

Abstract
ER/K α-helices are a subset of single alpha helical (SAH) domains, which exhibit unusual stability as isolated protein secondary structures. They adopt an elongated structural conformation, while regulating the frequency of interactions between proteins or polypeptides fused to their ends. Here we review recent advances on the structure, stability and function of ER/K α-helices as linkers (ER/K linkers) in native proteins. We describe methodological considerations in the molecular cloning, protein expression and measurement of interaction strengths, using sensors incorporating ER/K linkers. We highlight biological insights obtained over the last decade by leveraging distinct biophysical features of ER/K-linked sensors. We conclude with the outlook for the use of ER/K linkers in the selective modulation of dynamic cellular interactions.
Keywords: Single alpha helix (SAH), ER/K linker, Protein-protein interactions, FRET biosensors, signaling, GPCR, kinases
Structure, stability and functional characteristics of ER/K linkers
Seminal characterization of ER/K linker structure and function have been previously reviewed in Swanson et al. (Swanson and Sivaramakrishnan, 2014). This section highlights the significance of protein α-helices in folded proteins, followed by recent advances in our structural understanding of Single Alpha Helical (SAH) domains, resources that have emerged to identify SAH domains in native proteins, and concludes with three current studies that feature unique functional characteristics of ER/K linkers in cellular proteins.
α-helices are a dominant structural element in proteins –
α-helices, β-sheets and random coils are the most common elements of secondary structure in proteins. α-helices are formed and maintained by backbone interactions parallel to the primary axis of the helix. These interactions are hydrogen bonds between the carbonyl oxygen and amino nitrogen of the ith and i+4th amino acids. The side chains of all residues in the α-helix are directed outwards and away from the helical axis, and the occurrence of polar or charged side chains in the helix can facilitate additional interactions with other side chains in the helix or with other elements outside of the helical structure, imparting further stability (Pauling et al., 1951). Consequently, α-helices are the most commonly occurring secondary structure, representing 30% of the structure of the average globular protein (Pace and Scholtz, 1998). In globular domains, α-helices can also pack with β-sheets in different arrangements. Among these, orientation of the α-helix along the strands of β-sheets is energetically most favored, followed in stability by a perpendicular orientation of the helical axis to the β-strands (Chou et al., 1985). α-helices are also the core component of coiled-coil domains and of transmembrane bundles. Coiled-coil domains consist of two to seven α-helices, where the separate helices stabilize each other through hydrophobic patches of side-chain interactions following a “knobs-into-holes” motif (Crick, 1953). Multiple α-helices composed primarily of hydrophobic amino acids can also give rise to transmembrane bundles, through protein-protein and protein-lipid interactions (Lee, 2003).
Stability of a protein α-helix –
The stability of a protein α-helix is affected by the number of amino acids involved in backbone interactions and the helical propensity of each amino acid. The helix propensity (Pα) is denoted on a scale of 0 to 1 and expresses the tendency of an amino acid to occur in an α-helix. Not considering the ends of a helix, Alanine residues have the highest propensity of occurrence in the central, solvent exposed residues, and Glycine residues have the lowest helix propensity, while Proline residues tend to disrupt helices (Pace and Scholtz, 1998). Intra-helical interactions between side-chains also contribute to the helical state and their complete absence destabilizes the helix (Zangi, 2014). One extreme of the stabilization of an α-helix is a scenario in which the strength and number of intra-helical interactions are sufficient to maintain the helical state of a peptide, even when isolated from the tertiary folded context of a protein i.e., interactions with other secondary structural elements are not required to stabilize the α-helix. The C-peptide of ribonuclease A was observed to form such an α-helix when isolated in aqueous solution (Bierzynski et al., 1982). Intra-helical stabilization through side-chain interactions was tested by engineering Alanine-based peptides of 16 or 17 residues, containing three pairs of Glu-Lys (Marqusee and Baldwin, 1987). Based on the spacing between the Glu and Lys residues, the peptides were designated as either i+4 or i+3. i+4 denotes that the Glu and Lys are separated by 3 residues (EAAAK), with the subsequent Glu following immediately (i.e. EAAAKEAAAK…); while in the i+3 peptides, the Glu and Lys were separated by 2 residues with one Ala residue separating the subsequent Glu (EAAKA)3. This study revealed that the (i, i+4) spacing (i.e.(EAAAK)3) yielded significantly higher helicity as compared to (i, i+3) spacing ((EAAKA)3) between the Glu and Lys residues. It was hypothesized that the i+4 spacing provided better stability to the helix due to the Glu (E) and Lys (K) side chain rotamers (Ponder and Richards, 1987). Extending this observation, efforts were made to understand α-helix formation through charge interactions, independent of the greater helical propensity of Alanines (Lyu et al., 1992). 18-mer peptides were synthesized that contained repeating units of either E4K4 or E2K2. Though both peptides had the same chemical composition, only the E4K4 peptide formed a stable α-helix (Lyu et al., 1992). The periodicity of a protein α-helix (3.5 residues per turn) would limit stabilizing electrostatic interactions between E and K to the E4K4 but not E2K2 peptide, consistent with the observations from (i, i+4) helices.
Collectively, these studies on the intrinsic stability of α-helices led to the identification of other peptides that exist as isolated stable single α-helices or SAH domains. The high content of charged and polar residues in these peptides provides additional stability to the helix, and (i, i+4) spacing between side chains pairs provides optimal helicity. Such stable peptides are enriched in Glu (E), Lys (K) and Arg (R) arranged in a manner that they exhibit salt bridges between side-chains at the i, i+3 and i, i+4 position, in addition to the backbone hydrogen bonds. These intra-helix salt bridges provide additional stability allowing the SAH peptides to retain their secondary structure without relying on interactions with other regions of a larger protein structure (Scholtz and Baldwin, 1992). Even peptides as short as five to eight residues with i, i+3 and i, i+4 salt bridges were shown to exhibit high helicity (Sommese et al., 2010). Investigations into the differences in contributions of Glu-Lys and Glu-Arg pairs to α-helix stabilization were performed with short (Sommese et al., 2010) and long peptides (Wolny et al., 2017). Among the short peptides, E4R4 peptides were found to have higher helix content than E4K4 peptides (Sommese et al., 2010). More recently, a 98 residue polypeptide was created that was composed of repeating units of the 7-residue sequence AEEEXXX, where X is K or R. Only the Lys-rich sequences EK3 (AEEEKKK) or EK2R1 (AEEEKRK) were found to form a SAH in solution. Increasing the Arg content in the peptide (EK1R2 – AEEERKR, or ER3 – AEEERRR) resulted in aggregation of the synthesized domain. Molecular modeling and PDB analysis indicated that Glu and Arg were more dynamic than Glu-Lys pairs (Wolny et al., 2017). As a result, a greater fraction of the charged side-chains were involved in intra-molecular salt bridges, which dramatically reduced polar interactions between the peptide and water molecules. These factors combined to limit the solubility of the Glu-Arg peptides.
Similar SAH domains occur naturally in a variety of proteins, often as linkers of disparate domains (Peckham, 2011; Swanson and Sivaramakrishnan, 2014). To distinguish SAH domains in general from those that contain primarily E, R and K residues, the term ER/K α-helix was introduced (Sivaramakrishnan et al., 2008) and it is referred to as an ER/K linker when it is fused to polypeptides at both of its ends (Sivaramakrishnan et al., 2008). The relevance of Lys and Arg content is reflected in the sequences of native ER/K linkers, which contain either Lys or a mixture of Arg and Lys (Sivaraj Sivaramakrishnan and Spudich, 2011). Consistent with the increase in stability arising from Glu-Arg pairs, K→R substitutions in the naturally occurring myosin VI SAH, which constitutes an ER/K α-helix, led to an increase in thermostability (Wolny et al., 2017). Overall, increasing the R:K ratio of ER/K linkers enhances helix stability while potentially compromising protein function through aggregation.
Further experimental insights into the contribution of Arg and Lys residues to the stability of native SAH domains emerged from a recent NMR study (Batchelor et al., 2019). The authors validated that residues 858–935 of murine Myosin 7a (M7a) indeed formed a SAH in solution, as was originally predicted by Yang et al. (Yang et al., 2009). NMR experiments were used to understand the nature of bonds that contributed to conformational stability, and very few “through” hydrogen bond couplings were detected, indicating that the M7a SAH domain is stabilized either by a dynamic network of ion pairs, or by a preference for weaker, solvent-bridged ion pairs on the outside of the helix. Molecular dynamics (MD simulations) were used to test these mutually exclusive possibilities. The simulations revealed that each Arg residue had the ability to interact with a larger network of Glu side chains than a corresponding Lys residue. For example, in a representative sequence ExxRxxEE starting at the ith position, the central Arg residue (i+3) can form simultaneous bonds with Glu residues at both the i+6 and i+7 positions. The same Arg residue can dynamically alter its rotamer conformation to form simultaneous bonds with Glu at i and i+7 positions. This dynamic flexibility of the ion pair network arising from Glu-Arg interactions significantly enhances the stability of the SAH domain. Lys residues do not display this dynamic stability in their ion pair network, contributing to the stability of ER/K α-helices to a lesser extent.
Identifying SAH domains –
SAH domains have very distinct properties compared to other α-helices such as coiled-coil repeats. The functional differences between SAH domains and other α-helices can significantly affect interpretation of protein function. Hence, for biologists attempting functional characterization of a protein of interest, a quick check for the presence of SAH domains would be useful. However, many structural analysis tools that provide protein domain analysis misidentify SAH domains as either coiled-coil sequences or as being related to other protein families. For example, the MOTIF tool (https://www.genome.jp/tools-bin/search_motif_lib) indicates that the SAH domain of Myosin VI is related to the Macoilin family of transmembrane proteins. Two in silico tools – CSAH – for charged single alpha helices (Süveges et al., 2009) and Waggawagga-CLI (Simm and Kollmar, 2018) – have been created with the aim of predicting the propensity of an amino acid sequence to form SAH domains.
CSAH is applied to amino acid sequences to predict the length and probability that a contiguous stretch of amino acids could form a single α-helical segment. There are two methods available for this tool, SCAN4CSAH and FT_CHARGE. SCAN4CSAH detects ionic interactions between the ith and i+3rd residues, and the ith and i+4th residues to score the sequence for α-helix stabilization. The second method used by CSAH is FT_CHARGE, in which Fourier transformation is applied to the ionic state of individual side chains in the sequence to identify repeated charge patterns, separated by seven residues. Quality control for these methods involves checking the Chou-Fasman Helical propensity parameters (Pα) to eliminate hits that are unlikely to form α-helices. This tool has been provided as a web server (http://csahserver.itk.ppke.hu/cgi-bin/csahserver.cgi) that predicts and identifies SAH-containing proteins and analyzes full UniProt submissions to update the repertoire of proteins containing SAH domains on a monthly basis (Gáspári et al., 2012).
A second tool, Waggawagga-CLI provides a genome-wide prediction of SAH-containing proteins (Simm and Kollmar, 2018). This method first uses the helical net diagram which represents the peptide sequence and residue level interactions in a 2-dimensional format, along with the helical axis (Dunnill, 1968). Next, the interactions between residues are drawn and their effects are calculated. Three types of interactions are considered (1) binary interactions between residues at the (i, i+3) and (i, i+4) positions, (2) occurrence of pairs of hydrophobic amino acids in the (i+3, i+6), (i+3, i+7), (i+4, i+7) and (i+4, i+8) positions and (3) occurrence of pairs of oppositely charged amino acids at the positions listed above. The binary interactions are classified as having stabilizing, α-helix supporting or destabilizing effects. The stabilizing interactions are further differentiated as being strong, medium, or weak. The interactions are scored for defined window sizes, summed, and normalized to an ideal SAH sequence (e.g. EEEKKK). A default score is set as the cut-off to determine the likelihood of any amino acid being part of the SAH domain. For the entire SAH domain composed of multiple such windows, three criteria are evaluated to determine the possibility of SAH formation - (1) a sequence longer than 14 amino acids, (2) at least 80% amino acids have SAH score more than the cut-off and (3) amino acids at the beginning and end must have an SAH score greater than the cut-off value. This approach differs from the SCAN4CSAH algorithm in that it does not consider destabilizing interactions between identically charged residues in i+3, i+4 positions and between oppositely charged amino acids at i, i+1, i+2 positions. The Waggawagga-CLI approach has increased the frequency of detection of SAH domains from 0.2% by CSAH to between 0.5 and 3.5% of the total proteins in the genomes of different organisms. However, experimental validation of these computationally detected SAH domains will be necessary to compare the accuracy of the two methods.
