

Abstract
Sarcomas are rare tumors but comprise a wide histologic spectrum. Advances in technology have emerged to address the biologic complexity and challenging diagnosis and treatment of this disease. The diagnostic approach to sarcomas has historically been based on morphologic features, but technologic advances in immunohistochemistry and cytogenetic/molecular testing have transformed the interdisciplinary work-up of mesenchymal neoplasms in recent years. On the therapeutic side, technologic advances in the delivery of radiation have made it a linchpin in the treatment of localized and oligometastatic sarcoma. In this review, we discuss recent advances in the pathologic diagnosis of sarcomas and discuss select sarcoma types that illustrate how newly discovered diagnostic, prognostic, and predictive biomarkers have refined existing classification schemes and substantially shaped our diagnostic approach. Such examples include conventional and epithelioid malignant peripheral nerve sheath tumors (MPNSTs), emerging entities in the group of round cell sarcomas, and other mesenchymal neoplasms with distinct cytogenetic aberrations. Recent advances in radiation oncology, including intensity-modulated, stereotactic, MRI-guided, and proton radiotherapy (RT), will be reviewed in the context of neoadjuvant or adjuvant localized soft-tissue sarcoma and oligometastatic or oligoprogressive disease. Innovations in translational research are expected to be introduced into clinical practice over the next few years and will likely continue to affect the rapidly evolving field of sarcoma diagnostics and therapy.
INTRODUCTION
Sarcomas are rare tumors, accounting for approximately 1% of cancers, and comprise a diverse spectrum of mesenchymal neoplasms with varied prognosis, including approximately 70 with intermediate or malignant biologic potential.1,2 The number of tumors of soft tissue and bone included in the World Health Organization classification has been increasing over the past decades. This is attributed largely to continuously refined classification schemes and technical advances in the immunohistochemical and cytogenetic/molecular genetic work-up that enabled the discovery of biologically distinct entities with specific prognostic and/or predictive implications.
Technology has also affected the care of patients with sarcoma beyond diagnosis, with an imprint on the efficacy and safety of treatment approaches. RT is a technology-based therapeutic modality that has been a critical adjuvant to surgery since the emergence of limb-preservation approaches for soft-tissue sarcoma. Continued technologic advances in the areas of image guidance (including MRI guidance, intensity modulation, stereotactic RT, and proton-based RT) have improved the efficacy of RT while reducing its toxicity and expanding the role of RT for patients with oligometastatic or oligoprogressive disease.
Here, we will first cover select recent technologic advances and their implications for sarcoma diagnostics. We will then explore in detail the impact of technology on treatment, with an emphasis on therapeutic approaches in radiation oncology.
HOW THE RAPIDLY EVOLVING FIELD OF PATHOLOGY IS INTERFACING WITH THE COMPLEXITY OF SARCOMA
Brief Overview of Established Diagnostic Techniques
Given the complexity of the sarcoma histologic spectrum, thoughtful integration of clinical presentation, patient (and family) history, radiologic imaging (when available), gross and histomorphologic clues, immunohistochemical staining pattern, and (when appropriate) cytogenetic and molecular genetic testing are crucial to establish a correct diagnosis in most cases. As determined by rigorous classification schemes, the histopathologic diagnosis of sarcomas is largely based on morphology and distinguishes tumors with spindle cell, epithelioid/epithelial-like, round cell, myxoid, and pleomorphic appearances (Table 1). In fact, many entities with distinct features can be diagnosed on hematoxylin and eosin–stained sections alone.
TABLE 1.
Select Sarcomas With Spindle Cell, Epithelioid, Myxoid, Pleomorphic, and Round Cell Morphologies and Useful Diagnostic Features and/or Immunohistochemical Markers
| Morphologic Pattern | Sarcoma Type | Diagnostic Clues | Immunohistochemistry | |
|---|---|---|---|---|
| Positive Markers | Negative Markers | |||
| Spindle cell | Leiomyosarcoma | Fascicular growth, brightly eosinophilic cytoplasm, cigar-shaped nuclei | Desmin, SMA, caldesmon | |
| MPNST | Perivascular accentuation, alternation of hyper- and hypocellular areas, wavy nuclei | S100, SOX10, GFAP (< 50% each) | H3K27me3 (up to 80% of cases) | |
| Monophasic synovial sarcoma | Overlapping nuclei, monomorphic cytomorphology, hemangiopericytoma-like blood vessels | TLE1, EMA/keratins | ||
| Dedifferentiated liposarcoma | Adjacent well-differentiated liposarcoma | MDM2, CDK4, HMGA2 | ||
| GIST, spindle cell type | Location (GI tract); uniform cytomorphology, fibrillary/syncytial cytoplasm | DOG1, KIT | ||
| Spindle cell/sclerosing rhabdomyosarcoma | Presence of rhabdomyoblasts | Desmin, Myf4, MYOD1 | ||
| Solitary fibrous tumor | “Patternless” architecture, hemangiopericytoma-like blood vessels | STAT6, CD34 | ||
| Inflammatory myofibroblastic tumor | Fascicles of uniform plump spindle cells with ovoid to tapering nuclei, myxoid stroma, inflammatory infiltrate | ALK (50%), ROS1 (< 10%), smooth muscle markers (subset) | ||
| DFSP | Uniform spindle cells with storiform or whorled growth pattern, infiltration of adipose tissue (honeycomb appearance) | CD34 | ||
| Epithelioid/epithelial-like | Epithelioid sarcoma | Diffuse infiltration along fascial planes, pseudogranulomatous appearance | EMA/keratins, CD34 (55%) | SMARCB1 (90%) |
| Alveolar soft part sarcoma | Pseudo-alveolar/nested growth, large tumors cells with abundant granular eosinophilic cytoplasm | TFE3 | ||
| Epithelioid MPNST | Multinodular architecture, large nucleoli | S100, SOX10, GFAP (60%) | SMARCB1 (70%) | |
| GIST, epithelioid type | Location (GI tract); multinodular growth in gastric tumors suggestive of SDH deficiency | DOG1, KIT | SDHB in SDH-deficient GIST, additional SDHA loss in SDHA-mutant GIST; KIT often negative or limited in PDGFRA-mutant GIST | |
| Epithelioid hemangioendothelioma | Cords and strands of tumor cells embedded in (chondro-)myxoid stroma | ERG, CD31, CAMTA1 (90%) or TFE3 (5%) | ||
| Pseudomyogenic hemangioendothelioma | Sheets and loose fascicles of plump spindle cells with abundant eosinophilic cytoplasm, sometimes mimicking rhabdomyoblasts | CD31, ERG, keratin, FOSB (> 95%) | ||
| Round cell | Embryonal rhabdomyosarcoma | Monomorphic round to spindled cell, loose myxoid stroma | Desmin, Myf4 (limited to a subset of cells), MYOD1 | |
| Alveolar rhabdomyosarcoma | Monomorphic large cells with even chromatin and prominent nucleoli, pseudo-alveolar architecture | Desmin (diffuse), Myf4 (diffuse), MYOD1 | ||
| Desmoplastic small round cell tumor | Intra-abdominal location, young males | WT1 (C-terminal), keratin, EMA, desmin | ||
| Ewing sarcoma | Small amounts of cytoplasm, rare mitoses, monomorphic cytomorphology | CD99 (100%), NKX2.2 (> 90%) | ETV4, WT1, BCOR, CCNB3 | |
| CIC-rearranged sarcoma | Primitive round to ovoid and sometimes spindled cytomorphology, irregularly shaped vesicular nuclei, mitoses, necrosis, morphologic heterogeneity | CD99 (20%), ETV4 (> 90%), WT1 (> 90%) | NKX2.2 | |
| BCOR-rearranged sarcoma | Monomorphic or primitive appearing round to ovoid and occasionally spindled tumor cells arranged in intersecting fascicles or a patternless fashion | CD99 (80%), BCOR (> 90%), CCNB3 (90%) | NKX2.2 | |
| Myxoid | Myxofibrosarcoma | Curvilinear blood vessels, pleomorphic cells | None (can express focal MDM2) | |
| Low-grade fibromyxoid sarcoma | Abrupt transition between myxoid and nonmyxoid areas, bland and uniform spindle cell morphology | MUC4 | ||
| Extraskeletal myxoid chondrosarcoma | Lobulated growth, reticular architecture, uniform nuclear morphology | S100(< 50%), EMA (30%) | SMARCB1 (17%) | |
| Myxoid liposarcoma | Delicate plexiform vasculature (“crow’s feet” vessels), uni- or bivacuolated lipoblasts | None | ||
Added value of immunohistochemistry
The implementation of immunohistochemistry into the histopathologic work-up of soft-tissue tumors in the past decades enabled the detection of lineage-specific markers (Table 1). For instance, myogenic markers such as desmin, smooth muscle actin (SMA), and caldesmon are generally expressed in tumors exhibiting smooth muscle differentiation, such as leiomyoma and leiomyosarcoma.1 Another example are the neural markers S-100 protein and SOX10, expressed in benign and MPNSTs.1 However, none of these immunohistochemical stains is entirely specific or sensitive for a diagnosis per se.
Many sarcomas exhibit characteristic cytogenetic and/or molecular genetic aberrations (Table 2), with an exponentially growing number of newly discovered alterations, owing to increased sensitivity and more frequent application of molecular testing in routine diagnostics. Several immunohistochemical markers have been discovered that either directly or indirectly correspond to distinct genetic or epigenetic alterations, as will be discussed more in detail.
TABLE 2.
