

Abstract
Alzheimer's disease (AD) is the most common neurodegenerative disease and, owing to its increasing prevalence, represents one of the leading public health problems in aging populations. The molecular causes underlying the onset and progression of AD are manifold and hitherto still incompletely understood. Research over nearly four decades has clearly delineated genetics to play a crucial role in AD susceptibility, likely in concert with non‐genetic factors. The field has gained considerable momentum and novel insights over the past 10 years owing to the advent and application of high‐throughput genomics technologies in datasets of increasing size. In this contribution to the Mini‐Symposium on the Molecular Etiology of Alzheimer's Disease, we review the current status of genomics research in AD. To this end, we scrutinize and discuss the main findings from the two largest and most current genome‐wide association studies (GWAS) in the field, that is, the papers published by Jansen et al (Nat Genet 51:404–413) and Kunkle et al (Nat Genet 51:414–430). Particular focus is laid on genomics findings overlapping across both studies and on the novel insights they provide in terms of improving our understanding of the “genomic mechanisms” underlying this devastating disease.
Keywords: Alzheimer's disease, genomics, GWAS, risk genes
Introduction
Alzheimer's disease (AD) is the most common neurodegenerative disorder in humans and is characterized by progressive decline in cognitive functioning ultimately leading to dementia and death. Pathogenetically, AD is triggered by the aberrant deposition of β‐amyloid and tau protein, leading to the appearance of senile plaques, formation of neurofibrillary tangles, neuroinflammation, synaptic dysfunction, neuronal loss, and, ultimately, onset of cognitive decline. The molecular events causing neuronal cell death typically precede the onset of cognitive symptoms by a decade or more (23), implying that, once available, therapeutics targeting neuropathology will need to be administered very early, ideally prior to the onset of symptoms. Hence, the identification of genetic risk factors will not only be crucial for furthering our understanding of the molecular mechanisms causing the disease but will also be essential for an “early prediction—early detection—early intervention” approach to preventing the onset of AD‐related dementia, and aid in patient stratification schemes in clinical trials.
The Role of Genetics in AD Pathogenesis
After the first clinicopathological description of AD by German psychiatrist Alois Alzheimer in 1907 (2) it took more than half a century to realize that the disease shows familial aggregation consistent with genetic transmission (11). It took another decade until the use of genetic linkage analysis followed by positional cloning led to the discovery of rare mutations in three genes encoding the amyloid‐beta precursor protein (APP) and presenilins 1 and 2 (PSEN1/PSEN2) that cause fully penetrant monogenic forms of AD (22). While a large number of additional mutations in all three genes have been described following the original reports (for an up‐to‐date summary of monogenic findings in AD see the “AD & FTD Mutation Database” (9)), no additional genetic locus harboring clearly disease‐causing mutations has been established since. However, monogenic forms of AD only make up a small fraction of all cases (<5%) while the vast majority of patients suffer from “polygenic AD” (a.k.a. “non‐Mendelian AD” or “sporadic AD”). Susceptibility for this latter disease form is determined by the action (and interaction) of numerous independent genomic variants, likely in concert with non‐genetic factors, such as environmental exposures (eg, head trauma) and lifestyle choices (eg, alcohol consumption and cigarette smoking). A growing number of studies on a vast array of human disease and non‐disease traits has revealed that most—if not all—human phenotypes are driven by complex polygenic backgrounds (8). Thus, with its side‐by‐side instances of both monogenic and polygenic forms, AD represents a “genetically complex disorder” par excellence.
