

Abstract
Background
Hereditary retinopathy is a significant cause of blindness worldwide. Despite the discovery of many mutations in various retinopathies, a large number of patients remain genetically undiagnosed. Targeted next-generation sequencing of the human genome is a suitable approach for the molecular diagnosis of retinopathy.
Methods
We describe a cohort of 211 families from central China with various forms of retinopathy; 95 patients were investigated using multigene panel sequencing, and the other 116 with suspected Leber hereditary optic neuropathy (LHON) were tested by Sanger sequencing. The detected variation of targeted sequencing was verified by PCR-based Sanger sequencing. We performed a comprehensive analysis of the cases using sequencing data and ophthalmologic examination information.
Results
Potential causal mutations were identified in the majority of families with retinopathy (57.9% of 95 families) and suspected LHON (21.6% of 116 families). There were 68 variants of a certain significance distributed in 31 known disease-causing genes in the 95 families; 37 of the variants are novel and have not been reported to be related to hereditary retinopathy. The NGS panel solution provided a 45.3% potential diagnostic rate for retinopathy families, with candidate gene mutations of undefined pathogenicity revealed in another 12.6%of the families.
Conclusion
Our study uncovered novel mutations and phenotypic aspects of retinopathy and demonstrated the genetic and clinical heterogeneity of related conditions. The findings show the detection rate of pathogenic variants in patients with hereditary retinopathy in central China as well as the diversity and gene distribution of these variants. The significance of molecular genetic testing for patients with hereditary retinopathy is also highlighted.
Supplementary Information
The online version contains supplementary material available at 10.1186/s12920-021-00935-w.
Keywords: Hereditary retinopathy, Novel mutations, Targeted sequencing, Genetic testing
Background
Hereditary retinopathy is one category of the most common genetic retinal diseases causing blindness [1]. Hereditary retinopathy is characterized by heterogeneity of genetic variation and clinical manifestations. The main inheritance patterns include autosomal dominant, autosomal recessive inheritance and X-linked inheritance [2]. Hereditary retinopathy mainly includes retinitis pigmentosa, macular degeneration, Leber hereditary optic neuropathy (LHON) and retinal dysplasia. Retinitis pigmentosa (RP) comprises a group of blinding retinal diseases caused by abnormalities in photoreceptors [3], with main clinical features of progressive visual field defects, night blindness, bone spicules such as retinal pigmentation and abnormal electroretinograms [4]. LHON is a mitochondrial hereditary eye disease that involves retinal ganglion cells, and it eventually results in degeneration and atrophy of the optic nerve [5]. With the popularization and clinical application of gene sequencing technology, an increasing number of disease-causing genes and mutations have been discovered; these genes are mainly expressed in photoreceptor cells and retinal pigment epithelial cells [6]. Overall, a good understanding of retinopathy genes not only provides a theoretical basis for diagnosis and genetic counselling but also supports guidance for gene therapy [7–9].
The study of the genetics of retinopathy is important to enhance our understanding of the molecular aspects of eye development, disease and treatment. In this research, we chose a family-based strategy to determine the exact inheritance pattern and recurrence risk in offspring. Using such a family-based strategy, we can also determine whether phenotype and genotype co-segregate in a family, which helps to estimate the pathogenicity of candidate mutations. More than half of the patients in this study were suspected of having LHON; direct Sanger sequencing of mitochondrial DNA was performed for some, and next-generation sequencing (NGS) was carried out for the remainder. Despite the discovery of pathogenic mutations and genes of various types of retinopathy, many unknowns remain. Our study will increase knowledge of the mutations and phenotypes of diseases and provide more population information on pathogenic variants. Our research will also illustrate the importance of targeted NGS in the aetiological detection of hereditary eye diseases.
Methods
Subject recruitment
In total, 211 Chinese families with retinopathy from central China were recruited for this study, including 116 patients from different families with suspected monocular or binocular LHON and 95 families with other retinopathies. The inclusion criteria for LHON included (1) optic neuropathy and (2) a rapid decline in visual acuity for unknown reasons. The inclusion criteria for other retinopathies included (1) retinitis pigmentosa; (2) macular degeneration (MD); and (3) multiple fundus lesions or retinal dysplasia. Of the patients, 116 (Part A) with fundus optic atrophy were subjected to LHON Sanger sequencing, and targeted NGS for other retinopathies was performed for 95 (Part B); the patients were examined and diagnosed by the Ophthalmology Department. The specific clinical manifestations of the patients were recorded. Samples were obtained with written informed consent. The retinopathy patients sought medical and genetic consultations in the hospital during 2017 and 2019, and 4 mL peripheral blood was from individuals in 211 retinopathy families. The clinical data for the patients were collected at the outpatient clinic.
The subjects of Part A underwent Sanger sequencing that included specific pathogenic sites of mitochondrial DNA; the mother of a subject carrying pathogenic mitochondrial variants underwent the same test to explore the source of the variation. The proband of the families of Part B was screened by targeted NGS. Then, the parents of the proband and other members of the family were tested by Sanger sequencing to detect and verify the carrying status of candidate mutations screened through targeted NGS.
Targeted next-generation sequencing and sanger sequencing
Genomic DNA was extracted from EDTA-treated blood samples using a Blood DNA Midi Kit D3494 (Omega Bio-tek, USA) with nucleic acid automatic extraction equipment (Eppendorf epMotion 5075 m, Germany). A customized panel (MyGenostics Inc., China) capturing 463 known genes (Additional file 1: Table S1) related to retinal disease was designed to detect the genetic cause of the congenital retinopathy in the families. Panel sequencing was conducted using the Illumina NextSeq500 system in our clinical lab. The average sequencing depth of the target panel sequence was more than 100 × , and the coverage was 98%. Version GRCh37 is the human reference genome used for short-read mapping (https://www.gencodegenes.org/human/release_37lift37.html). The transcript RefSeq number was obtained from the Ensembl database (http://asia.ensembl.org) (Tables 1 and 2) [10]. PCR-based Sanger sequencing was used to validate disease-causing mutations based on NGS. The carrying status of a novel mutation in other family members was also assessed by Sanger sequencing. The primers used for PCR were designed by GeneTool software. A capillary electrophoresis apparatus (ABI 3130xl, USA) and dGTP BigDye® Terminator sequencing kit (ABI, USA) were used for Sanger sequencing.