Select Sarcomas With Diagnostically Relevant Distinct Cytogenetic and Molecular Alterations
| Sarcoma Type | Cytogenetic Alteration | Molecular Alteration |
|---|---|---|
| Alveolar rhabdomyosarcoma | t(2;13)(q35;q14) | PAX3-FOXO1 fusion |
| t(1;13)(p36;q14), double minutes | PAX7-FOXO1 fusion | |
| t(2;2)(q35;p23) | PAX3-NCOA1 fusion | |
| t(X;2)(q35;q13) | PAX3-AFX fusion | |
| Alveolar soft part sarcoma | t(X;17)(p11.2;q25) | TFE3-ASPSCR1 fusion |
| BCOR-rearranged sarcoma | Inv(X)(p11.4p11.22) | BCOR-CCNB3 fusion |
| t(X;4)(p11;q31) | BCOR-MAML3 fusion | |
| t(X;22)(p11;q13) | ZC3H7B-BCOR fusion | |
| CIC-rearranged sarcoma | t(4;19)(q35;q13) or t(10; 19)(q26;q13) | CIC-DUX4 fusion |
| t(X;19)(q13;q13.3) | CIC-FOXO4 fusion | |
| Clear cell sarcoma | t(12;22)(q13;q12) | EWSR1-ATF1 fusion |
| t(2;22)(q32.3;q12) | CREB1-EWSR1 fusion | |
| Dedifferentiated liposarcoma | Ring and giant marker chromosomes | Amplification of 12q13–15: MDM2, CDK4 (HMGA2) |
| Desmoplastic small round cell tumor | t(11;22)(p13;q12) | EWSR1-WT1 fusion |
| DFSP | Ring form of chromosomes 17 and 22 | COL1A1-PDGFB fusion |
| Endometrial stromal sarcoma, low grade | t(7;17)(p15;q21) | JAZF1-SUZ12 fusion |
| t(6;7)(p21;7p15) | PHF1-JAZF1 fusion | |
| t(6;10)(p21;p11) | EPC1-PHF1 fusion | |
| t(1;6)(p34;p21) | MEAF6-PHF1 fusion | |
| t(X;17)(p11;q21) | MBTD1-CXorf67 fusion | |
| Endometrial stromal sarcoma, high grade | t(10;17)(q22;p13) | YWHAE-NUT2 fusion |
| t(X;22)(p11;q13) | ZC3H7B-BCOR fusion | |
| Epithelioid hemangioendothelioma | t(1;3)(p36;q25) | WWTR1-CAMTA1 fusion |
| t(X;11)(p11;q22) | YAP1-TFE3 fusion | |
| Epithelioid sarcoma | Deletion 22q | SMARCB1 inactivation |
| t(8;22)(q22;q11) | ||
| t(10;22) | ||
| Ewing sarcoma | t(11;22)(q24;q12) | EWSR1-FLI1 fusion |
| t(21;22)(q12;q12) | EWSR1-ERG fusion | |
| Others | Others | |
| EWSR1/FUS-NFATC2 fusion sarcoma | t(20;22)(q13.2;q12.2) | EWSR1-NFATC2 fusion |
| t(16;20)(p11;q13.2) | FUS-NFATC2 fusion | |
| Extraskeletal myxoid chondrosarcoma | t(9;22)(q22;q12) | EWSR1-NR4A3 fusion |
| t(9;17)(q22;q11) | TAF2N-NR4A3 fusion | |
| t(9;15)(q22;q21) | TCF12-NR4A3 fusion | |
| t(3;9)(q11;q22) | TFG-NR4A3 fusion | |
| t(9;17)(q22;q11) | RBP56-NR4A3 fusion | |
| GIST | Deletion 14q, 22q, 1p, 15q | KIT or PDGFRA mutation (85% of cases); (NF1, SDH, BRAF mutation, SDHC hypermethylation, other) |
| Infantile fibrosarcoma | t(12;15)(p13;q25) | ETV6-NTRK3 fusion |
| t(2;15)(p21;q25) | EML4-NTRK3 fusions | |
| Inflammatory myofibroblastic tumor | t(1;2)(q22;p23) | TPM3-ALK fusion |
| t(2;19)(p23;p13) | TPM4-ALK fusion | |
| t(2;17)(p23;q23) | CLTC-ALK fusion | |
| t(2;2)(p23;q13) | RANBP2-ALK fusion | |
| t(2;2)(p23;q35) | ATIC-ALK fusion | |
| t(2;11)(p23;p15) | CARS-ALK fusion | |
| t(2;4)(p23;q21) | SEC31L1-ALK fusion | |
| t(2;12)(p23;p12) | PPFIBP1-ALK fusion | |
| t(3;6)(q12;q22) | TFG-ROS1 fusion | |
| t(6;17)(q22;p13)? | YWHAE-ROS1 fusion | |
| RRBP1-ALK fusion | ||
| Low-grade fibromyxoid sarcoma | t(7;16)(q33;p11) | FUS-CREB3L2 fusion |
| t(11;16)(p11;p11) | FUS-CREB3L1 fusion | |
| Mesenchymal chondrosarcoma | t(8;8)(q13;q21) | HEY1-NCOA2 fusion |
| MPNST | Complex changes | SUZ12 or EED mutation (80% of cases), NF1 inactivation |
| Myoepithelial tumor/carcinoma of soft tissue | t(6;22)(p21;q12) | EWSR1-POU5F1 fusion |
| t(19;22)(q13;q12) | EWSR1-ZNF444 fusion | |
| t(1;22)(q23;q12) | EWSR1-PBX1 fusion | |
| Myxoid liposarcoma | t(12;16)(q13;p11) | FUS-DDIT3 fusion |
| t(12;122)(q13;q12) | EWSR1-DDIT3 fusion | |
| Postradiation angiosarcoma | Gain 8q24 | MYC amplification |
| Sclerosing epithelioid fibrosarcoma | t(7;16)(q33;p11) | FUS-CREB3L2 fusion |
| Solitary fibrous tumor | Inv(12)(q13q13) | NAB2-STAT6 fusion |
| Synovial sarcoma | t(X;18)(p11;q11) | SS18-SSX1/SSX2 fusion |
Abbreviations: DFSP, dermatofibrosarcoma protuberans; GIST, gastrointestinal stromal tumor; MPNST, malignant peripheral nerve sheath tumor.
Ancillary cytogenetic and molecular genetic analysis
Cytogenetic and molecular genetic testing has substantially advanced the routine diagnostic work-up of sarcomas. This includes conventional karyotyping and fluorescence in situ hybridization (FISH) for detection of known genetic rearrangements as well as targeted next-generation sequencing (NGS), anchored multiplex polymerase chain reaction, targeted or whole transcriptome RNA sequencing for detection of unknown gene fusions, and/or reverse transcription polymerase chain reaction.3 These technologies are expected to further decrease in cost and effectively complement the morphology and immunohistochemistry-based diagnosis of sarcomas. In addition, they allow for the detection of hitherto unknown gene fusions, which may add to our current understanding of the genetic drivers of sarcoma and help inform and refine current classification schemes.
Recent Advances in Sarcoma Diagnostics
Recent advances in the discovery of distinct recurrent cyto-/genetic aberrations and development of associated immunohistochemical markers have had a substantial impact on the diagnostic approach to certain sarcomas and will be reviewed in detail.
Conventional and epithelioid malignant peripheral nerve sheath tumors
MPNSTs are aggressive sarcomas associated with high rates of distant metastases and 5-year survival rates of 35% to 50%.4 Their histopathologic diagnosis has been challenging because diagnostic features of (1) identifiable origin from a peripheral nerve or neurofibroma, (2) immunohistochemical/ultrastructural evidence of Schwann cell differentiation, and/or (3) association with neurofibromatosis type I are often absent. Histologically, MPNST appears as spindle cell sarcoma with alternation of hyper- and hypocellular areas and accentuation of tumors cells around blood vessels (Fig. 1A–C). However, these features are relatively nonspecific in isolation. Expression of nerve sheath markers (S-100 protein, SOX10, and GFAP) is detectable in only up to 40% of MPNSTs, and, if positive, usually limited in extent (Table 1). The discovery of characteristic loss-of-function mutations in SUZ12 or EED, encoding components of the polycomb repressive complex 2 (PRC2),5,6 and subsequent loss of trimethylation at lysine 27 of histone 3 (H3K27me3) led to the introduction of H3K27me3 immunohistochemistry as a useful diagnostic marker.7–9 H3K27me3 loss can be detected in up to 80% of MPNSTs, in particular in high-grade tumors, and has been shown to be quite specific in the distinction of MPNST from other spindle cell neoplasms.8 H3K27me3 loss by immunohistochemistry represents a biomarker that demonstrates the epigenetic consequences of genomic PRC2 inactivation characteristic of conventional MPNST.
FIGURE 1. Genetic and Immunohistochemical Characteristics in Conventional and Epithelioid Malignant Peripheral Nerve Sheath Tumors.
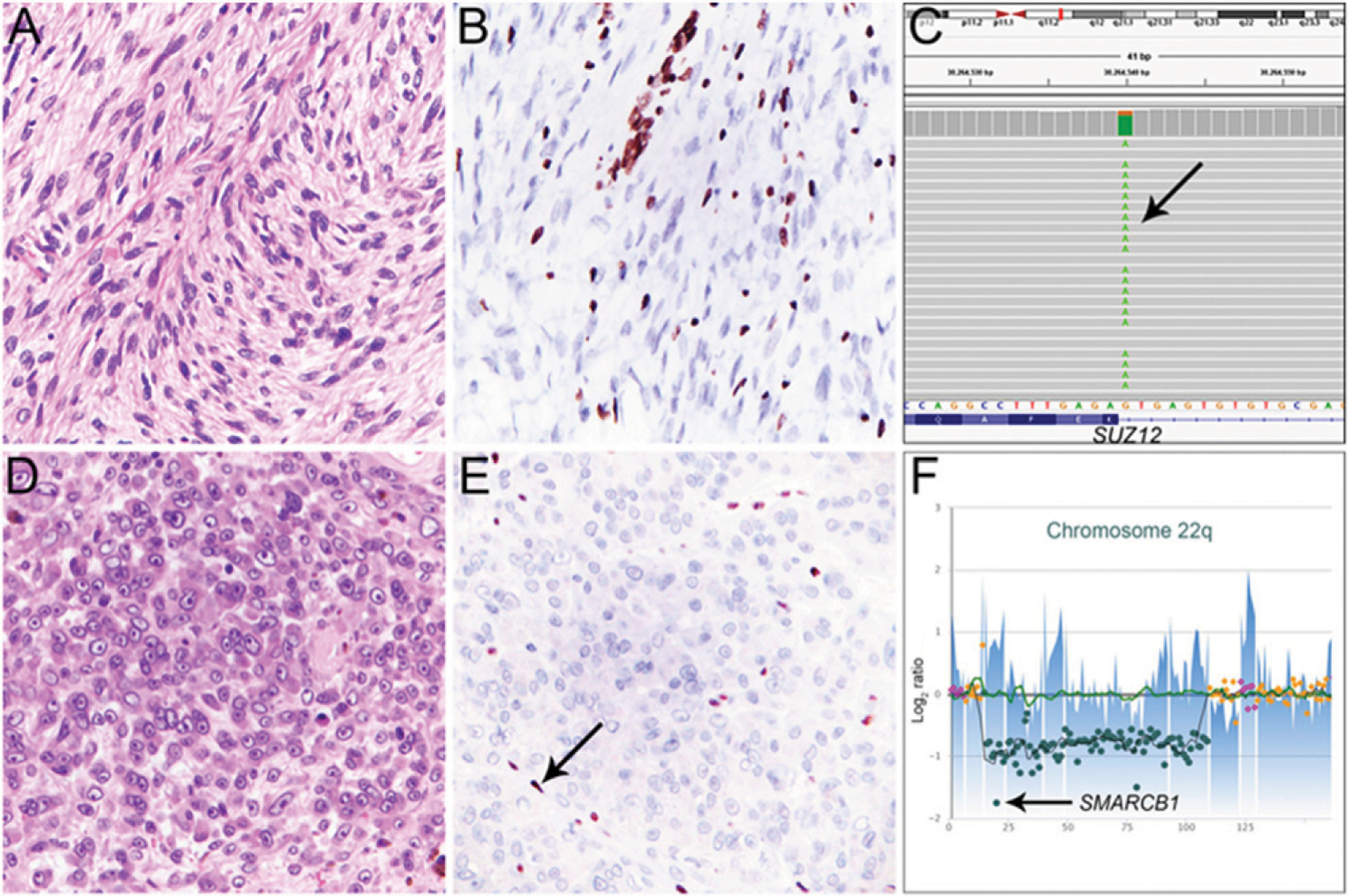
A high-grade malignant peripheral nerve sheath tumor (MPNST) of the psoas region (A), with loss of trimethylation at lysine 27 of histone 3 (H3K27me3) in tumor cells (B; vascular endothelial cells and inflammatory cells serve as positive internal control). This tumor shows a homozygous c.274+1G>A splice site mutation (allele fraction 80%) in SUZ12 (C, arrow), which leads to inactivation of the polycomb repressive complex 2 (PRC2) and loss of H3K27me3. This MPNST also had biallelic inactivation of NF1 (not shown). Epithelioid MPNST of the popliteal fossa with epithelioid morphology of tumor cells showing nuclear atypia, prominent nucleoli, and frequent mitoses (D) showing loss of SMARCB1 expression in tumor cells (E; inflammatory cells serve as positive internal control, arrow) resulting from a homozygous deletion affecting the entire coding region of SMARCB1 at 22q11.23 (F, arrow).
In contrast, epithelioid MPNST, which is considered a rare histologic variant of MPNST, follows a less aggressive clinical course, is generally not associated with neurofibromatosis type I, and lacks PRC2 genomic inactivation (Fig. 1D–F).5,8,10 Instead, these tumors arise sporadically or in association with conventional or epithelioid benign nerve sheath tumors and harbor loss of the tumor suppressor SMARCB1 (i.e., INI-1) by immunohistochemistry in 70% of cases (Table 1).10 As demonstrated recently, SMARCB1 loss in epithelioid MPNST results from inactivating mutations of SMARCB1 on chromosome 22.11 SMARCB1 encodes a key subunit of the SWI/SNF1 chromatin remodeling complex that orchestrates chromatin organization and accessibility and has, in part, biologic functions that oppose PRC2. SMARCB1 inactivation and SMARCB1 protein loss can be found in a wide range of benign and malignant mesenchymal neoplasms,12 including 40% of epithelioid schwannomas, which rarely give rise to epithelioid MPNST.13 SMARCB1 loss is therefore not specific for epithelioid MPNST, but when interpreted in the context of cytomorphology, the presence of necrosis and frequent mitoses, and strong and diffuse expression of S-100 protein and SOX10 support a diagnosis of epithelioid MPNST. In addition, SMARCB1 loss helps rule out other malignancies with overlapping histologic and immunohistochemical features, such as (metastatic) malignant melanoma, in which SMARCB1 expression is generally retained.14
Emerging entities in the group of round cell sarcomas
Added value of immunohistochemical markers has also been demonstrated in the differential diagnosis of recently discovered entities in the group of round cell sarcomas. First described in 2012 as a type of round cell sarcoma lacking EWSR1 rearrangement,15 CIC-rearranged sarcoma shows predilection for the soft tissue of trunk and extremities of younger male adults, follows a more aggressive clinical course compared with Ewing sarcoma with overall survival rates of 43% vs. 76%, and generally does not respond well to systemic therapies established for Ewing sarcoma16 (Fig. 2A–C). Distinction of CIC-rearranged sarcoma from Ewing sarcoma therefore has prognostic and predictive value. These tumors harbor characteristic t(4;19)(q35;q13) or t(10;19)(q26;q13), resulting mostly in CIC-DUX4 fusion (Table 2); rare cases with alternate CIC-FOXO4 fusion have been reported.17,18 Histologically, CIC-rearranged sarcomas consist of moderately pleomorphic round to ovoid tumor cells with frequent mitoses, apoptoses, and necrosis, which aides in the distinction from Ewing sarcoma, which usually comprises a monomorphic cell population and infrequent mitoses, apoptoses, or necrosis (Fig. 2D–F).