Genome‐Wide Screening in Polygenic Traits
DNA sequence variants underlying monogenic diseases, including those causing AD, are exceedingly rare in the population as a whole. This rarity is typically due to the large and highly penetrant molecular effects exerted by such variants resulting in being “selected against” from subsequent generations. DNA sequence variants exerting only moderate molecular effects lead to reduced overall penetrance and are much less subject to selective pressures allowing them to increase in frequency within populations over time. Once the minor allele, that is, the nucleotide with the lowest frequency, is present in 1% or more in the “general” (ie, non‐disease) population, a variant is defined as a DNA sequence “polymorphism.” Although the molecular effects and, hence, contribution to disease risk of any such polymorphism may be very small, it is via the combined action of many such polymorphisms that a genetically complex disease may eventually develop. Since many, that is, tens to hundreds to thousands, such polymorphisms can contribute to the genetic risk architecture of the same disease, these are said to be of a polygenic background. The identification of common genomic variants relevant for any given disorder is the focus of current gene findings efforts, including those ongoing in AD. The method of choice is genome‐wide screening, for example, in the context of genome‐wide association studies (GWAS), where the presence of specific alleles or genotypes at polymorphic sites is treated as exposure to predict a certain clinical outcome (here: onset of AD). By design, genome‐wide analyses afford a heavy multiple testing burden owing to the very large number of polymorphisms tested (typically several millions) which needs to be accounted for. In European populations it could be shown that approximately 1 million independent DNA sequence variants exist with a minor allele frequency (MAF) >5%. Bonferroni correction for this number of independent tests means that study‐wide significance is achieved at α = 0.05/1 000 000 = 5 × 10−8, a threshold now widely accepted and used in the field. For more details on this topic as well as a detailed account of the past (and possible future) achievements of GWAS in human genomics research we recommend consulting the review by Visscher et al (24).
In AD, about 60 GWAS have been published since 2007 (according to the “NHGRI‐EBI Catalog of published genome‐wide association studies” [GWAS catalog], URL: https://www.ebi.ac.uk/gwas/ (6)), although many of these are not independent as the same datasets were utilized in successive meta‐analyses. The two most recent and—by sample size—largest AD GWAS were published back‐to‐back in the March 2019 issue of Nature Genetics (14, 16), and represent the core “data” of this review. First, we begin by highlighting the main findings from each of these GWAS with particular emphasis on results overlapping across both studies. This will include a short excursion into rare‐variant‐based analyses and how data from those studies fit to the results from Jansen et al (14) and Kunkle et al (16). In the second part, we will discuss new putative “mechanistic” insights gained from these GWAS findings and their possible implications. We close by providing an outlook on the field for the next 10 years.
The Status of AD Genomics Research
Prior to spring of 2019, it had been over 5 years since the last bona‐fide GWAS was published for AD (17). That study, conducted by a group of researchers aligned under the auspices of the “International Genomics of Alzheimer's Disease Project” (IGAP), entailed the analysis of ~75 000 individuals allowing the identification of 20 genome‐wide significant (ie, P‐value < 5 × 10−8) AD risk loci, of which 11 were novel at the time. Since this IGAP‐2013 publication, the consortium continued collecting additional independent AD cases and controls culminating in an updated GWAS (16) on approximately 94 000 individuals of European descent (35K AD cases vs. 59K healthy controls; Table 1). The actual genome‐wide screening (“discovery phase”) in this IGAP‐2019 GWAS was performed in 64 000 individuals followed by validation analyses in another 30 000 individuals and led to the discovery of 25 genome‐wide significant loci, five of which were reported as “novel” (16). The second GWAS discussed in this review (14) was published (online) nearly 2 months before the IGAP follow‐up study and, overall, utilized nearly seven times as many samples as the IGAP‐2019 GWAS (total n ~ 635 000 individuals; Table 1). Partially owing to its much larger sample size, this AD GWAS identified 29 genome‐wide significant loci, of which nine were declared “novel” at the time of publication. Notwithstanding the identification of novel genetic risk loci in these studies it is worth noting that the overall number of genome‐wide significant findings in AD GWAS continues to be small compared to other GWAS of similar size, for example, Parkinson's disease or schizophrenia (see GWAS catalog for details (6)), possibly indicating that the degree of polygenicity underlying AD may be lower than that of other traits.
Table 1.