Table 1.
General situation of families with pathogenic or likely pathogenic mutations
| Fa | Np | Gene | Transcript RefSeq | Ex | NA Changes | AA changes | Hzyo | Pf | Reported | Gm | Disease | SPM | ACMG grade |
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| 14 | 2 | RHO | NM_000539 | 1 | c.251 T > C | p.L84P | Het | – | Novel | AD | RP, 4 | + + + | PS4 + PM1 + PM2 + PP1 + PP3 |
| 15 | 4 | RHO | NM_000539 | 2 | c.403C > T | p.R135W | Het | 0/ 1.082e−5 | Yes[36] | AD | RP, 4 | + + + | PS1 + PM1 + PP1 + PP3 |
| 48 | 1 | RHO | NM_000539 | 3 | c.541G > A | p.E181K | Het | – | Yes[37] | AD | RP, 4 | + + + | PS2 + PM2 + PP3 |
| 54 | 2 | RHO | NM_000539 | 2 | c.403C > T | p.R135W | Het | 0/ 1.082e−5 | Yes[38] | AD | RP, 4 | + + + | PS1 + PM1 + PP1 + PP3 |
| 18 | 3 | NDP | NM_000266 | 2 | c.124C > A | p.H42N | Hemi | – | Novel | XL | FEVR2[39] | + + + | PM2 + PM5 + PP1 + PP3 |
| 32 | 1 | NDP | NM_000266 | 3 | c.343C > T | p.R115X | Hemi | – | Yes[40] | XLR | Norrie | + + + | PVS1 + PS1 + PM2 + PP3 |
| 46 | 1 | NDP | NM_000266 | 3 | c.401_402delGA | p.*134Wfs*13 | Hemi | – | Novel | XL | FEVR2 | / / / | PVS1 + PS2 + PM2 |
| 55 | 3 | NDP | NM_000266 | 3 | c.268C > T | p.R90C | Hemi | – | Yes[41] | XLR | Norrie | + + + | PS1 + PM2 + PP1 + PP3 |
| 7 | 1 | USH2A | NM_206933 | 2 | c.99_100insT | p.R34Sfs*41 | Hom | 6.242 e−5/ 3.231e−5 | Yes[42] | AR | Usher 2A | / / / | PVS1 + PS1 + PM2 |
| 9 | 1 | USH2A | NM_206933 | 55 | c.10859 T > C | p.I3620T | Het | 1.16e−4/ 1.219e−5 | Yes[43] | AR | Usher 2A/RP, 39 | + + + | PS1 + PM2 + PM3 + PP3 |
| USH2A | NM_206933 | 13 | c.2802 T > G | p.C934W | Het | 2.441e−3/1.915e−4 | Yes[44] | AR | Usher 2A/RP, 39 | + + + | PS1 + PM2 + PM3 + PP3 | ||
| 47 | 1 | USH2A | NM_206933 | 63 | c.13596dupC | p.S4533Lfs*28 | Het | – | Novel | AR | Usher 2A | / / / | PVS1 + PM2 + PM3 |
| USH2A | NM_206933 | 56 | c.10962C > A | p.Y3654X | Het | – | Novel | AR | Usher 2A | + + + | PVS1 + PM2 + PM3 + PP3 | ||
| 27 | 1 | RS1 | NM_000330 | 6 | c.598C > T | p.R200C | Hemi | – | Yes[45] | XLR | Retinoschisis | + + + | PS1 + PM2 + PP3 |
| 38 | 1 | RS1 | NM_000330 | 4 | c.214G > A | p.E72K | Hemi | 0/ 1.678e−5 | Yes[45] | XLR | Retinoschisis | + + + | PS1 + PM2 + PP3 |
| 51 | 2 | RS1 | NM_000330 | 4 | c.206_207delTG | p.Leu69Argfs*16 | Hemi | – | Yes[46] | XLR | Retinoschisis | / / / | PVS1 + PS1 + PM2 + PP1 |
| 1 | 1 | MERTK | NM_006343 | 8 | c.1186G > T | p.E396X | Het | – | Yes[47] | AR | RP,38 | / / + | PVS1 + PS1 + PM2 |
| MERTK | NM_006343 | 3 | c.518A > G | p.Y173C | Het | 0/ 1.219e−5 | Novel | AR | RP,38 | + + + | PM2 + PM3 + PP3 + PP4 | ||
| 2 | 2 | MERTK | NM_006343 | 4 | c.754delC | p.P252Qfs*3 | Hom | – | Novel | AR | RP,38 | / / / | PVS1 + PM2 + PP1 |
| Fa | Np | Gene | Transcript ErfSeq | Ex | NA Changes | AA changes | Hzyo | Pf | Reported | Gm | Disease | SPM | ACMG grade |
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| 3 | 1 | CYP4V2 | NM_207352 | 7 | c.(802–8)_810delTCATACAGGTCATCGCTinsGC | ?p.268_270del/ Splicing | Hom | 7.963e−4/ 6.856e−5 | Yes[48] | AR | Bietti CCD | / / / | PVS1 + PS1 + PM2 |
| 4 | 2 | CYP4V2 | NM_207352 | 7 | c.(802–8)_810delTCATACAGGTCATCGCTinsGC | ?p.268_270del/ Splicing | Het | 7.963e−4/ 6.856e−5 | Yes[48] | AR | Bietti CCD | / / / | PVS1 + PS1 + PM2 + PM3 + PP1 |
| CYP4V2 | NM_207352 | 7 | c.958C > T | p.R320X | Het | 0/ 4.061e−6 | Yes[49] | AR | Bietti CCD | / / + | PVS1 + PS1 + PM2 + PM3 + PP1 | ||
| 5 | 4 | FSCN2 | NM_001077182 | 1 | c.72delG | p.T25Qfs*120 | Het | 0.01238/ 8.801e−4 | Yes | AD | RP, 30 | / / / | PVS1 + PS1 + PP1 |
| 6 | 2 | FSCN2 | NM_001077182 | 1 | c.72delG | p.T25Qfs*120 | Het | 0.01238/ 8.801e−4 | Yes[50] | AD | RP, 30 | / / / | PVS1 + PS1 + PP1 |
| 12 | 4 | PRPF31 | NM_015629 | 11 | c.(1074–8)_1079delGTCCCCAGGTACCG | ?p.358_360delRYRinsS/ Splicing | Het | – | Novel | AD | RP, 11 | / / / | PVS1 + PM2 + PP1 |
| 50 | 2 | PRPF31 | NM_015629 | 12 | c.1215delG | p.Val406fs*7 | Het | – | Yes[51] | AD | RP, 11 | / / / | PVS1 + PS1 + PM2 + PP1 |
| 33 | 1 | RPGR | NM_001034853 | 15 | c.2236_2237delGA | p.E746Rfs*23 | Hemi | – | Yes | XLR | MD | / / / | PVS1 + PS1 + PM2 |
| 52 | 2 | RPGR | NM_001034853 | 15 | c.2236_2237delGA | p.E746Rfs*23 | Hemi | – | Yes[52] | XLR | MD | / / / | PVS1 + PS1 + PM2 + PP1 |
| 43 | 5 | RP2 | NM_006915 | 3 | c.769–2A > G | splicing | Hemi | – | Yes[53] | XL | RP, 2 | / / + | PVS1 + PS1 + PM2 + PP1 |
| 44 | 4 | RP2 | NM_006915 | 2 | c.572_582dup11 | p.Pro190Profs*52 | Hemi | – | Novel | XL | RP, 2 | / / / | PVS1 + PM2 + PP1 |
| 36 | 1 | ABCA4 | NM_000350 | 29 | c.4352 + 1G > A | splicing | Het | 0/ 8.123e−6 | Yes[54] | AR | Stargardt 1 | / / + | PVS1 + PS1 + PM2 + PM3 |
| ABCA4 | NM_000350 | 13 | c.1804C > T | p.R602W | Het | 2.904e−4/ 4.477e−5 | Yes[55] | AR | Stargardt 1 | + + + | PS1 + PM2 + PM3 + PP3 | ||
| 11 | 1 | TULP1 | NM_003322 | 13 | c.1318C > T | p.R440X | Het | 0/ 1.145e−5 | Yes[56] | AR | LCA 15 | / / + | PVS1 + PS1 + PM2 |
| TULP1 | NM_003322 | 12 | c.1142 T > G | p.V381G | Het | – | Novel | AR | LCA 15 | + + + | PM2 + PM3 + PP3 + PP4 | ||
| 16 | 1 | CHM | NM_000390 | 5 | c.544delT | p.C182Vfs*14 | Hemi | – | Novel | XLD | choroideremia | / / / | PVS1 + PM2 |
| 28 | 1 | RPGRIP1 | NM_020366 | 16 | c.2662C > T | p.R888X | Hom | 0/ 1.68e−5 | Yes[57] | AR | LCA6 | + + + | PVS1 + PS1 + PM2 + PP3 |
| Fa | Np | Gene | Transcript RefSeq | Ex | NA Changes | AA changes | Hzyo | Pf | Reported | Gm | Disease | SPM | ACMG grade |
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| 17 | 2 | PRPF8 | NM_006445 | 36 | c.5792C > T | p.T1931M | Het | – | Novel | AD | RP, 13 | + + + | PM2 + PP1 + PP2 + PP3 + PP4 |
| 20 | 1 | TRPM1 | NM_0012 52,020 | 21 | c.2789 T > A | p.I930N | Het | – | Novel | AR | CSNB1C | + + + | PM2 + PM3 + PP2 + PP3 |
| TRPM1 | NM_0012 52,020 | 22 | c.3178 + 1G > A | splicing | Het | 6.889e−4/ 5.772e−5 | Yes[58] | AR | CSNB1C | / / + | PVS1 + PS1 + PM2 | ||
| 21 | 1 | NR2E3 | NM_014249 | 6 | c.925C > T | p.R309W | Hom | 0/ 8.34e−6 | Novel | AR | GF | / + / | PM2 + PM5 + PP2 + PP4 |
| 22 | 1 | PAX2 | NM_003990 | 2 | c.70dupG | p.V26Gfs*28 | Het | 0/ 1.237e−5 | Yes[59] | AD | RCS | / / / | PVS1 + PS1 |
| 34 | 2 | KCNV2 | NM_133497 | 1 | c.506_513delTGCTGCT | p.V169Gfs*40 | Het | – | Novel | AR | RCD3B | / / / | PVS1 + PM2 + PM3 + PP1 |
| KCNV2 | NM_133497 | 1 | c.137G > A | p.W46X | Het | – | Yes[60] | AR | RCD3B | + + + | PVS1 + PS1 + PM2 + PP1 + PP3 | ||
| 35 | 2 | FZD4 | NM_206933 | 2 | c.612 T > A | p.C204X | Het | – | Novel | AD | FEVR1 | + + + | PVS1 + PM2 + PP1 + PP3 |
| 37 | 2 | LRP5 | NM_002335 | 2 | c.485_488delACGG | p.H162Rfs*38 | Het | – | Novel | AD | FEVR4 | / / / | PVS1 + PM2 + PP1 |
