

ABSTRACT
Background
Neuroimaging has been used to support a diagnosis of possible multiple system atrophy (MSA). Only blood pressure changes upon standing are included in the second consensus criteria but other autonomic function tests (AFT) are also useful to diagnose widespread and progressive autonomic failure typical of MSA. Additional diagnostic tools are of interest to improve accuracy of MSA diagnosis.
Objectives
To assess the utility of diagnostic tools beyond brain imaging and AFT in enhancing a laboratory‐supported diagnosis of MSA to support the upcoming revision of the consensus criteria.
Methods
The International Parkinson and Movement Disorders Society MSA Study Group (MoDiMSA) performed a systematic review of original papers on biomarkers, sleep studies, genetic, neuroendocrine, neurophysiological, neuropsychological and other tests including olfactory testing and acute levodopa challenge test published before August 2019.
Results
Evaluation of history of levodopa responsiveness and olfaction is useful in patients in whom MSA‐parkinsonian subtype is suspected. Neuropsychological testing is useful to exclude dementia at time of diagnosis. Applicability of sphincter EMG is limited. When MSA‐cerebellar subtype is suspected, a screening for the common causes of adult‐onset progressive ataxia is useful, including spinocerebellar ataxias in selected patients. Diagnosing stridor and REM sleep behavior disorder is useful in both MSA subtypes. However, none of these tools are validated in large longitudinal cohorts of postmortem confirmed MSA cases.
Conclusions
Despite limited evidence, additional laboratory work‐up of patients with possible MSA beyond imaging and AFT should be considered to optimize the clinical diagnostic accuracy.
Keywords: multiple system atrophy, diagnostic tests
Multiple system atrophy (MSA) is an adult‐onset neurodegenerative disorder manifesting with autonomic failure, parkinsonism and cerebellar ataxia in any combination. Neuropathologically, MSA is a synucleinopathy characterized by abnormal aggregation of alpha‐synuclein in glial cytoplasmic inclusions and neurodegenerative changes in striatonigral or olivopontocerebellar structures. Clinical diagnosis of MSA is currently made according to the consensus criteria which combine clinical features and neuroimaging findings that reflect changes in putamen and infratentorial brain structures such as pons, middle cerebellar peduncle (MCP) and cerebellum. 1 In the current diagnostic criteria for MSA, brain MRI and [18F]FDG‐PET findings and dopamine transporter imaging contribute to the diagnosis of possible MSA, whereas the diagnosis of probable MSA is exclusively based on clinical features. 1 Two recent clinicopathological studies have shown that the accuracy of MSA diagnosis during lifetime against neuropathologically established diagnosis ranges between 62% and 79%. 2 , 3 The previous systematic review by the International Parkinson and Movement Disorder Society (MDS)‐endorsed MSA Study Group (MoDiMSA) focused on the utility of brain and cardiac imaging and autonomic function tests (AFT) for the early diagnosis of MSA. 4 Neuroimaging features characteristic of MSA may be absent in early disease stages suggesting their suboptimal sensitivity. Recent data suggest that the inclusion of diffusion‐weighted MRI sequences and automated volume segmentation in the conventional MRI protocols may allow for an earlier and more accurate diagnosis. 4 However, diagnosis of MSA based on imaging remains challenging due to overlap with Parkinson's disease (PD), dementia with Lewy bodies (DLB), progressive supranuclear palsy (PSP), sporadic adult onset ataxia (SAOA) and, less commonly, genetic disorders mimicking MSA. Cardiovascular autonomic tests (excluding blood pressure change upon standing), bladder ultrasonography and urodynamic tests, and [123]I‐metaiodobenzylguanidine (MIBG)‐scintigraphy are not recognized in the current consensus criteria, 1 although laboratory indices of early, progressive and severe autonomic failure can be useful to improve diagnostic accuracy in individual cases. 4 , 5 The MoDiMSA review on the utility of AFT for diagnosis of MSA suggested that incomplete bladder emptying or detrusor‐sphincter dyssynergia on urodynamic study, neurogenic OH (nOH) on head up tilt test, and normal myocardial sympathetic postgangionic innervation on [123]I‐MIBG scintigraphy may increase the accuracy of the revised MSA diagnostic criteria that are underway. 4 In some MSA patients, autonomic dysfunction may be mild or moderate or may appear later in the disease course, resembling that of Lewy body disorders. 6 Patients with PSP and genetic ataxia may occasionally present with urinary or cardiovascular autonomic dysfunction, suggesting overlapping AFT findings between MSA and related disorders. 4 Therefore, laboratory indices of autonomic failure should be regarded as a supportive feature of MSA with a diagnostic yield depending on the clinical context. Given the limitations of brain and cardiac imaging and AFT, inclusion of other diagnostic tools into the revised consensus criteria should be considered to enhance a laboratory‐supported diagnosis of MSA. 7 Therefore, the MoDiMSA Study Group conducted a systematic review of the literature to determine the accuracy, benefits and limitations of additional diagnostic tests in the work‐up of patients with MSA.
Methods
This systematic literature review was conducted by applying prespecified search terms (available for each domain in the Supplementary systematic evidence Tables [Link], [Link]) in Pubmed (Medline). Original articles published in extenso in English between 1989 and August 1, 2019 were included if the following inclusion criteria were met: at least 10 patients with MSA per study defined either by post‐mortem verification, or clinically probable, or clinically probable plus possible MSA according to current consensus criteria, 1 , 8 , 9 and at least one reference group of MSA‐related disorders, including PD, DLB, PSP, and SAOA. Due to the specific nature of biomarkers and genetic testing, we included studies with unclassified MSA (level of diagnostic accuracy not provided in the paper) and healthy controls as the only comparative group for these two sections.
Data were extracted using prespecified extraction forms including test domain, authors, publication year, number of patients with MSA and their disease duration, reference group(s), level of diagnostic accuracy, methods, sensitivity, specificity, positive and negative predictive values (PPV, NPV), study results and comments. Results are reported in seven systematic domain‐specific evidence tables, including biomarkers, genetic testing, neuroendocrine tests, neurophysiological tests, neuropsychological tests, sleep studies, and other tests including olfactory testing and acute levodopa challenge test. Relevant studies that fulfilled the inclusion criteria were critically analyzed by the MoDiMSA Study Group experts allocated to working groups on seven diagnostic domains. Working groups' statements on the assigned domains were summarized in the present manuscript.
Results
The search strategy identified 6531 publications including 1984 publications on biomarkers, 2144 on genetics, 196 on neuroendocrine tests, 356 on neurophysiological tests, 1396 on neuropsychological tests, and 455 on sleep studies. A total of 235 articles met the inclusion citeria (see Fig. 1 for an overview of numbers of publications and patients with MSA per diagnostic domain and Supplementary systematic evidence Tables [Link], [Link] for data from individual papers).
FIG. 1.
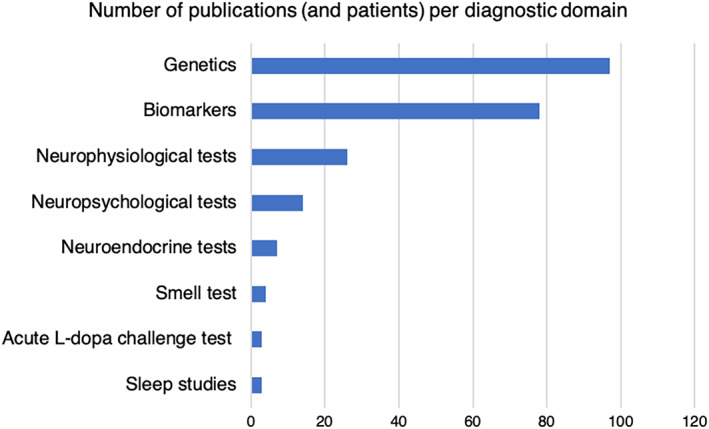
Number of publications and MSA patients studied (in brackets) per diagnostic domain.
Additional Tests In Patients With Parkinsonism Suggestive of MSA
Evaluation of Levodopa Responsiveness
Levodopa responsiveness should be reviewed in newly diagnosed patients with parkinsonism and at regular intervals afterwards. Parkinsonism that is poorly responsive to levodopa is considered a hallmark of MSA. 1 However, a transient, usually modest, levodopa response is documented in a considerable proportion of patients in clinicopathological and natural history studies, with occasional patients experiencing dramatic beneficial responses. 10 , 11 , 12 Levodopa unresponsiveness has usually been defined as either <30% improvement on the Movement Disorder Society Unified PD Rating Scale or Unified MSA Rating Scale motor examination on up to 1000 mg L‐dopa with a peripheral decarboxylase inhibitor daily for 1 month, if tolerated, or by applying an acute levodopa challenge test. This test showed poorer response with more frequent side effects upon levodopa administration (eg, nausea) in patients with MSA compared to patients with PD (Table 1). 13
TABLE 1.