FIGURE 2. Useful Diagnostic Markers for Round Cell Sarcomas.
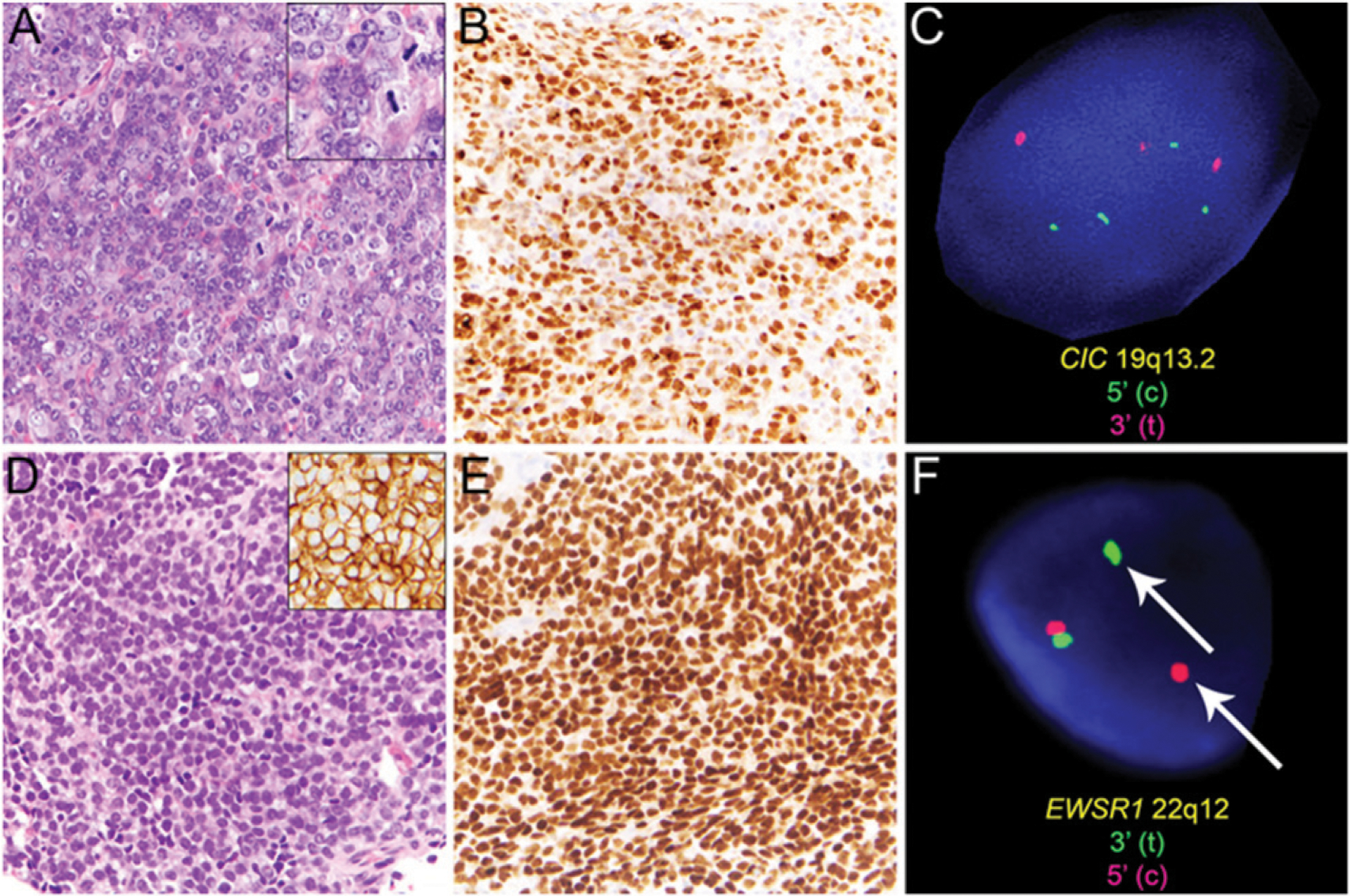
CIC-rearranged sarcoma of the perineal region is characterized by a morphologically heterogeneous population of primitive round to ovoid or spindled tumor cells (A) with frequent mitoses (A, inset) and nuclear expression of WT1 (B). Rearrangement of the CIC locus at 19q13.2 with several break-apart signals was detected by fluorescence in situ hybridization (FISH, C). In contrast, Ewing sarcoma comprises sheets of uniform tumor cells exhibiting rounded nuclei and inconspicuous nucleoli (D) with diffuse membranous expression of CD99 (D, inset) and diffuse nuclear expression of the transcription factor NKX2.2 (E). Rearrangement of EWSR1 at 22q12 can be detected by FISH (F; break-apart signal indicated by arrow).
Because access to cytogenetic analyses such as conventional karyotyping or FISH for detection of characteristic CIC-rearrangement and turnaround time can be limitations in some institutions, immunohistochemical expression of WT1 (> 90% of cases) and ETV4 (90% of cases)19,20 can be sufficient to support a diagnosis of CIC-rearranged sarcoma in the presence of unequivocal histologic features; CD99 staining is usually limited in CIC-rearranged sarcoma (Table 1). In contrast, diffuse membranous expression of CD99 and nuclear expression of the transcription factor NKX2.2 and the presence of EWSR1 rearrangement by FISH would favor a diagnosis of Ewing sarcoma (Fig. 2D–F; Tables 1 and 2).21–23
Another distinct type of round cell sarcoma lacking EWSR1 rearrangement initially reported in 201224 with predilection for bone and soft tissue of male children24,25 is characterized by BCOR-CCNB3 rearrangement resulting from inv(X)(p11) (i.e., X-chromosomal paracentric inversion; Tables 1 and 2). Rare cases harbor an alternate rearrangement of BCOR with MAML3 or ZC3H7B.26 BCOR-rearranged sarcomas follow an aggressive clinical course with 5-year overall survival rates of approximately 75%, similar to Ewing sarcoma but less aggressive than CIC-rearranged sarcoma.25,27 BCOR immunohistochemistry can aid in the diagnosis of BCOR-rearranged sarcomas, in particular when genetic testing for BCOR rearrangement is not available.25,28
Other mesenchymal neoplasms with distinct cytogenetic aberrations
Inflammatory myofibroblastic tumor (IMT), epithelioid vascular neoplasms, and postradiation angiosarcoma represent examples of mesenchymal neoplasms with characteristic genetic aberrations that have been ttranslated into useful diagnostic immunohistochemical markers.
IMT shows predilection for the visceral soft tissues of children and young adults, with a tendency for local recurrence but a small risk of distant metastasis.29 IMT comprises spindled tumor cells with fasciitis-like, compact spindle cells and hypocellular fibrous patterns, with minimal cytologic atypia and a scattered inflammatory infiltrate (Fig. 3A–C). Rearrangements of ALK at 2p23 are identified in about 50% of cases, particularly when arising in younger patients. TPM3-ALK fusion30 represents the most frequent aberration in IMT, but ALK fusions with various other partners have been reported (Table 2). ALK rearrangement results in upregulation of ALK expression, which is detectable by immunohistochemistry (Fig. 3A–C; Table 1).29 In the distinction from true smooth muscle tumors, this marker can be very useful; however, in older patients, ALK staining is often negative and does not rule out a diagnosis of IMT. In addition to ALK-positive anaplastic large cell lymphoma and ALK-positive large B-cell lymphoma,31,32 certain carcinomas of lung,33 thyroid,34 and kidney35 harbor ALK rearrangement. ALK staining has been reported in various ALK-rearranged mesenchymal neoplasms, such as epithelioid fibrous histiocytoma,36 Spitz nevus,37 so-called melanocytic myxoid spindle cell tumor with ALK rearrangement,38 and single cases of leiomyosarcoma,39 with potential implications for targeted therapies using ALK inhibitors. However, ALK expression is also found, for instance, in subsets of epithelioid and spindle cell rhabdomyosarcomas with TFCP2 fusion lacking ALK rearrangement,40 suggesting that ALK staining is not always associated with ALK rearrangement.
FIGURE 3. Mesenchymal Neoplasms With Distinct Cytogenetic Aberrations and Associated Immunohistochemical Markers.
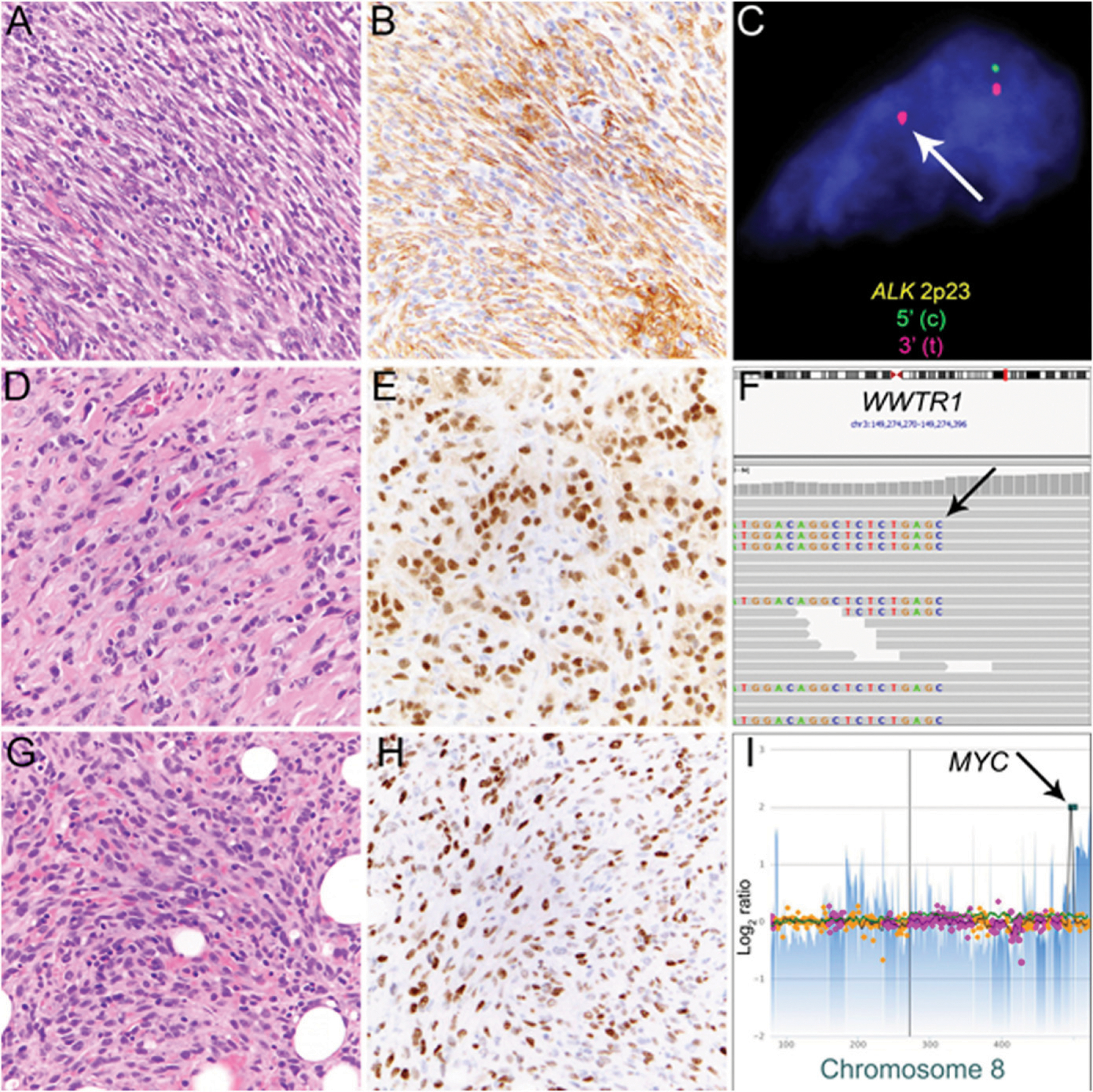
Inflammatory myofibroblastic tumor is characterized by a population of spindled tumor cells arranged in fascicles with scattered inflammatory cells (A) and expression of ALK in tumor cells (B). Rearrangement of the ALK locus at 2p23 can be detected by fluorescence in situ hybridization (C; break-apart signal indicated by arrow). Malignant epithelioid hemangioendothelioma (EHE) consisting of strands of tumor cells with epithelioid morphology, nuclear atypia, and glassy amphophilic cytoplasm embedded in a myxohyaline to collagenous stroma (D). Most cases of EHE have diffuse nuclear expression of CAMTA1 (E) resulting from WWTR1-CAMTA1 fusion. In this case, next-generation sequencing detected rearrangement involving the WWTR1 coding as evidenced by split reads (F, arrow) that match to CAMTA1 (not shown). Postradiation angiosarcoma of the breast consists of atypical endothelial cells growing in strands and sheets and diffusely infiltrating preexisting fat (G) is characterized by nuclear expression of MYC (H) resulting from high-level MYC amplification (I, arrow).
