
Figure 2.
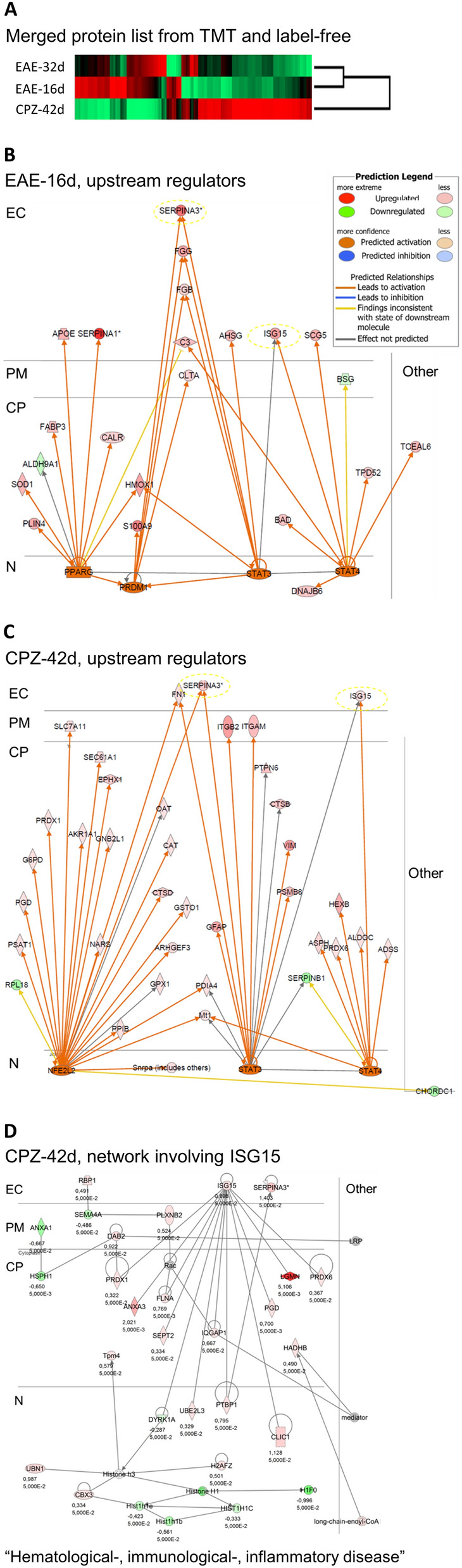
Clustering and pathway analyses of proteins quantified in EAE and CPZ. The protein ratios relative to respective controls for EAE-16d, EAE-32d and CPZ-42d from TMT and label-free were combined prior to analyses in Perseus and IPA. (A) Unsupervised hierarchical clustering of the TMT and label-free protein ratio list (Perseus). (B,C) The significance level of regulation (IPA “p-value”) was set to 0.005 if the protein was significantly regulated with more than 1.2-fold in both TMT and label-free experiments and 0.05 if only in one experiment. Predicted activated upstream regulators, such as STAT3 and STAT4, are illustrated as networks together with associated proteins from our uploaded dataset EAE-16d (B) and CPZ-42d (C). Predicted network in IPA for CPZ-42d “Hematological disease, Immunological disease, Inflammatory disease” involving ISG15 (upregulated in CPZ-42d, in EAE-16d and EAE-32d only in TMT), SERPINA3 (upregulated in CPZ-42d only in TMT), and LGMN (highly upregulated in CPZ-42d) (D). The average log2 ratio and the “IPA p-value” from the combined TMT and label-free protein list are shown for each protein node. The proteins significantly altered with a fold change higher than 20% in both TMT and label-free were considered more significant (IPA p-value 0.005) than the proteins regulated in only one of the experiments (IPA p-value 0.05). The IPA prediction legend is shown as an insert in (B). EC extracellular space, PM plasma membrane, CP cytoplasm, N nucleus.