Diagnostic modalities beyond imaging and autonomic function testing investigated in the work‐up of patients with MSA
| Diagnostic test | Reference group | Outcome measures | Total number of studies (number of studies providing test accuracy measures) a | Total number of MSA patients (number of MSA patients in studies providing test accuracy measures) a | Consistency of the results across studies (pro; contra) b | Test accuracy |
|---|---|---|---|---|---|---|
| Evaluation of responsiveness to levodopa | ||||||
| Acute levodopa challenge test (heterogenous levodopa doses and assessment methodology) | PD |
|
3 (3) | 80 (80) | Pro: 3; Contra: 0 |
Sensitivity: 75%–94% Specificity: 74%–100% |
| Olfactory testing | ||||||
| Smell test | PD |
|
3 (2) | 95 (75) | Pro: 3; Contra: 0 |
Sensitivity: 74%–77% Specificity: 86%–96% (for PD vs. MSA) |
| Sleep studies | ||||||
| vPSG‐confirmed RBD | PSP, SAOA |
|
1 (1) | 16 (16) (postmortem confirmed diagnosis) | ‐ | PPV: 98% (for synucleinopathy vs. non‐ synucleinopathy) |
| v‐PSG abnormalities other than RBD | PD, PSP |
|
2 (0) | 50 (0) | Pro: 2; Contra: 0 | ‐ |
| Neurophysiological tests | ||||||
| Anal sphincter EMG | PD |
|
11 (8) | 557 (518; 30 postmortem confirmed diagnosis) | Pro: 9; Contra: 2 |
Sensitivity: 35%–100% Specificity: 65%–100% (depending on criteria for abnormal EMG) |
| Mean MUP duration on sphincter EMG | PD |
|
8 (3) | 501 (424) | Pro: 6; Contra: 2 |
Sensitivity: 35%–93% Specificity: 54%–100% (depending on cut‐off value) |
| Proportion of prolonged MUP >10 ms on sphincter EMG | PD |
|
3 (1) | 294 (263) | Pro: 3; Contra 0 |
Sensitivity: 66% Specificity: 62% |
| Other abnormalities in sphincter EMG | PD |
|
7 (0) | 340 (0) | Pro: 6; Contra: 1 | ‐ |
| Bulbocavernosus reflex | PD |
|
2 (0) | 69 (0) | Pro: 2; Contra: 0 |
Sensitivity: 24% Specificity: 91–94% |
| Transcranial magnetic stimulation | PD |
|
3 (1) | 42 (15) | Pro: 2; Contra: 1 |
Sensitivity: 40% Specificity: 100% |
|
3 (0) | 37 (0) | Pro: 2; Contra: 1 | ‐ | ||
|
1 (0) | 14 (0) | ‐ |
Sensitivity: 50% Specificity: 100% |
||
| CBS |
|
1 (0) | 13 (0) | ‐ | ‐ | |
| Trigeminocervical reflexes | PSP |
|
1 (0) | 10 (0) | ‐ |
Sensitivity: 100% Specificity: 100% |
| Eyeblink classical conditioning | PD |
|
1 (0) | 11 (0) | ‐ |
Sensitivity: 91% Specificity: 75% |
| Neuropsychological tests | ||||||
| Dementia Rating Scale | PSP |
|
1 (1) | 44 (44) (postmortem confirmed diagnosis) | ‐ |
Sensitivity: 81% Specificity: 79% |
| PSP |
|
1 (1) | 372 (372) | ‐ |
Sensitivity: 80% Specificity: 60% |
|
| Frontal Assessment Battery | PSP |
|
3 (2) | 444 (383) | Pro: 3; Contra: 0 |
Sensitivity: 68%–92% Specificity: 61%–83% |
|
3 (2) | 444 (383) | Pro: 3; Contra: 0 |
Sensitivity: 58%–78% Specificity: 72%–78% |
||
| Verbal fluency + Luria series subitems from FAB | PSP |
|
1 (1) | 11 (11) | ‐ |
Sensitivity: 97% Specificity: 85% |
| Montreal Cognitive Assessment | PSP |
|
1 (1) | 35 (35) | ‐ | Sensitivity: 70% Specificity: 89% |
| Neuroendocrine testing | ||||||
| Arginine growth hormone stimulation test | PD |
|
2 (2) | 69 (69) | Pro: 2; Contra: 0 | Sensitivity: 92%–93% Specificity: 91%–96% |
| PSP |
|
1 (1) | 26 (26) | ‐ | Sensitivity: 78% Specificity: 96% | |
| SAOA |
|
1 (11) | 14 (14) | ‐ | ‐ | |
| Clonidine growth hormone stimulation test | PD |
|
2 (2) | 34 (34) | Pro: 2; Contra: 1 |
Sensitivity: 73%–78% Specificity: 57%–85% |
| Wet biomarkers (in CSF if not indicated otherwise) | ||||||
| α‐synuclein c | PD |
|
10 (0) | 277 (277) | Pro: 9; Contra (decreased in MSA): 1 | ‐ |
| DLB |
|
3 (0) | 20 (20) | Pro: 3; Contra: 0 | ||
| PSP |
|
3 (0) | 82 (0) | Pro: 2; Contra (decreased in MSA): 1 | ||
| α‐synuclein (serum) | PD |
|
2 (0) | 53 (0) | Pro: 1; Contra (similar in MSA): 1 | ‐ |
| Phosphorylated α‐synuclein | PD |
|
1 (1) | 25 (0) | ‐ |
Sensitivity: 64% Specificity: 86% |
| DJ‐1 | PD |
|
3 (0) | 64 (0) | Pro: 2; Contra (increased in MSA): 1 | ‐ |
| DLB |
|
1 (0) | 14 (0) | ‐ | ||
| PSP |
|
1 (0) | 14 (0) | ‐ | ||
| Total tau | PD |
|
11 (0) | 258 (258) | Pro: 6; Contra (similar in MSA): 5 | ‐ |
| DLB |
|
3 (0) | 73 (0) | Pro: 2; Contra (decreased in MSA): 1 | ||
| PSP |
|
5 (0) | 129 (0) | Pro: 5; Contra: 0 | ||
| SAOA |
|
1 (0) | 25 (0) | ‐ | ||
| Phosphorylated tau | PD |
|
7 (0) | 175 (0) | Pro: 6; Contra (decreased in MSA): 1 | ‐ |
| DLB |
|
2 (0) | 58 (0) | Pro: 1; Contra (similar in MSA): 1 | ||
| PSP |
|
5 (0) | Pro: 5; Contra: 0 | |||
| p‐tau/t‐tau | PD |
|
2 (0) | 57 (0) | Pro: 1; Contra (decreased in MSA): 1 | ‐ |
| t‐tau/Aβ42 | PD |
|
2 (0) | 47 (0) | Pro: 1; Contra (similar in MSA): 1 | ‐ |
| Neurofilament light chain | PD |
|
8 (2) | 200 (43) | Pro: 7; Contra (similar in MSA): 1 |
Sensitivity: 80%–83% Specificity: 90%‐ 97% |
| DLB |
|
1 (0) | 48 (0) | ‐ | ‐ | |
| PSP |
|
4 (0) | 98 (0) | Pro: 3; Contra (increased in MSA): 1 | ‐ | |
| SAOA |
|
1 (1) | 24 (24) | ‐ |
Sensitivity: 79% Specificity: 94% |
|
| Neurofilament light chain (serum) | PD |
|
2 (0) | 52 (0) | Pro: 2; Contra: 0 | ‐ |
| PSP |
|
2 (0) | 52 (0) | Pro: 2; Contra: 0 | ||
| SAOA |
|
1 (0) | 25 (0) | ‐ | ||
| Neurofilament heavy chain and phosphorylated neurofilament heavy chain | PD |
|
2 (2) | 39 (39) | Pro: 2; Contra: 0 |
Sensitivity: 61%–83% Specificity: 87%–89% |
| PSP |
|
1 (0) | 21 (0) | ‐ | ‐ | |
| SAOA |
|
1 (0) | 24 (0) | ‐ | ‐ | |
| Glial fibrillary acidic protein | PD |
|
4 (0) | 65 (0) | Pro: 4; Contra: 0 | ‐ |
| PSP |
|
3 (0) | 47 (0) | Pro: 3; Contra: 0 | ||
| SAOA |
|
1 (0) | 21 (0) | ‐ | ||
| Aβ42 | PD |
|
7 (0) | 201 (0) | Pro: 6; Contra (decreased in MSA): 1 | ‐ |
| DLB |
|
2 (0) | 58 (0) | Pro: 2; Contra: 0 | ||
| PSP |
|
5 (0) | 158 (0) | Pro: 4; Contra (decreased in MSA): 1 | ||
| SAOA |
|
1 (0) | 25 (0) | ‐ | ||
| Aβ42/Aβ40 | DLB |
|
1 (0) | 18 (0) | ‐ | ‐ |
| APPα and APPβ | PSP |
|
1 (0) | 31 (0) | ‐ | ‐ |
| UCH‐L1 | PD |
|
1 (0) | 34 (0) | ‐ | ‐ |
| FLT‐3 ligand | PD |
|
3 (0) | 89 (0) | Pro: 2; Contra (decreased in MSA): 1 | ‐ |
| PSP |
|
1 (0) | 30 (0) | ‐ | ||
| Complement C3/factor H | PD |
|
1 (1) | 32 (0) | ‐ |
Sensitivity: 80% Specificity: 87% |
| MCP‐1 | PD |
|
1 (0) | 31 (0) | ‐ | ‐ |
| PSP |
|
1 (0) | 31 (0) | ‐ | ||
| YKL‐40 | PD |
|
2 (0) | 68 (0) | Pro: 1; Contra (increased in MSA): 1 | ‐ |
| PSP |
|
1 (0) | 31 (0) | ‐ | ||
| Solubile CD14 receptor | PD, PSP, CBS |
|
1 (0) | 37 (0) | ‐ | ‐ |
| Urate (CSF and serum) | PD, PSP, CBS |
|
1 (0) | 13 (0) | ‐ | ‐ |
| Myelin basic protein | PD |
|
1 (0) | 28 (0) | ‐ | ‐ |
| SAOA |
|
1 (0) | 25 (0) | ‐ | ||
| S100B | PD |
|
1 (0) | 28 (0) | ‐ | ‐ |
| SAOA |
|
1 (0) | 25 (0) | ‐ | ||
| Uric adic (serum) | PD |
|
1 (0) | 40 (0) | ‐ | ‐ |
| Homocysteine (serum) | PD |
|
1 (0) | 47 (0) | ‐ | ‐ |
| CoQ10 | PD |
|
3 (0) | 78 (0) | Pro: 2; Contra (similar in MSA): 1 | ‐ |
| PSP |
|
1 (0) | 20 (0) | ‐ | ||
| α‐synuclein + p‐tau/t‐tau | PD |
|
1 (1) | 31 (32) | ‐ |
Sensitivity: 90% Specificity: 65% (for PD vs. MSA) |
| NFL + t‐tau | PD |
|
1 (1) | 25 (25) | ‐ |
Sensitivity: 76% Specificity: 97% |
| NFL + FLT3 ligand | PD |
|
1 (1) | 25 (25) | ‐ |
Sensitivity: 83% Specificity: 97% |
| NFL + FLT3 ligand + t‐tau | PD |
|
1 (1) | 25 (25) | ‐ |
Sensitivity: 82% Specificity: 97% |
| NFL + sAPPα + sAPPβ + Aβ42 + t‐tau + p‐tau + α‐synuclein + YKL‐40 + MCP‐1 | PSP |
|
1 (1) | 31 (31) | ‐ |
Sensitivity: 80% Specificity: 78% (for PSP vs. MSA) |
| DJ‐1 + t‐tau + p‐tau | PD |
|
1 (1) | 21 (21) | ‐ |
Sensitivity: 82% Specificity: 81% |
Refer to the Supplementary systematic evidence Tables [Link], [Link] for details on disease duration of MSA subjects, study design, methods, results and validity measures from individual studies that met the inclusion criteria for this review.
Pro: number of studies supporting the statement given in the outcome measures column; Contra: number of studies providing conflicting results and not supporting the statement given in the outcome measures column.
Excluding data on Real‐Time Quaking‐Induced Conversion assay and α‐synuclein‐protein misfolding cyclic amplification assay.
Abbreviations: APP, amyloid precursor protein; CD, carbidopa; CoQ10, coenzyme Q10; CSF, cerebrospinal fluid; DLB, dementia with Lewy bodies; EMG, electromyography; FAB, Frontal Assessment Battery; FLT3 ligand, fms‐like tyrosine kinase ligand; MCP‐1, monocyte chemoattractant protein‐1; MSA, multiple system atrophy; NFL, neurofilament light chain; p‐tau, phosphorylated tau; PD, Parkinson's disease; PSP, progressive supranuclear palsy; RBD, REM sleep behavior disorder; REM, rapid eye movements; SAOA, sporadic adult onset ataxia; t‐tau, total tau; TMS, transcranial magnetic stimulation; UCH‐L1, Ubiquitin carboxy‐terminal hydrolase L1; UPDRS, Unified Parkinson's Disease Rating Scale; vPSG, video‐polysomnography.
Olfactory Testing
Olfactory testing is easy, cost‐effective and non‐invasive. It aids in the differential diagnosis of MSA as most patients with PD have hyposmia in contrast to patients with MSA and PSP who have relatively preserved olfaction (Table 1). 14 A combination of hyposmia and abnormal cardiac MIBG‐scintigraphy, reflecting impaired norepinephrine analogue uptake due to degeneration of postganglionic myocardial fibers, should guide clinicians towards the diagnosis of PD versus MSA. 15 Fluctuations of olfactory performance that may affect the test's diagnostic value, especially at early disease stages, have been found in a small but relevant fraction of PD patients during observation periods of 4 to 5 years. 16 Common pitfalls of the smell test include the presence of allergic rhinitis and smoking habits.
Sleep Studies
Neuropathological studies have documented that 98% of patients with video‐polysomnography (vPSG)‐proven REM sleep behavior disorder (RBD) and a neurodegenerative syndrome (parkinsonism or cognitive impairment) have an underlying synucleinopathy. 17 Therefore, documentation of RBD can help to distinguish MSA from non‐synucleinopathy neurodegeneration such as PSP but cannot be used to distinguish MSA from Lewy body disorders. RBD can present before MSA onset; a multicenter prospective study found that 8% of patients with idiopathic RBD who develop neurodegenerative disease after 4 to 5 years, were diagnosed as clinically probable MSA. 18
Other sleep abnormalities are also common in MSA. They include general disruption of the sleep architecture, upper airway dysfunction (apnea and stridor), loss of REM atonia, and periodic leg movements during sleep. MSA patients have more severe loss of REM atonia compared to patients with PD and idiopathic RBD, 19 , 20 although an overlap between groups limits the diagnostic potential of REM atonia quantification. Evidence for diagnostic utility of other vPSG sleep parameters is limited. Compared to patients with PD and idiopathic RBD, patients with MSA have more periodic leg movements of sleep, more slow‐wave sleep, shorter overall sleep duration, and less wake after sleep onset (Table 1). 19 Apnea (ie, increased apnea/hypopnea index) is commonly observed on vPSG, but not clearly increased compared with other neurodegenerative conditions. 19 Increased snoring is specific for MSA and Lewy body disorders compared to PSP in a clinicopathological series. 3 Symptoms associated with restless legs syndrome are frequent in both patients with MSA (4.8%–28%) and PD (14%). 21
Inspiratory stridor is commonly observed in MSA and considered a red flag against the diagnosis of PD. 22 Home audio recording is sufficient to make a diagnosis of stridor. 23 Irregular arytenoid cartilage movements were observed on flexible endoscopic evaluation of swallowing in 91% of patients with MSA (of whom, 44% showed clinically overt laryngeal dysfunction with stridor), but in no patients with PD in a recent study published outside of the time window of this review. 24 Stridor onset within the first 3 years from disease onset was present in 16% of patients with MSA, indicating low sensitivity in early stages. 25 It has been only rarely documented in other degenerative parkinsonian disorders suggesting high specificity, although controlled studies are lacking.
