with the conserved blocks (Fig. 1A) shown. These blocks are organized in three groups colored differently.Blue, left-hand α-helix domain; green, right-hand β-sheet domain without counterparts of βA- and βB-strands; red, Zn2+ finger domain.Beneath the bar, the positions of the PL1pro domain, which is conserved among coronaviruses, and the HCoV minimal PL1pro domain determined by deletion analysis (41) are shown. The positions of mutations (Ref. 41 and Table II) are depicted withyellow vertical lines in thebar and yellow amino acid background in the alignments in A and C.