Functional significance of SAH domains in native proteins –
Characterization of the SAH domains of muscle caldesmon, apoptotic protein PDCD5, ribosomal protein L9 and myosin VI have been discussed previously (Swanson and Sivaramakrishnan, 2014). Subsequently, SAH domains have been identified in other proteins including Rpn12 subunit of the proteasome lid (Tomko et al., 2015), MFAP1 and Snu23 proteins that interact with Prp38 (Ulrich et al., 2016), Myosin X (Wolny et al., 2014), inner centromere protein INCENP (Samejima et al., 2015), and the Gag protein of Murine Leukemia Virus (MLV) (Doležal et al., 2016). Here we summarize the findings of mechanosensitivity of the SAH domain in Myosin X and INCENP, and the rotational flexibility enabled by the Gag protein.
Myosin X was shown to contain an ER/K-rich SAH domain, linking the motor domain at the N-terminus of the protein to a coiled-coil, and a FERM domain at the C-terminus (Knight et al., 2005). A recent study by Wolny et al., provides experimental evidence that the SAH domain of myosin X behaves like a constant force spring, undergoing mechanical extension. A 97-residue peptide from myosin X was verified to be an SAH based on helicity, non-cooperative thermal unfolding and high efficiency of refolding (Wolny et al., 2014). Atomic force microscopy (AFM) was used to record the changes in the peptide upon application of a constant force. Using this technique, the SAH domain unfolded at forces less than 30 pN in a non-cooperative manner. Simulations suggested that the application of a deforming force leads to mechanical extension of the SAH domain at constant force. Further, simulations also indicated that the domain would refold when the force was removed and the structure allowed to shorten. In the cell, myosin X functions as dimer and plays a key role in the biogenesis of filopodia through actin convergence, contributing to the formation of characteristic parallel actin bundles observed in filopodia (He et al., 2017), and through cargo transport into the filopodia (Kerber and Cheney, 2011). Unfolding of the SAH domains would allow both myosin heads in the dimer to stay attached to separate actin filaments as they are being pulled apart during actin remodeling. In the absence of this feature, the force experienced during this actin reorganization could cause either the motor domain to unfold and detach, or the coiled-coil dimerization domain to unwind, dissociating the functional dimer. Physical extension of the SAH domain would also allow myosin X to overcome physical obstructions introduced by actin cross-linking proteins during cargo transport along parallel actin bundles in the filopodia. The constant force spring characteristic identified for the myosin X SAH domain is likely to be relevant for other ER/K-linkers in mechanosensitive cytoskeletal proteins.
The SAH domain of INCENP regulates Aurora-B kinase activation during mitosis. INCENP and Aurora-B function together in the Chromosomal Passenger Complex (CPC), which regulates mitosis by removing improper attachments at the kinetochore, and also by activating the spindle assembly checkpoint (Carmena et al., 2012). A central domain of INCENP (503–715) was previously suggested to form a coiled-coil (Mackay et al., 1993). However, coiled-coil formation in INCENP would enforce Aurora-B dimerization, leading to unregulated kinase activation through transphosphorylation (Sessa et al., 2005). Further, given the elongated structure of INCENP, the rigidity of a coiled-coil would restrict the ability of Aurora-B kinase to access its substrates in the outer kinetochore. These disagreements were resolved by a recent study demonstrating that the putative coiled-coil in INCENP is in fact a SAH domain (Samejima et al., 2015). The monomeric nature of the SAH domain would not lead to spontaneous formation of Aurora-B dimers. Further, the N-terminus of the INCENP SAH domain binds to microtubules, and the authors hypothesize that forces arising from microtubule – kinetochore interaction could cause mechanical unfolding of the SAH domain. This would allow Aurora-B to access substrates in the outer kinetochore, further away from its CPC tethering site. Thus, the presence of a SAH domain in the central region of INCENP prevents the formation of obligate Aurora-B dimers, and indicates a mechanism for force/tension-dependent phosphorylation of substrates in the outer kinetochore. However, the factors that limit transphosphorylation of Aurora-B following the proximity induced recruitment to microtubule-bound INCENP-SAH remain unclear.
In select retroviruses, a SAH domain is critical for virion assembly, where its rotational flexibility facilitates the conformational changes associated with capsid maturation. The retroviral genome of murine leukemia virus (MLV) is enclosed by nucleocapsid domains (NC), which are connected to the capsid domain (CA) by a stretch of amino acids termed the charged assembly helix (CAH) (Cheslock et al., 2003). The structure-function relationships between these domains are critical for successful viral assembly, since deletion of CAH drastically reduces the number of assembled virions. A previous model assumed that the CAH would assemble as a coiled-coil and facilitate virus assembly. However, this model does not explain critical features in the maturation of the MLV capsid. In MLV, the interfaces of CA domains are largely constant between the immature and mature capsids (Qu et al., 2018). Hence, the assumption that the CAH segment constitutes a coiled-coil was evaluated through biophysical characterization by Doležal et al. This region of 41 amino acids exhibits helicity, thermal unfolding and refolding, and conformation consistent with a SAH, while remaining monomeric in solution (Doležal et al., 2016). A coiled-coil conformation of the CAH would have greater rigidity whereas a SAH linking CA and NC domains is theorized to facilitate rotation of the protein domains enabling capsid maturation.
ER/K-linked sensors – Mechanism, molecular cloning, protein expression, and measurement of interaction strengths
A subset of SAH domains contain a characteristic motif, which comprises approximately four glutamic acid residues (E4) followed by approximately four basic residues that are either arginine or lysine (R/K)4. A repeating sequence of this E4(R/K)4 motif creates a SAH termed an ER/K-helix. Naturally occurring ER/K helices of known length have been repurposed to tether two protein/polypeptide domains together (Sivaramakrishnan and Spudich, 2011). In this context, the ER/K helix is termed the ER/K linker. In this section, we describe the use of ER/K linkers to detect and modulate existing protein-protein interactions through the use of engineered sensors.
Sensor mechanism –
The ER/K α-helix functions as a worm-like chain (WLC) with rare, stochastic breaks proportional to helix length (Sivaramakrishnan et al., 2008; Sivaramakrishnan and Spudich, 2011). Initial biophysical characterization of the ER/K α-helix using circular dichroism and small-angle x-ray scattering (SAXS) revealed an elongated structure consistent with an isolated, stable, extended α-helix (Sivaramakrishnan et al., 2008). Subsequent single molecule optical tweezers measurements, combined with biophysical modeling suggested that the ER/K α-helix behaves like a worm-like chain (WLC) with persistence length (LP ~ 15 nm) (Sivaramakrishnan et al., 2009). For an ideal WLC with lengths comparable to LP the probability of proteins at the ends interacting with each other is negligible (probability ~ 10−8) (Mehraeen et al., 2008). Consequently, if ER/K linkers of length 10–30 nm behaved as an ideal WLC, it would function simply as a spacer for proteins/peptides at its ends. However, tethering a well-characterized interacting pair (CaM-peptide) at the ends of ER/K α-helix demonstrated a robust calcium-dependent interaction as measured by a fluorophore FRET pair fused to the interacting proteins (eCFP-CaM; Citrine-C15W CaM interacting peptide) (Figure 1A, B). Control constructs that lacked the peptide displayed insignificant FRET regardless of Ca2+ concentration. The dependence of FRET upon an interaction between the moieties at the ends of the linker is consistent with linker lengths substantially greater than the Förster’s radius of the FRET pair (Ro for CFP/YFP ~ 5 nm). Hence, while the ER/K linker does function as a spacer, it can also facilitate protein-protein interactions with proteins/peptides tethered to its ends. Given this divergence from an ideal WLC, it was proposed that rare stochastic breaks in the elongated α-helix could enable interactions. Regardless of underlying mechanism, subsequent studies have repeatedly demonstrated the ability of the ER/K linker to facilitate interactions.
Figure 1 – ER/K linkers modulate the effective concentration of protein-protein interactions.
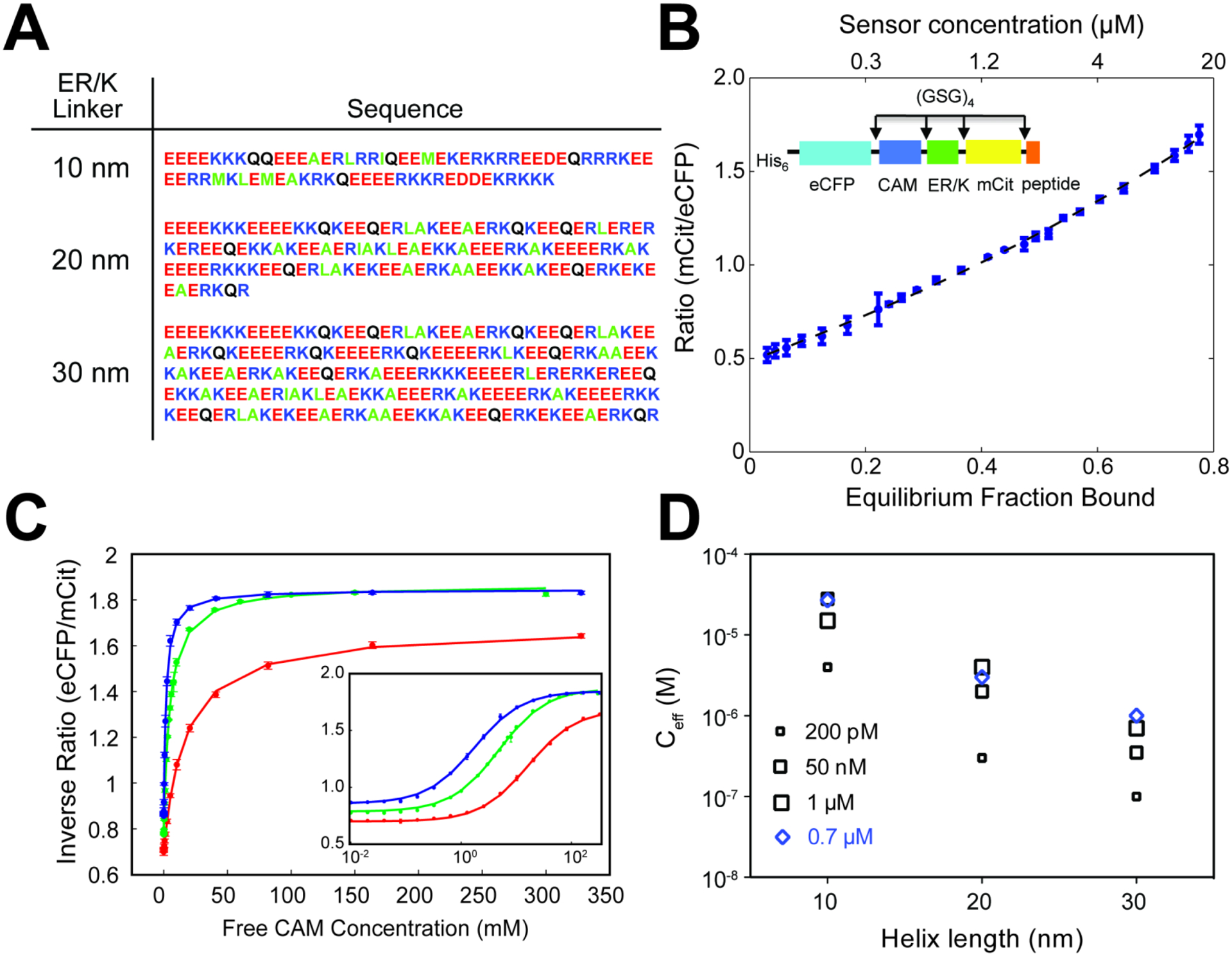
A) Sequence of 10, 20 and 30 nm linkers used in ER/K-linked sensors.
B) FRET ratio (mCit acceptor/eCFP donor) of ER/K-linked CaM-peptide sensor correlates linearly with fraction of sensors with proteins in the bound state.
C, D) Effective concentrations (Ceff) engineered by 10, 20 and 30 nm ER/K linkers determined from (C) concentration-dependent quenching of FRET by unlabeled, free CaM protein. (D) Ceff for CaM interaction with peptides of indicated binding affinity (KD listed) and 10, 20 and 30 nm ER/K linker length.
For all experiments: N = 4, and data are shown as mean ± SD.
Panels A-D adapted from Sivaramakrishnan and Spudich, Systematic control of protein interaction using a modular ER/K α-helix linker. PNAS 2011. 108(51):20467–20472.