Pelvic Neurophysiology
Evaluation of bladder function in patients with MSA comprises simple (ie, post‐void ultrasonography and uroflowmetry) and advanced methods such as urodynamic tests and sphincter electromyography (EMG). Post‐void bladder ultrasonography is a non‐invasive, widely available, highly specific tool for diagnosing MSA versus PD. However, in early disease stages when symptoms of overactive bladder may be present in both MSA and PD patients, the sensitivity of bladder ultrasonography is suboptimal. 4 Urodynamic testing is useful for investigation of the pathophysiology of both urinary incontinence and retention in patients with MSA. 4
EMG recordings from the external anal and urethral sphincters are commonly abnormal in MSA. 26 , 27 In a series of 30 definite MSA cases, 24 had abnormal sphincter EMG, five had borderline results, and only one was normal. 28 Neurogenic changes in MSA occur as a result of involvement of anterior horn cells in the Onuf's nucleus of the sacral spinal cord, and the most consistent abnormalities are prolonged duration of motor unit potentials (MUPs) compared to PD, suggestive of chronic reinnervation (Table 1). 29 , 30 The value of sphincter EMG in the differential diagnosis of parkinsonism has been debated over the years and a false‐negative result can arise if the Onuf's nucleus is yet to be involved. Moreover, machine‐automated MUP analysis tends to exclude long‐duration polyphasic potentials with satellite potentials, so that additional manual MUP analysis is advisable in cases where MSA is suspected. 31 Changes of chronic reinnervation similar to those seen in MSA, may be found, though usually to a lesser degree, in long standing PD and other parkinsonian syndromes such as PSP (which also affects Onuf's nucleus), DLB and spinocerebellar ataxia (SCA) type 3, following cauda equina injury, and following damage to the sphincter muscle such as haemorrhoid surgery and obstetric pelvic floor tears. The prevalence of neurogenic changes increases with duration of disease and worsening neurological disability. 32 A highly abnormal EMG in the absence of other obvious causes in a patient with suspected MSA in the first 5 years is significant. In contrast, an entirely normal result after 5 years makes the diagnosis of MSA very unlikely; thus, the test is of limited usefulness in between. 28 Lower elicitation rates and prolonged latencies of the bulbocavernosus reflex were observed in patients with MSA compared to patients with PD with early urogenital symptoms (Table 1). 33
Among other neurophysiological tests, auditory startle reflex has occasionally been used for distinguishing PSP (absent or reduced due to pathology in the reticular formation) from MSA (normal response) in small unblinded studies. 34
Neuropsychological Tests
Despite prevalence rates of cognitive impairment of up to 32% in clinical and autopsy confirmed MSA series, 35 , 36 neuropsychological testing is valuable in the differential diagnosis of MSA and other dementia disorders such as DLB, PD dementia (PDD) and PSP. 37 , 38 Disproportionate deficits in attention, executive functions and visual processing relative to memory and naming are typical for DLB. 38 PDD, characterized by frontal‐executive dysfunction, initially is mild but often evolves after a mean of 10 years from the onset of motor symptoms. 39 In comparative studies patients with DLB and PDD performed worse than patients with MSA across all cognitive domains. 40 In patients with PSP, global cognitive performance is poor compared with MSA patients 4 years after symptom onset, with more profound executive dysfunction and more rapid progression. 36 However, in early stages the difference in cognitive performance may not be present. One study reported that the Dementia Rating Scale might separate autopsy confirmed MSA from PSP patients with moderate sensitivity and specificity. 41 The Frontal Assessment Battery (FAB) 42 and Montreal Cognitive Assessment 43 also showed good discriminative power that was even better for the verbal fluency and Luria series subitems of the FAB (Table 1). 42 More severe deterioration also occurs in other cognitive domains in patients with PSP than in those with MSA. 41 , 44 , 45
A test battery specific for the cognitive screening of patients with MSA has not yet been developed. Level‐1 examination of the diagnostic procedures for PDD (cognitive deficits severe enough to impact daily living, MMSE<26 and impairment in at least two of the following tests: months backward or serial 7 subtraction, lexical fluency or clock drawing, MMSE pentagons, 3‐word recall) 39 showed excellent specificity of 96.9% and a negative predictive value of 94.1% for detecting dementia in MSA, while a sensitivity of 84.6% was achieved by applying a cut‐off MMSE score of 27 instead of 26. 46 Neuropsychological testing is of limited value in the differentiation of patients presenting with ataxia.
Biomarkers
Alpha‐Synuclein
Decreased α‐synuclein levels have been reported in cerebrospinal fluid (CSF) of patients with MSA, but most studies have failed to discriminate between patients with MSA and PD. 47 , 48 , 49 , 50 , 51 , 52 Most recently and outside of the time window of this review, a Real‐Time Quaking‐Induced Conversion (RT‐QuIC) assay was reported to accurately detect α‐synuclein seeding activity across Lewy body synucleinopathies but not in MSA. 53 Further research has shown that α‐synuclein aggregates associated with PD and MSA corresponded to different conformational strains of α‐synuclein 54 and that an α‐synuclein‐protein misfolding cyclic amplification (PMCA) assay can discriminate between these disorders with an excellent overall sensitivity. 55 In addition, α‐synuclein oligomers detected by PMCA analysis together with CSF neurofilament light chain (NfL) were able to discriminate patients with early MSA from those with Lewy body synucleinopathies. 56 Ultrasensitive single molecule array ELISA is another quantification method with the potential to detect plasma and CSF or exosomal α‐synuclein. The lower number of oligodendrogial‐derived plasma exosomes in MSA compared to PD (sensitivity: 62%, specificity: 81%) was reported in another very recent study. 57 Inconclusive results on plasma α‐synuclein levels in MSA have been reported; some of the variability may be ascribed to the influence of blood contamination, age and different detection procedures (Table 1). 47 , 58
Markers of Axonal and Glial Damage
Neurofilament light and heavy chain (NfH) concentrations in CSF are increased in patients with atypical parkinsonism including MSA compared to patients with PD (Table 1). 50 , 59 Higher NfL levels were also found in serum in patients with atypical parkinsonism including MSA compared to patients with PD, showing good discriminative power in the detection (sensitivity: 82%, specificity: 92%) and validation cohorts (sensitivity: 80%, specificity: 92%), as well as in the cohort of patients with disease duration less than 3 years (sensitivity: 70%, specificity: 80%), and a strong correlation with CSF levels of NfL. 60
Amyloid Markers
Decreased CSF levels of Aβ1‐42, a 42‐amino acid long peptide that forms toxic β‐amyloid aggregates, and a lower ratio of Aβ1‐42/Aβ1‐40, may be used to discriminate patients with DLB from patients with MSA (Table 1). 61 , 62
Panels of Biomarkers
Combining different wet biomarkers is a promising approach to increase diagnostic accuracy. A set of 9 CSF biomarkers (NfL, sAPPα, sAPPβ, Aβ1‐42, total tau, phosphorylated tau, α‐synuclein, YKL‐40, MCP‐1), as well as disease duration and severity were shown to differentiate patients with PD from those with atypical parkinsonism with a sensitivity and specificity of 91%. Among them NfL, α‐synuclein and sAPPα independently predicted the diagnosis of PD versus atypical parkinsonism. The same panel was able to differentiate between MSA and PSP patients (Table 1). 48 However, the applied methodology needs standardization of the procedures and validation in future prospective studies. Serum miR‐24, miR‐34b, and miR‐148b were upregulated in MSA compared to PD in one study. 63
Arginine Stimulation Test
The arginine stimulation test is based on the ability of this amino acid to induce growth hormone (GH) secretion through the inhibition of somatostatin release, which is possibly mediated by the cholinergic system. In small unblinded studies it was reported that the GH response to arginine is blunted in patients with MSA, and relatively preserved in patients with PD and PSP. 64
Genetic Screening
An increasing number of heredodegenerative syndromes that can occasionally mimic MSA have been described (Table 2). Among these, a combination of parkinsonism and ataxia may be observed in SCA2, SCA3, SCA6 and SCA17. A complex phenotype with L‐dopa unresponsive parkinsonism and central hypoventilation requires attention towards DCTN1 mutation. In patients of European ancestry, screening for the C9orf72 hexanucleotide repeat expansion should be considered, especially in cases with a family history of amyotrophic lateral sclerosis or frontotemporal dementia.
TABLE 2.
Neurogenetic MSA mimic syndromes (adapted from Stankovic I, et al.) 5
| Genetic characteristics | Clinical characteristics | ||||
|---|---|---|---|---|---|
| Gene | Locus | Inh. | Typical presentation | Typical AAO | Red flag(s) |
| ABCD1 | Xq28 | XR | ALD | 1st–3rd decade | Adrenal insufficiency, leukodystrophy on MRI, psychiatric symptoms, elevated very long chain fatty acids |
| LMNB1 | 5q23 | AD | ALD | 4th–6th decade | Extensive, U‐fiber sparing white matter lesions on MRI, cognitive impairment in advanced stages |
| ATXN1 | 6p22.3 | AD | SCA1 | 3rd–4th decade | Axonal sensory neuropathy, hyporeflexia, loss of vibration/proprioception |
| ATXN2 | 12q24.1 | AD | SCA2 | 4th decade | Chorea, dystonia, cognitive impairment, slow saccades |
| ATXN3 | 14q21 | AD | SCA3 | 4th decade | Upper motor neuron signs, executive dysfunction, ophthalmoparesis |
| ATXN7 | 3p21.1‐p12 | AD | SCA7 | 3rd–4th decade | Retinal degeneration |
| ATXN8 | 13q21 | AD | SCA8 | 4th decade | Slowly progressive, hyperreflexia |
| C9orf72 | 9p21.2 | AD | ALS/FTD | 4th‐7th decade | Motor neuron signs, cognitive impairment, psychiatric symptoms |
| CACNA1A | 19p13.13 | AD | SCA6 | 5th–6th decade | Family members can present with episodic ataxia or hemiplegic migraine |
| CYP27A1 | 2q35 | AR | CTX | 2nd–3rd decade | Diarrhea, cataracts, xanthomas, cognitive and psychiatric symptoms |
| DCTN1 | 2p13.1 | AD | Perry syndrome | 5th decade | Hypoventilation, weight loss, psychiatric symptoms |
| ATN1 | 12p13.31 | AD | DRPLA | 4th decade | Choreoathetosis, dementia, epilepsy, psychiatric symptoms |
|
FMR1 |
Xq27.3 |
XR |
FXTAS |
6th–7th decade | Female pre‐mutation carriers can present with primary ovarian insufficiency, family history of fragile‐X syndrome |
| FXN | 9q21.11 | AR | FRDA | 2nd–3rd decade | Hyporeflexia, loss of vibration/position sense, cardiomyopathy, diabetes |
|
GBA |
1q22 |
AR AD |
GD PD/LBD |
1st–2nd decade 6th decade |
Cognitive impairment, common in patients of Ashkenazi Jewish ancestry |
| LRRK2 | 12q12 | AD | PD | 6th decade | Family history of L‐dopa‐responsive Parkinson disease |
| NOP56 | 20p13 | AD | SCA36 | 5th–6th decade | Hearing loss, motor neuron involvement, slow progression |
| PDYN | 20p13 | AD | SCA23 | 5th–6th decade | Slow progression |
| POLG1 | 15q15 | AR, AD | Mitochondriopathy | 1st–4th decade | Ophthalmoplegia, hearing loss, neuropathy, epilepsy, dementia |
| RFC1 | 4p14 | AR | CANVAS | 4th–6th decade | Late‐onset ataxia, sensory neuronopathy, bilateral vestibulopathy, chronic cough, and autonomic dysfunction |
| PRNP | 20p13 | AD | Prion disease | 3rd–9th decade | Myoclonus, dementia, psychiatric symptoms, seizures, rapid progression |
| PPP2R2B | 5q32 | AD | SCA12 | 4th decade | Cerebellar ataxia, hyperreflexia, tremor |
| SNCA | 4q22.1 | AD | PD, LBD | 3rd–4th decade | Dementia, early‐onset L‐dopa responsive parkinsonism |
| SPG7 | 16q24.3 | AD, AR | HSP | 3rd–4th decade | Spastic paraplegia, cerebellar ataxia, executive dysfunction |
| SPG11 | 15q21.1 | AR | HSP | 3rd–4th decade | Severe spastic paraplegia |
| TBP | 6q27 | AD | SCA17 | 2nd–5th decade | Psychiatric symptoms, dementia, chorea |
Abbreviations: Inh, inheritance; Mt. DNA, mitochondrial DNA; XR, X‐chromosomal recessive; AD, autosomal dominant; AR, autosomal recessive; AAO, age at onset; ALD, adrenoleukodystrophy; SCA, spinocerebellar ataxia; ALS, amyotrophic lateral sclerosis; FTD, frontotemporal dementia; CTX, cerebrotendinous xanthomatosis; DRPLA, dentatorubral pallidoluysian atrophy; FXTAS, fragile X tremor ataxia syndrome; FRDA, Friedreich ataxia; GD, Gaucher disease; PD, Parkinson disease; LBD, Lewy body dementia; HSP, hereditary spastic paraplegia.