| 39 | 1 | SLC38A8 | NM_001080442 | 7 | c.927_928delCT | p.Y310Pfs*57 | Het | – | Novel | AR | FH2 | / / / | PVS1 + PM2 + PM3 |
| SLC38A8 | NM_001080442 | 6 | c.697G > A | p.E233K | Het | 2.778e−4/ 6.886e−5 | Yes[61] | AR | FH2 | + + + | PS1 + PM2 + PP3 | ||
| 40 | 1 | AIPL1 | NM_001285399 | 3 | c.385C > T | p.Q129X | Hom | – | Novel | AR | LCA4 | + + + | PVS1 + PM2 + PP3 |
| 41 | 1 | FRMD7 | NM_194277 | 10 | c.910C > T | p.R304X | Hemi | 0/ 5.608e−6 | Yes[62] | XLR | Nystagmus 1 | + + + | PVS1 + PS1 + PM2 + PP3 |
| 42 | 1 | GUCY2D | NM_000180 | 18 | c.3177_3178delAC | p.R1060Rfs*11 | Hom | 0/ 4.935e−6 | Novel | AR | LCA4 | / / / | PVS1 + PM2 |
| 45 | 1 | CNGA1 | NM_001142564 | 5 | c.472delC | p.L158Ffs*4 | Het | 0.0012/ 6.455e−5 | Novel[63] | AR | RP, 49 | / / / | PVS1 + PM2 |
| CNGA1 | NM_001142564 | 5 | c.453C > A | p.Y151X | Het | 5.798e−5/ 4.068e−6 | Novel | AR | RP, 49 | + + + | PVS1 + PM2 + PP3 | ||
| 49 | 3 | TSPAN12 | NM_012338 | 8 | c.731delT | p.L244Rfs*17 | Het | 0/ 4.064e−6 | Novel | AD | EV5 | / / / | PVS1 + PM2 + PP1 |
Fa denotes Family No.; Np denotes the number of patients; Ex denotes an exon; NA denotes nucleic acid; AA denotes amino acid; Hzyo denotes heterozygosity; Pf denotes the population frequency recorded in the gnomAD database; Gm denotes the genetic model; Disease denotes OMIM disease; SPM denotes SIFT, PolyPhen_2 and Mutation t@sting predicting, ‘ + ’denotes damaging, ‘-’denotes benign, and ‘/’ denotes no data. RP,4 denotes retinitis pigmentosa, type 4; FEVR2 denotes familial exudative vitreoretinopathy, type 2; Usher 2A denotes Usher syndrome, type 2A; RP,39 denotes retinitis pigmentosa, type 39; RP,38 denotes retinitis pigmentosa, type 38; Bietti CCD denotes Bietti crystalline corneoretinal dystrophy; RP, 30 denotes retinitis pigmentosa, type 30; RP, 11 denotes retinitis pigmentosa, type 11; MD denotes macular degeneration, X-linked atrophic; RP,2 denotes retinitis pigmentosa, type 2; Stargardt 1 denotes Stargardt's disease, type1; LCA 15 denotes Leber congenital amaurosis, type 15; LCA6 denotes Leber congenital amaurosis, type 6; RP,13 denotes retinitis pigmentosa, type 13; CSNB1C denotes congenital stationary night blindness, type 1C; GF denotes Goldmann-Favre syndrome; RCS denotes renal coloboma syndrome; RCD3B denotes retinal cone dystrophy, type 3B; FEVR1 denotes familial exudative vitreoretinopathy, type 1; FEVR4 denotes familial exudative vitreoretinopathy, type 4; FH2 denotes foveal hypoplasia, type 2; LCA4 denotes Leber congenital amaurosis, type 4; Nystagmus 1 denotes nystagmus, type 1, congenital, X-linked; RP, 49 denotes retinitis pigmentosa, type 49; EV 5 denotes exudative vitreoretinopathy, type 5
Table 2.
General situation of families with likely pathogenic mutations or related mutations of undetermined significance
| Fa | Np | Gene | Transcript RefSeq | Ex | NA Changes | AA changes | Hzyo | Pf | Reported | Gm | OMIM Disease | SPM | ACMG grade |
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| 8 | 1 | USH2A | NM_206933 | 41 | c.8002G > T | p.E2668X | Het | – | Novel | AR | Usher 2A/RP, 39 | / / + | PVS1 + PM2 |
| USH2A | NM_206933 | 13 | c.2802 T > G | p.C934W | Het | 2.441e−3/1.915e−4 | Yes[44] | AR | Usher 2A/RP, 39 | + + + | PS1 + PM2 + PP3 | ||
| 10 | 1 | USH2A | NM_206933 | 63 | c.12608A > G | p.Q4203R | Het | 9.457e−3/ 3.677e−3 | Novel | AR | Usher 2A/RP, 39 | – | PM2 + BP4 |
| USH2A | NM_206933 | 22 | c.4758 + 3A > G | Splicing | Het | 1.855e−2/ 1.457e−3 | Yes[64] | AR | Usher 2A/RP, 39 | / / / | PS1 + PM2 | ||
| 23 | 1 | USH2A | NM_206933 | 66 | c.14411G > A | p.G4804E | Het | – | Novel | AR | Usher 2A/RP, 39 | / / + | PM2 |
| USH2A | NM_206933 | 19 | c.4217C > A | p.S1406X | Het | – | Novel | AR | Usher 2A/RP, 39 | + + + | PVS1 + PM2 + PP3 | ||
| 53 | 1 | USH2A | NM_206933 | 65 | c.14287G > A | p.G4763R | Het | – | Yes[65] | AR | Usher 2A/RP, 39 | + + + | PS1 + PM2 + PP3 |
| USH2A | NM_206933 | 4 | c.784 + 2 T > G | Splicing | Het | – | Novel | AR | Usher 2A/RP, 39 | / / / | PVS1 + PM2 | ||
| 19 | 1 | USH1C | NM_153676 | 5 | c.407G > A | p.R136Q | Het | 1.16e−4/ 1.223e−4 | Novel | AR | Usher 1C | / / + | PM2 |
| USH1C | NM_153676 | 15 | c.1250C > T | p.T417I | Het | – | Novel | AR | Usher 1C | / / + | PM2 | ||
| 13 | 1 | BBS2 | NM_031885 | 6 | c.626 T > C | p.L209P | Het | – | Yes[66] | AR | RP, 74 | + + + | PS1 + PM2 + PP3 |
| BBS2 | NM_031885 | 1 | c.79A > C | p.T27P | Het | – | Novel | AR | RP, 74 | – | PM2 + BP4 | ||
| 25 | 1 | LRP5 | NM_002335 | 15 | c.3361A > G | p.N1121D | Het | 7.528e−3/ 5.616e−4 | Yes[67] | AR | FEVR4 | + + + | PS1 + PM2 + PP3 |
| LRP5 | NM_002335 | 18 | c.3901G > A | p.A1301T | Het | 2.403e−3/ 2.149e−4 | Novel | AR | FEVR4 | − | PM2 + BP4 | ||
| 56 | 1 | LRP5 | NM_002335 | 15 | c.3377 T > C | p.L1126P | Het | – | Novel | AR | FEVR4 | + + + | PM2 + PP3 |
| LRP5 | NM_002335 | 22 | c.4519G > T | p.D1507T | Het | – | Novel | AR | FEVR4 | + + + | PM2 + PP3 | ||
| 24 | 1 | ABCA4 | NM_000350 | 5 | c.553C > T | p.Q185X | Het | – | Yes[68] | AD | AMD2 | + + + | PVS1 + PS1 + PM2 + PP3 |
| 26 | 1 | RS1 | NM_000330 | 4 | c.240G > C | p.Q80H | Hemi | – | Novel | XLR | Retinoschisis | / / + | PM2 + PP4 |
| 29 | 1 | GPR143 | NM_000273 | 2 | c.263G > A | p.R88Q | Hemi | – | Novel | XL | Nystagmus 6 | + + + | PM2 + PP3 |
| 31 | 1 | FBN2 | NM_001999 | 30 | c.3923dupG | p.C1308Wfs*5 | Het | – | Novel | AD | EMD | / / / | PVS1 + PM2 |
Fa denotes Family No.; Np denotes the number of patients; Ex denotes an exon; NA denotes nucleic acid; AA denotes amino acid; Hzyo denotes heterozygosity; Pf denotes the population frequency recorded in the gnomAD database; Gm denotes the genetic model; Disease denotes OMIM disease; SPM denotes SIFT, PolyPhen_2 and Mutation t@sting predicting, ‘ + ’ denotes damaging, ‘-’ denotes benign, and ‘/’ denotes no data. Usher 2A denotes Usher syndrome, type 2A; RP,39 denotes retinitis pigmentosa, type 39; Usher 1C denotes Usher syndrome, type 1C; RP,74 denotes retinitis pigmentosa, type 74; FEVR4 denotes familial exudative vitreoretinopathy, type 4; AMD2 denotes age-related macular degeneration, type 2; Nystagmus 6 denotes nystagmus, type 6, congenital, X-linked; EMD denotes macular degeneration, early onset
The patients with LHON were evaluated using PCR-based Sanger sequencing, which included only 3 common mutant sites of mitochondrial DNA, namely, MTND1mt.3460, MTND4mt.11778 and MTND6mt.14484, and 10 rare mutant sites of mitochondrial DNA, namely, MTND1 (mt.3376, mt.3635, mt.3700, mt.3733), MTND6 (mt.14482, mt.14495, mt.14502, mt.14568, mt.14498, mt.14325). The pathogenicity of these mitochondrial mutations is known. The PCR primers used were designed with GeneTool software (refer to Additional file 1: Table S2).
Population control
The frequency of the detected mutations in the population was retrieved from Genome Aggregation Database (gnomAD, http://gnomad-old.broadinstitute.org/) because of its wide large-scale sequencing data. We chose the frequencies of mutation sites in all populations and in the East Asian population as controls.