To understand the relationship between FRET ratio and the interaction state of proteins at the ends of the ER/K linker, the interacting proteins and their associated fluorophores (eCFP-CaM; Citrine-C15W) were separated by TEV protease treatment targeting a TEV-site engineered at the N-terminus of the ER/K linker (Sivaramakrishnan and Spudich, 2011). A dilution series of the TEV-cleaved sensor was used to measure the concentration dependence of FRET ratio of the equivalent bi-molecular interaction (Figure 1B). Fitting this concentration-dependent measurement to the classic quadratic equation of bi-molecular association yields a linear relationship between sensor FRET ratio and the fraction of protein in the bound state (Figure 1B). Given that the elongated ER/K linker maintains minimal FRET in the absence of an interaction, these measurements demonstrate that the sensor FRET ratio is a surrogate for the fraction of sensors with the flanking protein/peptides in the interacting state.
To assess the effective concentration engineered by the ER/K linkers, increasing concentrations of unlabeled CaM were added to quench sensor FRET in a dose-dependent manner (Figure 1C). Given the linear relationship between sensor FRET ratio and fraction of protein in the interacting state, these dose-response curves were sufficient to measure the effective concentration (Ceff) of sensors engineered with different length ER/K linkers and CaM interacting peptides with varying affinity (Figure 1D). Regardless of the CaM-peptide interaction affinity, increasing ER/K linker length systematically decreased Ceff, from ~ 10 μM for the 10 nm linker, to ~1 μM for the 20 nm linker, and ~ 100 nM for the 30 nm linker. To dissect the role of the ER/K linker on the kinetics of the interaction (on versus off rates), the rate of dissociation of a high affinity peptide (Trp3) from CaM was measured by quenching the interaction with a vast excess of unlabeled CaM (300 μM >> Ceff). Sensors displayed very similar dissociation rates, regardless of ER/K linker length, demonstrating that the linker controls association (kon) rather than dissociation (koff) rates (Sivaramakrishnan and Spudich, 2011). While the interacting proteins have distinct shapes and sizes, resulting in distinct configurations of interactions, 12 residue-long GSG linkers were included between all protein domains (including between the fluorophores and ER/K linker) to allow rotational flexibility. Consequently, the limiting factor for the protein-protein interaction are the structural properties of the ER/K linker. Extensive biophysical characterization has established that the ensemble conformation of the ER/K α-helix is consistent with a worm-like chain with persistence length (Lp) ~ 15 nm (Sivaramakrishnan et al., 2009). For ER/K linkers with lengths comparable to Lp, the ER/K linker makes an effective spacer between proteins tethered at its ends. However, infrequent stochastic breaks in the ER/K α-helix can also facilitate an interaction between proteins tethered to its ends. Given the relative homogeneity of ER/K linker sequence (E4 – R4 / K4), the frequency of breaks is proportional to linker length, whereas the probability that the ends will be within 2 nm distance of each other, and thus allow an interaction, is inversely proportional to the square of the linker length, as the breaks are unlikely to be spatially coordinated. These factors together yield the observed inverse relationship between kon and Ceff and linker length (Figure 1D). Further, given that the ER/K linker length is significantly larger than most protein domains/polypeptides (> 10 nm compared to ~ 3 nm for a 66 kDa globular protein such as Bovine Serum Albumin), the linker length dependence of association rates and consequently effective concentrations is likely to persist across protein-protein interactions.
Sensor plasmid design –
ER/K-linked sensors are created by cloning distinct protein domains/polypeptide components into sequential sites of the multiple-cloning-site (MCS) of different vectors. The diversity of plasmid constructs available enables rapid generation of sensor plasmids for new protein-protein interactions using different expression systems. A range of sensor plasmids generated by the Protein Acrobatics Lab, along with detailed plasmid restriction maps are available through AddGene (https://www.addgene.org/Sivaraj_Sivaramakrishnan/). In all layouts of ER/K-linked sensors, the individual components are all separated from each other by 2–4 (Gly-Ser-Gly) linker segments. These short linkers impart rotational flexibility, distinct from the distance constraints programmed by the ER/K linker. This rotational flexibility is designed to allow the proteins tethered at either end of the linker to interact without being sterically limited by the other components.
Within the broad framework of sensor designs with the ER/K linker in the middle, flanked by a peptide-fluorophore combination on either side, multiple variations have been reported. In the earliest work that reported development of an ER/K-linked sensor, the ER/K linker was cloned between EcoRI and AscI, with an N-terminal TEV protease site (ENFLYQG/S) (Sivaramakrishnan and Spudich, 2011). Treatment with bacterially expressed TEV protease generates the equivalent bi-molecular interaction through selective proteolytic cleavage at the protease site. The TEV protease site can be used to compare FRET between the unimolecular context of the ER/K-linked sensor and the bi-molecular protein-protein interaction following TEV treatment. It can also be used as a purification tool, especially in context of catalytically active enzymes, whose activity and consequently expression are actively suppressed. For instance, the isolated kinase domain of constitutively active kinases such as PKCs express poorly by themselves, and are therefore expressed as part of an ER/K-linked sensor linked to a substrate peptide or pseudosubstrate domain (Sommese and Sivaramakrishnan, 2016). Following expression as a tandem protein, TEV treatment is used to cleave, separate, and purify the kinase domain for biochemical applications.
The repeating nature of the ER/K sequence makes plasmid DNA constructs susceptible to recombination during plasmid amplification in E. coli. Recombination deficient bacterial strains (Stbl2 and JM109) have been used selectively for DNA amplification, especially when plasmid recombination is detected in vector digests or DNA sequencing. To minimize recombination effects, most sensor plasmid clones have maintained the original EcoRI-AscI fragment of the ER/K linker with the N-terminal TEV site. Rather than PCR amplify the ER/K linker, restriction digests have been used to laterally transfer this fragment to other plasmid vectors. Nonetheless, plasmid designs containing BamHI/AscI sites as well as NotI/AvrII sites have subsequently been generated to avoid the limitations posed by the presence of EcoRI in either the plasmid vector backbone or the polypeptides being studied. EcoRI is a 6-cutter and a widely used restriction enzyme, whereas AscI is an 8-cutter and is relatively infrequent.
N- and C-terminal protein fusions to an ER/K linker involve a diversity of multiple cloning sites (MCS) to accommodate both the interacting polypeptide and fluorophores. The restriction site layout in the original ER/K-linked sensors was subsequently used to create constructs linking a variety of protein domains as depicted in Figure 2 and outlined next. Sensors for monitoring the conformational state of Focal Adhesion Kinase (FAK) were created by including from N-to-C an mCitrine (mCit), the FERM domain of FAK, the ER/K linker, an mCerulean (mCer) and the catalytic domain of FAK, in both pBiex1 (insect cell expression) and pcDNA/FRT (mammalian cell) expression vectors (Ritt et al., 2013). An alternate design incorporated (from N-to-C) a GPCR between HindIII-XbaI, followed by mCit between XbaI and EcoRI. As in the previous case, the ER/K linker was cloned between EcoRI-AscI, followed by mCer between AscI-PacI, and finally the Gα subunit or a C-terminal peptide encompassing the α5 helix of the Gα subunit (α5-peptide) between PacI-NotI (Malik et al., 2013). In the GPCR sensors, both fluorophores flanked the ER/K linker. An alternate ‘fluorophores-outside’ design was adopted in sensors linking the PKC catalytic domain to substrate peptides (Sommese and Sivaramakrishnan, 2016). Yet another variation incorporates two copies of the FRET acceptor, one on each side of the peptide moiety at the C-terminus of the ER/K linker (Ritt and Sivaramakrishnan, 2016).
Figure 2-. Schematics of sensors utilizing ER/K linkers.
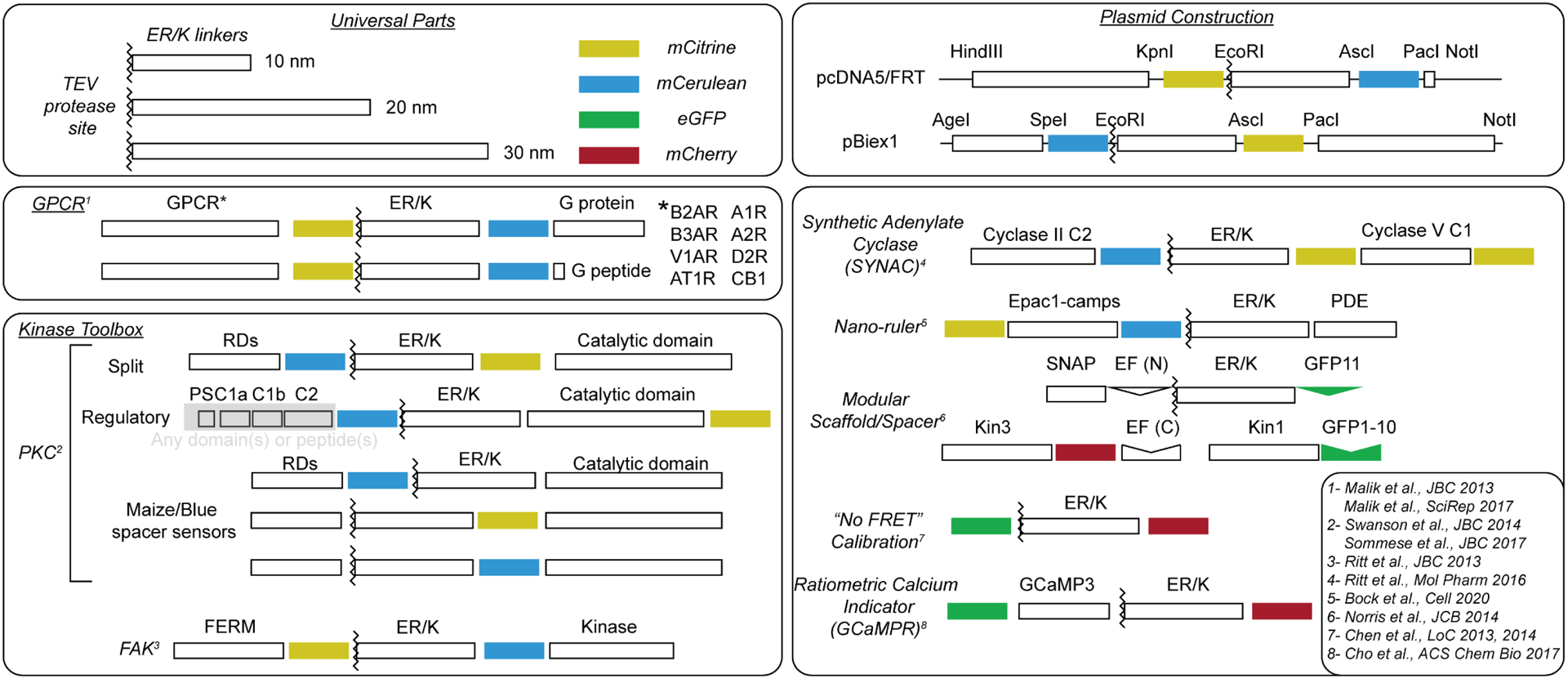
Generalized schematics of published sensor proteins that incorporate the ER/K linker. ER/K linkers have successfully been used to generate sensors employed to investigate cellular interactions involving GPCRs, kinases, motors, and second messenger signaling. ER/K-linked sensors are constructed so that the domains can be interchanged with relative ease during molecular cloning using restriction sites between protein elements (top right). Select citations for each sensor (superscript numbering) are also listed (bottom right).
Sensor expression and purification from bacterial and insect cells –
ER/K-linked sensors have been engineered in commonly used vectors for protein expression and purification including pET47b and pBiex1 that allow for expression and purification from bacteria and Sf9 cells. For purification purposes, the original ER/K linker based Ca2+ sensors were cloned with an N-terminal hexaHistidine-tag and purified using Ni-NTA chromatography (Sivaramakrishnan and Spudich, 2011). Ni-NTA purification requires multiple chromatography steps, given the non-specific binding of proteins containing more than one histidine to the Ni-NTA column (Bornhorst and Falke, 2000). The consequent trade-off between yield and protein quality necessitates large expression titers afforded by bacterial expression. Nonetheless, limitations in protein folding and post-translational modifications require sensor expression in insect and mammalian culture system. In contrast, the FLAG purification strategy yields relatively homogenous sensor protein (> 75% assessed by Coomasie staining) with modest expression titres (30 mL Sf9 cultures). A 30 mL culture of Sf9 cells, containing 3 – 5 × 107 cells at harvest can provide on average ~ 100 μL of 1 μM protein (yields are limited by the interacting polypeptides rather than the ER/K linker or fluorophores). Further, the high specificity of the anti-FLAG antibody compared to Ni-NTA provides relatively pure and intact protein (> 80% as assessed by fluorescence gel scans) in a single step, without the use of chromatography equipment. A fused FLAG-hexaHis sequence allows purification with FLAG and antibody labeling or detection with hexaHis (Hariadi et al., 2014). Both N-terminal (Ritt et al., 2013) and C-terminal (Ritt and Sivaramakrishnan, 2016) FLAG-tags have been used, with equal efficacy for purifying ER/K-linked sensors.