Additional Tests In Patients With Ataxia Suggestive of MSA
Sleep Studies
Adult onset progressive ataxia and autonomic failure, that initially presents with urogenital failure followed by nOH, with cerebellar, brainstem and MCP atophy on brain imaging is highly suggestive of MSA‐cerebellar type. 65 The presence of RBD in the ataxic patient may point towards the diagnosis of MSA‐C versus SAOA (Table 1). In a recent prospective study, probable RBD was present in 83% of MSA‐C patients and 11% of SAOA patients. 66 Sleep abnormalities can be also seen in patients with genetic ataxias. 67 , 68
Exclusion of Common Causes of Adult Onset Progressive Ataxia
A progressive course of ataxia starting in midlife requires screening for the common causes of cerebellar degeneration including toxic (ie, alcohol, phenytoin, lithium, barbiturates), metabolic (ie, vitamin B12, or B1 deficiency syndromes), paraneoplastic and non‐cancer related immune mediated disorders (ie, ataxia associated with antigliadin antibodies, or with anti‐glutamic acid decarboxylase antibodies), infections (ie, cerebellitis), parainfectious syndromes, brain mass lesions and multiple sclerosis.
Genetic Screening
Typically, MSA occurs sporadically in the community. Several pathologically confirmed MSA cases occurring in the same family have been reported. 69 , 70 The diagnostic value of genetic testing in MSA is evaluated in the setting of a suspected monogenic inheritance. Homozygous or compound heterozygous loss‐of‐function mutations in COQ2 gene, involved in the coenzyme Q10 (COQ10) biosynthesis, are the only monogenic mutations that have been suggested to cause MSA in two Japanese families. 70 Furthermore, the common COQ2 polymorphism V393A identified in the East Asian populations has been suggested as a possible risk variant. 71 This variant is extremely rare in the Caucasian population, which could explain the lack of disease associations in North American or European MSA cohorts. 72 , 73 Several additional heterozygous variants of unknown significance have been reported in COQ2, but their role in disease pathogenesis is unclear and requires further investigation. In addition, decreased concentrations of COQ2 in serum, CSF and cerebellum of MSA patients suggest that COQ10 deficiency may contribute to the pathogenesis of MSA (Table 1). 74
In patients presenting with ataxia, either the presence of a family history or non‐supportive features for MSA should guide the physician towards neurogenetic mimicry. These “red flags,” however, may not be present in a given case. Genetic screening, as a second tier after exclusion of the other common causes of midlife onset progressive ataxia, should be considered in selected cases to refine the clinical diagnosis by excluding of the most common mimicries due to the pathogenic mutations in the ATXN1, ATXN2, ATXN3, CACNA1A, ATXN7, PPP2R2B, FMR1, and TBP genes. Repeat expansion mutations in the RFC1 gene is an underrecognized cause and has been observed in 22% of patients with late‐onset cerebellar ataxia. 75 Fragile X–associated tremor/ataxia syndrome resulting from a premutation in the FMR1 gene more frequently poses a differential diagnosis challenge to MSA than the other syndromes due to overlapping clinical and MRI features including hyperintensities in the MCP. In Japanese patients, screening for DRPLA needs consideration. Notably, however, in a large postmortem series, none of the patients with a clinical diagnosis of MSA in life had a final diagnosis of neurogenetic mimic syndrome. 3 Other rare neurogenetic mimic syndromes have been described in the literature, but more detailed discussion goes beyond the scope of this article.
Biomarkers and Arginine Stimulation Test
Patients with MSA‐C have increased NfL and NfH levels in CSF compared to patients with SAOA (Table 1). 76
Arginine GH stimulation test failed to show efficacy in differentiating between patients with MSA‐cerebellar type from patients with SAOA, genetic ataxia and healthy controls (Table 1). 77
Conclusion And Test Limitations
Data assessed in this systematic review suggest that several diagnostic tools beyond imaging and AFT may support the diagnosis of MSA in individual cases. The diagnostic discrimination of each tool depends on the clinical context (ie, predominant clinical presentation and differential diagnosis) and changes over the disease course. The paucity of studies in patients with MSA presenting with isolated autonomic failure (ie, distinguishing premotor MSA from pure autonomic failure due to Lewy body disease) prevented us from analyzing the evidence in this specific population. Although severe, widespread and progressive autonomic failure is specific for MSA, there are very few comparative studies on diagnostic accuracy of autonomic function testing versus other diagnostic tests. A major limitation of the available evidence is an absence of postmortem diagnostic confirmation in the majority of studies. Because most studies were cross‐sectional, including patients with advanced disease stages, the evaluation of the test performance in the first 2 to 3 years from onset (when the sensitivity for a diagnosis of MSA is most required) is poor.
In patients presenting with parkinsonism, a history of the levodopa response is required, as a poor response to levodopa is characteristic of MSA compared to PD. Studies addressing the performance of an acute levodopa challenge test were difficult to interpret, as they analyzed different MSA populations, and used different levodopa doses, assessment methodologies and outcome measures. There is no information on the value of an acute levodopa challenge in de novo drug‐naïve MSA patients. Given the methodological diversity, the proportion of MSA patients in early disease stages with levodopa‐responsive parkinsonism, and the high number of false‐negative cases due to common peripheral side‐effects we conclude that the acute levodopa challenge test cannot assist in the earlier diagnosis of MSA. Consequently, a negative levodopa challenge – when available – should not deter clinicians from initiating chronic levodopa maintenance therapy, until a daily dose of 1000 mg has been tried for at least a month if needed and tolerated. The moderate discriminative power of olfactory testing in distinguishing MSA (where the test is normal) from PD (where olfaction is typically impaired) suggests that it might be useful to support a diagnosis of MSA, despite a lack of blinded data. There are no studies on the efficiency of combined olfactory testing and cardiac sympathetic imaging in differentiating MSA from PD+nOH. Otherwise unexplained neurogenic findings in sphincter EMG within a few years from disease onset are suggestive of MSA. However, due to overlapping denervation patterns between MSA and PD such changes may not support the diagnosis in individual patients. Given some denervation in healthy subjects, the test should be interpreted with caution. Limitations of the sphincter EMG include discomfort for the patient, difficulties in interpreting the results, effects of age, sex, multiple childbirths, and comorbidities such as prostate hypertrophy, bladder neck stenosis, or stress incontinence.
Careful neuropsychological screening is useful to exclude dementia, which is, based on current evidence, rare in MSA but is an essential feature of DLB and PDD. Assessment of global cognitive functions employing the Dementia Rating Scale, or executive functions by applying the FAB may help differentiate patients with MSA from patients with PSP. However, differentiation made on the basis of cognitive state is not likely to be helpful in early stages. There is a need to define a specific cognitive battery with tests whose performance would not be affected by motor disability. 78
VPSG‐documented RBD and severe loss of REM atonia are highly indicative of a neurodegenerative synucleinopathy such as MSA; hence, their absence makes a diagnosis of MSA unlikely. Documentation of inspiratory stridor by home audio recording or vPSG is very specific for MSA. 23 Based on a small number of relevant studies we conclude that vPSG is useful to distinguish patients with MSA from patients with tauopathies and sporadic, symptomatic and genetic ataxias (although RBD has been documented in selected disorders such as SCA3). VPSG cannot assist in the differential diagnosis of MSA vs. other synucleinopathies.
In patients presenting with progressive adult onset ataxia, immune mediated (including paraneoplastic and non‐cancer related disorders), metabolic, toxic, and infectious/postinfectious causes should be excluded. As a second tier, genetic screening for the most common SCAs is recommended, particularly in cases with positive family history or non‐supportive features for MSA. Other rare MSA neurogenetic mimicries have been described in the literature (Table 2) but broad genetic testing beyond the common SCAs is currently not recommended.
Although there are several promising biomarker candidates such as α‐synuclein (RT‐QuIC and PMCA assays allowed differentiation between MSA and Lewy body synucleinopathies) or NfL (that allowed differentiation between MSA and PD, but not MSA versus tauopathies) in CSF and plasma none of them is sufficiently robust to support a diagnosis of MSA. By applying panels with multiple biomarkers, diagnostic accuracy could be improved. The high variability of findings on fluid biomarkers across the literature highlights the need to standardize analytical methods and harmonize standard operating procedures. The validation of current biomarkers in large prospective studies is needed before any wet biomarker can be used for MSA diagnosis. The arginine growth hormone stimulation test provided conflicting results in different MSA cohorts. The role of this and other neuroendocrine tests remains to be defined in future, larger studies.
As with all systematic reviews, this study has several limitations. First, we may have missed original studies due to potential publication bias. Second, we did not report uncertainty of accuracy data, for example, 95% confidence intervals sensitivity and specificity. Third, PPV and NPV must be judged in the light of prevalence, which was not available for most settings. Fourth, data on the same patients could have been published in more than one study; hence, the cumulative number of patients in studies from which the range of diagnostic accuracy measures was derived (Table 1) may not be correct. Fifth, diagnostic test accuracy characteristics alone are not sufficient to inform clinical decision making. Further aspects including the benefits and harms to patients with false negative and false positive results as well as cost effectiveness must be included in the decision‐making process, which may require decision‐analytic modeling approaches. 79
In summary, current best evidence suggests that in patients with parkinsonism suggestive of MSA, evaluation of history of levodopa responsiveness and olfactory function is useful. Neuropsychological testing should be performed to exclude dementia at the time of diagnosis. When MSA‐cerebellar type is suspected, a screening for the common causes of adult onset progressive ataxia is useful. Genetic screening beyond the most common SCAs is not currently recommended. Diagnosing sleep abnormalities is useful in both motor MSA subtypes. The results of pelvic neurophysiology should be interpreted with caution, and the role of this testing is limited due to overlapping finding with PD and other common non‐neurological diseases. Based on current evidence, we conclude that the laboratory work‐up should be extended beyond brain and cardiac imaging and autonomic function tests in selected patients with MSA to improve diagnostic accuracy during lifetime. Cohort studies enrolling patients with MSA within the first 2 years after symptom onset with blinded test results and postmortem diagnostic confirmation are required to generate sufficient evidence on test accuracies in early disease stages.