Functional prediction analysis
Candidate pathogenic mutation sites were searched in public databases, including dbSNP (https://www.ncbi.nlm.nih.gov/snp/), 1000G (https://www.internationalgenome.org/) and ExAC (The Exome Aggregation Consortium, https://exac.hms.harvard.edu). Candidate sites in HGMD (The Human Gene Mutation Database at the Institute of Medical Genetics in Cardiff, http://www.hgmd.cf.ac.uk/ac/index.php), professional version, were also searched in to determine whether pathogenicity has been reported in the literature. PhyloP and PhastCons software were used to analyse the conservation of corresponding amino acid sequence of missense mutations [11]. Pathogenic analysis was conducted by SIFT (http://sift-dna.org), PolyPhen_2 (http://genetics.bwh.harvard.edu/pph2/) and Mutation t@sting online tools (http://mutationtaster.org/) [11–13]. We also analysed the secondary structure, disordered region and mutation effect of missense mutations by the PredictProtein online tool (https://predictprotein.org/). Three-dimensional structure construction of the target protein sequence was performed using Swiss-Model (https://swissmodel.expasy.org/) protein model structure simulation software [14]. The pathogenicity of candidate mutations was graded and judged according to the 2015 edition of the ACMG standard and guidelines [15].
Results
The genome variation results of different patients and their families are classified and summarized based on pathogenic genes.
Mutation distribution in patients with suspected LHON
In total, 116 patients with suspected optic neuropathy were examined, and 25 cases of LHON were diagnosed (Fig. 1).The diagnostic rate for LHON of the Part A was 21.6% in our optic atrophy group using the mtDNA Sanger sequencing panel (OPA1, WFS1, etc., not included). The ratio of males to females among patients with LHON was 4:1 in our investigation. The average age of the patients diagnosed with LHON was 19 years old, and their age ranged from 6 to 36 years. The three common mutant sitesmt.3460, mt.11778 and mt.14484were found to be the main (96%) causes of LHON, with MTND4 m.11778G > A being the most common pathogenic mutation, followed by MTND6 m.14484 T > C and MTND1 m.3460G > A. Only one rare mutant, MTND6 m.14502 T > C, was found in these Chinese patients from central China. Several LHON patients harboured incomplete mitochondrial mutations or two mutations.
Fig. 1.
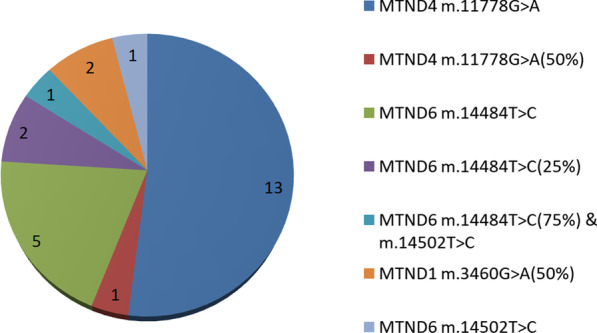
Variation distribution of 25 patients with Leber hereditary optic neuropathy (21.6%)
Pathogenic mutations in hereditary retinopathy
Ninety-five families were examined by using targeted sequencing technology and were suspected to have retinitis pigmentosa or congenital retinopathy. Partial genealogical trees are depicted in Additional file 1: Figure S25. We identified 68 distinct mutations in 31 known disease genes in the patients of these families; 37 mutations are novel. The results are grouped by related genes found in retinopathy patients. In this investigation, significant mutants were detected in 57.9% of the families tested (Tables 1 and 2). The mutations listed in Table 1 are predicted to be damaging or disease causing by function prediction software, and some of the mutations have been studied and reported. The phenotypes and mutations of these families co-segregate. Targeted sequencing of retinopathy-related genes for Part B provided a 45.3% diagnostic rate, and another 12.6% of the families in this study carried candidate gene mutations with undefined pathogenicity. The diagnostic rate of RP and MD was 45.5% (30/66), and the significant detection yield was 57.6% (38/66). The diagnostic rate of multiple fundus lesions or retinal dysplasia was 44.8% (13/29), and the significant detection yield was 58.6% (17/29).
Four families (families 14, 15, 48 and 54) developed retinitis pigmentosa caused by RHO mutations, and the patients in these families manifested night blindness in childhood, visual field defects or tubular visual fields and retinitis pigmentosa. The Sanger sequencing results for mutations in Family 14 and Family 15 are presented in Additional file 1: Figures S13 and S14, respectively. NDP mutations can lead to familial exudative vitreoretinopathy (FEVR) or Norrie disease. Two families (18 and 46) with FEVR2 carried two novel NDP mutations, c.124C > A (p.H42N) (Fig. 2) and c.401_402delGA (p.*134Wfs*13). The eyes of those with FEVR2 do not follow movement when they are a few months old, and no blood vessel area of the binocular fundus is detected by ophthalmoscopic examination. Male patients of the two families had no other serious visual problems. Two families (32 and 55) diagnosed with Norrie disease carried two known NDP mutations, c.343C > T (p.R115X) and c.268C > T (p.R90C). The two-month-old male patient in Family 32 had vitreous hyperplasia, right microphthalmos and microcorneas; the male patient in Family 55 had legal blindness and atrophy of the eyeballs. Mutation of USH2A can cause retinitis pigmentosa with or without sensorineural hearing loss. The patients of Family 7 and Family 47 with Usher syndrome type 2A presented with retinitis pigmentosa and hearing impairment, harbouring different mutations in the USH2A gene (Additional file 1: Figures S11, S17 and S18). The patient of Family 9 with USH2A mutation had nonsyndromic retinitis pigmentosa. Three families (families 27, 38 and 51) carried different RS1 hemizygous mutations in the retinoschisis patients. The results of fundus examination and optical coherence tomography of the patient with congenital retinoschisis in Family 38 are shown in Additional file 1: Figures S5 and S6. Patients from Family 1 and Family 2 maybe diagnosed with RP 38 caused by MERTK gene mutation. These cases are characterized by retinitis pigmentosa, night blindness and visual field loss.
Fig. 2.

NDP c.124C > A hemizygous mutation and the fundus avascular area of the FEVR2 patient in Family 18. In part a, fundus examination of the one-year-old patient showed an avascular area in both eyes. The temporal side of the blood vessel arch in the right eye fundus showed the epiretinal membrane and macular traction. Part b, NDP c.124C > A mutation of the mother and the child, respectively
A small deletion and nonsense mutation in CYP4V2 was found to be the cause of the Bietti crystalline corneoretinal dystrophy of the patients from Families 3 and 4. The visual electrophysiology results for Family 3 are shown in Additional file 1: Figure S1. The CYP4V2 c.(802–8)_810delTCATACAGGTCATCGCTinsGC and c.958C > T mutations in Family 4 are shown in FAdditional file 1: Figures S8 and S9. FSCN2 c.72delG was the cause of RP 30 in two unrelated families (families 5 and 6). The Sanger sequencing result for FSCN2 c.72delG is shown in Additional file 1: Figure S10. A small deletion and frameshift mutation in PRPF31 led to RP 11 in Family 12 (PRPF31 c.1074-8_1079delGTACCGGTCCCCAG novel mutation in Additional file 1: Figure S12) and Family 50. There were four RP patients from three generations in Family 12. In addition to the symptoms of retinitis pigmentosa, night blindness and tubular visual field, the proband and his father (Additional file 1: Figure S25) also underwent postoperative cataract extraction with intraocular lens implantation. There were two families (families 33 and 52) with a family history of RP and night blindness caused by the same mutation: RPGR c.2236_2237delGA. Two families (families 43 and 44) had a family history of RP and night blindness caused by different mutations of RP2, which included the reported splicing mutation c.769-2A > G and the novel frameshift mutation c.572_582dup11.
Seventeen families affected by different retinal diseases carried pathogenic or likely pathogenic mutations in17 different related genes. The patient of Family 36 with macular degeneration had poor eyesight. The patient of Family 16 had retinochoroidal coloboma, and his visual field examination and mutation sequencing results are provided in Additional file 1: Figure S4. Sanger sequencing results of the mutant site in Family 17 are shown in Additional file 1: Figure S15, and the RP proband also had cataracts when he was twenty-six years old. The patient of Family 20 was two years old (Sanger results in Additional file 1: Figure S20). Her full-field ERG (electroretinogram) showed that the rod cells had no waves, while scotopic ERG showed decreased amplitudes of α and β waves. The ophthalmoscopic image and sequencing results of RCS patients from Family 22 are presented in Additional file 1: Figure S19. The CNGA1 mutations in Family 45 were validated by Sanger sequencing (Additional file 1: Figure S16). The thirty-four-year-old mother and her daughter in Family 34 had macular degeneration. The forty-one-year-old patient of Family 35 experienced retinal detachment, primary vitreous hyperplasia and FEVR, and his mother with the same FZD4 c.612 T > A heterozygous mutation had the same manifestations. Both a thirty-three-year-old man and his mother with neurodystrophy and FEVR in Family 37 harboured the LRP5 c.485_488delACGG heterozygous mutation. A three-year-old girl in Family 39 with congenital horizontal nystagmus had compound heterozygous variation of SLC38A8, and her parents were heterozygous carriers of the variant. A two-year-old boy in Family 40 had Leber congenital amaurosis (LCA), and his parents were heterozygous carriers of an AIPL1 variant. The hemizygous FRMD7 c.910C > T (p.R304X) mutation led to Nystagmus of the boy in Family 41, and his mother was a heterozygous carrier of the mutation. A five-year-old boy was diagnosed with LCA caused by GUCY2D c.3177_3178delAC homozygosity inherited from his parents.