Sensor plasmids are transiently expressed in Sf9 cells using Escort IV transfection reagent (Sigma). Individual FRET measurements in a fluorimeter (Fluoromax 4; Horiba) require 50 μL of 5 – 50 nM sensor, when used in conjunction with quartz microcuvettes. Thus, one 30 mL culture is sufficient for ~ 40 – 400 individual readings. Alternately, fluorescence spectra can be measured in a plate reader format, which allows more conditions to be sampled in a shorter time, and with greater consistency (Lee et al., 2017). However, this method typically requires 50 μL sample per well at a concentration of 100 nM due to the relatively lower sensitivity of most plate reader instrumentation compared to individual cuvette measurements.
Sensor expression in mammalian cells –
For experiments involving the use of ER/K-linked sensors in mammalian cells, pcDNA5/FRT plasmids containing the mammalian pCMV promoter were used (Malik et al., 2013; Ritt et al., 2013). Fugene HD (Promega) and XtremeGene HP (Roche) were used in early studies to promote high levels of expression that can be tuned by a combination of expression time and plasmid/transfection reagent titers (Malik et al., 2013). Appropriate cellular localization of the sensors is sensitive to many variables including cell passage number, length of expression and level of sensor expression. Assessing sensor localization is especially important when dealing with GPCR signaling with extracellular water-soluble ligand application (Malik et al., 2013) and also in monitoring changes in sensor relocalization in response to stimuli (Sommese et al., 2017). GPCR sensors with α5 peptides derived from distinct Gα subunits displayed a wide variation in optimal expression time required to obtain significant expression, while maintaining plasma membrane localization (assessed by fluorescence microscopy of live cells). The variations in optimal expression times dictated that experiments be set-up at multiple overlapping time points, with subsequent selection of matched expression levels prior to experiments. As a cost-effective alternative to expensive transfection reagents, polyethyleneimine (PEI) mediated transfection was successfully used with ER/K-linked sensors (Gupte et al., 2019). While this requires removal of PEI at 4 hr post-transfection, the use of PEI routinely provides >80% transfection efficiency (fraction of cells expressing sensor assessed by fluorescence microscopy) with appropriate membrane localization of ER/K-linked GPCR sensors.
Sensor measurements –
Fluorescence emission spectra are routinely used to calculate a FRET ratio. For commonly used pairs of FRET probes, eCFP-mCitrine and mCerulean-mCitrine, the donor excitation wavelength is set to 430 nm and an emission scan spanning 450 nm to 600 nm is obtained (Sivaramakrishnan and Spudich, 2011). The fluorescence intensity is noted at the emission maxima of the donor (475 nm, designated emDonorMax) and at the emission maxima of the acceptor (525 nm, emAcceptorMax). FRET ratio is the ratio of emAcceptorMax to emDonorMax. The FRET ratio has been shown to linearly correlate with the fraction of sensors in which the polypeptides flanking the linker are in the bound (interacting) state (Sivaramakrishnan and Spudich, 2011). Addition of one of the partners as a non-fluorescent variant leads to a loss of FRET and has been used to estimate the dissociation constant of the interaction (Sivaramakrishnan and Spudich, 2011). An alternate method relies on the use of time-resolved FRET or TCSPC (time correlated single photon counting), rather than steady-state FRET measurements. Based on light sources and spectral overlap, ER/K-linked sensors incorporating eGFP and mCherry, as FRET donor and acceptor, were employed with a pulsed diode laser and photomultiplier module to calculate the amplitude weighted lifetime <τFRET> and the efficiency of energy transfer as 1-<τFRET>/τeGFP (Sommese and Sivaramakrishnan, 2016).
Biological insights using ER/K-linked sensors
The application of ER/K-linked sensors has provided numerous insights into protein interactions underlying signal transduction. Here, we highlight distinct features of ER/K-linked sensors that have been used to circumvent the limitations of traditional biophysical and cell biological techniques.
Pairwise comparison of the relative strengths of weak interactions in live cells –
The application of ER/K-linked sensors in detecting G protein selective GPCR conformations and engineering differential levels of FAK activation have been reviewed previously (Swanson and Sivaramakrishnan, 2014). While the prototypical GPCR, β2-AR, is conventionally thought of as a Gs-coupled receptor, various results indicate the possibility of Gi-coupling as well (Daaka et al., 1997; Pierce et al., 2002). It was therefore suggested that β2-AR could adopt multiple conformations, including a Gi-selective conformation (Kobilka, 2011). The α5 helix of the Gα subunit captures the majority of structural contacts (~ 75%) between receptor and G protein, and its engagement is essential for selective G protein activation (Chung et al., 2011). However, the receptor-Gα peptide interaction is inherently weak (KD > 10 μM) and transient (Gupte et al., 2019), and receptor conformation is influenced by the lipid microenvironment in live cells (Bruzzese et al., 2018; Strohman et al., 2019). Hence, control over effective concentration provided by the ER/K linker, enabled pair-wise comparison of the strength of the interaction of β2-AR with native peptides derived from α5 helices of distinct Gα subunits. As expected, the beta-blocker metoprolol diminished the β2-AR-Gαs peptide interaction (Figure 3A, B), consistent with its previously reported role as an inverse agonist (Galandrin and Bouvier, 2006). In contrast, metoprolol, but not the full agonist isoproterenol, stimulated the β2-AR-Gαi peptide interaction. Hence, the use of an ER/K-linked sensor provided support for an inverse agonist at β2-AR inducing/stabilizing a Gi-selective conformation (Malik et al., 2013).
Figure 3-. GPCR biology investigated through the use of ER/K linkers.
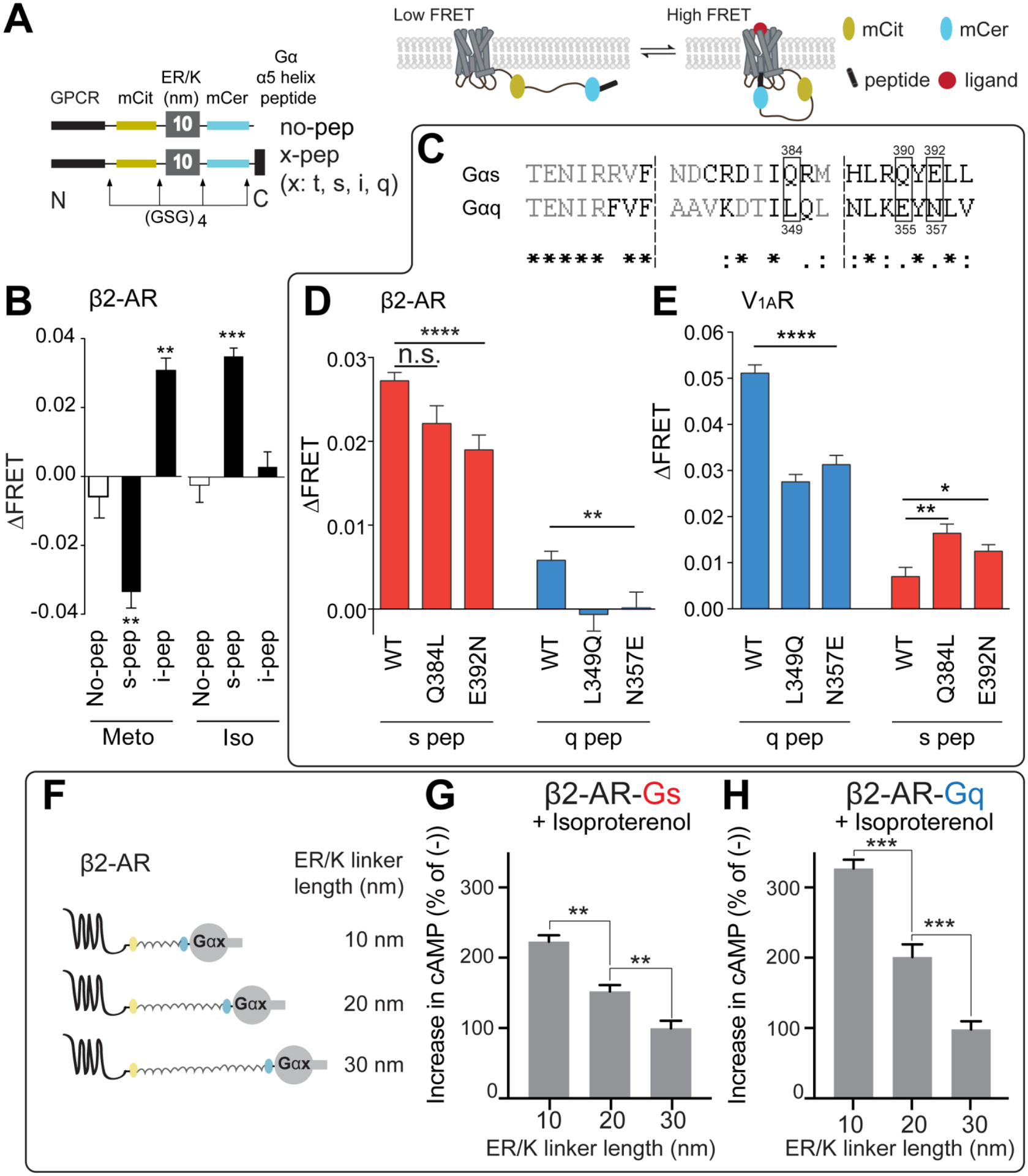
A) Detailed schematic design of GPCR-Gα α5-helix peptide (Gα peptide) sensors (left). Cartoon rendering of GPCR-Gα peptide sensor interactions (right).
B) Metoprolol stimulation of the indicated GPCR-Gα peptide sensors. Metoprolol (Meto), an inverse agonist of β1-AR signaling in the heart, suppresses the β2-AR-Gαs peptide interaction while stimulating interaction between β2-AR and Gαi peptide. In contrast, isoproterenol (Iso), a full agonist is shown to stimulate only the canonical β2-AR-Gαs peptide interaction.
C) Sequence alignment of Gαs and Gαq α5-helix, from which the Gα peptides are derived.
D,E) The indicated residues in panel C have been mutated to their counterpart residue from the other peptide. β2AR, is a Gαs coupled receptor (D) and shows diminished interaction with S-peptide mutated to look like Q-peptide. V1AR, a Gαq coupled receptor (E) shows enhanced interaction to S-peptide mutated to look like Q-peptide.
F) Cartoon rendering of GPCR-Gα protein sensors with different length linkers. These protein sensors have the full-length G protein instead of the Gα α5-helix alone.
G,H) β2-AR sensors tethered to either full length Gαs (G) or full length Gαq (H) with varying length ER/K linkers. Both sensors show decreases in cAMP production with increasing length of ER/K linkers.
For all experiments: N ≥ 3, *, p≤0.05; **, p≤0.01; ***, p≤0.001; ****, p≤0.0001; n.s. not significant. Significance determined using Student’s t-test or ANOVA with Tukey’s post-hoc test where appropriate.
Panels A (left) and B adapted from Malik et al., JBC 2013. This research was originally published in the Journal of Biological Chemistry. Detection of G Protein-selective G Protein-coupled Receptor (GPCR) Conformations in Live Cells. J Biol Chem. 2013; 288(24):17167–78. © the American Society for Biochemistry and Molecular Biology.
Panels A(right), and C-E adapted from Semack et al., JBC 2016. This research was originally published in the Journal of Biological Chemistry. Structural elements in the Gαs and Gαq C-termini that mediate selective GPCR signaling. J Biol Chem. 2016; 19;291(34):17929–40. © the American Society for Biochemistry and Molecular Biology
Panels F-H adapted from Gupte et al., Priming GPCR signaling through the synergistic effect of two G proteins. PNAS 2017. 114(14):3756–3761.
Identifying structural interaction hot-spots through site-directed-mutagenesis –
While most GPCRs signal primarily through a single cognate G protein, the mechanistic basis for selectively activating the cognate versus structurally similar non-cognate G proteins remains an outstanding challenge. A seminal crystal structure of β2-AR in complex with cognate Gs provided high-resolution information about the interface for this interaction (Rasmussen et al., 2011). More recently, high-resolution structures for other cognate GPCR-G protein interactions have emerged from a combination of x-ray crystallography and cryo-electron microscopy (García-Nafría and Tate, 2019). However, given that interactions with non-cognate G proteins are significantly weaker (Gupte et al., 2019), there are currently no high-resolution structures of non-cognate GPCR-G protein interactions. In the absence of such structural information, molecular dynamics (MD) simulations were used to gain comparative insights into a cognate Gs receptor, β2-AR and a cognate Gq receptor, V1aR. MD simulations identified distinct orientations of the α5 helix between cognate and non-cognate interactions. To gain insights into potential structural hot-spots that drive G protein selectivity, ER/K-linked sensors were used to examine the effects of switching hot-spots from cognate to non-cognate using site-directed mutagenesis (Sandhu et al., 2019; Semack et al., 2016). The pairwise control over effective concentration provided by the ER/K linker, revealed three hot-spot residues that are necessary and sufficient to tune selectivity between cognate and non-cognate G proteins (Figure 3C–E). While the α5 helix has been extensively reported as a determinant of G protein selectivity, the combination of MD simulations and ER/K-linked sensors was able to precisely identify a structural basis of this phenomenon.