Appendix
MoDiMSA Study Group: Angelo Antonini (Department of Neuroscience, University of Padua, Italy); Sweta Bajaj (Department of Neurosurgery, Medical University of Innsbruck, Austria), Jee Bang (Department of Neurology, Johns Hopkins University School of Medicine, Baltimore, Maryland, USA), Paolo Barone (Neuroscience Section, Department of Medicine, Surgery and Dentistry “Scuola Medica salernitana,” University of Salerno, Italy), Alfredo Berardelli (IRCCS Neuromed, Pozzilli, IS, Italy; Department of Human Neurosciences, Sapienza University of Rome, Rome, Italy), Daniela Berg (Department of Neurodegeneration and Hertie‐Institute for Clinical Brain Research, University of Tübingen, Tübingen, Germany; Department of Neurology, Christian‐Albrechts‐University Kiel, Kiel, Germany), Italo Biaggioni (Departments of Medicine and Pharmacology, Vanderbilt University Medical Center, Nashville, Tennessee, USA), Bastiaan Bloem (Radboud University Medical Center; Donders Institute for Brain, Cognition and Behaviour; Department of Neurology; Center of Expertise for Parkinson & Movement Disorders; Nijmegen, The Netherlands), David J. Brooks (Neurology Imaging Unit, Department of Medicine, Imperial College London, London, United Kingdom; Department of Nuclear Medicine, Aarhus University, Aarhus, Denmark; Institute of Neuroscience, University of Newcastle upon Tyne, Newcastle University Campus for Ageing and Vitality, Newcastle, United Kingdom), Giovanna Calandra‐Buonaura (IRCCS Istituto delle Scienze Neurologiche di Bologna, Bologna, Italy; DIBINEM, Alma Mater Studiorum, University of Bologna, Bologna, Italy), Carlo Colosimo (Department of Neurological Sciences, Santa Maria University Hospital, Terni, Italy), Pietro Cortelli (IRCCS Istituto delle Scienze Neurologiche di Bologna, Bologna, Italy; DIBINEM, Alma Mater Studiorum, University of Bologna, Bologna, Italy), Joaquim Ferreira (Instituto de Medicina Molecular, Faculdade de Medicina, Universidade de Lisboa, Portugal), Susan Fox (Edmond J. Safra Program in Parkinson's Disease and the Morton and Gloria Shulman Movement Disorders Clinic, Toronto Western Hospital, University Health Network, Toronto, Canada; Division of Neurology, University of Toronto, Toronto, Canada; Krembil Brain Institute, Toronto, Ontario, Canada), Birgit Frauscher (Montreal Neurological Institute and Hospital, McGill University, Montreal, Quebec, Canada), Roy Freeman (Department of Neurology, Beth Israel Deaconess Medical Center, Harvard Medical School, Boston, Massachusetts, USA), Victor Fung (Movement Disorders Unit, Neurology Department, Westmead Hospital, Westmead, NSW, Australia; Sydney Medical School, University of Sydney, Sydney, NSW, Australia), Thomas Gasser (Department of Neurology, Christian‐Albrechts‐University Kiel, Kiel, Germany; German Center for Neurodegenerative Diseases, Tübingen, Germany), Alexander Gerhard (Wolfson Molecular Imaging Centre, University of Manchester, Manchester, United Kingdom; Departments of Nuclear Medicine and Geriatric Medicine, University Hospital Essen, Germany), David Goldstein (Clinical Neurocardiology Section, Clinical Neurosciences Program, Division of Intramural Research, National Institute of Neurological, Disorders and Stroke, National Institutes of Health, Bethesda, Maryland, USA), Mark Hallett (Human Motor Control Section, Medical Neurology Branch, National Institute of Neurological Disorders and Stroke, National Institutes of Health, Bethesda, Maryland, USA), Glenda Halliday (Brain and Mind Centre, Faculty of Medicine and Health, School of Medical Sciences, The University of Sydney, Sydney, NSW, Australia), Günter U. Höglinger (Department of Neurology, Hanover Medical School, Hanover, Germany; German Center for Neurodegenerative Diseases, München, Germany), Henry Houlden (Department of Neuromuscular Disease, UCL Queen Square Institute of Neurology and The National Hospital for Neurology and Neurosurgery, London, United Kingdom; Neurogenetics Laboratory, UCL Queen Square Institute of Neurology and The National Hospital for Neurology and Neurosurgery, London, United Kingdom); Valeria Iodice (Autonomic Unit, National Hospital for Neurology and Neurosurgery, Queen Square/Division of Clinical Neurology, Institute of Neurology, University College London, London, United Kingdom), Horacio Kaufmann (Department of Neurology, Dysautonomia Center, Langone Medical Center, New York University School of Medicine, New York, New York, USA), Thomas Klockgether (Department of Neurology, University of Bonn, Germany; German Center for Neurodegenerative Diseases, Bonn, Germany), Anthony Lang (Division of Neurology, University of Toronto, Toronto, Canada), Helen Ling (Reta Lila Weston Institute of Neurological Studies, UCL Queen Square Institute of Neurology, 1 Wakefield Street, London, United Kingdom; Queen Square Brain Bank for Neurological Disorders, UCL Queen Square Institute of Neurology, London, United Kingdom); Phillip Low (Department of Neurology, Mayo Clinic, Rochester, Minnesota, USA), Irene Litvan (Department of Neurosciences, Parkinson and Other Movement Disorders Center, University of California, San Diego, California, USA), Yasuo Miki (Queen Square Brain Bank for Neurological Disorders, UCL Queen Square Institute of Neurology, London, United Kingdom; Department of Neuropathology, Institute of Brain Science, Hirosaki University Graduate School of Medicine, Hirosaki, Japan), Takashi Nomura (Nomura Neuro Sleep Clinic, Tottori, Japan), Satoshi Orimo (Department of Neurology, Kanto Central Hospital, Tokyo, Japan), Tetsutaro Ozawa (Department of Neurology, Uonuma Institute of Community Medicine, Niigata University Medical and Dental Hospital, Japan), Alexander Pantelyat (Department of Neurology, Johns Hopkins University School of Medicine, Baltimore, Maryland, USA), Maria Teresa Pellecchia (Neuroscience Section, Department of Medicine, Surgery and Dentistry “Scuola Medica salernitana”, University of Salerno, Italy), Ron Postuma (Department of Neurology, Montreal General Hospital, Montreal, Quebec, Canada), Niall Quinn (UCL Institute of Neurology, Queen Square, London, United Kingdom), Olivier Rascol (Centre Référence Maladie Rare AMS, CIC 1436, ToNIC UMR 1214, Department of Neurosciences, Centre COEN NeuroToul; University Hospital of Toulouse, INSERM, University of Toulouse 3; Toulouse; FRANCE), Milos Sabanovic (Neurology Clinic, Clinical Center of Serbia, School of Medicine, University of Belgrade, Serbia), Ryuji Sakakibara (Neurology, Internal Medicine, Sakura Medical Center, Toho University, Sakura, Japan), Christina Sampaio (Cure Huntington's Disease InitiativeEl (CHDI) Management/CHDI Foundation, Princeton, New Jersey, USA and Laboratorio de Farmacologia Clinica, Faculdade de Medicinade Lisboa, Portugal), Jeremy D. Schmahmann (Ataxia Center, Laboratory for Neuroanatomy and Cerebellar Neurobiology, Department of Neurology, Massachusetts General Hospital, Harvard Medical School, Boston, Massachusetts, USA), Sonja Scholz (Department of Neurology, Johns Hopkins University School of Medicine, Baltimore, Maryland, USA; Neurodegenerative Diseases Research Unit, National Institute of Neurological Disorders and Stroke, National Institutes of Health, Bethesda, Maryland, USA), Jean‐Michel Senard (Institut des maladies métaboliques et cardiovasculaires, université de Toulouse, CHU Rangueil, Toulouse, France; Pharmacology department, Faculty of Medicine, Toulouse, France), Manu Sharma (Centre for Genetic Epidemiology, Institute for Clinical Epidemiology and Applied Biometry, University of Tubingen, Germany), Wolfgang Singer (Department of Neurology, Mayo Clinic, Rochester, Minnesota, USA), Maria Stamelou (Parkinson's disease and Movement Disorders Department, HYGEIA Hospital, and Aiginiteion Hospital, University of Athens, Greece; Philipps University Marburg, Germany and European University of Cyprus, Nicosia, Cyprus), Atsushi Takeda (Department of Neurology, National Hospital Organization Sendai Nishitaga Hospital, Miyagi, Japan), Eduardo Tolosa (Centrode Investigación Biomédica en Red sobre Enfermedades Neurodegenerativas (CIB ERNED) Hospital Clínic, IDIBAPS, Universitat de Barcelona, Catalonia, Span; Movement Disorders Unit, Neurology Service, Hospital Clínic de Barcelona, Catalonia, Spain), Shoji Tsuji (Department of Molecular Neurology, The University of Tokyo, Graduate School of Medicine, Tokyo, Japan; International University of Health and Welfare, Chiba, Japan), Luca Vignatelli (DIBINEM, Alma Mater Studiorum, University of Bologna, Bologna, Italy), Uwe Walter (69 Department of Neurology, University of Rostock, Rostock, Germany; German Center for Neurodegenerative Diseases, Rostock, Germany), Tom Warner (Queen Square Brain Bank for Neurological Disorders, UCL Queen Square Institute of Neurology, London, United Kingdom), Hirohisa Watanabe (Department of Neurology, Nagoya University Graduate School of Medicine, Nagoya, Japan), Daniel Weintraub (Department of Psychiatry, University of Pennsylvania, Philadelphia, Pennsylvania, USA; Parkinson's Disease and Mental Illness Research, Education and Clinical Centers (Philadelphia Parkinson's Disease Research, Education and Clinical Center (PADRECC) and Mental Illness Research Education Clinical, Centers of Excellence (MIRECC)), Philadelphia Veterans Affairs Medical Center, Philadelphia, Pennsylvania, USA), Uwe Siebert (Department of Public Health, Health Services Research and Health Technology Assessment, Institute of Public Health, Medical Decision Making and Health Technology Assessment, UMIT‐University for Health Sciences, Medical Informatics and Technology, Hall in Tirol, Austria; Department of Health Policy and Management, Center for Health Decision Science, Harvard Chan School of Public Health, Boston, Massachusetts, USA; Department of Radiology, Institute for Technology Assessment, Massachusetts General Hospital, Harvard Medical School, Boston, Massachusetts, USA),Werner Poewe (Department of Neurology, Medical University of Innsbruck, Austria).
Author Roles
(1) Research project: A. Conception, B. Organization, C. Execution; (2) Manuscript: A. Writing of the first draft, B. Review and Critique.
I.S.: 1A,B,C; 2A
A.F.: 1A,B,C; 2B
V.K.: 1A,B; 2B
F.K.: 1A,B,C; 2B
W.G.M.: 1A; 2B
J.A.P.: 1A,B; 2B
J.N.P.: 1A,B; 2B
K.S.: 1A,B; 2B
G.K.W.: 1A,B,C; 2B
A.A.: 1A,B; 2B
S.B.: 1B,C; 2B
J.B.: 1A,B; 2B
P.B.: 1A,B; 2B
A.B.: 1A,B; 2B
D.B.: 1A,B; 2B
I.B.: 1A,B; 2B
B.B.: 1A,B; 2B
D.J.B.: 1A,B; 2B
G.C.‐B.: 1A,B; 2B
C.C.: 1A,B; 2B
P.C.: 1A,B; 2B
J.F.: 1A,B; 2B
S.F.: 1A,B; 2B
B.F.: 1A,B; 2B
R.F.: 1A,B; 2B
V.F.: 1A,B; 2B
T.G.: 1A,B; 2B
A.G.: 1A,B; 2B
D.G.: 1A,B; 2B
M.H.: 1A,B; 2B
G.H.: 1A,B; 2B
G.U.H.: 1A,B; 2B
H.H.: 1A,B; 2B
V.I.: 1A,B; 2B
H.K.: 1A,B; 2B
T.K.: 1A,B; 2B
A.L.: 1A,B; 2B
H.L.: 1A,B; 2B
P.L.: 1A,B; 2B
I.L.: 1A,B; 2B
Y.M.: 1A,B; 2B
T.N.: 1A,B; 2B
S.O.: 1A,B; 2B
T.O.: 1A,B; 2B
A.P.: 1A,B; 2B
M.T.P.: 1A,B; 2B
R.P.: 1A,B; 2B
N.Q.: 1A,B; 2B
O.R.: 1A,B; 2B
M.S.: 1B,C; 2B
R.S.: 1A,B; 2B
C.S.: 1A,B; 2B
J.D.S.: 1A,B; 2B
S.S.: 1A,B,C; 2B
J.M.S.: 1A,B; 2B
M.S.: 1A,B; 2B
W.S.: 1A,B; 2B
M.S.: 1A,B; 2B
A.T.: 1A,B; 2B
E.T.: 1A,B; 2B
S.T.: 1A,B; 2B
L.V.: 1A,B; 2B
U.W.: 1A,B; 2B
T.W.: 1A,B; 2B
H.W.: 1A,B; 2B
D.W.: 1A,B; 2B
U.S.: 1A,B; 2B
W.P.: 1A,B; 2B
Disclosures
Ethical Compliance Statement: The authors confirm that the approval of an institutional review board was not required for this work. No informed consent was required. We confirm that we have read the Journal's position on issues involved in ethical publication and affirm that this work is consistent with those guidelines.
Funding Sources and Conflict of Interest: No specific funding was received for this work. The authors declare that there are no conflicts of interest relevant to this work.