Summary of key aspects of the two GWAS discussed in this review.
| Jansen et al (2019) | Kunkle et al (2019) | |
|---|---|---|
| Sample size | ||
| Discovery | 455K (72K vs. 383K) | 64K (22K vs. 42K) |
| Follow‐up | 180K (6.6K vs. 174K) | 30K (13K vs. 17K) |
| Total | 636K (79K vs. 557K) | 94K (35K vs. 59K) |
| Results | ||
| # variants analyzed | 13 367 300 | 11 480 633 |
| MAF threshold | No constraint | No constraint |
| # gw sign. loci | 29 | 15 |
| # gw sign. genes | 169 | 95 |
Of these two most recent AD GWAS, the study published by Jansen et al (14) is considered the more “remarkable” by us based on several grounds. First, not only was it submitted and published before the IGAP‐2019 (16) study, but it was also the first bona fide AD GWAS to make all its results (main and supplementary) publicly available as preprint (on bioRxiv at https://doi.org/10.1101/258533) prior to peer‐review and publication in Nature Genetics. Some 6 weeks later, the IGAP‐2019 group published their results as preprint as well (https://doi.org/10.1101/294629). Second, Jansen et al (14) drastically increased the overall sample size (and along with it: statistical power) by nearly an order of magnitude compared to all other previous GWAS in the AD field. This was made possible by utilizing genome‐wide data from nearly 48 000 AD (proxy; see below) cases and 330 000 non‐AD controls of the UK biobank (UKB) project. UKB is a unique prospective cohort study with deep genetic and phenotypic data collected on ~500 000 individuals from across the United Kingdom (7). At baseline, UKB participants were aged between 49 and 69 years, and therefore mostly too young for having developed polygenic AD, which peaks after the age of 65. To circumvent this problem, and this is the third reason for being a truly remarkable study, Jansen et al utilized a method based on “proxy phenotyping” which makes use of parental AD status as recorded in UKB medical records. This approach was recently proposed (18) to be a valid approximation of future AD status in UKB individuals for whom genotype data were available but who had not (yet) developed AD themselves.
Main Results From the GWAS by Jansen et al (14)
Some parts of the ensuing results summary were already highlighted in a “News and Views” article which we published earlier this year (5). In their discovery phase, Jansen et al (14) combined the data from UKB, IGAP‐2013 and two smaller case‐control datasets from Europe and the US to arrive at an overall sample size of ~455 000 individuals. As expected owing to the partial sample overlap, the loci reported to show genome‐wide significance in (14) included many also highlighted in the IGAP‐2013 GWAS (17), but also pinpointed evidence for genome‐wide significant (P‐value < 5 × 10−8) association at nine additional and novel loci (Table 2), all of which were subsequently tested in an independent replication sample of 180 000 individuals from Iceland (Table 1). At the same time, the data by Jansen et al (14) did not confirm several of those reported in the IGAP‐2013 study (ie, MEF2C, NME8, CELF1, and FERMT2) and renewed association evidence at one locus (ie, CD33; originally identified in a GWAS by our group more than 10 years ago (3)) showing clear genome‐wide significant (P‐value < 5 × 10−8) association with AD risk (Table 2). Also new on the list is ADAM10, encoding the key enzyme cleaving APP to preclude Aβ (the core constituent of β‐amyloid and, hence, senile plaques) generation, which has previously been shown to contain rare variants segregating with AD status in AD families (21), and APH1B, whose encoded protein, Aph‐1 homolog B, together with the presenilins is a component of the gamma secretase complex, responsible for cleaving APP to produce Aβ. The other novel loci identified in the GWAS by Jansen et al (14) include ADAMTS4 (located on chromosome 1), CLNK (chr. 4), KAT8 (chr. 16), ALPK2 (chr. 18), AC074212.3 (chr. 19), HESX1 (chr. 3), and CNTNAP2 (chr. 7). All but the last two loci were pinpointed with common polymorphisms (ie, those with an MAF > 1%; Table 2), while the latter two loci showed their lead signals with rare variants (defined here as MAF < 1%; Table 3).
Table 2.