Determine the cause-effect relationships in GPCR priming –
The cellular environment allows interactions of GPCRs with cognate G proteins, which are canonically activated by the ligand-bound GPCR. The GPCR can also interact with non-cognate G proteins, though the outcome of this weak, transient interaction is not known. Using an ER/K linker based sensor, we showed that facilitating non-cognate GPCR-G protein interactions paradoxically stimulated signaling downstream of the canonical pathway, a phenomenon that was termed GPCR priming (Gupte et al., 2017). However, GPCR networks are notorious for cross-talk, as diverse upstream activation events can converge on similar downstream factors. For example, activation of Gq and Gi pathways can result in an increase in cytosolic calcium. While the Gαq directly stimulates PLC-β, to influence inositol triphosphate (IP3) and IP3-receptor (IP3R), the Gi effect is mediated by the Gβγ dimer that functionally dissociates from its active Gαi counterpart, and interacts with IP3R (Zeng et al., 2003). Similarly, while the activation of Gα12/13 subunits exhibits cytoskeletal reorganization through Rho proteins, an increase in cAMP through Gαs activation can also lead to changes in cytoskeletal architecture (Ganguly et al., 2011). Hence, there was a possibility that the increase in cAMP observed from tethering non-cognate Gαq to β2-AR (i.e. GPCR priming), arose from the convolution of downstream events. To address potential cross-talk between signaling pathways, the ER/K linker-length dependence of effective concentration of the intra-molecular interaction was leveraged. Specifically, three different length linkers (10, 20 and 30 nm) were used to engineer apparent concentrations of ~ 10 μM, 1 μM, and 100 nM respectively, while maintaining the stoichiometry of expression of GPCR and G protein (Sivaramakrishnan and Spudich, 2011). While high apparent concentrations (~ 10 μM) resulted in enhanced cAMP upon tethering Gαq, compared to the Gαs subunit, low concentrations (~ 100 nM) resulted in comparable responses that are indistinguishable from β2-AR sensors without any Gα subunit (Figure 3F–H). Such experiments indicate that the modulation of downstream signaling results from an intra-molecular interaction between β2-AR and the tethered Gα subunit, and is not a consequence of the convolution of downstream signaling (Gupte et al., 2017).
Quantifying intra-molecular interactions in multi-domain proteins –
Classical PKCs (cPKC) are lipid and Ca2+-dependent AGC kinases that play critical roles in modulating a number of diverse physiological and pathophysiological processes, including cardiovascular performance and tumor generation (Newton, 2018). Despite their therapeutic significance, there are currently no isoform-specific inhibitors that can be used to selectively target disease states. The lack of isoform selectivity stems from inhibitor binding to a structurally conserved ATP-binding site within the kinase (Huse and Kuriyan, 2002). In contrast, biological modulation of kinase activity is often achieved through regulatory interactions. Multi-domain kinases, such as PKC have evolved intra-molecular regulatory domain interactions that are isoform-specific, with sequence-divergent binding interfaces (Steinberg, 2008). However, therapeutic targeting of these binding interfaces is limited by the knowledge of inter-domain interactions under basal and activated conditions. The kinase and regulatory domains are serially connected by flexible linkers that contribute to the conformational heterogeneity of the full-length protein, and have stymied efforts to obtain high-resolution structural information on inter-domain interactions. In the absence of structures of full-length PKCs, there has been significant confusion on the relative significance of intra-molecular regulatory interactions necessary to maintain basal auto-inhibition (Antal et al., 2015; Leonard et al., 2011; Sommese et al., 2017). Further, regulatory interactions can be interdependent and multiplexed, clouding interpretation of biochemical measurements of binary interactions between individual domains. Given these limitations, ER/K-linked sensors were used to probe the relative strengths of interactions between combinations of domains of PKCα under basal conditions (Sommese et al., 2017) (Figure 4A). Sensor measurements revealed that the C1a regulatory domain functioned cooperatively with the pseudosubstrate domain to suppress basal kinase activity. The use of ER/K-linked sensors provided the added advantage of enabling recombinant kinase expression for measurement of protein interactions. The kinase domain alone expresses poorly in insect cells, whereas tethering it to regulatory domains, in particular the pseudosubstrate, using the ER/K linker substantially enhanced protein expression (~ 100 nM versus > 2 μM from a 30 ml Sf9 culture). These ER/K-linked proteins were digested with TEV-protease to cleave an engineered protease site at the N-terminus of the ER/K linker, yielding a bi-molecular pair of kinase and regulatory domains fused respectively to a FRET acceptor and donor. Given the equal stoichiometry derived from the co-expression of the bi-molecular combination, measuring the FRET ratio in a serial dilution of the TEV digested fragments was sufficient to quantify an equilibrium dissociation constant for the kinase-regulatory domain combination (Sivaramakrishnan and Spudich, 2011; Sommese et al., 2017) (Figure 4B).
Figure 4 – Investigating PKC structure and function using ER/K linkers.
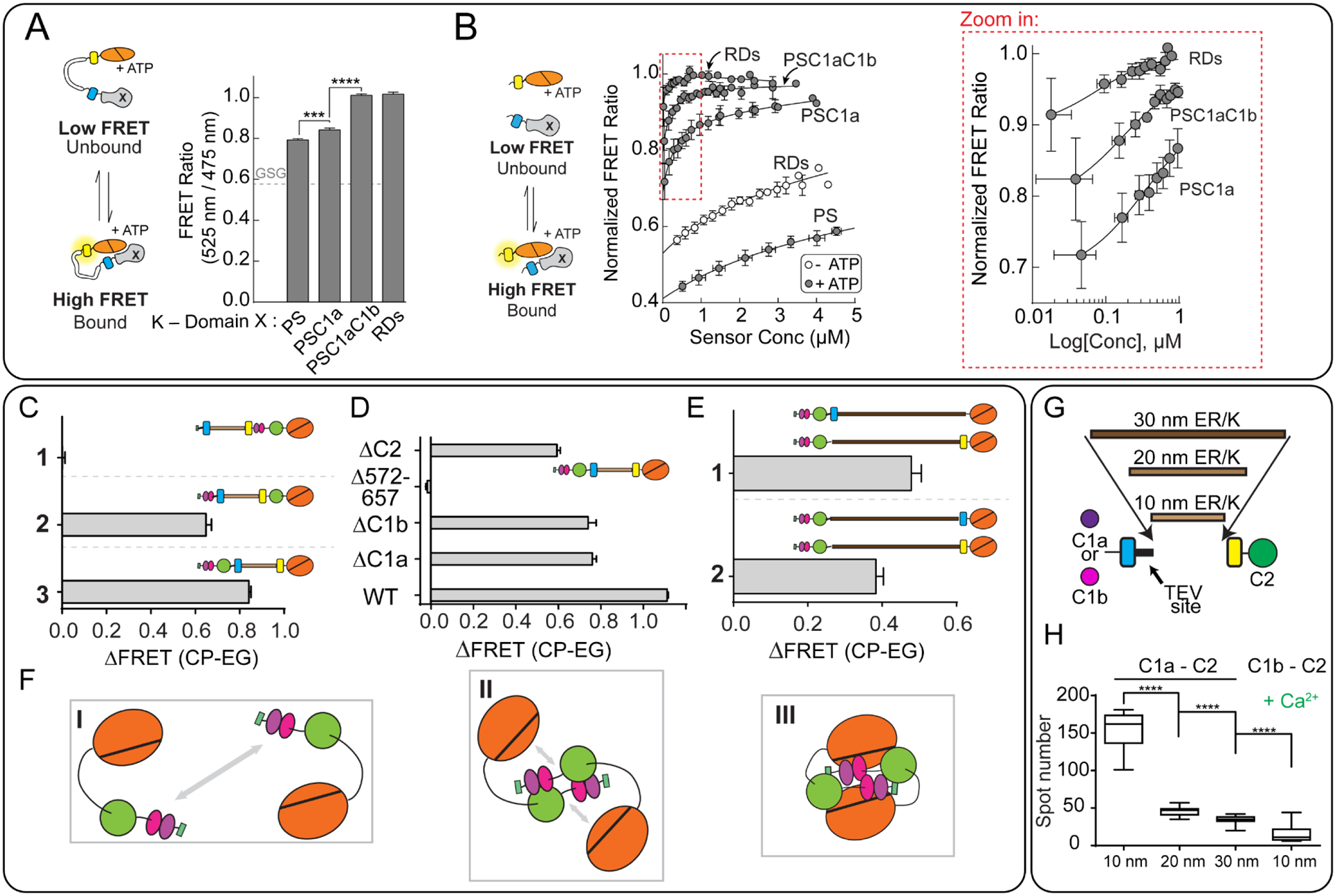
A) Cartoon diagram depicting interactions between regulatory domains and kinase domains when tethered with the ER/K linker (left). The observed FRET ratio for sensors with the kinase domain (K) and the indicated domains (right). PS – pseudosubtrate, C1a, C1b – diacylglycerol-sensitive regulatory domains, RDs – includes all regulatory domains (PS-C1a-C1b-C2).
B) Cartoon diagram depicting bi-molecular interactions between the regulatory domains and kinase domain derived by proteolytic cleavage of a TEV-protease site at the N-terminus of the ER/K linker (left). FRET ratio of the indicated pairings between regulatory domains and kinase domain (center). The region highlighted by the red box is depicted in the zoom-in on the right.
C-E) Change in FRET ratio in the indicated PKC sensors (ΔFRET) following activation. CP (calcium and PMA stimulation); EG (buffer containing EGTA).
C) By inserting the ER/K linker at various points in the protein, the domains that participate in the interaction can be discerned. 1- Sensor with pseudo-substrate separated from the regulatory domains. 2- pseudo-substrate and C1a/b separated from the C2 and kinase domains. 3- regulatory domains separated from the kinase domain.
D) PKC sensors with various truncations. The changes in ΔFRET following PKC activation indicated the relative importance of each excluded domain to the interaction. For instance, residues 572–657 that constitute the C-terminus of the kinase domain are essential to stimulate interactions upon PKC activation.
E) The change in FRET ratio for a pair of PKC sensors, each containing 30 nm linkers, and either an mCitrine or mCerulean fluorophore. By locating the fluorophores at opposite ends of the ER/K linker (1), a greater change in FRET is observed upon calcium and PMA stimulation than when they are located on the same side of the linker (2), indicating that the interaction between molecules likely involves close interaction between the regulatory and kinase domains on at least two separate proteins.
F) A proposed model for the interactions probed by the experiments in panels C-E
G) Diagram of the sensors used in panel H. These sensors contain either the C1a or C1b domains linked to C2 with varying length ER/K linkers.
H) Decrease in observed spots in a microscopy assay using the indicated sensors depicted in (G). C1a, but not C1b aggregates in response to calcium stimulation in a manner proportional to the length of the ER/K linker. For all experiments: N ≥ 3, **, p≤0.01; ****, p≤0.0001. Significance determined using ANOVA with Tukey’s post-hoc test.
