
Fig. 1.
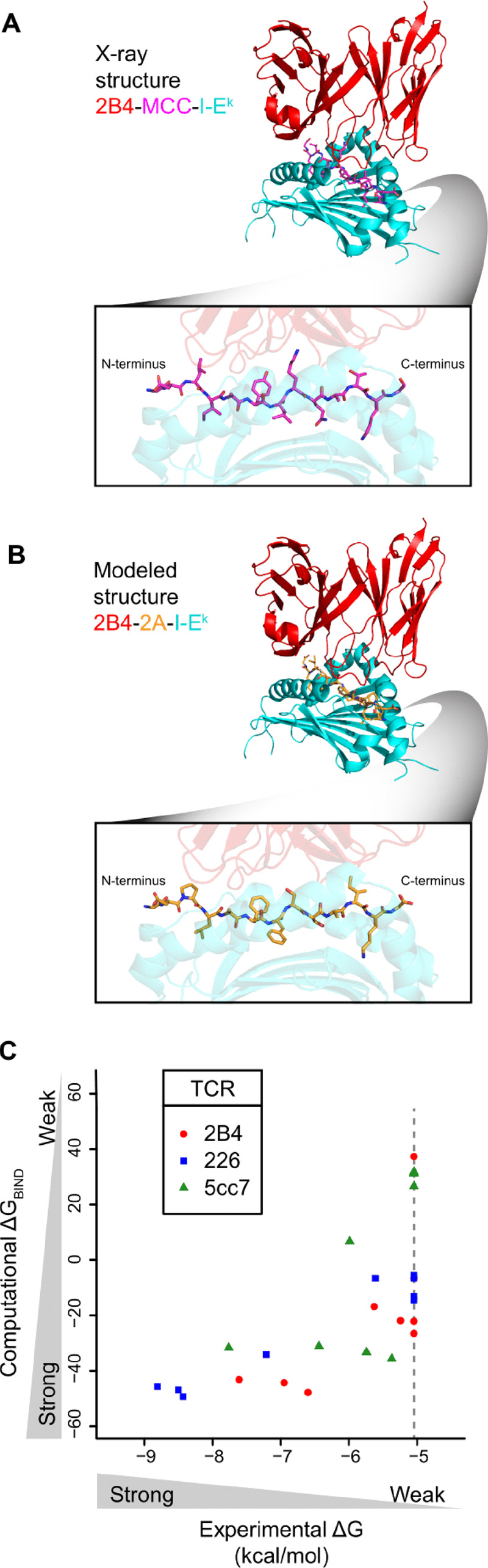
Prediction of TCR-pMHC binding free energies. (A) Crystal structure of TCR-pMHC interface for the 2B4 TCR (red) interacting with the MCC peptide (magenta) displayed by the I-Ek MHC (cyan). Bottom box: profile of the MCC peptide. (B) Fixed backbone model structure of TCR-pMHC interface for the 2B4 TCR (red) interacting with the 2A peptide (orange) displayed by the I-Ek MHC (cyan). 2A peptide was modeled onto the backbone of the MCC peptide in (A), TCR and MHC protein structures remain identical to (A). Bottom box: profile of the 2A peptide. (C) Scatter plot of ΔGBIND from computational modeling and scoring versus ΔG from experimental binding energies. Each point represents a peptide in Supplementary Table S1, where experimental ΔG is determined by binding affinity of pMHC with the indicated TCR (red, blue, green) via surface plasmon resonance from Birnbaum et al., and computational ΔGBIND is determined from modeling and scoring of peptide in TCR-pMHC complex. Pearson correlations are 0.67, 0.97 and 0.67 for TCRs 2B4, 226 and 5cc7, respectively. Correlation across the entire set of peptides is 0.69. Peptides with unreliable KDs due to weak or non-binding interactions were assigned a KD of 200 μM (ΔG = -5.05 kcal/mol, gray dotted line). If we assigned a ΔG of 0 kcal/mol for these weak/non-binding peptides the correlation across the entire set of peptides was similar (r = 0.74). (Color version of this figure is available at Bioinformatics online.)