Variants of undetermined significance in retinopathy families
The mutations listed in Table 2 are predicted to be damaging or associated with the clinical phenotypes of the families and can be considered candidate mutations. The families included in Table 2 generally had no family history of hereditary diseases. Four families (8, 10, 23 and 53) showed different compound heterozygous mutations of USH2A, and the mutations were associated with the nonsyndromic retinitis pigmentosa of these patients but without obvious hearing impairment. The mutations found in the four families are likely pathogenic. A four-year-old boy in Family 19 carried a compound heterozygous mutation of USH1C; mutation of this gene can cause Usher syndrome-type 1C characterized as severe hearing impairment and retinitis pigmentosa. The boy with RP and night blindness had bilateral secretory otitis media, but his bilateral hearing was basically normal. He passed an TEOAE (transient evoked otoacoustic emissions) examination and DPOAE (distortion product otoacoustic emissions) test at acoustic frequencies of 1 k, 2 k, 4 k and 8 k Hz, but his left ear did not pass DPOAE at 0.5 k Hz. In addition, I-wave latency was slightly longer after 80 dBnHL short-tone stimulation in the ABR (auditory brainstem response) test, though other waves were normal. Therefore, the USH1C mutation is associated with these phenotypes but has undetermined significance. The other patients from different families (13, 25, 56, 24, 29 and 31) (Table 2) carried candidate gene mutations and corresponding phenotypes. The Sanger sequencing results for Family 56 are shown in Additional file 1: Figure S21 and S22. It should be noted that the RS1 c.240G > C (p.Q80H) mutation did not co-segregate with the phenotype and genotype in Family 26.
Specific cases
Family 2
There were two RP patients in Family 2. The thirteen-year-old sister had patchy defects of the visual field and abnormal ERG, and she harboured a homozygous mutation of MERTK c.754delC (Fig. 3). Herten-year-old brother’s symptoms were milder, but he also had defects in the visual field (Additional file 1: Figure S7) and carried the same homozygous mutation. They all had night blindness and visual impairment. Their parents were heterozygous carriers of MERTK c.754delC. According to ACMG guidelines, the novel frameshift mutation of MERTK c.754delC should be considered pathogenic, and its grade (PVS1) is high. A healthy boy was born into this family through three generations of IVF technology (pre-implantation genetic diagnosis).
Fig. 3.

In Part a, P-VEP examination of the older sister with a binocular patchy visual field in Family 2 showed bilateral P100 wave latency delay with normal amplitude. F-ERG examination showed binocular light adaptation, moderately or severely decreased 30 Hz response amplitude, and moderately decreased other response amplitudes; a binocular dark response could not be induced, OPS wavelets could not be separated, other waves could be induced, and the amplitude decreased moderately. Part b shows the Sanger sequencing results of the mutated site
Family 3
The patient with homozygosity of CYP4V2 c.(802-8)_810delTCATACAGGTCATCGCTinsGC developed retinitis pigmentosa and visual impairment. This mutation is known to be pathogenic for Bietti crystalline corneoretinal dystrophy (Bietti CCD), and it involves small deletions and insertions in splicing regions. The patient had typical fundus and visual electrophysiological symptoms (Additional file 1: Figure S24 and Figure S1). Therefore, she can be diagnosed with Bietti CCD according to ocular manifestations and gene mutations.
Family 5
The RP patients in Family 5 all carried the known pathogenic mutation FSCN2 c.72delG. The proband had typical fundus and visual electrophysiological symptoms (Additional file 1: Figure S23 and Figure S2). This mutation was the same genetic cause as found for Family 6, and it is a common pathogenic mutation for RP 30.
Family 11
A three-year-old boy, one of fraternal twins, was given medical advice for night blindness. The boy’s clinical manifestations also included retinal abnormalities, lateral nystagmus and finger-stimulation eyeball phenomena. He carried the TULP1 compound heterozygous mutation c.1318C > T (p.R440X) and c.1142 T > G (p.V381G); his parents are heterozygous carriers of each of the mutations. The nonsense mutation c.1318C > T (p.R440X) is known to be pathogenic for LCA, type 15, and the missense mutation c.1142 T > G (p.V381G) is novel. c.1142 T > G can lead to amino acid substitution and affects the protein’s function. The OCT (optical coherence tomography) image, fundus photography and mutations are presented in Fig. 4. The boy was diagnosed with LCA 15 according to his clinical manifestations and gene mutations.
Fig. 4.

TULP1 mutations and clinical manifestation of the LCA 15 patient in Family 11. Part a, optical coherence tomography (OCT) shows that the temporal retinal neuroepithelium of macula of both eyes were thinning, with the central fovea of macula forming a backward concave. Part b, ophthalmoscopic examination shows that the boundary of optic disc is blurred and the retina dark. Part c, the compound heterozygous mutation TULP1 c.1318C > T p.R440X (above) and c.1142 T > G p.V381G (below). Part d, Secondary structure change of the novel mutation TULP1 c.1142 T > G
Family 18
The one-year-old boy’s fundus photographs and mutation sequencing results are shown in Fig. 2. The cornea of both eyes was clear, the anterior chamber was preserved, and the lens was transparent. Fundus photography showed no blood vessel area in either eye. The temporal epiretinal membrane of the right fundus vascular arch pulled the macula. NDP mutation can lead to FEVR2, and c.124C > A (p.H42N) is a novel mutation causing FEVR2. There is one known pathogenic mutation of c.125A > G (p.H42R) at the same location of the polypeptide chain of this novel variant. According to ACMG guidelines and related prediction software, c.124C > A (p.H42N) should be pathogenic. FEVR2 is characterized by no blood vessel area of the fundus, but the severity of the disease varies. There three persons with c.124C > A (p.H42N) mutation in this family showed no blood vessel area in either fundus.
Family 21
This family of Chinese Hui nationality (a Chinese minority) involved a consanguineous marriage. The patient presented with retinoschisis, macular oedema and night blindness, and was a homozygous carrier of NR2E3 c.925C > T (p.R309W). The ophthalmological examination and mutation sequencing results of the patient are shown in Fig. 5 and Additional file 1: Figure S3. The missense mutation c.925C > T of NR2E3 is a novel mutation for Goldmann-Favre syndrome, but the c.925C > G (p.R309G) at the same location of mRNA and polypeptide chain is known to be pathogenic for Goldmann-Favre syndrome and enhanced S-cone syndrome [16]. Some scholars believe that Goldmann-Favre syndrome is the severe type of enhanced S-cone syndrome [17]. The patient's condition worsened over the past 10 years, and he was diagnosed with Goldmann-Favre syndrome according to his phenotype and genotype.
Fig. 5.

Fluorescein fundus angiography examination of the patient in family 21 showed prolonged filling time of bilateral arteriovenous fluorescence. In the early stage, inhomogeneous strong fluorescence and occluded fluorescence was seen in the posterior pole of both eyes. Strong fluorescence and inhomogeneous fluorescence was also seen in the periphery. In the late stage, inhomogeneous strong fluorescence was seen in the periphery of both eyes, and no obvious fluorescence leakage was observed.OCT examination showed a split nerve cortex in the macular area of the left eye, the fovea in the macular area were not seen, and the pigmented epithelium was rough; the fovea in the macular area of the right eye were not obvious, the pigmented epithelium in the macular area was rough, and the nasal retinal layer was split
Discussion
Using targeted NGS technology and Sanger sequencing, we investigated the mutation profile and clinical features of 211 Chinese families with hereditary retinopathy over three years. Ninety-five families were evaluated by targeted next-generation sequencing, and fifty-five had meaningful positive findings. One hundred and sixteen patients from different families were tested by Sanger sequencing, and twenty-five members carried related mitochondrial mutations. Hereditary retinopathy covers a group of genetically and clinically highly heterogeneous disorders [1]. Targeted NGS analysis is a valuable method for molecular genetics diagnostics of these diseases, as supported by previous studies [18–20]. These studies show that the potential molecular genetics diagnostic rate of targeted sequencing is between 38% [19] and 76% [20]. Jespersgaard's report indicated a detection rate of related genotypes of 72%, whereas the detection rate of causative variants was 48% [18]. Our study attained a 45.3% potential diagnostic rate of hereditary retinopathy families and a 58% meaningful detection rate of families. The diagnostic rate of the genetic tests in this study is in the middle of the range in Europe [19, 20]. Although our detection rate was lower than that is a Japanese study [18], we implemented strict standards to achieve diagnosis. Our research results also support that DNA sequencing is a powerful diagnostic tool for hereditary retinal disease.