Panels A and B adapted from Sommese et al., JBC 2017 This research was originally published in the Journal of Biological Chemistry. The role of regulatory domains in maintaining auto-inhibition in the multi-domain kinase PKCα. J Biol Chem. 2017; 292(7):2873–2880. © the American Society for Biochemistry and Molecular Biology
Panels C-F adapted from Swanson et al., JBC 2014 This research was originally published in the Journal of Biological Chemistry. Conserved modular domains team up to latch-open Active protein kinase Cα. J Biol Chem. 2014; 20;289(25):17812–29. © the American Society for Biochemistry and Molecular Biology
Panels G and H adapted from Swanson et al., PLoSone 2016
Mapping inter-molecular interactions in PKC oligomers –
Lipid/Ca2+ activation of PKCs results in their activation through membrane translocation. Studies using cross-linking, enzyme activity, and bi-molecular FRET sensors both in vitro and in live cells have shown that classical PKCs self-associate with high apparent affinity following activation (~ 5 nM) (Bonny et al., 2016; Slater et al., 2002; Swanson et al., 2014). Further, dynamic light scattering and fluorescence microscopy measurements revealed that they form distinct self-assembly states depending on the activating stimuli (Ca2+, DAG, PIP2) (Swanson et al., 2016). Macromolecular self-assembly has been observed for other signaling proteins including Ras (nano-clusters) (Tian et al., 2007) and Lck (Rossy et al., 2013). In each instance, the amorphous nature of self-assembly precludes the use of traditional high-resolution structural biology techniques to characterize inter-molecule interactions. Further, potential synergies between weak inter-domain interactions complicate interpretation of domain-deletion experiments. In this context, a toolbox of ER/K-linked sensors provided a coarse map of key inter-domain interactions that drive distinct assembly states of activated PKCα. PKCα contains five conserved domains (PS, C1a, C1b, C2, and kinase) separated by four variable linker domains (V1–V4) (Steinberg, 2008). An ER/K linker flanked by mCerulean and mCitrine fluorophores was inserted individually, after each of the first four conserved domains (Swanson et al., 2014). FRET ratio measurements of recombinant sensor protein, expressed and purified from Sf9 insect cells, reported on the strength of the interaction between combinations of domains flanking the ER/K linker. These combinations capture potential synergies between interacting domains, such as PS and C1a/C1b interacting with the kinase domain (Swanson et al., 2014) (Figure 4C). The use of fluorophores at both ends of the ER/K linker does not distinguish between intra- and inter-molecular interactions. Hence, bi-molecular pairs of sensors containing only mCerulean or mCitrine, either at the same or opposite ends of the ER/K-linker can be used to assess intra- vs inter-domain interactions in a protein oligomer. For instance, such a strategy revealed a folded-back interaction within an activated PKC oligomer (Swanson et al., 2014) (Figure 4D–F). Last but not least, varying ER/K linker length tunes intra-molecular but not inter-molecular interaction and distinguish their relative roles in the context of an oligomer. Varying ER/K linker length from 10–30 nm, systematically reduces the concentration of an intra-molecular C1a-C2 interaction and diminishes oligomerization assessed by the number of fluorescent C1a-C2 punctae visualized by fluorescence microscopy (Swanson et al., 2016) (Figure 4E, G). Taken together, these examples highlight features of ER/K-linked sensors that uniquely enable structural characterization of amorphous oligomeric assemblies.
Allosteric effects of small-molecule inhibitors on weak protein-protein interactions –
Small-molecule inhibitors of enzymes such as protein kinases often target the nucleotide-binding site. While high affinity competitive displacement of nucleotide effectively shuts down kinase activity, this targeting mode suffers from a lack of inhibitor-kinase selectivity due to the structural conservation of the kinase domain for efficient catalysis (Taylor and Kornev, 2011). Further, while each individual kinase phosphorylates a range of protein substrates, only those substrates responsible for disease-associated signaling should be targeted to avoid side effects from disrupting normal physiological functions. Kinases bind substrates though a kinase-specific recognition motif surrounding the phosphorylated residue (Miller and Turk, 2018). Hence, small-molecule inhibitors that disrupt the kinase-substrate interaction have the potential to enhance both kinase and pathway specificity. Nonetheless, the kinase-substrate interaction is inherently weak and transient and involves an intrinsically disordered peptide sequence on the substrate. Consequently, the kinase-substrate interaction is not easily accessible to biophysical approaches. In this context, the ER/K-linked sensors have provided a robust assay to profile the relative interaction strengths of distinct substrate peptides for PKCα. Measurements using ER/K-linked sensors demonstrated an inverse correlation between kinase-substrate interaction strength and specific activity (Sommese and Sivaramakrishnan, 2016) (Figure 5A, B). The sensors were also leveraged using site-directed mutagenesis to identify key interaction hot-spots on the substrate peptide that were predicted by molecular dynamics (MD) simulations (Lee et al., 2017). By integrating MD simulations and FRET measurements, we were able to delineate distinct mechanisms that dictate substrate binding strength and kinase activity.
Figure 5 – Substrate peptide-kinase interactions and their allosteric modulation by small molecules probed using ER/K linkers.
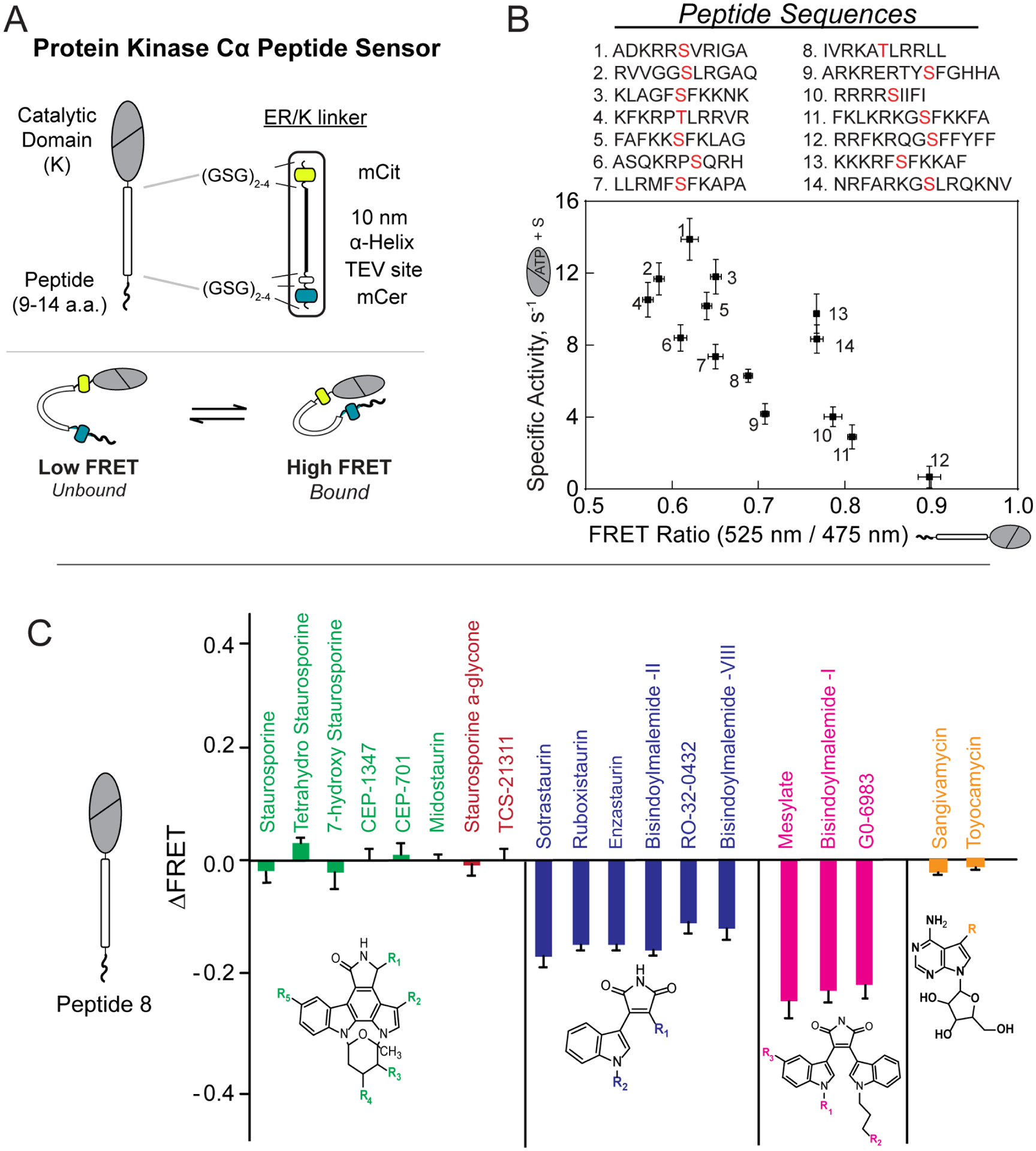
A) Cartoon schematic of PKC substrate peptide biosensor (top) and mechanism of action (bottom)
B) Correlation between kinase activity and FRET ratio observed for 14 different peptides derived from phosphorylated substrates of PKC (phosphorylated Ser/Thr residue is highlighted in red). Lower FRET ratios (affinities) for peptides were observed to correlate with higher activity.
C) Families of structurally similar small molecule kinase inhibitors led to similar observed changes in FRET ratio in PKC biosensors. Small molecules with a purported bitopic mode of binding (navy, magenta) were more effective in disrupting the kinase/substrate interaction.
For all experiments: N ≥ 3, and data are shown as mean ± SE.
Panels A and B adapted from Sommese et al., JBC 2016 This research was originally published in the Journal of Biological Chemistry. Substrate Affinity Differentially Influences Protein Kinase C Regulation and Inhibitor Potency. J Biol Chem. 2016; 291(42):21963–21970. © the American Society for Biochemistry and Molecular Biology
Panel C adapted with permission from (Ma et al., Biochemistry 2018). Copyright (2018) American Chemical Society
ER/K-linked FRET sensors that probe the kinase-substrate interaction have been used to profile the allosteric effects of chemically distinct small-molecule kinase inhibitors. Specifically, butyl-derivatives of staurosporine, such as Bisindolylmaleimde I (BimI), were found to allosterically disrupt the kinase-substrate interaction (Ma et al., 2018). In contrast to ATP analog inhibitors such as sangivamycin, BimI and sotrastaurin function as bitopic inhibitors that competitively displace ATP and substrate peptide (Figure 5C). The ability of ER/K-linked sensors to profile the comparative effects of SMKIs on the weak kinase-substrate peptide interaction, combined with their utility in plate reader format (Lee et al., 2017) make them suitable for screening efforts to identify allosteric modulators of enzymes such as protein kinases.
ER/K linker as a FRET-free spacer in Ca2+ biosensors –
Monitoring changes in the ubiquitous intracellular second messenger Ca2+ is of broad interest in cell biology. Ratiometric Ca2+ sensing dyes can be limited by the need to selectively deliver them to cells of interest, cellular toxicity through the chelation of endogenous Ca2+ by high affinity dyes, and the use of phototoxic UV excitation wavelengths. These limitations drove the development of genetically encoded Ca2+ sensors, which rely on a Ca2+-sensing module from calmodulin (CaM) to undergo conformational changes in the presence of calcium, resulting in an increase in fluorescence from a circularly permuted GFP (cpGFP) (Nakai et al., 2001; Ohkura et al., 2012). The change in fluorescence intensity from such sensors, termed GCaMP, is quantitatively related to the change in cytosolic Ca2+. However, the lack of basal fluorescence complicates distinction between changes in Ca2+ and sensor expression levels. Hence, a red-shifted fluorophore, mCherry was incorporated in the sensors to quantify expression (Al-Osta et al., 2018). Nonetheless, FRET between mCherry and cpGFP partially quenches GFP fluorescence, complicating quantitative interpretation of Ca2+ changes from the intensity data. Thus, there was a need to visualize the fluorescence of the Ca2+ sensor while preventing FRET between cpGFP and mCherry. In the absence of a significant interaction between proteins fused to the ends of an ER/K linker, its ensemble extended conformation minimizes FRET (Sivaramakrishnan et al., 2008). This property was used to design GCaMP-R, a Ca2+ biosensor in which the GCaMP module is linked to mCherry through a 30 nm ER/K linker (Cho et al., 2017). While minimizing FRET, tethering GCaMP to mCherry with the ER/K linker ensures 1:1 stoichiometry of expression enabling ratiometric imaging (GFP/mCherry) of changes in Ca2+. Avoiding FRET also significantly increased sensor brightness (notably at low calcium concentrations), the signal-to-noise ratio, and sensor dynamic range. This example illustrates the utility of the ER/K linker as an extended spacer to minimize FRET in the absence of a significant interaction, while maintaining stoichiometry of expression.
ER/K-linked sensors measure endogenous cellular activity –
In general, ER/K-linked sensors have been used to report on the interaction of proteins tethered to the ends of the ER/K linker. A recent report from Maziarz et al. describes an ER/K-linked sensor designed to report the activity of endogenous G proteins (Maziarz et al., 2020). The sensor uses the ER/K linker as a spacer to separate a bioluminescent donor (NLuc) and a resonance energy acceptor (YFP). This BRET module with ER/K linker and YFP (BERKY) was inserted between a lipid anchor and KB-1753, a synthetic peptide that selectively binds activated (GTP loaded) Gαi subunits. BERKY sensors transfected into cells report a significantly faster activation kinetics of endogenous G proteins (t1/2 ~ 250 ms) compared to exogenously expressed counterparts (t1/2 ~ 1300 ms). The faster activation potentially stems from the rapid access of the GPCR to a localized endogenous G protein pool, whereas exogenous G protein activation is limited by the progressive disengagement of their endogenous counterparts. The BERKY sensors illustrate the utility and significance of ER/K linkers in measuring endogenous cellular activity, with broader application to other cellular interactions.