With the discovery of recurrent fusions in the group of vascular neoplasms, highly specific and sensitive immunohistochemical markers have been introduced into the routine diagnostic setting over the past few years. Although they are classified as low-grade malignant endothelial neoplasm, some cases of epithelioid hemangioendothelioma (EHE) behave in a frankly malignant fashion, and their distinction from other mesenchymal and nonmesenchymal neoplasms with overlapping histologic appearances, such as metastatic carcinoma, is important for patient management. EHEs occur over a wide age range and show predilection for the soft tissue of extremities and trunk, often arising in association with a large vein. Occasionally, these tumors present as multifocal disease in lung, liver, and bone. Local recurrence occurs in 15%, distant metastasis in 30% (in soft-tissue sites), and mortality ranges from 15% for soft-tissue sites to 50% for tumors arising in liver and lung. Histologically, EHE comprises cords and strands of round to epithelioid endothelial cells with characteristic glassy to palely eosinophilic cytoplasm and intracytoplasmic vacuoles, surrounded by myxohyaline or collagenous stroma (Fig. 3D–F; Table 1). The discovery of recurrent t(1;3)(p36.3; q25) translocation in 201141 in approximately 90% of cases resulting in WWTR1-CAMTA1 fusion,42,43 led to the development of a highly specific and sensitive CAMTA1 immunohistochemical stain for the diagnosis of EHE in distinction from histologic mimics (Tables 1 and 2).44 A small subset (< 10% of cases) lack WWTR1-CAMTA1 fusion and instead harbor alternate YAP1-TFE3 fusion resulting from t(X;11)(p11;q22) (Tables 1 and 2).45 This subset of EHE has recently been shown to follow a less aggressive clinical course compared with those with canonical WWTR1-CAMTA1 fusion, with 5-year overall survival rates of 86% versus 59%,46 and exhibits distinct morphologic features, such as tumor cells with prominent, voluminous eosinophilic cytoplasm and focally well-formed vascular channels. This subset of EHE is negative for CAMTA1 immunohistochemistry and instead shows nuclear expression of TFE3.
Pseudomyogenic hemangioendothelioma (PHE), another vascular neoplasm of intermediate biologic potential, was recently found to harbor recurrent SERPINE1-FOSB fusion resulting from t(7;19)(q22;q13)47; rare cases of PHE with alternate ACTB-FOSB48 fusion have been described.FOSB rearrangement leading to FOSB overexpression can be detected by recently introduced FOSB immunohistochemistry, which is positive in 96% of cases of PHE (Table 1).49 However, subsets of other vascular neoplasms may harbor FOSB rearrangement, such as the “cellular variant” of epithelioid hemangioma (i.e., ZFP36-FOSB or WWTR1-FOSB),50,51 with positive FOSB staining demonstrated in approximately 50% of cases.49
Secondary postradiation angiosarcoma of the breast develops with a median latent interval of 5 to 6 years after radiation following breast-conserving surgery with increasing incidence over the past years and is mostly cutaneous in location.52–54 Presenting as small erythematous to violaceous papules, nodules, or large plaques with skin discoloration, postradiation angiosarcoma infiltrates the reticular dermis, often with subtle radial extension of individual neoplastic vessels at a distance from the primary lesion and deeper infiltration into subcutis. In contrast to conventional mammary angiosarcoma, which arises in breast parenchyma, postradiation angiosarcoma of the skin is characterized by high-level MYC amplification55,56 (Fig. 3G–I), which can be detected by immunohistochemical staining for MYC and by genomic evidence of high-level copy number gain at 8q24.21. Diffuse nuclear MYC expression in postradiation angiosarcoma enables distinction from atypical postradiation vascular proliferation and can be particularly useful in cases of postradiation angiosarcoma with deceptively bland cytomorphology.
Newly Emerging Entities and Predictive Biomarkers
With increased use of molecular analyses in the diagnostic work-up of soft-tissue tumors, newly discovered genomic aberrations have led to the identification of novel entities defined by distinct gene fusions: these include NTRK-rearranged spindle cell neoplasms57,58 and EWSR1-SMAD3–positive fibroblastic tumors,59,60 which are being included as “emerging entities” in the upcoming World Health Organization Classification of Tumors. However, biologic potential and clinical implications of any newly reported entity defined by a recurrent genomic event remain to be defined. As exemplified by the notorious “promiscuity” of EWSR1 and ALK as common fusion partners, many genetic alterations initially considered “tumor-specific” or even “disease-defining” evolve to be quite nonspecific over time and must be carefully evaluated before they can add diagnostic, predictive, or prognostic value to existing classification schemes.
As an example, various solid tumors (including rare benign and malignant mesenchymal neoplasms) harbor fusions of NTRK1, NTRK2, or NTRK3 encoding neurotrophic tyrosine kinases NTRK1–3 (i.e., TRKA-C), which are generally believed to be mutually exclusive with other genomic aberrations. A classic example of a mesenchymal neoplasm with canonical NTRK rearrangement is infantile fibrosarcoma, a pediatric spindle cell sarcoma with characteristic ETV6-NTRK3 fusion in most cases and rare alternate EML4-NTRK3 fusion (Fig. 4A and B; Table 2).61,62 A recently introduced “pan-TRK” immunohistochemical stain has been shown to be highly sensitive63 but not entirely specific for tumors with NTRK fusions and can be expressed, for instance, in ALK-rearranged tumors.
FIGURE 4. Examples of Mesenchymal Neoplasms Showing Pan-TRK Expression by Immunohistochemistry.
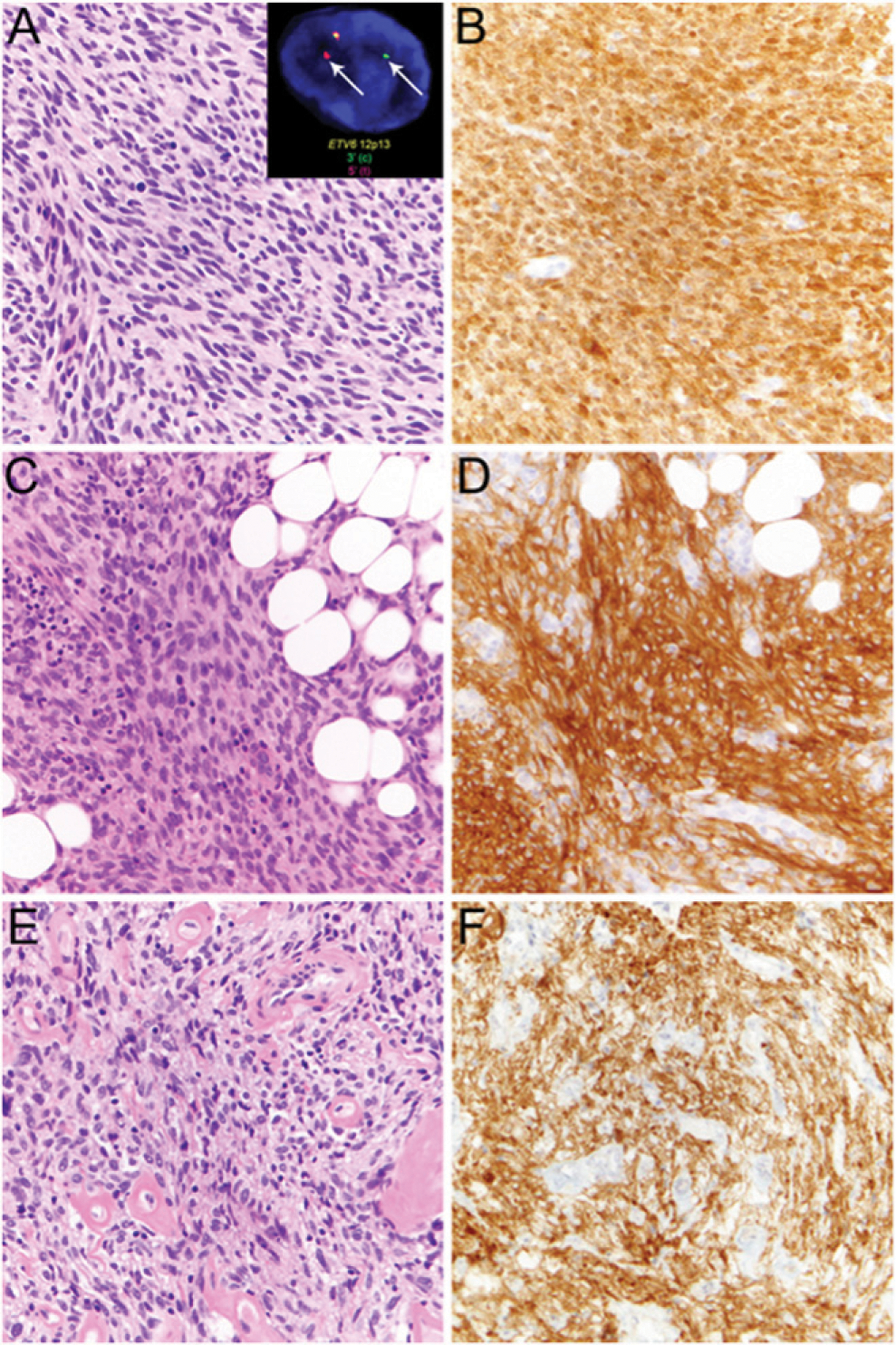
Infantile fibrosarcoma comprises monotonous population of spindle cells (A) with ETV6-NTRK3 rearrangement, detected by ETV6 break-apart fluorescence in situ hybridization (A, inset, arrows), and shows diffuse staining by pan-TRK immunohistochemistry (B). Lipofibromatosis-like neural tumor with spindled tumor cells lacking nuclear atypia containing wavy neural-like nuclei and eosinophilic cytoplasm with diffuse infiltration of adjacent fat (C) shows diffuse pan-TRK staining (D). Unclassified sarcoma of the endocervix comprising ovoid to spindled tumor cells with nuclear atypia scattered in a collagenous stroma with prominent hyalinized blood vessels (E) showing positive pan-TRK staining (F) suggestive of underlying NTRK rearrangement.