Summary of current common variant (MAF ≥ 0.01) AD genomics findings from the GWAS by Jansen et al (14) and Kunkle et al (16)
| Chr | Pos | LeadSNP | A1 vs. A2 | MAF‡ | P‐value Jansen et al | P‐value Kunkle et al | AD effect | Nearest gene | AD pathway | Potential link to AD pathogenesis |
|---|---|---|---|---|---|---|---|---|---|---|
| 1 | 161155392 | rs4575098 | A vs. G | 0.240 | 2.05E‐10 | 2.34E‐02* | Risk | ADAMTS4 | None | Neuroprotection: Extracellular Matric Protease |
| 1 | 207786828 | rs2093760 | A vs. G | 0.225 | 1.10E‐18 | 1.66E‐15* | Risk | CR1 | Immune | Innate Immunity; Neuroinflammation |
| 2 | 127891427 | rs4663105 | A vs. C | 0.412 | 3.38E‐44 | 2.16E‐26* | Risk | BIN1 | Lipid | Cellular Protein Trafficking |
| 2 | 233981912 | rs10933431 | G vs. C | 0.240 | 8.92E‐10 | 3.42E‐09** | Protection | INPPD5 | None | Autophagy; Viral Infection |
| 4 | 11026028 | rs6448453 | A vs. G | 0.228 | 1.93E‐09 | 4.90E‐05* | Risk | CLNK | None | Innate Immunity; Neuroinflammation |
| 6 | 32583357 | rs9469112 | T vs. A | 0.153 | 8.41E‐11 | 2.32E‐07** | Protection | HLA‐DRB1 | Immune | Adaptive Immunity |
| 6 | 47432637 | rs9381563 | C vs. T | 0.344 | 2.52E‐10 | 3.57E‐10** | Risk | CD2AP | None | Blood Brain Barrier; Aβ Transcytosis |
| 7 | 99971834 | rs4727449rs1859788 | A vs. G | 0.323 | 2.22E‐15 | 1.22E‐09** | Protection | ZCWPW1 | None | Innate Immunity; Neuroinflammation |
| 7 | 143108158 | rs7810606 | T vs. C | 0.425 | 3.59E‐11 | 1.13E‐06** | Protection | EPHA1 | None | Signal Transduction |
| 8 | 27464929 | rs28834970/rs4236673 | A vs. G | 0.390 | 2.61E‐19 | 5.60E‐23** | Protection | CLU/PTK2B | Immune; lipid; tau | Aβ clearance/SignalTransduction |
| 10 | 11717397 | rs11257238 | C vs. T | 0.382 | 1.26E‐08 | 2.61E‐07** | Risk | ECHDC3 | None | Lipid Metabolism |
| 11† | 47380340 | rs3740688 | G vs. T | 0.458 | 4.50E‐05 | 5.46E‐13** | Protection | SPI1/CELF1 | Immune | Potential false‐positive result (not replicated in UKB) |
| 11 | 59958380 | rs2081545 | A vs. C | 0.342 | 1.55E‐15 | 5.35E‐17** | Protection | MS4A6A | Immune | Innate Immunity; Neuroinflammation |
| 11 | 85776544 | rs867611 | G vs. A | 0.342 | 2.19E‐18 | 3.41E‐19** | Protection | PICALM | APP | Blood Brain Barrier; Aβ Transcytosis |
| 11 | 121435587 | rs11218343 | C vs. T | 0.035 | 1.09E‐11 | 2.88E‐12** | Protection | SORL1 | Lipid; APP | Cellular Protein Trafficking |
| 14† | 53391680 | rs17125924 | G vs. A | 0.099 | 5.26E‐06 | 1.42E‐09** | Protection | FERMT2 | n.a. | Potential false‐positive result (not replicated in UKB) |
| 14 | 92938855 | rs12590654 | A vs. G | 0.347 | 1.65E‐10 | 8.73E‐09* | Protection | SLC24A4 | None | Calcium Homeostasis |
| 15 | 59022615 | rs442495 | C vs. T | 0.334 | 1.31E‐09 | 2.51E‐7** | Protection | ADAM10 | Immune | Sheddase; APP Processing |
| 15 | 63569902 | rs117618017 | T vs. C | 0.132 | 3.35E‐08 | 2.38E‐04* | Risk | APH1B | None | γ‐secretase; APP Processing |