Visualizing cyclic AMP (cAMP) compartmentalization using ER/K linkers as ‘nanorulers’ –
cAMP is a universal second messenger that activates numerous signaling pathways. To address signaling specificity, cAMP signaling is thought to be compartmentalized through its hydrolysis by phosphodiesterases (PDEs) (Buxton and Brunton, 1983; Nikolaev et al., 2005; Omori and Kotera, 2007). However, compartmentalized signaling is inconsistent with the rapid diffusion of small molecules such as cAMP combined with the relatively low activity of PDEs. To address the potential for nanometer-scale confinement of cAMP, Bock et al. fused the catalytic domain of PDEs to an EPAC biosensor using 10 and 30 nm ER/K linkers (Bock et al., 2020). The ER/K linker serves as a nanoruler that reports on the cAMP concentration at a defined distance from the PDE anchors. As a control, a direct fusion of PDE and EPAC was expressed in cells and did not report any measurable signal from the EPAC sensor following activation of cellular cAMP. In contrast, tethering EPAC to PDE4A1 with a 10 nm ER/K linker resulted in a sensor response comparable with that elicited by co-expression of EPAC and PDE4A1. This result suggests that PDE4A1 is capable of sustaining a nanometer scale compartment devoid of cAMP. Interestingly, the degree of confinement is isoform-specific, as PDE2 is capable of suppressing sensor response even when tethered to it with a 30 nm ER/K linker. These observations using the ER/K linker as a nanoruler, combined with both computational and experimental measurements of cAMP diffusion in cells suggest a “buffered diffusion” model, wherein under basal conditions cAMP is buffered by cytosolic components resulting in slow diffusion. In turn, PDEs acts on cAMP in their immediate vicinity and in conjunction with buffering by cytosolic cAMP binding sites create nanometer-sized cAMP compartments.
Future perspectives
Cell signaling processes contain transient, selective protein-protein interactions that together transduce diverse extracellular stimuli to distinct physiological responses. Disrupting pathophysiological signaling pathways, without perturbing normal cell function, requires the development of small-molecule or biologic-based therapies that can selectively target pathological interactions. However, the similarity in sequence and structure of the human members of signaling protein superfamilies such as GPCRs (~ 800), G proteins (~ 20), and kinases (~ 500) has confounded our understanding of the structural determinants of signaling specificity and challenges ongoing efforts to selectively target them. Hence, drug discovery efforts are increasingly focused on identifying allosteric modulators that target distal, non-conserved sites to gain specificity. In parallel, reducing the harmful side-effects of target-selective therapies requires tuning responses rather than switching on/off signaling pathways. Addressing these challenges requires technologies that can robustly monitor and modulate weak, transient interactions in cells. In this context, the natively occurring ER/K α-helical linker provides an accessible, genetically encoded approach to control the stoichiometry and effective concentration of protein-protein interactions both in isolated recombinant preparations and in live cells. Over the last decade, ER/K-linked sensors that monitor and modulate protein-protein interactions have repeatedly demonstrated their niche in profiling modest changes in protein interactions in response to perturbations induced by mutagenesis, post-translational modifications, and exogenous small-molecules or biologics. Technical advances in the dynamic range of sensor readout, cell-free assays that capture the complexity of the cellular interaction milieu, and integration with protein evolution strategies will further enhance the utility of the ER/K-linker technology in drug discovery efforts to identify selective modulators that alleviate pathologic cellular processes.
Acknowledgements
We acknowledge funding from the NIH (R35-GM126940 and R01-GM117923 to S.S.)
References
- 1.Al-Osta I, Mucha M, Pereda D, Piqué-Gili M, Okorocha AE, Thomas R, et al. Imaging Calcium in Hippocampal Presynaptic Terminals With a Ratiometric Calcium Sensor in a Novel Transgenic Mouse. Front Cell Neurosci 2018;12:209. 10.3389/fncel.2018.00209. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Antal CE, Callender JA, Kornev AP, Taylor SS, Newton AC. Intramolecular C2 Domain-Mediated Autoinhibition of Protein Kinase C βII. Cell Rep 2015;12:1252–60. 10.1016/j.celrep.2015.07.039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Batchelor M, Wolny M, Baker EG, Paci E, Kalverda AP, Peckham M. Dynamic ion pair behavior stabilizes single -helices in proteins. J Biol Chem 2019;294:3219–34. 10.1074/jbc.RA118.006752. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Bierzynski A, Kim PS, Baldwin RL. A salt bridge stabilizes the helix formed by isolated C-peptide of RNase A. Proc Natl Acad Sci U S A 1982;79:2470–4. 10.1073/pnas.79.8.2470. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Bock A, Annibale P, Konrad C, Hannawacker A, Anton SE, Maiellaro I, et al. Optical Mapping of cAMP Signaling at the Nanometer Scale. Cell 2020;182:1–12. 10.1016/j.cell.2020.07.035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Bonny M, Hui X, Schweizer J, Kaestner L, Zeug A, Kruse K, et al. C2-domain mediated nano-cluster formation increases calcium signaling efficiency. Sci Rep 2016;6:1–11. 10.1038/srep36028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Bornhorst JA, Falke JJ. Purification of proteins using polyhistidine affinity tags. Methods Enzymol 2000;326:245–54. 10.1016/s0076-6879(00)26058-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Bruzzese A, Gil C, Dalton JAR, Giraldo J. Structural insights into positive and negative allosteric regulation of a G protein-coupled receptor through protein-lipid interactions. Sci Rep 2018;8:1–14. 10.1038/s41598-018-22735-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Buxton I, Brunton L. Compartments of cyclic AMP and protein kinase in mammalian cardiomyocytes - PubMed. J Biol Chem 1983:10233–9. https://pubmed.ncbi.nlm.nih.gov/6309796/(accessed September 3, 2020). [PubMed] [Google Scholar]
- 10.Carmena M, Wheelock M, Funabiki H, Earnshaw WC. The chromosomal passenger complex (CPC): From easy rider to the godfather of mitosis. Nat Rev Mol Cell Biol 2012;13:789–803. 10.1038/nrm3474. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Cheslock SR, Poon DTK, Fu W, Rhodes TD, Henderson LE, Nagashima K, et al. Charged Assembly Helix Motif in Murine Leukemia Virus Capsid: an Important Region for Virus Assembly and Particle Size Determination. J Virol 2003;77:7058–66. 10.1128/jvi.77.12.7058-7066.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Cho JH, Swanson CJ, Chen J, Li A, Lippert LG, Boye SE, et al. The GCaMP-R Family of Genetically Encoded Ratiometric Calcium Indicators. ACS Chem Biol 2017;12:1066–74. 10.1021/acschembio.6b00883. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Chou KC, Némethy G, Rumsey S, Tuttle RW, Scheraga HA. Interactions between an α-helix and a β-sheet. Energetics of α β packing in proteins. J Mol Biol 1985;186:591–609. 10.1016/0022-2836(85)90133-0. [DOI] [PubMed] [Google Scholar]
- 14.Chung KY, Rasmussen SGF, Liu T, Li S, Devree BT, Chae PS, et al. Conformational changes in the G protein Gs induced by the β 2 adrenergic receptor. Nature 2011;477:611–7. 10.1038/nature10488. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Crick FHC. The packing of α-helices: simple coiled-coils. Acta Crystallogr 1953;6:689–97. . [DOI] [Google Scholar]
- 16.Daaka Y, Luttrell LM, Lefkowitz RJ. Switching of the coupling of the β2-adrenergic receptor to different g proteins by protein kinase A. Nature 1997;390:88–91. 10.1038/36362. [DOI] [PubMed] [Google Scholar]
- 17.Doležal M, Hadravová R, Kožíšek M, Bednárová L, Langerová H, Ruml T, et al. Functional and structural characterization of novel type of linker connecting capsid and nucleocapsid protein domains in murine leukemia virus. J Biol Chem 2016;291:20630–42. 10.1074/jbc.M116.746461. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Dunnill P The Use of Helical Net-Diagrams to Represent Protein Structures. Biophys J 1968;8:865–75. 10.1016/S0006-3495(68)86525-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Galandrin S, Bouvier M. Distinct signaling profiles of β1 and β2 adrenergic receptor ligands toward adenylyl cyclase and mitogen-activated protein kinase reveals the pluridimensionality of efficacy. Mol Pharmacol 2006;70:1575–84. 10.1124/mol.106.026716. [DOI] [PubMed] [Google Scholar]
- 20.Ganguly S, Saxena R, Chattopadhyay A. Reorganization of the actin cytoskeleton upon G-protein coupled receptor signaling. Biochim Biophys Acta - Biomembr 2011;1808:1921–9. 10.1016/j.bbamem.2011.04.001. [DOI] [PubMed] [Google Scholar]
- 21.García-Nafría J, Tate CG. Cryo-EM structures of GPCRs coupled to G s , G i and G o. Mol Cell Endocrinol 2019;488:1–13. 10.1016/j.mce.2019.02.006. [DOI] [PubMed] [Google Scholar]
- 22.Gáspári Z, Süveges D, Perczel A, Nyitray L, Tóth G. Charged single alpha-helices in proteomes revealed by a consensus prediction approach. Biochim Biophys Acta - Proteins Proteomics 2012;1824:637–46. 10.1016/j.bbapap.2012.01.012. [DOI] [PubMed] [Google Scholar]
- 23.Gupte TM, Malik RU, Sommese RF, Ritt M, Sivaramakrishnan S. Priming GPCR signaling through the synergistic effect of two G proteins. Proc Natl Acad Sci U S A 2017;114:3756–61. 10.1073/pnas.1617232114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Gupte TM, Ritt M, Dysthe M, Malik RU, Sivaramakrishnan S. Minute-scale persistence of a GPCR conformation state triggered by non-cognate G protein interactions primes signaling. Nat Commun 2019;10:1–15. 10.1038/s41467-019-12755-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Hariadi RF, Cale M, Sivaramakrishnan S. Myosin lever arm directs collective motion on cellular actin network. Proc Natl Acad Sci U S A 2014;111:4091–6. 10.1073/pnas.1315923111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.He K, Sakai T, Tsukasaki Y, Watanabe TM, Ikebe M. Myosin X is recruited to nascent focal adhesions at the leading edge and induces multi-cycle filopodial elongation. Sci Rep 2017;7:1–12. 10.1038/s41598-017-06147-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Huse M, Kuriyan J. The conformational plasticity of protein kinases. Cell 2002;109:275–82. 10.1016/S0092-8674(02)00741-9. [DOI] [PubMed] [Google Scholar]
- 28.Kerber ML, Cheney RE. Myosin-X: A MyTH-FERM myosin at the tips of filopodia. J Cell Sci 2011;124:3733–41. 10.1242/jcs.023549. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Knight PJ, Thirumurugan K, Xu Y, Wang F, Kalverda AP, Stafford WF, et al. The Predicted Coiled-coil Domain of Myosin 10 Forms a Novel Elongated Domain That Lengthens the Head. J Biol Chem 2005;280:34702–8. 10.1074/jbc.M504887200. [DOI] [PubMed] [Google Scholar]
- 30.Kobilka BK. Structural insights into adrenergic receptor function and pharmacology. Trends Pharmacol Sci 2011;32:213–8. 10.1016/j.tips.2011.02.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Lee AG. Lipid-protein interactions in biological membranes: A structural perspective. Biochim Biophys Acta - Biomembr 2003;1612:1–40. 10.1016/S0005-2736(03)00056-7. [DOI] [PubMed] [Google Scholar]
- 32.Lee S, Devamani T, Song HD, Sandhu M, Larsen A, Sommese R, et al. Distinct structural mechanisms determine substrate affinity and kinase activity of protein kinase Cα. J Biol Chem 2017;292:16300–9. 10.1074/jbc.M117.804781. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Leonard TA, Róycki B, Saidi LF, Hummer G, Hurley JH. Crystal structure and allosteric activation of protein kinase C βiI. Cell 2011;144:55–66. 10.1016/j.cell.2010.12.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Lyu PC, Gans PJ, Kallenbach NR. Energetic contribution of solvent-exposed ion pairs to alpha-helix structure. J Mol Biol 1992;223:343–50. 10.1016/0022-2836(92)90735-3. [DOI] [PubMed] [Google Scholar]