The testing approach for detection of NTRK fusions in sarcoma remains to be determined. A recent retrospective analysis evaluated the performance of immunohistochemistry and DNA-based NGS to detect NTRK fusions relative to RNA-based NGS.64 Among a total of 33,997 patients, the authors identified 87 patients with oncogenic NTRK1–3 fusions in solid tumors.64 The reported sensitivity and specificity for detection of NTRK fusions were 81.1% and 99.9% for DNA-based sequencing and 87.9% and 81.1% for immunohistochemistry, respectively.64 Specifically, immunohistochemistry showed 96% to 100% sensitivity for fusions of NTRK1 and NTRK2 but only 79% sensitivity for NTRK3. Both sensitivity and specificity were found to be poor in sarcomas.64
Lipofibromatosis-like neural tumor, which harbors morphologic resemblance to lipofibromatosis but exhibits locally aggressive behavior in children and young adults, has recurrent NTRK1 rearrangement65 and expresses pan-TRK (Fig. 4C and D).63 In addition, rare cases of unclassified sarcomas with positive pan-TRK staining are increasingly recognized (Fig. 4E and F); however, their biologic potential, clinical course, and potential for response to inhibitors of the TRK family of kinases, such as larotrectinib and entrectinib, which have recently been U.S Food and Drug Administration-approved for the treatment of NTRK-rearranged solid tumors,66–70 remain to be defined.
RADIATION ONCOLOGY: ADVANCEMENTS AND NEW APPROACHES FOR THE TREATMENT OF SARCOMA
RT in the Neoadjuvant or Adjuvant Setting for Extremity and Trunk Soft-Tissue Sarcoma
Here, we focus on the use of RT in the treatment of extremity and trunk soft-tissue sarcoma, a common scenario for the use of RT in sarcoma care. However, the concepts discussed here apply to RT for the treatment of sarcoma in other scenarios, such as the neoadjuvant treatment of retroperitoneal sarcoma, treatment of oligometastatic disease, and in palliative settings.
Despite early proclamations that sarcoma was a “radioresistant” tumor type, RT has been a part of the care of patients with sarcoma for nearly a century.71 RT began to take hold as a critical adjuvant therapy for primary soft-tissue sarcoma in the second half of the 20th century along with conservative surgery.72 Ultimately, randomized evidence emerged to illustrate that limb salvage surgery combined with RT could provide a less morbid alternative to amputation for extremity soft-tissue sarcoma.73
At that time, RT was delivered to patients in what today would be considered medieval fashion. The target area for RT was delineated clinically or using two-dimensional radiographs, without any refined capacity to control the distribution of the radiation dose, avoid or reduce radiation dose to nearby organs at risk, maximize radiation dose to the target structures, or account for daily variability in treatment setup. Two major advances in technology in the late 20th and early 21st century revolutionized RT for all disease sites including sarcoma.
The major advances in RT technology have been the use of more refined imaging for treatment planning and image guidance prior to daily treatments. Radiation treatment plans are now based on three-dimensional imaging of tumor and normal anatomy. In some cases, four-dimensional imaging is used to account for changes in anatomy over time (e.g., during the respiratory cycle). With this imaging, radiation treatment plans can account for differences in the densities of patients’ tissues (air, soft tissue, bone) that directly affect the distribution of radiation dose. Furthermore, radiation target volumes can be defined more accurately, minimizing unnecessary radiation dose to normal tissue. Furthermore, just prior (or even during) each radiation treatment, two-dimensional and/or three-dimensional imaging of a patient can be acquired to confirm patient and target positioning and ensure treatment accuracy within as few as 2 to 3 mm of error.
RTOG 0630 was an early example of the potential for image-guided RT to improve the care of patients with sarcoma.74 This multi-institutional phase II study was designed to assess the frequency and severity of late toxicities in patients receiving preoperative RT for extremity soft-tissue sarcoma. With the availability of image guidance, RT was delivered to a reduced volume using smaller margins around the primary tumor. Predictably, important late toxicities related to radiation of the extremity (joint stiffness, fibrosis, and edema) occurred in 5% or less of the patients, compared with rates between 15% and 30% in patients using older techniques without image guidance.75
The second key advance in the technology of RT was the introduction of intensity-modulated radiation therapy (IMRT). With conventional three-dimensional conformal RT, beams are arranged to maximize coverage of the target structure while minimizing radiation dose to surrounding organs at risk. The radiation dose from each beam can be modulated to a certain extent, but this process is limited by human capacity for calculation. With IMRT, the modulation of each radiation beam can be performed by computer-based calculations, which are derived from constraints to normal structures and organs at risk determined and prioritized a priori by the radiation oncologist. Thus, radiation oncologists have greater ability to control the dose of radiation from a particular radiation beam over time, resulting in improved accuracy and homogeneity of the radiation dose with respect to target volumes and improved avoidance of normal tissues.
In a phase II study of preoperative image-guided RT for soft-tissue sarcoma exclusively using IMRT, reconstructive tissue flaps were less frequently required to manage wound complications, and late toxicities (joint stiffness, fibrosis, edema) were improved compared with historical rates using conventional three-dimensional conformal RT techniques.76 Subsequent retrospective data have further supported low late toxicity rates with the use of IMRT as well as potentially lower local recurrence rates.77,78 According to the National Cancer Database, the use of IMRT in the neoadjuvant or adjuvant treatment of soft-tissue sarcoma has risen sharply over the past 10 years (V. Reddy, et al, unpublished data, 2019).
To maximize the benefit of image-guided RT and IMRT, there has been increased use of preoperative compared with postoperative RT for extremity and trunk soft-tissue sarcoma. Although randomized data provided evidence for reduced late toxicities with preoperative RT, there has also been a competing concern for an increase in perioperative wound complications in patients receiving preoperative RT. Improvements in image guidance and IMRT have provided further impetus to shift practice patterns toward the use of preoperative RT, where the presence of a tumor target allows for more discrete target delineation to maximize the benefit of these technologies.79 Still, there are advantages to both preoperative and postoperative approaches that must be considered on a case-by-case basis (Table 3).
TABLE 3.
Comparison of Preoperative and Postoperative RT for Soft-Tissue Sarcoma
| Advantage of Preoperative RT | Advantage of Postoperative RT |
|---|---|
| Reduced late toxicities (e.g., fibrosis, lymphedema, joint stiffness) for extremity tumors | Pathologic evaluation of an untreated specimen in cases of diagnostic uncertainty |
| Smaller RT volumes | Reduced wound-complication rate |
| Shorter course of RT (5 vs. 6 weeks) | Pathologic evaluation of tumor extent to define RT volume |
| Maximize benefits of image-guided RT due to presence of tumor target | Earlier removal of tumor can reduce patient stress/anxiety |
Abbreviation: RT, radiotherapy.
Stereotactic RT for the Treatment of Oligometastatic or Oligoprogressive Sarcoma
Another common scenario for the use of RT in sarcoma that has benefited from advances in technology has been the setting of oligometastatic or oligoprogressive disease, in which a patient may have a small number of progressing or metastatic lesions that can be addressed with tumor-directed therapies. The lung and spine are two common areas of metastases arising in patients with primary soft-tissue and bone sarcoma. Although systemic therapy (e.g., chemotherapy, targeted therapy, or immunotherapy) is the mainstay of overall disease control in the metastatic setting, individual tumor lesions may be poorly controlled by systemic therapy and may require additional tumor-directed therapy (oligoprogressive disease). As improvements in systemic therapies allow patients to live longer with disease, tumor-directed therapy can allow for a much-needed hiatus from the toxicities of systemic therapies. In other cases, where patients develop oligometastatic disease after a long disease-free interval, local therapies can obviate the need for immediate systemic therapy.
Several options are available for tumor-directed therapy. Among these, stereotactic body RT (SBRT) and stereotactic surgery (SRS) are two modern RT technologies that have been widely adopted and refined for the treatment of patients with metastatic disease of all histologies over the past 20 years. SRS and SBRT refer to cases where a high dose of RT is delivered per fraction (treatment) to a tumor target in one (SRS) or up to five (SBRT) total fractions (treatments). SRS and SBRT are predicated on the use of the advanced immobilization devices to ensure accuracy of patient setup reproducibility, high-resolution imaging for target delineation, optimal image guidance for treatment delivery, and advanced treatment planning techniques (e.g., inverse planning).
Because of the unique biology of delivering high radiation doses per fraction, the antitumor effect of SRS or SBRT is potent and can result in long-term disease control or cure of the irradiated tumor. In two separate studies of patients with metastatic high-grade sarcoma in the lung treated with SBRT at two separate institutions, over 86% and 94% of irradiated tumors (25 patients and 39 patients) were controlled after 24 and 43 months of follow-up, respectively.80,81 In a separate study of metastatic sarcoma to the spine, the actuarial rate of local control among surviving patients treated with SRS or SBRT was over 85%.82 Another consideration is that these are noninvasive procedures performed on an outpatient basis, are typically well tolerated (depending on the anatomic location of the target lesion), and can be safely combined with some systemic therapies. However, radiation oncologists should always discuss less common but serious toxicities, including radiation neuritis or plexopathy (nerve injury resulting in pain syndrome, weakness, and/or muscle atrophy), radiation pneumonitis, or radiation-induced bowel injury (ulceration, bleeding, perforation). Ultimately, the appropriate tumor-directed therapy (e.g., metastasectomy, interventional ablation) should be considered in a multidisciplinary setting and on a case-by-case basis.
MRI-Guided RT
Most modern radiation planning is based on CT scans of the target area. Although some soft-tissue sarcomas are well visualized on CT, others can be difficult to localize without the improved soft-tissue resolution of MRI.83 One approach to circumvent this issue has been to obtain diagnostic MRI that can be fused with CT-based treatment planning scans. This has limited utility because of the difficulty of obtaining diagnostic imaging with the patient in the precise setup used for treatment delivery, thus making the fusion of MRI and CT scans imprecise. The adoption of MRI-based simulation, in which patients undergo MRI-based imaging in the treatment position, can facilitate fusion with CT-based planning scans and improve accuracy of target delineation.84 MRI-only treatment planning may also be a future alternative, circumventing CT planning entirely.85
Identification of the tumor on daily image guidance prior to treatment delivery is also essential for accurate treatment; in some cases, the tumor is not visible using the on-board cone-beam CT scans used for setup verification. In these cases, MRI-guided RT allows for imaging of patients prior to and during delivery of RT.86 Imaging just prior to treatment delivery ensures accurate treatment delivery for fixed lesions. In cases of mobile tumors that are susceptible to respiratory motion, such as those in the abdomen or thorax near the diaphragm or even cardiac tumors susceptible to cardiac motion, cine imaging of tumors during RT can allow for target tracking and better accuracy throughout the treatment delivery. As MRI-based linear accelerators become more widely adopted, their value in selected cases of soft-tissue sarcoma treatment will be difficult to ignore (Fig. 5).
FIGURE 5. MRI-Guided Radiotherapy for Sarcoma Liver Metastasis.