| 16 * | 19808163 | rs7185636 | C vs. T | 0.156 | 1.40E‐01 | 2.4E‐08 *** | Protection | IQCK | n.a. | Likely false‐positive result (not replicated in Jansen) |
| 16 | 31133100 | rs59735493 | A vs. G | 0.324 | 3.98E‐08 | 7.42‐03* | Protection | KAT8 | None | Transcriptional Regulation |
| 16 * | 79355857 | rs62039712 | G vs. A | 0.094 | 7.66E‐01 | 3.70E08 * | Risk | WWOX | n.a. | Likely false‐positive result (not replicated in Jansen) |
| 17 | 5138980 | rs113260531 | A vs. G | 0.118 | 9.16E‐10 | 3.70E‐04** | Risk | SCIMP | None | Innate Immunity; Neuroinflammation |
| 17 | 47450775 | rs28394864 | A vs. G | 0.471 | 1.87E‐08 | 4.85E‐03* | Risk | ABI3 | None | Innate Immunity; Neuroinflammation |
| 17 | 61538148 | rs138190086 | A vs. G | 0.017 | 2.65E‐04 | 5.30E‐09*** | Risk | ACE | Immune | Aβ degradation; blood pressure regulation |
| 18 | 56189459 | rs76726049 | C vs. T | 0.011 | 3.30E‐08 | 1.76E‐01* | Risk | ALPK2 | None | Signal Transduction |
| 19 | 1039323 | rs111278892 | G vs. C | 0.165 | 7.93E‐11 | 1.10E‐07* | Risk | ABCA7 | Lipid; APP | Lipid Metabolism; Innate Immunity |
| 19 | 45411941 | rs429358 | C vs. T | 0.155 | <1E‐900 | 1.17E‐881* | Risk | APOE | Lipid; APP; tau | Aβ clearance/Lipid Metabolism |
| 19 | 46241841 | rs76320948 | T vs. C | 0.059 | 4.64E‐08 | 1.22E‐04* | Risk | AC074212.3 | None | ? |
| 19 | 51727962 | rs3865444 | A vs. C | 0.336 | 6.34E‐09 | 5.27E‐06** | Protection | CD33 | None | Innate Immunity; Neuroinflammation |
| 20 | 54998544 | rs6014724 | G vs. A | 0.089 | 6.56E‐10 | 3.65E‐07* | Protection | CASS4 | None | Signal Transduction |
| 21 * | 28156856 | rs2830500 | A vs. C | 0.336 | 1.65E‐02 | 2.60E‐08 *** | Protection | ADAMTS1 | n.a. | Likely false‐positive result (not replicated in Jansen) |
“Chr,” “Pos,” “A1 vs. A2,” “AD effect” taken from Jansen et al. “Nearest gene” determined from UCSC genome browser (GRCh37; hg19).
Blue highlight indicates "novel" finding in GWAS by Jansen et al, grey highlight indicates "novel" findings in GWAS by Kunkle et al, no highlight indicates previously described GWAS findings.
AD pathway as determined in either or both GWAS: “immune” = “immune system response”; “lipid” = “lipid metabolism”, “APP” = “APP metabolism”, “tau” = “tau protein binding” (see text for more details).
“Potential link to AD pathogenesis” = based on the authors' review (and interpretation) of the literature.
Stage 1 result from Kunkle et al.
Stage 2 result from Kunkle et al.
Stage 3 result from Kunkle et al.
SPI1/CELF and FERMT2 represent results featured in Kunkle et al that poorly replicate in the UKB portion of Jansen et al; these are interpreted as possibly false‐positives here.
MAF = minor allele frequency; from European controls (non‐Finnish) as provided on GnomAD [v.2.1.1.; https://gnomad.broadinstitute.org/].