Financial Disclosures for the Previous 12 months: Dr J. Bang reports serving on the Advisory Board of Wave Life Science. Dr P. Barone reports funding from Lundbeck, UCB, Zambon, Chiesi, Roche, Bial, and Biogen (scientific advisory boards, speakter activities). Dr D. Berg reports consultancies/advisory boards fees from Biogen, BIAL, UCB Pharma GmbH and Zambon, honoraria from AbbVie, Biogen, BIAL, UCB Pharma GmbH, Zambon, Desitin and GE and grants from Deutsche Forschung sgemeinschaft (DFG), German Parkinson's Disese Association (dPV), Bundesministerium für Bildung und Forschung (BMBF), Parkinson Fonds Deutschland GmbH, UCB Pharma GmbH, EU, Novartis Pharma GmbH, Ludbeck, and Damp foundation. Dr I. Biaggioni reports consultancy fees from Theravance Biopharma. Dr B. Bloem reports consultancy fees from Biogen, AbbVie, Walk and Path and UCB, grants/research support from Netherlands Organization for Scientific research, Michael J Fox Foundation, Parkinson Vereniging, Parkinsons's Foundation, Gatsby Foundation, Verily Life Sciencies, Horizon 2020, Topsector Life Sciencies and Health, Stichting Parkinson Fonds, UCB and AbbVie and fees for speaking at conferences from AbbVie, Zambon, BIAL and Roche. Dr C. Colosimo reports grant from Ipsen. Dr A. Fanciulli reports royalties from Springer Nature and Thieme Verlag, speaker fees and honoraria from the Ordensklinikum Linz, Austrian Parkinson Society, International Parkinson Disease and Movement Disorder Society and Theravance Biopharma and research grants from the Stichting ParkinsonFond, the MSA Coalition and the Österreichischer Austauschdienst. Dr J. Ferreira reports grants from GlaxoSmithKline, Grunenthal, Fundacao MSD (Portugal), TEVA, MSD, Allergan, Novartis, and Medtronic, serving at advisory board for BIAL, testimony to Novartis and consultancy fees from GlaxoSmithKline, Novartis, TEVA, Lundbeck, Solvay, BIAL, Merck‐Serono, Mez, Ipsen, Biogen, Acadia, Allergan, AbbVie, and Sunovion Pharma. Dr S. Fox reports research Funding from Michael J Fox Foundation for Parkinson Research, NIH (Dystonia Coalition), CIHR, and Parkinson Canada, honoraria from the International Parkinson and Movement Disorder Society and American Academy of Neurology, Site PI for clinical trials for Biotie, Cynapsus, Eisai, and Revance, consultancy/speaker fees from Acadia, Atuka, Sunovion, Teva, and Paladin and royalties from Oxford University Press. Dr B. Frauscher reports advisory board fees and speaker engagements from Eisai and UCB Pharma, discovery grant from NSERC, and infrastructure grant from CFI. Dr V. Fung reports unrestricted research grants from Michael J. Fox Foundation, Abbvie and Merz. He is on Advisory Boards and/or has received travel grants from Abbvie, Allergan, Cavion, Ipsen, Merz, Praxis, Seqirus, Stada, Teva and UCB, and receives royalties from Health Press Ltd. Dr T. Gasser reports grants from Bundesministerium für Bildung und Forschung (BMBF), Deutsche Forschung sgemeinschaft (DFG), speaker honoraria from UCB Pharma, Novartis, Teva and MedUpdate and serving as a chairman of the scientific advisory board of the “Joint Programming for Neurodegenerative Diseases” program, funded by the European Commission. Dr A. Gerhard reports research grant from MRC UK. Dr M. Hallett is an inventor of patents held by NIH for an immunotoxin for the treatment of focal movement disorders and the H‐coil for magnetic stimulation; in relation to the latter, he has received license fee payments from the NIH (from Brainsway). He is on the Medical Advisory Boards of CALA Health and Brainsway. He has research grants from Allergan for studies of methods to inject botulinum toxins, Medtronic, Inc. for a study of DBS for dystonia, and CALA Health for studies of a device to suppress tremor. Dr G. Halliday is supported in part by funding from the National Health and Medical Research of Australia, MJ Fox Foundation, Shake‐it‐up Australia, MSA Coalition, and the University of Sydney. Dr H. Kaufmann reports compensation as a consultant/advisory board member for Lundbeck and Theravance Biopharma Inc. Dr. Klockgether receives/has received research support from the Deutsche Forschungsgemeinschaft (DFG), the Bundesministerium für Bildung und Forschung (BMBF), the Bundesministerium für Gesundheit (BMG), the Robert Bosch Foundation, the European Union (EU), and the National Institutes of Health (NIH). He has received consulting fees from Biohaven, Roche, UBC, Uniqure, and Vico Therapeutics. He has received a speaker honorarium from Novartis and Bayer. Dr V. Kostic has received speaker honoraria from Roche and Alkaloid and receives research supports from the Swiss Pharm, Serbian Ministry of Education, Science and Development and Serbian Academy of Sciences and Art. Dr. F. Krismer reports a research grant from the MSA Coalition. Dr. Lang has served as an advisor for Abbvie, AFFiRis, Biogen, Janssen, Lilly, Lundbeck, Merck, Paladin, Roche, Sun Pharma, Theravance, and Corticobasal Degeneration Solutions, received honoraria from Sun Pharma, AbbVie and Sunovion, received grants from Brain Canada, Canadian Institutes of Health Research, Edmond J Safra Philanthropic Foundation, Michael J. Fox Foundation, the Ontario Brain Institute, National Parkinson Foundation, Parkinson Society Canada, and W. Garfield Weston Foundation and received publishing royalties from Saunders, Wiley‐Blackwell, Johns Hopkins Press, and Cambridge University Press. Dr H. Ling reports research grant from CBD Solutions. Dr I. Litvan research is supported by the National Institutes of Health grants: 5P50AG005131–33, 2R01AG038791‐06A, U01NS090259, U01NS100610, U01NS80818, R25NS098999, P20GM109025, U19 AG063911‐1, 1R21NS114764‐01A1, Parkinson Study Group, Michael J Fox Foundation, Parkinson Foundation, Lewy Body Association, Roche, Abbvie, Biogen, EIP‐ Pharma and Biohaven Pharmaceuticals. She was member of a Lundbeck Advisory Board and Corticobasal Degeneration Solutions. She is a Chief Editor of Frontiers in Neurology. Dr W. G. Meissner has received fees for editorial activities with Springer Nature and Elsevier, has served as advisor for Lundbeck and Biohaven, and has received teaching honoraria from UCB. Dr Y. Miki reports grant from the Sakurai Memorial Fund for Medical Research. Dr A. Palma reports salary from Novartis Gene Therapies, research funding from the NIH, Michael J. Fox Foundation, MSA Coalition, Familial Dysautonomia Foundation, FDA, and Biogen. He is advisory board member for Lundbeck, Biogen, PTC Therapeutics, Astellas, and Dr. Reddy's Laboratories, managing editor of Clinical Autonomic Research and principal investigator in MSA studies funded by Biohaven Pharmaceuticals, Theravance, and Biogen. Dr J. Panicker is supported in part by funding from the United Kingdom's Department of Health NIHR Biomedical Research Centres. Dr W. Poewe reports personal fees from Alterity, AbbVie, Affiris, Astra Zeneca, BIAL, Biogen, Britannia, Lilly, Lundbeck, Neuroderm, Neurocrine, Denali Pharmaceuticals, Novartis, Orion Pharma, Roche, Takeda, Teva, UCB and Zambon (consultancy and lecture fees in relation to clinical drug development programmes for PD), royalties from Thieme, Wiley Blackwell, Oxford University Press and Cambridge University Press and grant support from MJFF, EU FP7 and Horizon 2020. Dr R. Postuma reports grants and personal fees from Fonds de la Recherche en Sante, the Canadian Institute of Health Research, Parkinson Canada, the Weston‐Garfield Foundation, the Michael J. Fox Foundation, the Webster Foundation, and personal fees from Takeda, Roche/Prothena, Teva Neurosciences, Novartis, Biogen, Boehringer Ingelheim, Theranexus, GE HealthCare, Jazz Pharmaceuticals, Abbvie, Jannsen, Curasen and Otsuko. Dr C. Sampaio reports consultancy honoraria from Kyowa Kirin, Pinteon Pharmaceuticals, Neuraly, Green Valley Pharmaceuticals, and Pfizer. Dr J. Schmahmann reports clinical trial support from Biohaven Pharmaceuticals and consultancy to Cadent Therapeutics. Dr K. Seppi reports personal fees from Teva, UCB, Lundbeck, AOP Orphan Pharmaceuticals AG, Roche, Grünenthal and Abbvie, honoraria from the International Parkinson and Movement Disorders Society, research grants from FWF Austrian Science Fund, Michael J. Fox Foundation, and International Parkinson and Movement Disorder Society. Dr M. Stamelou reports serving on the advisory board for Biogen and PD neurotechnology, advisory services to UCB, speaker honoraria from MDS, EAN, UCB, Abbvie, research funding from MJFF (genetic PPMI), and honoraria from Oxford and Cambridge University Press. She is an associate editor in MDJ. Dr E. Tolosa reports honoraria for consultancy from TEVA, Bial, Prevail, Boehringer Ingelheim, Roche and Biogen and funding for research from Spanish Network for Research on Neurodegenerative Disorders (CIBERNED) Instituto Carlos III (ISCIII), and The Michael J. Fox Foundation for Parkinson's Research (MJFF). Dr S. Tsuji reports grant from Nobelpharma Co., Ltd. and consultancies from i‐TRS, Ono Pharmaceutical, Daiichi Sankyo Company, Sanofi K. K., and Aeglea Biotherapeutics. Dr U. Walter reports grants from Bundesministerium für Bildung und Forschung, Germany, and personal fees from Merz Pharma, Bristol‐Myers Squibb, Daiichi Sankyo, Pfizer, Thieme, and Elsevier Press. Dr T. Warner reports grants received from Medical Research Council, Reta Lila Weston Medical Trust, Corticobasal Degeneration Solutions Inc., UCLH BRC, ABN and RoseTrees. Dr D. Weintraub reports research funding or support from Michael J. Fox Foundation for Parkinson's Research, Alzheimer's Therapeutic Research Initiative (ATRI), Alzheimer's Disease Cooperative Study (ADCS), the International Parkinson and Movement Disorder Society (IPMDS), and National Institute on Aging (NIA), honoraria for consultancy from Acadia, Aptinyx, CHDI Foundation, Clintrex LLC (Alkahest, Aptinyx, Avanir, Otsuka), Eisai, Great Lake Neurotechnologies, Janssen, Sage, Scion, Signant Health and Sunovion and license fee payments from the University of Pennsylvania for the QUIP and QUIP‐RS. Dr G. K. Wenning reports grants from Biogen, Biohaven, Minoryx, Lundbeck and Theravance. Drs A. Antonini, Dr S. Bajaj, A. Berardelli, Dr D. Brooks, Dr G. Calandra Buonaura, Dr P. Cortelli, Dr R. Freeman, Dr D. Goldstein, Dr G. Hoglinger, Dr H. Houlden, Dr V. Iodice, Dr P. Low, Dr T. Nomura, Dr S. Orimo, Dr T. Ozawa, Dr A. Pantelyat, Dr M. T. Pellecchia, Dr N. Quinn, Dr O. Rascol, Dr M. Sabanovic, Dr R. Sakakibara, Dr S. Scholz, Dr J. M. Senard, Dr M. Sharma, Dr U. Siebert, Dr W. Singer, Dr I. Stankovic, Dr A. Takeda, Dr L. Vignatelli, Dr H. Watanabe report no financial disclosures for the past 12 months.
Supporting information
Table S1. Summary of studies on biomarkers in MSA. Search terms: (“multiple system atrophy” OR MSA OR “olivopontocerebellar atrophy” OR OPCA OR “striatonigral degeneration” OR SND OR “shy drager syndrome”) AND (blood OR plasma OR “cerebrospinal fluid” OR CSF). A total of 1984 papers were identified in Pubmed using the search terms on July 20th, 2019. A total of 77 relevant papers were included in the analysis.