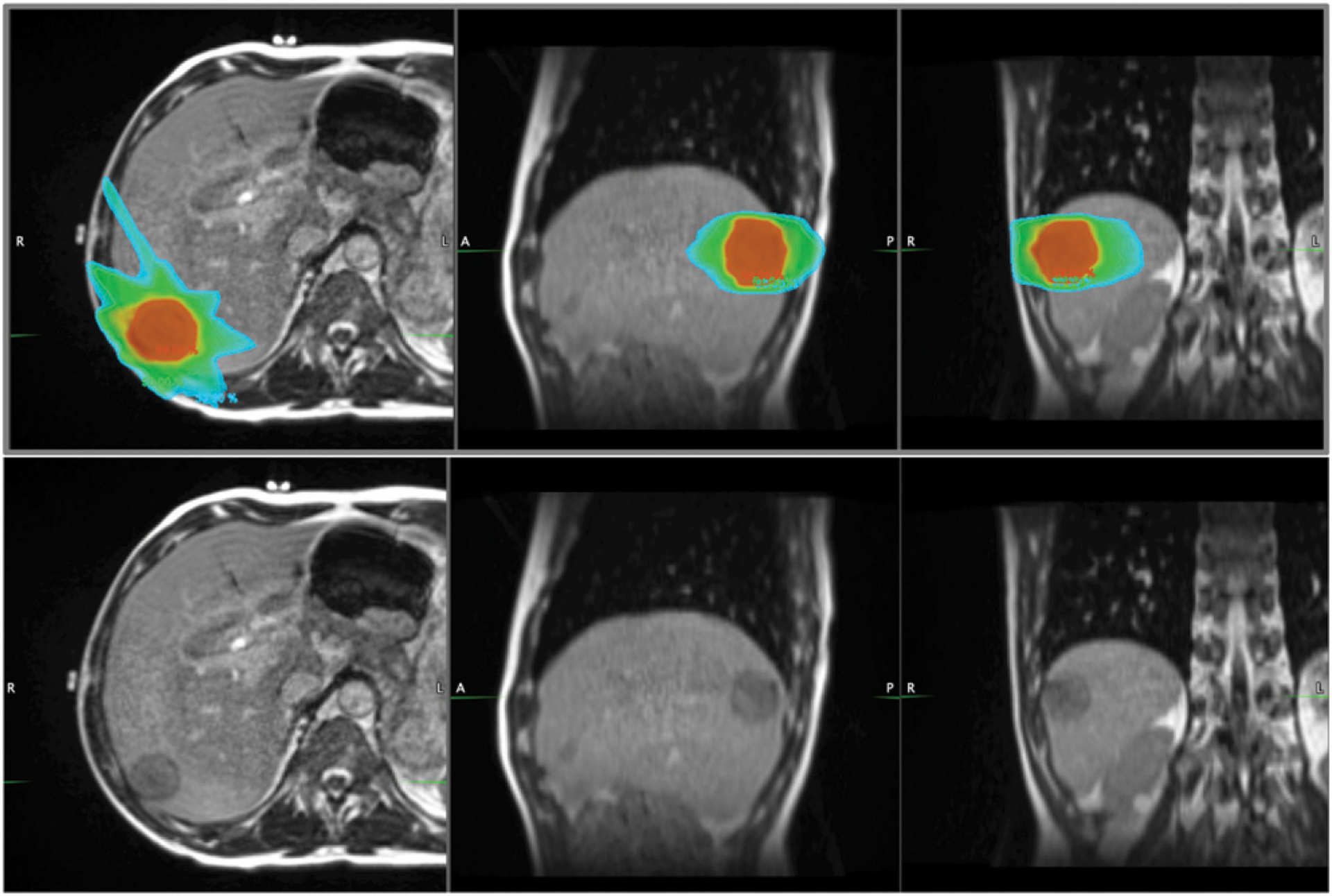
Radiation dose distribution (top panel) for a patient receiving magnetic resonance (MR)-guided stereotactic body radiotherapy for oligometastatic spindle cell sarcoma of the liver. A 172s balance steady-state free precession MR sequence was used to acquire three-dimensional anatomic MRI with 1.5 mm isotropic resolution using MRIdian’s 0.35T on-board MRI. There are distinct advantages of MR-guided radiotherapy for sarcoma at simulation, treatment planning, and patient setup prior to treatment delivery. In this case, the target lesion was not identifiable on CT imaging. The ability to visualize the tumor just prior to treatment allowed safe reduction of the planning margin from 1 cm to 5 mm, reducing radiation dose to surrounding normal tissue. Given the challenging treatment setup for sarcoma extremity lesions, the added imaging allowed more reproducible patient positioning. From left to right in both top and bottom panels: axial, sagittal, and coronal MR images. Isodose colors (top panel): red, 99%; green, 50%, light blue, 33%.
Proton Therapy
Proton-based RT is an alternative approach to delivery radiation. Protons are heavy, positively charged particles with distinct physical characteristics compared with photons, which are particles more commonly used to deliver RT and have negligible mass and no charge. Although proton therapy has been available regionally at select institutions for several decades, it has emerged as an alternative to conventional photon-based RT over the past 15 years. This is the result of an advance in proton therapy that allowed its delivery on advanced isocentric gantry systems, resulting in more flexibility for its use. Proton therapy provides an advantage to photon therapy in that the dose of RT has a finite penetration in tissue and delivers the majority of its energy at a specific depth window. As a result, in certain scenarios, proton therapy may be able to reduce the exposure of low to medium doses of RT in the vicinity of the tumor target. Furthermore, pound-for-pound, proton therapy may also deliver a slightly higher biologic effect compared with photons.87
This advantage is particularly relevant when the tumor target is located on one side of a critical normal structure. For example, tumors located posterior to the spinal cord and anterior to the mediastinum may maximally benefit from the use of proton therapy, which could significantly reduce radiation dose to the structures immediately beyond the tumor target (e.g., spinal cord, mediastinum). As another example, proton therapy has been proposed as an alternative approach to deliver dose-escalated RT to the high-risk margin of retroperitoneal sarcoma due to its ability to spare tissue immediately adjacent small bowel.88
The use of proton therapy is particularly encouraged in pediatric tumors, including pediatric sarcoma, to limit putative long-term effects of low- and medium-dose exposure to nearby normal structures. Specific use cases include rhabdomyosarcoma, especially those arising in the head and neck, including parameningeal and orbital rhabdomyosarcoma, where protons may allow sparing of the optic apparatus, central nervous system structures, and salivary glands.89,90 Other pediatric sarcoma cases where proton therapy may provide an advantage include osteosarcoma of the spine or skull base91 and pelvic Ewing sarcoma.92
In some classically radioresistant tumors, including chordoma and chondrosarcoma, the putative higher biologic potency of proton therapy may result in improved tumor control.93 Given the location of these tumors in the spine and skull base, the radiation dose characteristics of proton beam therapy are important in sparing the optic apparatus and other central nervous system structures. It should be noted that all proton therapy is not created equal; more advanced delivery using pencil beam scanning and intensity-modulated proton beam therapy provide the most conformal therapy and the greatest potential therapeutic advantage.
Thus, although there is a paucity of clinical data at this point to support the superiority of proton therapy in terms of reduced late toxicities or improved disease outcomes, the accumulation of clinical case series supports the selected use of proton therapy on a case-by-case scenario for the treatment of patients with sarcoma.
Active Investigations to Improve RT as a Therapeutic Modality for Sarcoma
Condensed radiation treatment regimens (SBRT and SRS) for metastatic sarcoma and other cancers are common-place. However, the standard neoadjuvant or adjuvant treatment of primary soft-tissue sarcoma involves a 5- to 6- week course of daily radiation that takes place Monday through Friday. This is a burdensome therapy that is logistically and socioeconomically burdensome for patients, especially those who wish to be treated at tertiary sarcoma centers (where there is an association with improved outcomes), but these centers are often inconveniently located at a greater distance than community oncology practices.
Shorter treatment regimens may soon challenge this treatment paradigm. For decades, a neoadjuvant approach using an eight-treatment neoadjuvant radiation regimen in combination with neoadjuvant chemotherapy has resulted in acceptable (but not ideal) local control and toxicity.94 More recently, we evaluated a condensed 5-day radiation regimen for primary high-risk soft-tissue sarcoma of the trunk and extremity in a single-institution phase II study.95 Early toxicity and disease control outcomes were encouraging, and the 5-day approach also improved utilization and access of neoadjuvant RT at our high-volume sarcoma center. Other studies using shorter RT regimens (5–15 fractions) for soft-tissue sarcoma are ongoing (Table 4).
TABLE 4.
Active Clinical Trials Using Modified Radiation Regimens in Preoperative or Postoperative Treatment of STS
| NCT No. | Study Title | Patients | Radiotherapy Regimen |
|---|---|---|---|
| NCT02106312 | Dose Reduction of Preoperative Radiotherapy in Myxoid Liposarcomas (DOREMY) | Adults with myxoid liposarcoma | 2 Gy × 18 fractions = 36 Gy total (3.5 weeks) |
| NCT02701153 | Phase II Study of 5-Day Hypofractionated Preoperative Radiation Therapy for Soft Tissue Sarcomas: Expansion Cohort | Adults with extremity and trunk STS | 5–6 Gy × 5 fractions =30 Gy (1 week) |
| NCT02634710 | Hypofractionated Pre-operative Radiation Therapy for Soft Tissue Sarcomas of the Extremity and Chest-wall | Adults with extremity and chest-wall STS | 7 Gy × 5 fractions = 35 Gy (2 weeks) |
| NCT03819985 | Shorter Course, Hypofractionated Pre-Surgery Radiation Therapy in Treating Patients With Localized, Resectable Soft Tissue Sarcoma of the Extremity of Superficial Trunk | Adults with extremity and trunk STS | 2.85 Gy × 15 fractions = 42.75 Gy (3 weeks) |
| NCT02565498 | Preoperative vs. Postoperative IMRT for Extremity/Truncal STS | Adults with extremity and trunk STS | 2 Gy × 25 fractions = 50 Gy preoperatively or postoperatively (5 weeks) |
Abbreviations: IMRT, intensity-modulated radiation therapy; STS, soft-tissue sarcoma.
Traditionally, radiation for soft-tissue sarcoma has been agnostic of histologic subtype, but this is also poised to change. Myxoid liposarcoma has long been known to be a well-defined histologic subtype with a unique chromosomal aberration and a clearly radiosensitive phenotype. Local control for myxoid liposarcoma after surgery and radiation can reportedly exceed 95%.96 A multi-institutional study evaluating radiation dose reduction in the preoperative treatment of myxoid liposarcoma is ongoing (Table 4).
How Technology Can Fill the Gap
Technologic advances will be necessary to continue to improve the impact of RT on patients with sarcoma over the next few decades. In particular, better understanding of the biology of radiation responses in tumor and normal tissue will permit the personalization of RT with respect to toxicities, secondary malignancies, and disease control. Biomarkers to identify a priori patients most at risk for RT-associated toxicities, such as wound complications or late extremity complications, or even RT-associated malignancies will improve the therapeutic window. Tailoring radiation dose and volumes to tumor subtypes—as defined not only by histology but also by biology—will further amplify the potential impact of RT on patients with sarcoma.
CONCLUSIONS
The pathologic work-up of sarcomas relies mainly on the thoughtful integration of clinical information (i.e., patient age, sex), anatomic location, radiologic features, gross and histomorphologic appearances, as well as immunohistochemical staining and ancillary cytogenetic and molecular genetic analyses. With the development of targeted therapies directed against distinct oncogenic aberrations and tumor-specific signatures that predict sensitivity to immunotherapy, the accurate and timely identification of subsets of patients most likely to benefit from systemic therapies gains importance. Whereas some cytogenetic/molecular genetic aberrations or immunohistochemical markers are considered tumor specific, others can be present in various benign and malignant mesenchymal neoplasms and require careful evaluation in the diagnostic context.
Despite improvements in these diagnostic tools and the expanding armamentarium of systemic therapies, effective local therapy involving surgery and radiation therapy remains the primary curative approach. Advances in image guidance, intensity modulation, and proton beam technology have expanded the therapeutic window of radiation therapy for local disease. Furthermore, for limited-burden metastatic disease refractory to systemic therapy, stereotactic approaches offer a reliable, noninvasive and well-tolerated tool for local disease control.
We await the outcomes of ongoing efforts in the translational research setting because we anticipate these will further promote discoveries that expand our understanding of sarcoma biology, development, and progression. These discoveries will lead to refined classification systems and novel therapeutic approaches that, in the context of a multidisciplinary approach, will continue to raise the bar for the care of patients with sarcoma.
PRACTICAL APPLICATIONS.
Although rare, sarcomas comprise a wide histologic spectrum of diseases characterized by unique prognostic and therapeutic implications.
Initial sarcoma diagnostic work-up may require ancillary immunohistochemical, cytogenetic, and/or molecular genetic testing as appropriate to confirm or further refine a diagnosis based on morphology alone.
For localized extremity or trunk soft-tissue sarcoma, advanced radiation techniques, including CT or MRI guidance and IMRT, should be considered standard.
Selective use of stereotactic RT for oligometastatic and oligoprogressive disease is a well-tolerated and effective approach to bridge patients between systemic therapies.
ACKNOWLEDGMENT
Dr. Schaefer and Dr. Kalbasi contributed equally to this article. The authors thank Dr. Christopher D. M. Fletcher, MD, FRCPath, Department of Pathology, Brigham and Women’s Hospital, for sharing representative cases; and the Center for Advanced Molecular Diagnostics and Division of Cytogenetics, Department of Pathology, Brigham and Women’s Hospital, and Dr. Yingli Yang, PhD, Department of Radiation Oncology, University of California Los Angeles for providing select images.