IQCK, WWOX, and ADAMTS1 represent results featured in Kunkle et al that are not replicated at P‐value < 0.01 in Jansen et al; these are interpreted as possibly false‐positives here.
Table 3.
Summary of current rare variant (MAF ≤ 0.01) AD genomics findings from the GWAS by Jansen et al (14) and Kunkle et al (16)
| Chr | Pos | LeadSNP | A1 vs. A2 | MAF‡ | P‐value Jansen | P‐value Kunkle | AD effect | Nearest gene | AD pathway | Potential link to AD pathogenesis |
|---|---|---|---|---|---|---|---|---|---|---|
| 3 | 57226150 | rs184384746 | T vs. C | 0.002 | 1.24E‐08 | n.a. | Risk | HESX1 | None | Homoebox Gene; Development |
| 6 | 41129252 | rs75932628 | T vs. C | 0.002 | 2.95E‐15 | 2.95E‐12* | Risk | TREM2 | Immune system response | Innate Immunity; neuroinflammation |
| 7 | 145950029 | rs114360492 | T vs. C | 0.0003 | 2.10E‐09 | n.a. | RISK | CNTNAP2 | None | Neuronal Development |
| 16† | 81942028 | rs72824905 | G vs. C | 0.01 | 2.11E‐03 | 7.92E‐03* | Protection | PLCG2 | None | Microglial activation; neuroinflammation |
| 17† | 47297297 | rs616338 | T vs. C | 0.01 | 7.81E‐07 | n.a. | Risk | ABI3 | None | Microglial activation; neuroinflammation |
“Chr,” “Pos,” “A1 vs. A2,” “AD effect” taken from Jansen et al. “Nearest gene” determined from UCSC genome browser (GRCh37; hg19).
Blue highlight indicates “novel” AD locus in GWAS by Jansen et al, grey highlight indicates “novel” AD locus in GWAS by Kunkle et al, no highlight indicates previously described GWAS findings.
AD pathway as determined in either or both GWAS: “immune” = “immune system response”; “lipid” = “lipid metabolism”, “APP” = “APP metabolism”, “tau” = “tau protein binding” (see text for more details).
“Potential link to AD pathogenesis” = based on the authors' review (and interpretation) of the literature.
Stage 1 result from Kunkle et al.
Stage 2 result from Kunkle et al.
Stage 3 result from Kunkle et al.
Variants in PLCG2, and ABI3 were highlighted in a rare variant GWAS by the IGAP group (Sims et al, 2018) (20) published prior to the Kunkle et al, 2019 (16) study and are listed here for reasons of completeness.