Table S2. Summary of studies on genetic screening in MSA. Search terms: (“multiple system atrophy” OR MSA OR “olivopontocerebellar atrophy” OR OPCA OR “striatonigral degeneration” OR SND OR “shy drager syndrome”) AND (genetic OR genetics OR familial OR mutation). A total of 2144 papers were identified in Pubmed using the search terms on July 20th, 2019. A total of 95 relevant papers were included in the analysis.
Table S3. Summary of studies on neuroendocrine tests in MSA. Search terms: (“multiple system atrophy” OR MSA OR “olivopontocerebellar atrophy” OR OPCA OR “striatonigral degeneration” OR SND OR “shy drager syndrome”) AND (neuroendocrine OR arginine OR “growth hormone” OR GH OR clonidine). A total of 196 papers were identified in Pubmed using the search terms on July 20th, 2019. A total of 7 relevant papers were included in the analysis.
Table S4. Summary of studies on neurophysiological tests in MSA. Search terms: (“multiple system atrophy” OR MSA OR “olivopontocerebellar atrophy” OR OPCA OR “striatonigral degeneration” OR SND OR “shy drager syndrome”) AND (neurophysiology OR “evoked potentials” OR EVP OR electromyography OR EMG). A total of 356 papers were identified in Pubmed using the search terms on July 20th, 2019. A total of 26 relevant papers were included in the analysis.
Table S5. Summary of studies on neuropsychological tests in MSA. Search terms: (“multiple system atrophy” OR MSA OR “olivopontocerebellar atrophy” OR OPCA OR “striatonigral degeneration” OR SND OR “shy drager syndrome”) AND (neuropsychology OR neuropsychological OR dementia OR cognition OR cognitive OR frontal‐executive OR memory). A total of 1396 papers were identified in Pubmed using the search terms on July 20th, 2019. A total of 14 relevant papers were included in the analysis.
Table S6. Summary of sleep studies in MSA. Search terms: (“multiple system atrophy” OR MSA OR “olivopontocerebellar atrophy” OR OPCA OR “striatonigral degeneration” OR SND OR “shy drager syndrome”) AND (sleep OR “REM sleep behavior disorder” OR RBD OR REM sleep without atonia). A total of 455 papers were identified in Pubmed using the search terms on July 20th, 2019. A total of 3 relevant papers were included in the analysis.
Table S7. Summary of other tests in MSA. Relevant papers on other tests were searched manually. A total of 13 relevant papers were included in the analysis.
Acknowledgments
One of the coauthors of this research (Dr S. Scholz) was supported in part by the Intramural Research Program of the National Institutes of Health (National Institute of Neurological Disorders and Stroke; project number: 1ZIANS003154).
Members of the MoDiMSA Study Group are listed in the Appendix.
References
- 1. Gilman S, Wenning GK, Low PA, et al. Second consensus statement on the diagnosis of multiple system atrophy. Neurology 2008;71(9):670–676. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Koga S, Aoki N, Uitti R, et al. When DLB, PD, and PSP masquerade as MSA. Neurology 2015;85(5):404–412. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Miki Y, Foti SC, Asi YT, et al. Improving diagnostic accuracy of multiple system atrophy: a clinicopathological study. Brain 2019;142:3294–3305. [DOI] [PubMed] [Google Scholar]
- 4. Pellecchia MT, Stankovic I, Fanciulli A, et al. Can autonomic testing and imaging contribute to the early diagnosis of MSA? A systematic review and recommendations by the MDS multiple system atrophy (MoDiMSA) study group. Mov Disord Clin Pract 2020;7(7):750–762. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Fanciulli A, Goebel G, Lazzeri G, et al. Early distinction of Parkinson‐variant multiple system atrophy from Parkinson's disease. Mov Disord 2019;34(3):440–441. [DOI] [PubMed] [Google Scholar]
- 6. Petrovic IN, Ling H, Asi Y, Ahmed Z, et al. Multiple system atrophy – Parkinsonism with slow progression and prolonged survival: a diagnostic catch. Mov Disord 2012;27(9):1186–1190. [DOI] [PubMed] [Google Scholar]
- 7. Stankovic I, Quinn N, Vignatelli L, et al. A critique of the second consensus criteria for multiple system atrophy. Mov Disord 2019;34(7):975–984. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Quinn N. Multiple system atrophy – the nature of the beast. J Neurol Neurosurg Psychiatry 1989;52:78–89. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Gilman S, Low PA, Quinn N, et al. Consensus statement on the diagnosis of multiple system atrophy. J Neurol Sci 1999;163(1):94–98. [DOI] [PubMed] [Google Scholar]
- 10. Low PA, Reich SG, Jankovic J, et al. Natural history of multiple system atrophy in the USA: a prospective cohort study. Lancet Neurol 2015;14(7):710–719. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Wenning GK, Geser F, Krismer F, et al. The natural history of multiple system atrophy: a prospective European cohort study. Lancet Neurol 2013;12(3):264–274. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Meissner WG, Laurencin C, Tranchant C, et al. Outcome of deep brain stimulation in slowly progressive multiple system atrophy: a clinico‐pathological series and review of the literature. Parkinsonism Relat Disord 2016;24:69–75. [DOI] [PubMed] [Google Scholar]
- 13. Vasta R, Nicoletti A, Mostile G, et al. Side effects induced by the acute levodopa challenge in Parkinson's disease and atypical parkinsonisms. PLoS One. 2017;12(2):e0172145. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Krismer F, Pinter B, Mueller C, et al. Sniffing the diagnosis: olfactory testing in neurodegenerative parkinsonism. Parkinsonism Relat Disord 2017;35:36–41. [DOI] [PubMed] [Google Scholar]
- 15. Fujita H, Suzuki K, Numao A, et al. Usefulness of cardiac MIBG scintigraphy, olfactory testing and substantia nigra hyperechogenicity as additional diagnostic markers for distinguishing between Parkinson's disease and atypical parkinsonian syndromes. PLoS One. 2016;11(11):e0165869. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Herting B, Schulze S, Reichmann H, Haehner A, Hummel T. A longitudinal study of olfactory function in patients with idiopathic Parkinson's disease. J Neurol 2008;255(3):367–370. [DOI] [PubMed] [Google Scholar]
- 17. Boeve B, Silber M, Ferman T, et al. Clinicopathologic correlations in 172 cases of rapid eye movement sleep behavior disorder with or without a coexisting neurologic disorder. Sleep Med 2013;14(8):754–762. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Postuma RB, Iranzo A, Hogl B, et al. Risk factors for neurodegeneration in idiopathic rapid eye movement sleep behavior disorder: a multicenter study. Ann Neurol 2015;77(5):830–839. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Iranzo A, Santamarıa J, Rye DB, et al. Characteristics of idiopathic REM sleep behavior disorder and that associated with MSA and PD. Neurology 2005;65(2):247–252. [DOI] [PubMed] [Google Scholar]
- 20. Ferri R, Manconi M, Plazzi G, et al. A quantitative statistical analysis of the submentalis muscle EMG amplitude during sleep in normal controls and patients with REM sleep behavior disorder. J Sleep Res 2008;17(1):89–100. [DOI] [PubMed] [Google Scholar]
- 21. Alonso‐Navarro H, Garcia‐Martin E, Agubdez JA, Jimenez‐Jimenez FJ. Association between restless legs syndrome and other movement disorders. Neurology 2019;92(20):948–964. [DOI] [PubMed] [Google Scholar]
- 22. Postuma RB, Berg D, Stern M, et al. MDS clinical diagnostic criteria for Parkinson's disease. Mov Disord 2015;30(12):1591–1601. [DOI] [PubMed] [Google Scholar]
- 23. Cortelli P, Calandra‐Buonaura G, Benarroch EE, et al. Stridor in multiple system atrophy consensus statement on diagnosis, prognosis, and treatment. Neurology 2019;93(14):630–639. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Gandor F, Vogel A, Claus I, et al. Laryngeal movement disorders in multiple system atrophy: a diagnostic biomarker? Mov Disord 2020;35(12):2174–2183. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Giannini G, Calandra‐Buonaura G, Mastrolilli F, et al. Early stridor onset and stridor treatment predict survival in 136 patients with MSA. Neurology 2016;87(13):1375–1383. [DOI] [PubMed] [Google Scholar]
- 26. Pramstaller PP, Wenning GK, Smith SJM, Beck R, Quinn NP, Fowler CJ. Nerve conduction studies, skeletal muscle EMG, and sphincter EMG in multiple system atrophy. J Neurol Neurosurg Psychiatry 1995;58(5):618–621. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Sakakibara R, Hattori T, Uchiyama T, Yamanishi T. Videourodynamic and sphincter motor unit potential analyses in Parkinson's disease and multiple system atrophy. J Neurol Neurosurg Psychiatry 2001;71(5):600–606. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Paviour DC, Williams D, Fowler CJ, Quinn NP, Lees AJ. Is sphincter electromyography a helpful investigation in the diagnosis of multiple system atrophy? A retrospective study with pathological diagnosis. Mov Disord 2005;20(11):1425–1430. [DOI] [PubMed] [Google Scholar]
- 29. Tison F, Arne P, Sourgen C, Chrysostome V, Yeklef F. The value of external anal sphincter electromyography for the diagnosis of multiple system atrophy. Mov Disord 2000;15(6):1148–1157. [DOI] [PubMed] [Google Scholar]
- 30. Yamamoto T, Asahina M, Yamanaka Y, et al. The utility of post‐void residual volume versus sphincter electromyography to distinguish between multiple system atrophy and Parkinson's disease. PLoS One 2017;12(1):e0169405. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Podnar S, Fowler CJ. Sphincter electromyography in diagnosis of multiple system atrophy: Technical issues. Muscle Nerve 2004;29(1):151–156. [DOI] [PubMed] [Google Scholar]
- 32. Yamamoto T, Sakakibara R, Uchiyama T, et al. When is Onuf's nucleus involved in multiple system atrophy? A sphincter electromyography study. J Neurol Neurosurg Psychiatry 2005;76(12):1645–1649. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Cai Z‐Y, Niu X‐T, Pan J, Ni P‐Q, Wang X, Shao B. The value of the bulbocavernosus reflex and pudendal nerve somatosensory evoked potentials in distinguishing between multiple system atrophy and Parkinson's disease at an early stage. Acta Neurol Scand 2016;136(3):195–203. [DOI] [PubMed] [Google Scholar]
- 34. Kofler M, Muller J, Wenning GK, et al. The auditory startle reaction in parkinsonian disorders. Mov Disord 2001;16(1):62–71. [DOI] [PubMed] [Google Scholar]
- 35. Koga S, Parks A, Uitti RJ, van Gerpen JA , Cheshire WP, Wszolek ZK, Dickson DW. Profile of cognitive impairment and underlying pathology in multiple system atrophy. Mov Disord 2017;32(3):405–413. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Stankovic I, Krismer F, Jesic A, et al. Cognitive impairment in multiple system atrophy: a position statement by the neuropsychology task force of the MDS multiple system atrophy (MoDiMSA) Study Group. Mov Disord 2014;29(7):857–867. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Hoglinger G, Respondek G, Stamelou M, et al. Clinical diagnosis of progressive supranuclear palsy: the Movement Disorder Society criteria. Mov Disord 2017;32(6):853–864. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Mckeith IG, Boeve BF, Dickson DW, Halliday G, et al. Diagnosis and management of dementia with Lewy bodies fourth consensus report of the DLB consortium. Neurology 2017;89(1):88–100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Dubois B, Burn D, Goetz C, et al. Diagnostic procedures for Parkinson's disease dementia: recommendations from the Movement Disorder Society task force. Mov Disord 2007;22(16):2314–2324. [DOI] [PubMed] [Google Scholar]