Twenty-five patients with positive mitochondrial gene test results in our study were 19 years old on average, with a male-to-female ratio of 4:1. This indicates that retinopathy patients in the Chinese population have a younger age and higher sex ratio than those in Europe and America [21, 22]. However, our study did not find new mitochondrial variants and showed that mt.11778 and mt.14484 are the most common pathogenic mutations for LHON. The possibility of LHON diagnosis over representation may be due to the small scale and single-centre collection.
Due to clinical heterogeneity, many subjects did not have a definite ophthalmological diagnosis before NGS examination; thus, we subdivided them into two larger subtypes: multiple fundus lesions or retinal dysplasia. Overall, our genetic test results may help ophthalmologists make diagnoses or even indicate unobserved lesions confirmed by further clinical examination. Our impression is a high positive rate of genetic testing for rare and severe ocular lesions in this study. One recent study of visual impairment gene detection in a large Dutch cohort provided meaningful information, including various types of inherited eye disorders [23]. Four main types, RP, cataract, developmental eye defects and optic atrophy, were investigated in this previous research, and the detection rates were 63%, 50%, 33% and 17%, respectively. Due to imbalance in the number of subjects with different types, the detection rate of several types with few subjects may need more independent analysis. In contrast, we focused more on the genetic variation of fundus lesions such as RP, MD and specialoptic atrophy.
Retinitis pigmentosa is a hereditary progressive retinopathy. It is the most common blinding disease and is characterized by nocturnal blindness and progressive visual field defects caused by degeneration of retinal photoreceptor cells and pigment epithelial cells [24]. Its inheritance modes include autosomal dominant, autosomal recessive and X-linked recessive inheritance. Thirty families (families 14, 15, 48, 54, 7, 9, 47, 1, 2, 3, 4, 5, 6, 12, 50, 33, 52, 43, 44, 36, 17, 45, 8, 10, 23, 53, 19, 13, 24 and 31) with RP (or MD) had positive meaningful findings of gene mutations. With the help of targeted NGS, these patients were diagnosed with various types of retinitis pigmentosa. In some of these families, healthy offspring were born through genetic prenatal diagnosis or third-generation test-tube infant technology (pre-implantation genetic diagnosis). RP accounted for a large proportion of hereditary retinopathy in our study. Overall, it is difficult to distinguish cone-rod dystrophy from retinitis pigmentosa only through ophthalmic examination because of the similarity in clinical manifestations [25]. Molecular genetic tests help in making accurate diagnoses for patients with cone-rod dystrophy (Family 34). Moreover, in some patients, it is difficult to differentiate choroideremia from RP, and the detection of CHM gene mutations has noteworthy diagnostic value. Choroideremia has a worse prognosis than RP [26], and the patient of Family 16 with choroideremia was diagnosed through targeted sequencing. Congenital stationary night blindness (CSNB) is similar to RP in clinical presentation, but its prognosis is better. We identified a case (Family 20) of CSNB, type 1C, through genetic targeting sequencing in this study.
Vitreoretinopathy is another major type of hereditary retinopathy. In nine families (families 18, 32, 46, 55, 35, 37, 49, 25, 56), gene mutations related to such diseases were detected. FEVR is a retinal vascular structural abnormality with different inheritance patterns. The clinical symptoms of the disease vary greatly, even in the same family [27]. For example, mild cases have no symptoms; the only disease-related abnormality is a circular arc without vascular retina at the periphery of the terminal temporal area. These characteristics were observed in this study. Non-syndromic retinoschisis is an X-linked hereditary retinopathy, and its known pathogenic gene is RS1 [28]. We observed three different known pathogenic mutations of RS1 in three different families (Families 27, 38 and 51). The new mutation RS1 c.240G > C found in Family 26 may be benign because both the patient and his normal maternal grandfather carried it. Goldmann-Favre syndrome is an ocular syndrome with clinical symptoms, including retinoschisis (Family 21).
LCA involves early onset and serious impairment of visual function [29]. Most children become blindness. Parents can usually observe visual abnormalities within one year of the child's birth. Children from four different families (families 11, 28, 40 and 42) were diagnosed with LCA in our investigation, showing that targeted sequencing is of great significance for the diagnosis of hereditary ophthalmopathy and that it will become part of our eye health management. We also detected two cases of fundus developmental disease: renal coloboma syndrome (Family 22) and foveal hypoplasia (Family 39). These two diseases are general lyuntreatable and have a general prognosis, but families with the disease might avoid high-risk offspring according to genetic rules. Two families (families 41 and 29) with nystagmus carried two different mutations, GPR143 and FRMD7. GPR143-orFRMD7-related nystagmus shows X-linked inheritance, with or without obvious retinal abnormalities [30]. The genetic causes can guide these two families in having healthy offspring.
Previous studies have used the Sanger method to sequence only one or several genes for the molecular diagnosis of patients with different retinal diseases [31–34]. One Bietti crystalline corneoretinal dystrophystudy showed an 84% detection rate by CYP4V2 sequencing alone [31], but it is likely to be a single example of over representation due to the only known pathogenic gene being CYP4V2. The related detection rates of two FEVR studies in which three and six genes were sequenced were just 23% [32] and 38.7% [33] respectively. Meaningful results of genetic testing usually require a high degree of accurate clinical diagnosis. Nevertheless, retinal diseases have complex clinical manifestations and genetic heterogeneity. The clinical symptoms of some diseases are difficult to distinguish, and some diseases are related to multiple genes. As the number of genes needed to be detected increases, the efficiency of the Sanger sequencing method decreases, and targeted sequencing becomes a better choice. Analysis of exon copy number variants in targeted gene was also executed in this study using panel sequencing data, though there were no positive findings. Exon duplication of OCRL was found in Lowe syndrome in our previous work [35].
In summary, we report 37 novel related meaningful mutations and 31 known pathogenic variants for retinopathy in 31 different genes, leading to different relevant phenotypes of eye diseases. The diagnostic rate of LHON was 21.6% in our study, but no new mitochondrial pathogenic mutations were found. To our knowledge, this is a larger-scale medical genetic study of retinal diseases in the Chinese population than previously reported. The innovation of this research is that we report new variants and phenotypes of diseases as well as the important role of sequencing results in diagnosis and differential diagnosis. New research advances suggest that molecular genetic tests may be used not only to clarify diagnoses and to direct counselling but also to move the field of 'incurable' and 'blinding' inherited retinal diseases substantially forward [1]. Our study demonstrates the importance of examining a large collection of families with hereditary retinopathy because of the clinical manifestations and genetic heterogeneity of the diseases, with guiding significance for this disease diagnosis and aristogenesis.
Conclusion
The targeted NGS of the human genome in related Chinese families in this study expands the mutational spectrum and deepens our understanding of the mechanism of disease. This investigation also increases knowledge of the heterogeneity of clinical manifestations of diseases and enriches the phenotypic spectrum of diseases. Our study contributes novel mutations and the phenotypic aspects of retinopathy and reveals the genetic and clinical heterogeneity of related conditions. Our results illustrate the significance of molecular genetic testing for patients with hereditary retinopathy.
Supplementary Information
Additional file 1. Supplementary material of tables and figures.
Acknowledgements
We would like to thank all the participants for their help and willingness to participate in this study. We thank the reviewers for their comments. We also thank the editors of this manuscript.
Abbreviations
- NGS
Next-generation sequencing
- gnomAD
Genome Aggregation Database
- dbSNP
The Single Nucleotide Polymorphism Database
- 1000G
The 1000 Genomes Project
- ExAC
The Exome Aggregation Consortium
- HGMD
The Human Gene Mutation Database at the Institute of Medical Genetics in Cardiff
- ACMG
The American College of Medical Genetics and Genomics
- LHON
Leber hereditary optic neuropathy
- RP
Retinitis pigmentosa
- MD
Macular degeneration
Authors' contributions
ZB and XK contributed to the conception and design of the study. YX and LL carried out the experiments. ZB, JS and YL performed the data analysis. ZB wrote and revised the manuscript. All authors read and approved the final manuscript.
Funding
This work was funded in part by the National Key R&D Program of China (2018YFC1002206-2). The role of the funding body—This study is the authors’ independent work, and the funding agency only provides relevant financial support. The design of the study and collection, analysis, and interpretation of data and writing of the manuscript were completed by all authors independently.
Availability of data and materials
The raw datasets used and analysed during the current study are not deposited in publicly available repositories because of considerations about the security of human genetic resources. The transcript RefSeq number (Tables 1 and 2) was obtained from the Ensembl database (http://asia.ensembl.org) [10]. Any questions should be directed to the corresponding author. We provide conclusive variant information without identifying/confidential patient data in the paper or its appendix. For other details of the availability of data and material, please refer to the methods section of the article.
Declarations
Ethics approval and consent to participate
The clinical investigations were conducted according to the Declaration of Helsinki, and the study was approved by the institutional review board of the Medical Ethics Committee of the First Affiliated Hospital of Zhengzhou University(Grant No: KS-2018-KY-36). The Medical Ethics Committee of the First Affiliated Hospital of Zhengzhou University authorized our team to access the clinical patient data used in this research. For all minor patients, parents agreed to study participation, and written consent was obtained. Adult patients consented to participate in this study, and written consent was obtained.