MAF = minor allele frequency; from European controls (non‐Finnish) as provided on GnomAD [v.2.1.1.; https://gnomad.broadinstitute.org/].
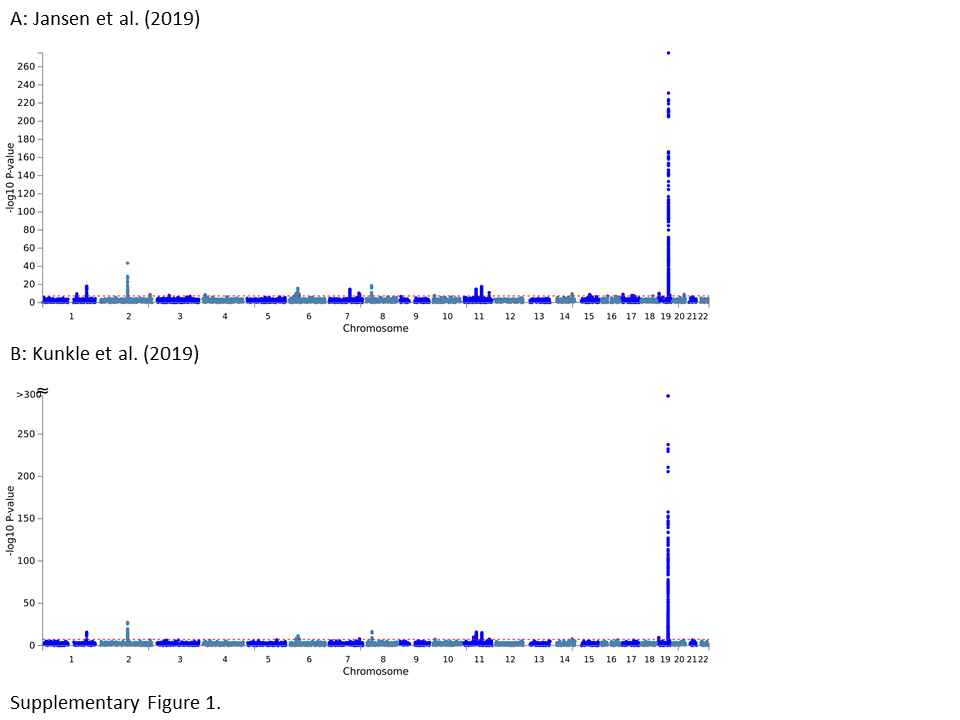
For a visual summary of the genome‐wide association results by Jansen et al (14) as a “Manhattan plot” see Figure 1; for a detailed summary of top AD loci see Tables 2 and 3. To facilitate comparison, both sets of results are depicted next to the discovery‐phase GWAS findings by Kunkle et al (16) in Figure 1. Despite there being a total of 29 loci showing genome‐wide significance, it needs to be emphasized that one locus stands out both in terms of statistical support and exerted effect size, that is, locus #26 in (14), located in the APOE region on the long arm of chromosome 19. The “standing out” nature of this locus can be grasped from Figure S1 where we plot non‐truncated association P‐values in the APOE region for both GWAS: with P‐values at 1 × 10−300 and below, markers in the APOE region are hundreds of orders of magnitude more significantly associated with AD risk than any other locus in the genome (the next best association is observed with a variant in the BIN1 locus showing a P‐value = 3.38 × 10−44; Table 2). Interestingly, of all currently established AD genetic risk factors, APOE represents the only locus to emerge from the pre‐GWAS “candidate gene era” (4); all other AD loci in Table 2 where established to represent bona fide AD genes by genome‐wide screening.
Figure 1.
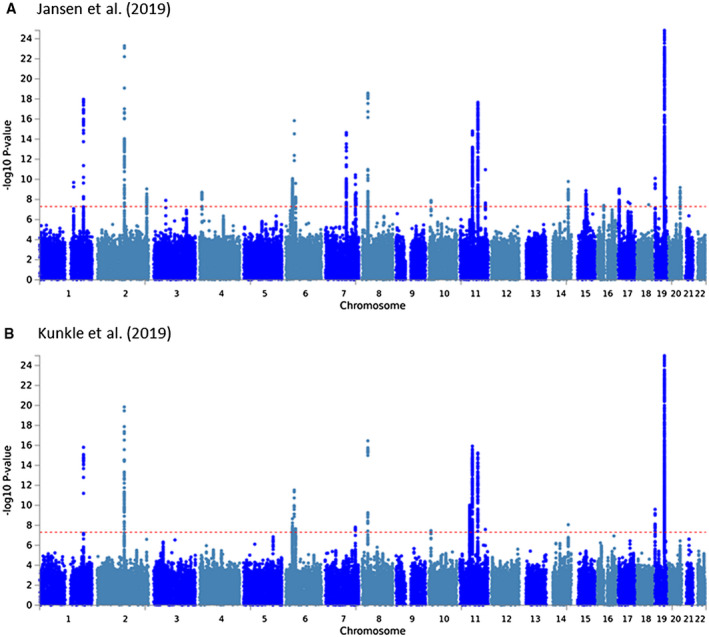
Manhattan plots of “discovery phase” GWAS findings from the two GWAS discussed in this review. A. Jansen et al (14); B. Kunkle et al (16). Results are based on data distributed by the two respective studies (see “Data Availability Statement” section for more details). P‐values are truncated at 1E‐25 for didactic reasons. For Manhattan plots of non‐truncated results see Figure S1.