- 40. Kao A, Racine C. Cognitive and neuropsychiatric profile of the synucleinopathies: Parkinson's disease, dementia with Lewy bodies and multiple system atrophy. Alzheimer Dis Assoc Disord 2009;23(4):365–370. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Brown RG, Lacomblez L, Landwehrmeyer BG, et al. Cognitive impairment in patients with multiple system atrophy and progressive supranuclear palsy. Brain 2010;133(8):2382–2393. [DOI] [PubMed] [Google Scholar]
- 42. Stamelou M, Diehl‐Schmid J, Hapfelmeier A, Kontaxopoulou D, Stefanis L. The frontal assessment battery is not useful to discriminate progressive supranuclear palsy from frontotemporal dementias. Parkinsonism Relat Disord 2015;21(10):1264–1268. [DOI] [PubMed] [Google Scholar]
- 43. Fiorenzato E, Weis L, Falup‐Pecurariu C, et al. Montreal Cognitive Assessment (MoCA) and Mini‐Mental State Examination (MMSE) performance in progressive supranuclear palsy and multiple system atrophy. J Neural Transm 2016;123(12):1435–1442. [DOI] [PubMed] [Google Scholar]
- 44. Santangelo G, Cuoco S, Pellecchia MT, Erro R, Barone P, Picillo M. Comparative cognitive and neuropsychiatric profiles between Parkinson's disease, multiple system atrophy and progressive supranuclear palsy. J Neurol 2018;265(11):2602–2613. [DOI] [PubMed] [Google Scholar]
- 45. Santangelo G, Cuoco S, Picillo M, et al. Evolution of neuropsychological profile in motor subtypes of multiple system atrophy. Parkinsonism Relat Disord 2020;70:67–73. [DOI] [PubMed] [Google Scholar]
- 46. Auzou N, Dujardin K, Biundo R, et al. Diagnosing dementia in multiple system atrophy by applying Movement Disorder Society diagnostic criteria for Parkinson's disease dementia. Parkinsonism Relat Disord 2015;21(10):1273–1277. [DOI] [PubMed] [Google Scholar]
- 47. Mollenhauer B, Locascio JJ, Schulz‐Schaeffer W, Sixel‐Döring F, Trenkwalder C, Schlossmacher MG. α‐Synuclein and tau concentrations in cerebrospinal fluid of patients presenting with parkinsonism: A cohort study. Lancet Neurol 2011;10(3):230–240. [DOI] [PubMed] [Google Scholar]
- 48. Magdalinou NK, Paterson RW, Schott JM, et al. A panel of nine cerebrospinal fluid biomarkers may identify patients with atypical parkinsonian syndromes. J Neurol Neurosurg Psychiatry 2015;86(11):1240–1247. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Shi M, Bradner J, Hancock AM, et al. Cerebrospinal fluid biomarkers for Parkinson disease diagnosis and progression. Ann Neurol 2011;69(3):570–580. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Hall S, Öhrfelt A, Constantinescu R, et al. Accuracy of a panel of 5 cerebrospinal fluid biomarkers in the differential diagnosis of patients with dementia and/or parkinsonian disorders. Arch Neurol 2012;69(11):1445–1452. [DOI] [PubMed] [Google Scholar]
- 51. Aerts MB, Esselink RAJ, Abdo WF, Bloem BR, Verbeek MM. CSF α‐synuclein does not differentiate between parkinsonian disorders. Neurobiol Aging 2012;33(2):430.e1–430.e3. [DOI] [PubMed] [Google Scholar]
- 52. Herbert MK, Eeftens JM, Aerts MB, et al. CSF levels of DJ‐1 and tau distinguish MSA patients from PD patients and controls. Parkinsonism Relat Disord 2014;20(1):112–115. [DOI] [PubMed] [Google Scholar]
- 53. Rossi M, Candelise N, Baiardi S, et al. Ultrasensitive RT‐QuIC assay with high sensitivity and specificity for Lewy body‐associated synucleinopathies. Acta Neuropathol 2020;140(1):49–62. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Yamasaki XTR, Holmes BB, Furman JL, et al. Parkinson's disease and multiple system atrophy have distinct alpha‐synuclein seed characteristics. J Biol Chem 2019;294(3):1045–1058. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Shahnawaz M, Mukherjee A, Pritzkow S, et al. Discriminating α‐synuclein strains in Parkinson's disease and multiple system atrophy. Nature 2020;578(7794):273–277. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Singer W, Schmeichel AM, Shahnawaz M, et al. Alpha‐synuclein oligomers and neurofilament light chain in spinal fluid differentiate multiple system atrophy from Lewy body synucleinopathies. Ann Neurol 2020;88:503–512. 10.1002/ana.25824. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Yu Z, Shi M, Stewart T, et al. Reduced oligodendrocyte exosome secretion in multiple system atrophy involves SNARE dysfunction. Brain 2020;143(6):1780–1797. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Lee PH, Lee G, Park HJ, Bang OY, Joo IS, Huh K. The plasma alpha‐synuclein levels in patients with Parkinson's disease and multiple system atrophy. J Neural Transm 2006;113(10):1435–1439. [DOI] [PubMed] [Google Scholar]
- 59. Holmberg B, Rosengren L, Karlsson JE, Johnels B. Increased cerebrospinal fluid levels of neurofilament protein in progressive supranuclear palsy and multiple system atrophy compared with Parkinson's disease. Mov Disord 1998;13(1):70–77. [DOI] [PubMed] [Google Scholar]
- 60. Hansson O, Janelidze S, Hall S, Magdalinou N, Lees AJ. Blood‐based NfL: a biomarker for differential diagnosis of parkinsonian disorder. Neurology 2017;88(10):930–937. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61. Bech S, Hjermind LE, Salvesen L, et al. Amyloid‐related biomarkers and axonal damage proteins in parkinsonian syndromes. Parkinsonism Relat Disord 2012;18(1):69–72. [DOI] [PubMed] [Google Scholar]
- 62. Mollenhauer B, Bibl M, Esselmann H, Steinacker P, Trenkwalder C, Wiltfang J, Otto M. Tauopathies and synucleinopathies: do cerebrospinal fluid beta‐amyloid peptides reflect disease‐specific pathogenesis? J Neural Transm 2007;114(7):919–927. [DOI] [PubMed] [Google Scholar]
- 63. Vallelunga A, Ragusa M, Di Mauro S, et al. Identification of circulating microRNAs for the differential diagnosis of Parkinson's disease and multiple system atrophy. Front Cell Neurosci 2014;8:156. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64. Pellecchia MT, Longo K, Pivonello R, et al. Multiple system atrophy is distinguished from idiopathic Parkinson's disease by the arginine growth hormone stimulation test. Ann Neurol 2006;60(5):611–615. [DOI] [PubMed] [Google Scholar]
- 65. Lin D, Hermann K, Schmahmann J. The diagnosis and natural history of multiple system atrophy, cerebellar type. Cerebellum 2017;15(6):663–679. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66. Teive H, Arruda W, Moro A, Moscovich M, Munhoz R. Differential diagnosis of sporadic adult‐onset ataxia: the role of REM sleep behavior disorder. Parkinsonism Relat Disord 2015;21(6):640–643. [DOI] [PubMed] [Google Scholar]
- 67. Boesch SM, Frauscher B, Brandauer E, Wenning G, Hogl B, Poewe W. Disturbance of rapid eye movement sleep in spinocerebellar ataxia type 2. Mov Disord 2006;21(10):1751–1754. [DOI] [PubMed] [Google Scholar]
- 68. Boesch SM, Frauscher B, Brandauer E, Wenning GK, Poewe W, Hogl B. Restless legs syndrome and motor activity during sleep in spinocerebellar ataxia type 6. Sleep Med 2006;7(6):529–532. [DOI] [PubMed] [Google Scholar]
- 69. Itoh K, Kasai T, Tsuji Y, et al. Definite familial multiple system atrophy with unknown genetics. Neuropathology 2014;34(3):309–313. [DOI] [PubMed] [Google Scholar]
- 70. The Multiple‐System Atrophy Research Collaboration . Mutations in COQ2 in familial and sporadic multiple system atrophy. N Engl J Med 2013;369(3):233–244. [DOI] [PubMed] [Google Scholar]
- 71. Zhao Q, Yang X, Tian S, An R, Zheng J. Association of the COQ2 V393A variant with risk of multiple system atrophy in East Asians: a case–control study and meta‐analysis of the literature. Neurol Sci 2016;37:423–430. [DOI] [PubMed] [Google Scholar]
- 72. Jeon BS, Farrer MJ, Bortnick SF, Korean Canadian Alliance on Parkinson's Disease and Related Disorders . Mutant COQ2 in multiple system atrophy. N Engl J Med 2014;371(1):80. [DOI] [PubMed] [Google Scholar]
- 73. Sharma M, Wenning G, Kruger R, European Multiple‐System Atrophy Study Group (EMSA‐SG) . Mutant COQ2 in multiple system atrophy. N Engl J Med 2014;371(1):80–81. [DOI] [PubMed] [Google Scholar]
- 74. Compta Y, Giraldo D, Munoz E, et al. Cerebrospinal fluid levels of coenzyme Q10 are reduced in multiple system atrophy. Parkinsonism Relat Disord 2018;46:16–23. [DOI] [PubMed] [Google Scholar]
- 75. Cortese A, Simone R, Sullivan R, et al. Biallelic expansion of an intronic repeat in RFC1 is a common cause of late‐onset ataxia. Nat Genet 2019;51(4):649–658. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76. Abdo WF, Van De Warrenburg BPC, Munneke M, et al. CSF analysis differentiates multiple‐system atrophy from idiopathic late‐onset cerebellar ataxia. Neurology 2006;67(3):474–479. [DOI] [PubMed] [Google Scholar]
- 77. Gardner RC, Schmahmann JD. Arginine test is not reliable for diagnosing cerebellar multiple system atrophy. Ann Neurol 2010;67(3):404–408. [DOI] [PubMed] [Google Scholar]
- 78. Fiorenzato E, Antonini A, Wenning G, Biundo R. Cognitive impairment in multiple system atrophy. Mov Disord 2017;32(9):1338–1339. [DOI] [PubMed] [Google Scholar]
- 79. Trikalinos TA, Siebert U, Lau J. Decision‐analytic modeling to evaluate benefits and harms of medical tests: uses and limitations. Med Decis Making 2009;29(5):E22–E29. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Table S1. Summary of studies on biomarkers in MSA. Search terms: (“multiple system atrophy” OR MSA OR “olivopontocerebellar atrophy” OR OPCA OR “striatonigral degeneration” OR SND OR “shy drager syndrome”) AND (blood OR plasma OR “cerebrospinal fluid” OR CSF). A total of 1984 papers were identified in Pubmed using the search terms on July 20th, 2019. A total of 77 relevant papers were included in the analysis.
Table S2. Summary of studies on genetic screening in MSA. Search terms: (“multiple system atrophy” OR MSA OR “olivopontocerebellar atrophy” OR OPCA OR “striatonigral degeneration” OR SND OR “shy drager syndrome”) AND (genetic OR genetics OR familial OR mutation). A total of 2144 papers were identified in Pubmed using the search terms on July 20th, 2019. A total of 95 relevant papers were included in the analysis.
Table S3. Summary of studies on neuroendocrine tests in MSA. Search terms: (“multiple system atrophy” OR MSA OR “olivopontocerebellar atrophy” OR OPCA OR “striatonigral degeneration” OR SND OR “shy drager syndrome”) AND (neuroendocrine OR arginine OR “growth hormone” OR GH OR clonidine). A total of 196 papers were identified in Pubmed using the search terms on July 20th, 2019. A total of 7 relevant papers were included in the analysis.
Table S4. Summary of studies on neurophysiological tests in MSA. Search terms: (“multiple system atrophy” OR MSA OR “olivopontocerebellar atrophy” OR OPCA OR “striatonigral degeneration” OR SND OR “shy drager syndrome”) AND (neurophysiology OR “evoked potentials” OR EVP OR electromyography OR EMG). A total of 356 papers were identified in Pubmed using the search terms on July 20th, 2019. A total of 26 relevant papers were included in the analysis.
Table S5. Summary of studies on neuropsychological tests in MSA. Search terms: (“multiple system atrophy” OR MSA OR “olivopontocerebellar atrophy” OR OPCA OR “striatonigral degeneration” OR SND OR “shy drager syndrome”) AND (neuropsychology OR neuropsychological OR dementia OR cognition OR cognitive OR frontal‐executive OR memory). A total of 1396 papers were identified in Pubmed using the search terms on July 20th, 2019. A total of 14 relevant papers were included in the analysis.
Table S6. Summary of sleep studies in MSA. Search terms: (“multiple system atrophy” OR MSA OR “olivopontocerebellar atrophy” OR OPCA OR “striatonigral degeneration” OR SND OR “shy drager syndrome”) AND (sleep OR “REM sleep behavior disorder” OR RBD OR REM sleep without atonia). A total of 455 papers were identified in Pubmed using the search terms on July 20th, 2019. A total of 3 relevant papers were included in the analysis.
Table S7. Summary of other tests in MSA. Relevant papers on other tests were searched manually. A total of 13 relevant papers were included in the analysis.