Consent for publication
Written informed consent for publication of clinical details and/or clinical images was obtained from all participants. Written informed consent for individuals younger than the age of 18 was obtained from their parents.
Competing interests
The authors report no conflicts of interest.
Footnotes
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
References
- 1.Garafalo AV, et al. Progress in treating inherited retinal diseases: Early subretinal gene therapy clinical trials and candidates for future initiatives. Prog Retin Eye Res. 2020;77:100827. doi: 10.1016/j.preteyeres.2019.100827. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Tsang SH, Sharma T. Autosomal dominant retinitis pigmentosa. Adv Exp Med Biol. 2018;1085:69–77. doi: 10.1007/978-3-319-95046-4_15. [DOI] [PubMed] [Google Scholar]
- 3.Xu L, et al. Prevalence of retinitis pigmentosa in urban and rural adult Chinese: the Beijing Eye Study. Eur J Ophthalmol. 2006;16(6):865–866. doi: 10.1177/112067210601600614. [DOI] [PubMed] [Google Scholar]
- 4.Tsang SH, Sharma T. Retinitis pigmentosa (non-syndromic) Adv Exp Med Biol. 2018;1085:125–130. doi: 10.1007/978-3-319-95046-4_25. [DOI] [PubMed] [Google Scholar]
- 5.Dimopoulos, I.S. and M. Xu, Re: Feuer et al.: Gene therapy for Leber hereditary optic neuropathy: initial results (Ophthalmology 2016;123:558–570). Ophthalmology, 2017. 124(3): p. e22. [DOI] [PubMed]
- 6.Petrs-Silva H, et al. Suppression of rds expression by siRNA and gene replacement strategies for gene therapy using rAAV vector. Adv Exp Med Biol. 2012;723:215–223. doi: 10.1007/978-1-4614-0631-0_29. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Ghazi NG, et al. Treatment of retinitis pigmentosa due to MERTK mutations by ocular subretinal injection of adeno-associated virus gene vector: results of a phase I trial. Hum Genet. 2016;135(3):327–343. doi: 10.1007/s00439-016-1637-y. [DOI] [PubMed] [Google Scholar]
- 8.Rodrigues GA, et al. Pharmaceutical development of AAV-based gene therapy products for the eye. Pharm Res. 2018;36(2):29. doi: 10.1007/s11095-018-2554-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.DiCarlo JE, Mahajan VB, Tsang SH. Gene therapy and genome surgery in the retina. J Clin Invest. 2018;128(6):2177–2188. doi: 10.1172/JCI120429. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Yates AD, et al. Ensembl 2020. Nucleic Acids Res. 2020;48(D1):D682–d688. doi: 10.1093/nar/gkz966. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Schwarz JM, et al. MutationTaster2: mutation prediction for the deep-sequencing age. Nat Methods. 2014;11(4):361–362. doi: 10.1038/nmeth.2890. [DOI] [PubMed] [Google Scholar]
- 12.Sim N-L, et al. SIFT web server: predicting effects of amino acid substitutions on proteins. Nucleic Acids Res. 2012;40(W1):W452–W457. doi: 10.1093/nar/gks539. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Adzhubei IA, et al. A method and server for predicting damaging missense mutations. Nat Methods. 2010;7(4):248–249. doi: 10.1038/nmeth0410-248. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Waterhouse A, et al. SWISS-MODEL: homology modelling of protein structures and complexes. Nucleic Acids Res. 2018;46(W1):W296–w303. doi: 10.1093/nar/gky427. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Richards S, et al. Standards and guidelines for the interpretation of sequence variants: a joint consensus recommendation of the American College of Medical Genetics and Genomics and the Association for Molecular Pathology. Genet Med. 2015;17(5):405–424. doi: 10.1038/gim.2015.30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Haider NB, et al. Mutation of a nuclear receptor gene, NR2E3, causes enhanced S cone syndrome, a disorder of retinal cell fate. Nat Genet. 2000;24(2):127–131. doi: 10.1038/72777. [DOI] [PubMed] [Google Scholar]
- 17.Marmor MF. A teenager with nightblindness and cystic maculopathy: enhanced S cone syndrome (Goldmann-Favre syndrome) Doc Ophthalmol. 2006;113(3):213–215. doi: 10.1007/s10633-006-9031-z. [DOI] [PubMed] [Google Scholar]
- 18.Jespersgaard C, et al. Molecular genetic analysis using targeted NGS analysis of 677 individuals with retinal dystrophy. Sci Rep. 2019;9(1):1219. doi: 10.1038/s41598-018-38007-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Martin-Merida I, et al. Genomic landscape of sporadic retinitis pigmentosa: findings from 877 Spanish Cases. Ophthalmology. 2019;126(8):1181–1188. doi: 10.1016/j.ophtha.2019.03.018. [DOI] [PubMed] [Google Scholar]
- 20.Stone EM, et al. Clinically focused molecular investigation of 1000 consecutive families with inherited retinal disease. Ophthalmology. 2017;124(9):1314–1331. doi: 10.1016/j.ophtha.2017.04.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Carelli V, et al. Parsing the differences in affected with LHON: genetic versus environmental triggers of disease conversion. Brain. 2016;139(Pt 3):e17. doi: 10.1093/brain/awv339. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Black GC, et al. Leber's hereditary optic neuropathy: implications of the sex ratio for linkage studies in families with the 3460 ND1 mutation. Eye (Lond) 1995;9(Pt 4):513–516. doi: 10.1038/eye.1995.117. [DOI] [PubMed] [Google Scholar]
- 23.Haer-Wigman L, et al. Diagnostic exome sequencing in 266 Dutch patients with visual impairment. Eur J Hum Genet. 2017;25(5):591–599. doi: 10.1038/ejhg.2017.9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Berson, E.L., Retinitis pigmentosa. The Friedenwald Lecture. Invest Ophthalmol Vis Sci, 1993. 34(5): p. 1659–76. [PubMed]
- 25.Daiger SP, Sullivan LS, Bowne SJ. Genes and mutations causing retinitis pigmentosa. Clin Genet. 2013;84(2):132–141. doi: 10.1111/cge.12203. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Chan SC, et al. Choroideremia research: Report and perspectives on the second international scientific symposium for choroideremia. Ophthalmic Genet. 2016;37(3):267–275. doi: 10.3109/13816810.2015.1088958. [DOI] [PubMed] [Google Scholar]
- 27.Seo SH, et al. Molecular CHARacterization of FZD4, LRP5, and TSPAN12 in familial exudative vitreoretinopathy. Invest Ophthalmol Vis Sci. 2015;56(9):5143–5151. doi: 10.1167/iovs.14-15680. [DOI] [PubMed] [Google Scholar]
- 28.Molday RS, Kellner U, Weber BH. X-linked juvenile retinoschisis: clinical diagnosis, genetic analysis, and molecular mechanisms. Prog Retin Eye Res. 2012;31(3):195–212. doi: 10.1016/j.preteyeres.2011.12.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Kumaran N, et al. Leber congenital amaurosis/early-onset severe retinal dystrophy: clinical features, molecular genetics and therapeutic interventions. Br J Ophthalmol. 2017;101(9):1147–1154. doi: 10.1136/bjophthalmol-2016-309975. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Zhao H, et al. Molecular genetic analysis of patients with sporadic and X-linked infantile nystagmus. BMJ Open. 2016;6(4):e010649. doi: 10.1136/bmjopen-2015-010649. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Yin X, et al. Identification of CYP4V2 mutation in 36 Chinese families with Bietti crystalline corneoretinal dystrophy. Exp Eye Res. 2016;146:154–162. doi: 10.1016/j.exer.2016.03.007. [DOI] [PubMed] [Google Scholar]
- 32.Tang M, et al. Mutation spectrum of the LRP5, NDP, and TSPAN12 genes in chinese patients with familial exudative vitreoretinopathy. Invest Ophthalmol Vis Sci. 2017;58(13):5949–5957. doi: 10.1167/iovs.17-22577. [DOI] [PubMed] [Google Scholar]
- 33.Rao, F.Q., et al., Mutations in LRP5,FZD4, TSPAN12, NDP, ZNF408, or KIF11 Genes Account for 38.7% of Chinese Patients With Familial Exudative Vitreoretinopathy. Invest Ophthalmol Vis Sci, 2017. 58(5): p. 2623–2629. [DOI] [PubMed]
- 34.Alapati A, et al. Molecular diagnostic testing by eyeGENE: analysis of patients with hereditary retinal dystrophy phenotypes involving central vision loss. Invest Ophthalmol Vis Sci. 2014;55(9):5510–5521. doi: 10.1167/iovs.14-14359. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Bai Z, et al. Clinical and genetic analysis of an infant with lowe syndrome caused by exonic duplication of OCRL gene. Zhonghua Yi Xue Yi Chuan Xue Za Zhi. 2020;37(1):28–32. doi: 10.3760/cma.j.issn.1003-9406.2020.01.008. [DOI] [PubMed] [Google Scholar]