- 35.Ma N, Lippert LG, Devamani T, Levy B, Lee S, Sandhu M, et al. Bitopic Inhibition of ATP and Substrate Binding in Ser/Thr Kinases through a Conserved Allosteric Mechanism. Biochemistry 2018;57:6387–90. 10.1021/acs.biochem.8b00729. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Mackay AM, Eckley DM, Chue C, Earnshaw WC. Molecular analysis of the INCENPs (inner centromere proteins): Separate domains are required for association with microtubules during interphase and with the central spindle during anaphase. J Cell Biol 1993;123:373–85. 10.1083/jcb.123.2.373. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Malik RU, Ritt M, DeVree BT, Neubig RR, Sunahara RK, Sivaramakrishnan S. Detection of G protein-selective G protein-coupled receptor (GPCR) conformations in live cells. J Biol Chem 2013;288:17167–78. 10.1074/jbc.M113.464065. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Marqusee S, Baldwin RL. Helix stabilization by Glu-…Lys+ salt bridges in short peptides of de novo design. Proc Natl Acad Sci U S A 1987;84:8898–902. 10.1073/pnas.84.24.8898. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Maziarz M, Park JC, Leyme A, Marivin A, Garcia-Lopez A, Patel PP, et al. Revealing the Activity of Trimeric G-proteins in Live Cells with a Versatile Biosensor Design. Cell 2020;182:770–785.e16. 10.1016/j.cell.2020.06.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Mehraeen S, Sudhanshu B, Koslover EF, Spakowitz AJ. End-to-end distribution for a wormlike chain in arbitrary dimensions. Phys Rev E - Stat Nonlinear, Soft Matter Phys 2008;77. 10.1103/PhysRevE.77.061803. [DOI] [PubMed] [Google Scholar]
- 41.Miller CJ, Turk BE. Homing in: Mechanisms of Substrate Targeting by Protein Kinases. Trends Biochem Sci 2018;43:380–94. 10.1016/j.tibs.2018.02.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Nakai J, Ohkura M, Imoto K. A high signal-to-noise ca2+ probe composed of a single green fluorescent protein. Nat Biotechnol 2001;19:137–41. 10.1038/84397. [DOI] [PubMed] [Google Scholar]
- 43.Newton AC. Protein kinase C: perfectly balanced. Crit Rev Biochem Mol Biol 2018;53:208–30. 10.1080/10409238.2018.1442408. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Nikolaev VO, Gambaryan S, Engelhardt S, Walter U, Lohse MJ. Real-time monitoring of the PDE2 activity of live cells: Hormone-stimulated cAMP hydrolysis is faster than hormone-stimulated cAMP synthesis. J Biol Chem 2005;280:1716–9. 10.1074/jbc.C400505200. [DOI] [PubMed] [Google Scholar]
- 45.Ohkura M, Sasaki T, Sadakari J, Gengyo-Ando K, Kagawa-Nagamura Y, Kobayashi C, et al. Genetically Encoded Green Fluorescent Ca2+ Indicators with Improved Detectability for Neuronal Ca2+ Signals. PLoS One 2012;7:e51286. 10.1371/journal.pone.0051286. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Omori K, Kotera J. Overview of PDEs and their regulation. Circ Res 2007;100:309–27. 10.1161/01.RES.0000256354.95791.f1. [DOI] [PubMed] [Google Scholar]
- 47.Pace CN, Scholtz JM. A helix propensity scale based on experimental studies of peptides and proteins. Biophys J 1998;75:422–7. 10.1016/s0006-3495(98)77529-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Pauling L, Corey RB, Branson HR. The structure of proteins; two hydrogen-bonded helical configurations of the polypeptide chain. Proc Natl Acad Sci U S A 1951;37:205–11. 10.1073/pnas.37.4.205. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Peckham M Coiled coils and SAH domains in cytoskeletal molecular motors. Biochem Soc Trans 2011;39:1142–8. 10.1042/BST0391142. [DOI] [PubMed] [Google Scholar]
- 50.Pierce KL, Premont RT, Lefkowitz RJ. Seven-transmembrane receptors. Nat Rev Mol Cell Biol 2002;3:639–50. 10.1038/nrm908. [DOI] [PubMed] [Google Scholar]
- 51.Ponder JW, Richards FM. Tertiary templates for proteins. Use of packing criteria in the enumeration of allowed sequences for different structural classes. J Mol Biol 1987;193:775–91. 10.1016/00222836(87)90358-5. [DOI] [PubMed] [Google Scholar]
- 52.Qu K, Glass B, Dolezal M, Schur FKM, Murciano B, Rein A, et al. Structure and architecture of immature and mature murine leukemia virus capsids. Proc Natl Acad Sci U S A 2018;115:E11751–60. 10.1073/pnas.1811580115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Rasmussen SGF, Devree BT, Zou Y, Kruse AC, Chung KY, Kobilka TS, et al. Crystal structure of the β 2 adrenergic receptor-Gs protein complex. Nature 2011;477:549–57. 10.1038/nature10361. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Ritt M, Guan JL, Sivaramakrishnan S. Visualizing and manipulating Focal Adhesion Kinase regulation in live cells. J Biol Chem 2013;288:8875–86. 10.1074/jbc.M112.421164. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Ritt M, Sivaramakrishnan S. Correlation between activity and domain complementation in adenylyl cyclase demonstrated with a novel fluorescence resonance energy transfer sensor. Mol Pharmacol 2016;89:407–12. 10.1124/mol.115.101626. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Rossy J, Owen DM, Williamson DJ, Yang Z, Gaus K. Conformational states of the kinase Lck regulate clustering in early T cell signaling. Nat Immunol 2013;14:82–9. 10.1038/ni.2488. [DOI] [PubMed] [Google Scholar]
- 57.Samejima K, Platani M, Wolny M, Ogawa H, Vargiu G, Knight PJ, et al. The inner centromere protein (INCENP) coil is a single α-helix (SAH) domain that binds directly to microtubules and is important for chromosome passenger complex (CPC) localization and function in mitosis. J Biol Chem 2015;290:21460–72. 10.1074/jbc.M115.645317. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Sandhu M, Touma AM, Dysthe M, Sadler F, Sivaramakrishnan S, Vaidehi N. Conformational plasticity of the intracellular cavity of GPCR–G-protein complexes leads to G-protein promiscuity and selectivity. Proc Natl Acad Sci U S A 2019;116:11956–65. 10.1073/pnas.1820944116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Scholtz JM, Baldwin RL. The Mechanism of alpha-Helix Formation by Peptides. Annu Rev Biophys Biomol Struct 1992;21:95–118. 10.1146/annurev.bb.21.060192.000523. [DOI] [PubMed] [Google Scholar]
- 60.Semack A, Sandhu M, Malik RU, Vaidehi N, Sivaramakrishnan S. Structural elements in the Gαs and Gβq C termini that mediate selective G Protein-coupled Receptor (GPCR) signaling. J Biol Chem 2016;291:17929–40. 10.1074/jbc.M116.735720. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Sessa F, Mapelli M, Ciferri C, Tarricone C, Areces LB, Schneider TR, et al. The allosteric activators of Aurora B activation by INCENP and inhibition by hesperadin. Mol Cell 2005;18:379–91. 10.1016/j.molcel.2005.03.031. [DOI] [PubMed] [Google Scholar]
- 62.Simm D, Kollmar M. Waggawagga-CLI: A command-line tool for predicting stable single α-helices (SAH-domains), and the SAH-domain distribution across eukaryotes. PLoS One 2018;13:e0191924. 10.1371/journal.pone.0191924. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Sivaramakrishnan S, Spink BJ, Sim AYL, Doniach S, Spudich JA. Dynamic charge interactions create surprising rigidity in the ER/K α-helical protein motif. Proc Natl Acad Sci U S A 2008;105:13356–61. 10.1073/pnas.0806256105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Sivaramakrishnan Sivaraj, Spudich JA. Systematic control of protein interaction using a modular ER/K α-helix linker. Proc Natl Acad Sci U S A 2011;108:20467–72. 10.1073/pnas.1116066108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Sivaramakrishnan S, Spudich JA. Systematic control of protein interaction using a modular ER/K α-helix linker. Proc Natl Acad Sci 2011;108:20467–72. 10.1073/pnas.1116066108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Sivaramakrishnan S, Sung J, Ali M, Doniach S, Flyvbjerg H, Spudich JA. Combining Single-Molecule Optical Trapping and Small-Angle X-Ray Scattering Measurements to Compute the Persistence Length of a Protein ER/K α-Helix. Biophys J 2009;97:2993. 10.1016/J.BPJ.2009.09.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Slater SJ, Seiz JL, Cook AC, Buzas CJ, Malinowski SA, Kershner JL, et al. Regulation of PKCα activity by C1–C2 domain interactions. J Biol Chem 2002;277:15277–85. 10.1074/jbc.M112207200. [DOI] [PubMed] [Google Scholar]
- 68.Sommese RF, Ritt M, Swanson CJ, Sivaramakrishnan S. The role of regulatory domains in maintaining autoinhibition in the multidomain kinase PKCα. J Biol Chem 2017;292:2873–80. 10.1074/jbc.M116.768457. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Sommese RF, Sivaramakrishnan S. Substrate affinity differentially influences protein kinase C regulation and inhibitor potency. J Biol Chem 2016;291:21963–70. 10.1074/jbc.M116.737601. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Sommese RF, Sivaramakrishnan S, Baldwin RL, Spudich JA. Helicity of short E-R/K peptides. Protein Sci 2010;19:2001–5. 10.1002/pro.469. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Steinberg SF. Structural basis of protein kinase C isoform function. Physiol Rev 2008;88:1341–78. 10.1152/physrev.00034.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Strohman MJ, Maeda S, Hilger D, Masureel M, Du Y, Kobilka BK. Local membrane charge regulates β2 adrenergic receptor coupling to Gi3. Nat Commun 2019;10:1–10. 10.1038/s41467-019-10108-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Süveges D, Gáspári Z, Tóth G, Nyitray L. Charged single α-helix: A versatile protein structural motif. Proteins Struct Funct Bioinforma 2009;74:905–16. 10.1002/prot.22183. [DOI] [PubMed] [Google Scholar]
- 74.Swanson CJ, Ritt M, Wang W, Lang MJ, Narayan A, Tesmer JJ, et al. Conserved modular domains team up to latch-open active protein kinase Cα. J Biol Chem 2014;289:17812–29. 10.1074/jbc.M113.534750. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Swanson CJ, Sivaramakrishnan S. Harnessing the unique structural properties of isolated α-helices. J Biol Chem 2014;289:25460–7. 10.1074/jbc.R114.583906. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Swanson CJ, Sommese RF, Petersen KJ, Ritt M, Karslake J, Thomas DD, et al. Calcium Stimulates Self-Assembly of Protein Kinase C α In Vitro. PLoS One 2016;11:e0162331. 10.1371/journal.pone.0162331. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Taylor SS, Kornev AP. Protein kinases: Evolution of dynamic regulatory proteins. Trends Biochem Sci 2011;36:65–77. 10.1016/j.tibs.2010.09.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Tian T, Harding A, Inder K, Plowman S, Parton RG, Hancock JF. Plasma membrane nanoswitches generate high-fidelity Ras signal transduction. Nat Cell Biol 2007;9:905–14. 10.1038/ncb1615. [DOI] [PubMed] [Google Scholar]
- 79.Tomko RJ, Taylor DW, Chen ZA, Wang HW, Rappsilber J, Hochstrasser M. A Single α Helix Drives Extensive Remodeling of the Proteasome Lid and Completion of Regulatory Particle Assembly. Cell 2015;163:432–44. 10.1016/j.cell.2015.09.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Ulrich AKC, Seeger M, Schütze T, Bartlick N, Wahl MC. Scaffolding in the Spliceosome via Single α Helices. Structure 2016;24:1972–83. 10.1016/j.str.2016.09.007. [DOI] [PubMed] [Google Scholar]
- 81.Wolny M, Batchelor M, Bartlett GJ, Baker EG, Kurzawa M, Knight PJ, et al. Characterization of long and stable de novo single alpha-helix domains provides novel insight into their stability. Sci Rep 2017;7:44341–44341. 10.1038/srep44341. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Wolny M, Batchelor M, Knight PJ, Paci E, Dougan L, Peckham M. Stable single α-Helices are constant force springs in proteins. J Biol Chem 2014;289:27825–35. 10.1074/jbc.M114.585679. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Yang Y, Baboolal TG, Siththanandan V, Chen M, Walker ML, Knight PJ, et al. A FERM domain autoregulates Drosophila myosin 7a activity. Proc Natl Acad Sci U S A 2009;106:4189–94. 10.1073/pnas.0808682106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Zangi R Side-chain-side-chain interactions and stability of the helical state. Phys Rev E - Stat Nonlinear, Soft Matter Phys 2014;89:012723. 10.1103/PhysRevE.89.012723. [DOI] [PubMed] [Google Scholar]
- 85.Zeng W, Mak DOD, Li Q, Shin DM, Foskett JK, Muallem S. A new mode of Ca2+ signaling by G protein-coupled receptors: Gating of IP3 receptor Ca2+ release channels by Gβγ. Curr Biol 2003;13:872–6. 10.1016/S0960-9822(03)00330-0. [DOI] [PubMed] [Google Scholar]