Footnotes
AUTHORS’ DISCLOSURES OF POTENTIAL CONFLICTS OF INTEREST AND DATA AVAILABILITY STATEMENT
Disclosures provided by the authors and data availability statement (if applicable) are available with this article at DOI https://doi.org/10.1200/EDBK_280729.
REFERENCES
- 1.Fletcher C, Bridge JA, Hogendoorn PCW, et al. In Fletcher CDM, (ed). WHO Classification of Tumours of Soft Tissue and Bone. Lyon: IARC Press; 2013. [Google Scholar]
- 2.Siegel RL, Miller KD, Jemal A. Cancer statistics, 2016. CA Cancer J Clin. 2016;66:7–30. [DOI] [PubMed] [Google Scholar]
- 3.Dickson BC, Swanson D. Targeted RNA sequencing: a routine ancillary technique in the diagnosis of bone and soft tissue neoplasms. Genes Chromosomes Cancer. 2019;58:75–87. [DOI] [PubMed] [Google Scholar]
- 4.Zou C, Smith KD, Liu J, et al. Clinical, pathological, and molecular variables predictive of malignant peripheral nerve sheath tumor outcome. Ann Surg. 2009; 249:1014–1022. [DOI] [PubMed] [Google Scholar]
- 5.Lee W, Teckie S, Wiesner T, et al. PRC2 is recurrently inactivated through EED or SUZ12 loss in malignant peripheral nerve sheath tumors. Nat Genet. 2014; 46:1227–1232. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.De Raedt T, Beert E, Pasmant E, et al. PRC2 loss amplifies Ras-driven transcription and confers sensitivity to BRD4-based therapies. Nature. 2014;514:247–251. [DOI] [PubMed] [Google Scholar]
- 7.Prieto-Granada CN, Wiesner T, Messina JL, et al. Loss of H3K27me3 expression is a highly sensitive marker for sporadic and radiation-induced MPNST. Am J Surg Pathol. 2016;40:479–489. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Schaefer IM, Fletcher CD, Hornick JL. Loss of H3K27 trimethylation distinguishes malignant peripheral nerve sheath tumors from histologic mimics. Mod Pathol. 2016;29:4–13. [DOI] [PubMed] [Google Scholar]
- 9.Cleven AH, Sannaa GA, Briaire-de Bruijn I, et al. Loss of H3K27 tri-methylation is a diagnostic marker for malignant peripheral nerve sheath tumors and an indicator for an inferior survival. Mod Pathol. 2016;29:582–590. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Jo VY, Fletcher CD. Epithelioid malignant peripheral nerve sheath tumor: clinicopathologic analysis of 63 cases. Am J Surg Pathol. 2015;39:673–682. [DOI] [PubMed] [Google Scholar]
- 11.Schaefer IM, Dong F, Garcia EP, et al. Recurrent SMARCB1 inactivation in epithelioid malignant peripheral nerve sheath tumors. Am J Surg Pathol. 2019; 43:835–843. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Hollmann TJ, Hornick JL. INI1-deficient tumors: diagnostic features and molecular genetics. Am J Surg Pathol. 2011;35:e47–e63. [DOI] [PubMed] [Google Scholar]
- 13.Jo VY, Fletcher CDM. SMARCB1/INI1 loss in epithelioid schwannoma: a clinicopathologic and immunohistochemical study of 65 cases. Am J Surg Pathol. 2017; 41:1013–1022. [DOI] [PubMed] [Google Scholar]
- 14.Hornick JL, Dal Cin P, Fletcher CD. Loss of INI1 expression is characteristic of both conventional and proximal-type epithelioid sarcoma. Am J Surg Pathol. 2009; 33:542–550. [DOI] [PubMed] [Google Scholar]
- 15.Italiano A, Sung YS, Zhang L, et al. High prevalence of CIC fusion with double-homeobox (DUX4) transcription factors in EWSR1-negative undifferentiated small blue round cell sarcomas. Genes Chromosomes Cancer. 2012;51:207–218. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Antonescu CR, Owosho AA, Zhang L, et al. Sarcomas with CIC-rearrangements are a distinct pathologic entity with aggressive outcome: a clinicopathologic and molecular study of 115 cases. Am J Surg Pathol. 2017;41:941–949. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Sugita S, Arai Y, Tonooka A, et al. A novel CIC-FOXO4 gene fusion in undifferentiated small round cell sarcoma: a genetically distinct variant of Ewing-like sarcoma. Am J Surg Pathol. 2014;38:1571–1576. [DOI] [PubMed] [Google Scholar]
- 18.Solomon DA, Brohl AS, Khan J, et al. Clinicopathologic features of a second patient with Ewing-like sarcoma harboring CIC-FOXO4 gene fusion. Am J Surg Pathol. 2014;38:1724–1725. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Specht K, Sung YS, Zhang L, et al. Distinct transcriptional signature and immunoprofile of CIC-DUX4 fusion-positive round cell tumors compared to EWSR1-rearranged Ewing sarcomas: further evidence toward distinct pathologic entities. Genes Chromosomes Cancer. 2014;53:622–633. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Hung YP, Fletcher CD, Hornick JL. Evaluation of ETV4 and WT1 expression in CIC-rearranged sarcomas and histologic mimics. Mod Pathol. 2016; 29:1324–1334. [DOI] [PubMed] [Google Scholar]
- 21.Yoshida A, Sekine S, Tsuta K, et al. NKX2.2 is a useful immunohistochemical marker for Ewing sarcoma. Am J Surg Pathol. 2012;36:993–999. [DOI] [PubMed] [Google Scholar]
- 22.Hung YP, Fletcher CD, Hornick JL. Evaluation of NKX2–2 expression in round cell sarcomas and other tumors with EWSR1 rearrangement: imperfect specificity for Ewing sarcoma. Mod Pathol. 2016;29:370–380. [DOI] [PubMed] [Google Scholar]
- 23.Shibuya R, Matsuyama A, Nakamoto M, et al. The combination of CD99 and NKX2.2, a transcriptional target of EWSR1-FLI1, is highly specific for the diagnosis of Ewing sarcoma. Virchows Arch. 2014;465:599–605. [DOI] [PubMed] [Google Scholar]
- 24.Pierron G, Tirode F, Lucchesi C, et al. A new subtype of bone sarcoma defined by BCOR-CCNB3 gene fusion. Nat Genet. 2012;44:461–466. [DOI] [PubMed] [Google Scholar]
- 25.Kao YC, Owosho AA, Sung YS, et al. BCOR-CCNB3 fusion positive sarcomas: a clinicopathologic and molecular analysis of 36 cases with comparison to morphologic spectrum and clinical behavior of other round cell sarcomas. Am J Surg Pathol. 2018;42:604–615. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Specht K, Zhang L, Sung YS, et al. Novel BCOR-MAML3 and ZC3H7B-BCOR gene fusions in undifferentiated small blue round cell sarcomas. Am J Surg Pathol. 2016;40:433–442. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Cohen-Gogo S, Cellier C, Coindre JM, et al. Ewing-like sarcomas with BCOR-CCNB3 fusion transcript: a clinical, radiological and pathological retrospective study from the Société Française des Cancers de L’Enfant. Pediatr Blood Cancer. 2014;61:2191–2198. [DOI] [PubMed] [Google Scholar]
- 28.Kao YC, Sung YS, Zhang L, et al. BCOR overexpression is a highly sensitive marker in round cell sarcomas with BCOR genetic abnormalities. Am J Surg Pathol. 2016;40:1670–1678. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Coffin CM, Hornick JL, Fletcher CD. Inflammatory myofibroblastic tumor: comparison of clinicopathologic, histologic, and immunohistochemical features including ALK expression in atypical and aggressive cases. Am J Surg Pathol. 2007;31:509–520. [DOI] [PubMed] [Google Scholar]
- 30.Lawrence B, Perez-Atayde A, Hibbard MK, et al. TPM3-ALK and TPM4-ALK oncogenes in inflammatory myofibroblastic tumors. Am J Pathol. 2000; 157:377–384. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Morris SW, Kirstein MN, Valentine MB, et al. Fusion of a kinase gene, ALK, to a nucleolar protein gene, NPM, in non-Hodgkin’s lymphoma. Science. 1994; 263:1281–1284. [DOI] [PubMed] [Google Scholar]
- 32.Pan Z, Hu S, Li M, et al. ALK-positive large B-cell lymphoma: a clinicopathologic study of 26 cases with review of additional 108 cases in the literature. Am J Surg Pathol. 2017;41:25–38. [DOI] [PubMed] [Google Scholar]
- 33.Mano H. Non-solid oncogenes in solid tumors: EML4-ALK fusion genes in lung cancer. Cancer Sci. 2008;99:2349–2355. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Chou A, Fraser S, Toon CW, et al. A detailed clinicopathologic study of ALK-translocated papillary thyroid carcinoma. Am J Surg Pathol. 2015;39:652–659. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Mariño-Enríquez A, Ou WB, Weldon CB, et al. ALK rearrangement in sickle cell trait-associated renal medullary carcinoma. Genes Chromosomes Cancer. 2011; 50:146–153. [DOI] [PubMed] [Google Scholar]
- 36.Dickson BC, Swanson D, Charames GS, et al. Epithelioid fibrous histiocytoma: molecular characterization of ALK fusion partners in 23 cases. Mod Pathol. 2018; 31:753–762. [DOI] [PubMed] [Google Scholar]
- 37.Kiuru M, Jungbluth A, Kutzner H, et al. Spitz tumors: comparison of histological features in relationship to immunohistochemical staining for ALK and NTRK1. Int J Surg Pathol. 2016;24:200–206. [DOI] [PubMed] [Google Scholar]
- 38.Perron E, Pissaloux D, Charon Barra C, et al. Melanocytic myxoid spindle cell tumor with ALK rearrangement (MMySTAR): report of 4 cases of a nevus variant with potential diagnostic challenge. Am J Surg Pathol. 2018;42:595–603. [DOI] [PubMed] [Google Scholar]
- 39.Davis LE, Nusser KD, Przybyl J, et al. Discovery and characterization of recurrent, targetable ALK fusions in leiomyosarcoma. Mol Cancer Res. 2019;17:676–685. [DOI] [PubMed] [Google Scholar]
- 40.Le Loarer F, Cleven AHG, Bouvier C, et al. A subset of epithelioid and spindle cell rhabdomyosarcomas is associated with TFCP2 fusions and common ALK upregulation. Mod Pathol. 2020;33:404–419. [DOI] [PubMed] [Google Scholar]
- 41.Mendlick MR, Nelson M, Pickering D, et al. Translocation t(1;3)(p36.3;q25) is a nonrandom aberration in epithelioid hemangioendothelioma. Am J Surg Pathol. 2001;25:684–687. [DOI] [PubMed] [Google Scholar]
- 42.Errani C, Zhang L, Sung YS, et al. A novel WWTR1-CAMTA1 gene fusion is a consistent abnormality in epithelioid hemangioendothelioma of different anatomic sites. Genes Chromosomes Cancer. 2011;50:644–653. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Tanas MR, Sboner A, Oliveira AM, et al. Identification of a disease-defining gene fusion in epithelioid hemangioendothelioma. Sci Transl Med. 2011;3:98ra82. [DOI] [PubMed] [Google Scholar]
- 44.Doyle LA, Fletcher CD, Hornick JL. Nuclear expression of CAMTA1 distinguishes epithelioid hemangioendothelioma from histologic mimics. Am J Surg Pathol. 2016;40:94–102. [DOI] [PubMed] [Google Scholar]
- 45.Antonescu CR, Le Loarer F, Mosquera JM, et al. Novel YAP1-TFE3 fusion defines a distinct subset of epithelioid hemangioendothelioma. Genes Chromosomes Cancer. 2013;52:775–784. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Rosenbaum E, Jadeja B, Xu B, et al. Prognostic stratification of clinical and molecular epithelioid hemangioendothelioma subsets. Mod Pathol. Epub 2019. September 19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Walther C, Tayebwa J, Lilljebjörn H, et al. A novel SERPINE1-FOSB fusion gene results in transcriptional up-regulation of FOSB in pseudomyogenic hae-mangioendothelioma. J Pathol. 2014;232:534–540. [DOI] [PubMed] [Google Scholar]
- 48.Zhu G, Benayed R, Ho C, et al. Diagnosis of known sarcoma fusions and novel fusion partners by targeted RNA sequencing with identification of a recurrent ACTB-FOSB fusion in pseudomyogenic hemangioendothelioma. Mod Pathol. 2019;32:609–620. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Hung YP, Fletcher CD, Hornick JL. FOSB is a useful diagnostic marker for pseudomyogenic hemangioendothelioma. Am J Surg Pathol. 2016;41:596–606. [DOI] [PubMed] [Google Scholar]