- 36.Sung CH, et al. Rhodopsin mutations in autosomal dominant retinitis pigmentosa. Proc Natl Acad Sci USA. 1991;88(15):6481–6485. doi: 10.1073/pnas.88.15.6481. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Dryja TP, et al. Mutation spectrum of the rhodopsin gene among patients with autosomal dominant retinitis pigmentosa. Proc Natl Acad Sci USA. 1991;88(20):9370–9374. doi: 10.1073/pnas.88.20.9370. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Bunge S, et al. Molecular analysis and genetic mapping of the rhodopsin gene in families with autosomal dominant retinitis pigmentosa. Genomics. 1993;17(1):230–233. doi: 10.1006/geno.1993.1309. [DOI] [PubMed] [Google Scholar]
- 39.Shastry BS, Hejtmancik JF, Trese MT. Identification of novel missense mutations in the Norrie disease gene associated with one X-linked and four sporadic cases of familial exudative vitreoretinopathy. Hum Mutat. 1997;9(5):396–401. doi: 10.1002/(SICI)1098-1004(1997)9:5<396::AID-HUMU3>3.0.CO;2-2. [DOI] [PubMed] [Google Scholar]
- 40.Liu D, et al. A novel nonsense mutation in the NDP gene in a Chinese family with Norrie disease. Mol Vis. 2010;16:2653–2658. [PMC free article] [PubMed] [Google Scholar]
- 41.Smith, S.E., et al., Norrie disease: extraocular clinical manifestations in 56 patients. Am J Med Genet A, 2012. 158a(8): p. 1909–17. [DOI] [PubMed]
- 42.Dai H, et al. Identification of five novel mutations in the long isoform of the USH2A gene in Chinese families with Usher syndrome type II. Mol Vis. 2008;14:2067–2075. [PMC free article] [PubMed] [Google Scholar]
- 43.Katagiri S, et al. Whole exome analysis identifies frequent CNGA1 mutations in Japanese population with autosomal recessive retinitis pigmentosa. PLoS ONE. 2014;9(9):e108721. doi: 10.1371/journal.pone.0108721. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Xu W, et al. Seven novel mutations in the long isoform of the USH2A gene in Chinese families with nonsyndromic retinitis pigmentosa and Usher syndrome Type II. Mol Vis. 2011;17:1537–1552. [PMC free article] [PubMed] [Google Scholar]
- 45.Functional implications of the spectrum of mutations found in 234 cases with X-linked juvenile retinoschisis. The Retinoschisis Consortium. Hum Mol Genet, 1998. 7(7): p. 1185–92. [DOI] [PubMed]
- 46.Huang Y, et al. A novel deletion mutation in RS1 gene caused X-linked juvenile retinoschisis in a Chinese family. Eye (Lond) 2014;28(11):1364–1369. doi: 10.1038/eye.2014.196. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Xu Y, et al. Mutations of 60 known causative genes in 157 families with retinitis pigmentosa based on exome sequencing. Hum Genet. 2014;133(10):1255–1271. doi: 10.1007/s00439-014-1460-2. [DOI] [PubMed] [Google Scholar]
- 48.Wada Y, et al. Screening for mutations in CYP4V2 gene in Japanese patients with Bietti's crystalline corneoretinal dystrophy. Am J Ophthalmol. 2005;139(5):894–899. doi: 10.1016/j.ajo.2004.11.065. [DOI] [PubMed] [Google Scholar]
- 49.Xiao X, et al. Identification of CYP4V2 mutation in 21 families and overview of mutation spectrum in Bietti crystalline corneoretinal dystrophy. Biochem Biophys Res Commun. 2011;409(2):181–186. doi: 10.1016/j.bbrc.2011.04.112. [DOI] [PubMed] [Google Scholar]
- 50.Wada Y, et al. Mutation of human retinal fascin gene (FSCN2) causes autosomal dominant retinitis pigmentosa. Invest Ophthalmol Vis Sci. 2001;42(10):2395–2400. [PubMed] [Google Scholar]
- 51.Dong B, et al. Two novel PRP31 premessenger ribonucleic acid processing factor 31 homolog mutations including a complex insertion-deletion identified in Chinese families with retinitis pigmentosa. Mol Vis. 2013;19:2426–2435. [PMC free article] [PubMed] [Google Scholar]
- 52.Yang L, et al. Novel mutations of RPGR in Chinese retinitis pigmentosa patients and the genotype-phenotype correlation. PLoS ONE. 2014;9(1):e85752. doi: 10.1371/journal.pone.0085752. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Hosono K, et al. Molecular diagnosis of 34 Japanese families with leber congenital amaurosis using targeted next generation sequencing. Sci Rep. 2018;8(1):8279. doi: 10.1038/s41598-018-26524-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Ernest PJ, et al. Outcome of ABCA4 microarray screening in routine clinical practice. Mol Vis. 2009;15:2841–2847. [PMC free article] [PubMed] [Google Scholar]
- 55.Lewis, R.A., et al., Genotype/phenotype analysis of a photoreceptor-specific ATP-binding cassette transporter gene, ABCR, in Stargardt disease. Am J Hum Genet, 1999. 64(2): p. 422-34. [DOI] [PMC free article] [PubMed]
- 56.Wang H, et al. Comprehensive molecular diagnosis of a large Chinese leber congenital amaurosis cohort. Invest Ophthalmol Vis Sci. 2015;56(6):3642–3655. doi: 10.1167/iovs.14-15972. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Gerber S, et al. Complete exon-intron structure of the RPGR-interacting protein (RPGRIP1) gene allows the identification of mutations underlying Leber congenital amaurosis. Eur J Hum Genet. 2001;9(8):561–571. doi: 10.1038/sj.ejhg.5200689. [DOI] [PubMed] [Google Scholar]
- 58.van Genderen MM, et al. Mutations in TRPM1 are a common cause of complete congenital stationary night blindness. Am J Hum Genet. 2009;85(5):730–736. doi: 10.1016/j.ajhg.2009.10.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Negrisolo S, et al. PAX2 gene mutations in pediatric and young adult transplant recipients: kidney and urinary tract malformations without ocular anomalies. Clin Genet. 2011;80(6):581–585. doi: 10.1111/j.1399-0004.2010.01588.x. [DOI] [PubMed] [Google Scholar]
- 60.Lenis TL, et al. Novel compound heterozygous mutations resulting in cone dystrophy with supernormal rod response. JAMA Ophthalmol. 2013;131(11):1482–1485. doi: 10.1001/jamaophthalmol.2013.4681. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Poulter JA, et al. Recessive mutations in SLC38A8 cause foveal hypoplasia and optic nerve misrouting without albinism. Am J Hum Genet. 2013;93(6):1143–1150. doi: 10.1016/j.ajhg.2013.11.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Li N, et al. Five novel mutations of the FRMD7 gene in Chinese families with X-linked infantile nystagmus. Mol Vis. 2008;14:733–738. [PMC free article] [PubMed] [Google Scholar]
- 63.Chen X, et al. Targeted sequencing of 179 genes associated with hereditary retinal dystrophies and 10 candidate genes identifies novel and known mutations in patients with various retinal diseases. Invest Ophthalmol Vis Sci. 2013;54(3):2186–2197. doi: 10.1167/iovs.12-10967. [DOI] [PubMed] [Google Scholar]
- 64.Aparisi MJ, et al. Targeted next generation sequencing for molecular diagnosis of Usher syndrome. Orphanet J Rare Dis. 2014;9:168. doi: 10.1186/s13023-014-0168-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.McGee TL, et al. Novel mutations in the long isoform of the USH2A gene in patients with Usher syndrome type II or non-syndromic retinitis pigmentosa. J Med Genet. 2010;47(7):499–506. doi: 10.1136/jmg.2009.075143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Redin C, et al. Targeted high-throughput sequencing for diagnosis of genetically heterogeneous diseases: efficient mutation detection in Bardet-Biedl and Alstrom syndromes. J Med Genet. 2012;49(8):502–512. doi: 10.1136/jmedgenet-2012-100875. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Qin M, et al. Complexity of the genotype-phenotype correlation in familial exudative vitreoretinopathy with mutations in the LRP5 and/or FZD4 genes. Hum Mutat. 2005;26(2):104–112. doi: 10.1002/humu.20191. [DOI] [PubMed] [Google Scholar]
- 68.Jiang F, et al. Screening of ABCA4 gene in a chinese cohort with stargardt disease or cone-rod dystrophy with a report on 85 novel mutations. Invest Ophthalmol Vis Sci. 2016;57(1):145–152. doi: 10.1167/iovs.15-18190. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Additional file 1. Supplementary material of tables and figures.
Data Availability Statement
The raw datasets used and analysed during the current study are not deposited in publicly available repositories because of considerations about the security of human genetic resources. The transcript RefSeq number (Tables 1 and 2) was obtained from the Ensembl database (http://asia.ensembl.org) [10]. Any questions should be directed to the corresponding author. We provide conclusive variant information without identifying/confidential patient data in the paper or its appendix. For other details of the availability of data and material, please refer to the methods section of the article.