- 50.Antonescu CR, Chen HW, Zhang L, et al. ZFP36-FOSB fusion defines a subset of epithelioid hemangioma with atypical features. Genes Chromosomes Cancer. 2014;53:951–959. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Huang SC, Zhang L, Sung YS, et al. Frequent FOS gene rearrangements in epithelioid hemangioma: a molecular study of 58 cases with morphologic reappraisal. Am J Surg Pathol. 2015;39:1313–1321. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Brenn T, Fletcher CD. Radiation-associated cutaneous atypical vascular lesions and angiosarcoma: clinicopathologic analysis of 42 cases. Am J Surg Pathol. 2005;29:983–996. [PubMed] [Google Scholar]
- 53.Strobbe LJ, Peterse HL, van Tinteren H, et al. Angiosarcoma of the breast after conservation therapy for invasive cancer, the incidence and outcome: an unforseen sequela. Breast Cancer Res Treat. 1998;47:101–109. [DOI] [PubMed] [Google Scholar]
- 54.Billings SD, McKenney JK, Folpe AL, et al. Cutaneous angiosarcoma following breast-conserving surgery and radiation: an analysis of 27 cases. Am J Surg Pathol. 2004;28:781–788. [DOI] [PubMed] [Google Scholar]
- 55.Manner J, Radlwimmer B, Hohenberger P, et al. MYC high level gene amplification is a distinctive feature of angiosarcomas after irradiation or chronic lymphedema. Am J Pathol. 2010;176:34–39. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Guo T, Zhang L, Chang NE, et al. Consistent MYC and FLT4 gene amplification in radiation-induced angiosarcoma but not in other radiation-associated atypical vascular lesions. Genes Chromosomes Cancer. 2011;50:25–33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Suurmeijer AJ, Dickson BC, Swanson D, et al. The histologic spectrum of soft tissue spindle cell tumors with NTRK3 gene rearrangements. Genes Chromosomes Cancer. 2019;58:739–746. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Miettinen M, Felisiak-Golabek A, Luiña Contreras A, et al. New fusion sarcomas: histopathology and clinical significance of selected entities. Hum Pathol. 2019; 86:57–65. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Kao YC, Flucke U, Eijkelenboom A, et al. Novel EWSR1-SMAD3 gene fusions in a group of acral fibroblastic spindle cell neoplasms. Am J Surg Pathol. 2018; 42:522–528. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Michal M, Berry RS, Rubin BP, et al. EWSR1-SMAD3-rearranged fibroblastic tumor: an emerging entity in an increasingly more complex group of fibroblastic/myofibroblastic neoplasms. Am J Surg Pathol. 2018;42:1325–1333. [DOI] [PubMed] [Google Scholar]
- 61.Knezevich SR, McFadden DE, Tao W, et al. A novel ETV6-NTRK3 gene fusion in congenital fibrosarcoma. Nat Genet. 1998;18:184–187. [DOI] [PubMed] [Google Scholar]
- 62.Church AJ, Calicchio ML, Nardi V, et al. Recurrent EML4-NTRK3 fusions in infantile fibrosarcoma and congenital mesoblastic nephroma suggest a revised testing strategy. Mod Pathol. 2018;31:463–473. [DOI] [PubMed] [Google Scholar]
- 63.Hung YP, Fletcher CDM, Hornick JL. Evaluation of pan-TRK immunohistochemistry in infantile fibrosarcoma, lipofibromatosis-like neural tumour and histological mimics. Histopathology. 2018;73:634–644. [DOI] [PubMed] [Google Scholar]
- 64.Solomon JP, Linkov I, Rosado A, et al. NTRK fusion detection across multiple assays and 33,997 cases: diagnostic implications and pitfalls. Mod Pathol. 2020; 33:38–46. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Agaram NP, Zhang L, Sung YS, et al. Recurrent NTRK1 gene fusions define a novel subset of locally aggressive lipofibromatosis-like neural tumors. Am J Surg Pathol. 2016;40:1407–1416. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Doebele RC, Drilon A, Paz-Ares L, et al. Entrectinib in patients with advanced or metastatic NTRK fusion-positive solid tumours: integrated analysis of three phase 1–2 trials. Lancet Oncol. 2020;21:271–282. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Laetsch TW, DuBois SG, Mascarenhas L, et al. Larotrectinib for paediatric solid tumours harbouring NTRK gene fusions: phase 1 results from a multicentre, open-label, phase 1/2 study. Lancet Oncol. 2018;19:705–714. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Drilon A, Laetsch TW, Kummar S, et al. Efficacy of larotrectinib in TRK fusion-positive cancers in adults and children. N Engl J Med. 2018;378:731–739. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Farago AF, Le LP, Zheng Z, et al. Durable clinical response to entrectinib in NTRK1-rearranged non-small cell lung cancer. J Thorac Oncol. 2015;10:1670–1674. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Drilon A. TRK inhibitors in TRK fusion-positive cancers. Ann Oncol. 2019;30 (suppl 8):viii23–viii30. [DOI] [PubMed] [Google Scholar]
- 71.Sarcoma and radiotherapy. Edinb Med J. 1930;37:707–708. [PMC free article] [PubMed] [Google Scholar]
- 72.Lindberg RD, Martin RG, Romsdahl MM, et al. Conservative surgery and postoperative radiotherapy in 300 adults with soft-tissue sarcomas. Cancer. 1981; 47:2391–2397. [DOI] [PubMed] [Google Scholar]
- 73.Rosenberg SA, Tepper J, Glatstein E, et al. The treatment of soft-tissue sarcomas of the extremities: prospective randomized evaluations of (1) limb-sparing surgery plus radiation therapy compared with amputation and (2) the role of adjuvant chemotherapy. Ann Surg. 1982;196:305–315. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Wang D, Zhang Q, Eisenberg BL, et al. Significant reduction of late toxicities in patients with extremity sarcoma treated with image-guided radiation therapy to a reduced target volume: results of Radiation Therapy Oncology Group RTOG-0630 trial. J Clin Oncol. 2015;33:2231–2238. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.O’Sullivan B, Davis AM, Turcotte R, et al. Preoperative versus postoperative radiotherapy in soft-tissue sarcoma of the limbs: a randomised trial. Lancet. 2002; 359:2235–2241. [DOI] [PubMed] [Google Scholar]
- 76.O’Sullivan B, Griffin AM, Dickie CI, et al. Phase 2 study of preoperative image-guided intensity-modulated radiation therapy to reduce wound and combined modality morbidities in lower extremity soft tissue sarcoma. Cancer. 2013;119:1878–1884. [DOI] [PubMed] [Google Scholar]
- 77.Folkert MR, Casey DL, Berry SL, et al. Femoral fracture in primary soft-tissue sarcoma of the thigh and groin treated with intensity-modulated radiation therapy: observed versus expected risk. Ann Surg Oncol. 2019;26:1326–1331. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Folkert MR, Singer S, Brennan MF, et al. Comparison of local recurrence with conventional and intensity-modulated radiation therapy for primary soft-tissue sarcomas of the extremity. J Clin Oncol. 2014;32:3236–3241. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Haas RLM, Miah AB, LePechoux C, et al. Preoperative radiotherapy for extremity soft tissue sarcoma; past, present and future perspectives on dose fractionation regimens and combined modality strategies. Radiother Oncol. 2016;119:14–21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Baumann BC, Nagda SN, Kolker JD, et al. Efficacy and safety of stereotactic body radiation therapy for the treatment of pulmonary metastases from sarcoma: a potential alternative to resection. J Surg Oncol. 2016;114:65–69. [DOI] [PubMed] [Google Scholar]
- 81.Mehta N, Selch M, Wang PC, et al. Safety and efficacy of stereotactic body radiation therapy in the treatment of pulmonary metastases from high grade sarcoma. Sarcoma. 2013;2013:360214. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Leeman JE, Bilsky M, Laufer I, et al. Stereotactic body radiotherapy for metastatic spinal sarcoma: a detailed patterns-of-failure study. J Neurosurg Spine. 2016; 25:52–58. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Wu JS, Hochman MG. Soft-tissue tumors and tumorlike lesions: a systematic imaging approach. Radiology. 2009;253:297–316. [DOI] [PubMed] [Google Scholar]
- 84.Paulson ES, Erickson B, Schultz C, et al. Comprehensive MRI simulation methodology using a dedicated MRI scanner in radiation oncology for external beam radiation treatment planning. Med Phys. 2015;42:28–39. [DOI] [PubMed] [Google Scholar]
- 85.Owrangi AM, Greer PB, Glide-Hurst CK. MRI-only treatment planning: benefits and challenges. Phys Med Biol. 2018;63:05tr1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Massaccesi M, Cusumano D, Boldrini L, et al. A new frontier of image guidance: organs at risk avoidance with MRI-guided respiratory-gated intensity modulated radiotherapy: technical note and report of a case. J Appl Clin Med Phys. 2019;20:194–198. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Ilicic K, Combs SE, Schmid TE. New insights in the relative radiobiological effectiveness of proton irradiation. Radiat Oncol. 2018;13:6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.DeLaney TF, Chen YL, Baldini EH, et al. Phase 1 trial of preoperative image guided intensity modulated proton radiation therapy with simultaneously integrated boost to the high risk margin for retroperitoneal sarcomas. Adv Radiat Oncol. 2017;2:85–93. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Leiser D, Calaminus G, Malyapa R, et al. Tumour control and quality of life in children with rhabdomyosarcoma treated with pencil beam scanning proton therapy. Radiother Oncol. 2016;120:163–168. [DOI] [PubMed] [Google Scholar]
- 90.Doyen J, Jazmati D, Geismar D, et al. Outcome and patterns of relapse in childhood parameningeal rhabdomyosarcoma treated with proton beam therapy. Int J Radiat Oncol Biol Phys. 2019;105:1043–1054. [DOI] [PubMed] [Google Scholar]
- 91.DeLaney TF, Park L, Goldberg SI, et al. Radiotherapy for local control of osteosarcoma. Int J Radiat Oncol Biol Phys. 2005;61:492–498. [DOI] [PubMed] [Google Scholar]
- 92.Rombi B, DeLaney TF, MacDonald SM, et al. Proton radiotherapy for pediatric Ewing’s sarcoma: initial clinical outcomes. Int J Radiat Oncol Biol Phys. 2012; 82:1142–1148. [DOI] [PubMed] [Google Scholar]
- 93.Weber DC, Malyapa R, Albertini F, et al. Long term outcomes of patients with skull-base low-grade chondrosarcoma and chordoma patients treated with pencil beam scanning proton therapy. Radiother Oncol. 2016;120:169–174. [DOI] [PubMed] [Google Scholar]
- 94.Pennington JD, Eilber FC, Eilber FR, et al. Long-term outcomes with ifosfamide-based hypofractionated preoperative chemoradiotherapy for extremity soft tissue sarcomas. Am J Clin Oncol. 2018;41:1154–1161. [DOI] [PubMed] [Google Scholar]
- 95.Kalbasi A, Kamrava M, Chu FI, et al. A phase II trial of 5-day neoadjuvant radiation therapy for patients with high-risk primary soft tissue sarcoma. Clin Cancer Res. Epub 2020. February 13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Guadagnolo BA, Zagars GK, Ballo MT, et al. Excellent local control rates and distinctive patterns of failure in myxoid liposarcoma treated with conservation surgery and radiotherapy. Int J Radiat Oncol Biol Phys. 2008;70:760–765. [DOI] [PubMed] [Google Scholar]