Main Results From the GWAS by Kunkle et al (16) (IGAP‐2019)
Owing to its smaller sample size, the IGAP‐2019 GWAS identified fewer loci at genome‐wide significance than the study by Jansen et al (14). Overall, there were 25 loci highlighted by Kunkle et al (16) as AD risk factors, five of which were deemed “novel” by the authors, namely ADAM10 (on chromosome 15), IQCK and WWOX (both chr. 16), ACE (chr. 17), and ADAMTS1 (chr. 21; Table 2). Technically, ADAM10 was first published as a genome‐wide significant AD locus 2 months prior in the GWAS by Jansen et al (14) to which it is, hence, credited for the purpose of this review. The other four novel IGAP‐2019 AD GWAS loci either show no or only very modest evidence of association with AD risk in the larger datasets by Jansen et al (14), which is why these are interpreted only as “potential” or “possibly false positive” findings by us (Table 2). Conversely, Kunkle et al (16) also did not replicate some of the novel signals in the Jansen GWAS (namely: ADAMTS4 and ALPK2), although this may simply be attributed to the much smaller sample size (and, hence, reduction in power) of the IGAP‐2019 study. While these could therefore also represent false‐positive findings and more data are needed to more conclusively address this possibility, we consider the likelihood of such an outcome as relatively low, and therefore count these latter two signals as genuine AD genetic loci for the purpose of this review.
For a visual summary of the genome‐wide association results by Kunkle et al (16) as a “Manhattan plot” see Figure 1 (and Figure S1); for a detailed summary of top AD loci see Tables 2 and 3. To facilitate comparison, the results of the IGAP‐2019 GWAS are depicted next to the discovery‐phase GWAS findings by Jansen et al (14) in Figure 1. As for the results by Jansen et al (14), markers in the APOE region outshine all other genome‐wide significant AD loci by a large margin, again emphasizing the predominant role of this locus in AD pathogenesis (Figure S1B).
Overlapping Findings Across GWAS by Jansen et al and Kunkle et al
Before engaging in considerations on the potential “mechanistic” implications of the current AD genomics findings, one needs to derive a subset of results that stands a large likelihood of surviving the test of time, that is, likely representing genuine AD loci. As outlined above, Jansen et al (14) reported to have identified nine novel loci at genome‐wide significance. Two of these, that is, HESX1 and CNTNAP2, were elicited by rare‐variants (MAF < 1%; Table 3) for which IGAP‐2019 did not compute or make available summary association statistics at the time of writing. Based on the strength of the reported association evidence, we still consider them as genuine AD risk loci here. Furthermore, five of the remaining seven novel AD loci elicited by common variants (MAF > 1%) highlighted by Jansen et al (14) showed the same direction of effect and association P‐values < 0.01 in IGAP‐2019, namely CLNK, ADAM10, APH1B, KAT8, and AC074212.3 (Table 2). Therefore, for the purpose of this review they are also considered as genuine AD loci. Interestingly, the reverse is not true for most loci: of all 4 novel IGAP‐2019 loci, only ACE on chromosome 17 replicates in Jansen et al (14) and, thus, appears to represent a genuine AD finding. Finally, there remain two loci with somewhat unclear association evidence, that is, SPI1/CELF1 (on chromosome 11) and FERMT2 (chr. 14). Both of these were originally identified in the IGAP‐2013 study (17) and continue to show genome‐wide significant association with AD risk in IGAP‐2019 (Table 2). This is not surprising given that 80% of individuals in IGAP‐2019 were also included in IGAP‐2013. However, Jansen et al (14) did not consider these loci further due to the “lower association signals in the UKB data set” (ie, P‐values of 0.02 and 0.004 for SPI1/CELF1 and FERMT2, respectively). Given the extremely modest association evidence for these two loci in UKB, we tend to agree with the notion of Jansen et al (14), and will also not consider these two loci further in the remainder of this review.
In summary, taking all the available evidence together, most genome‐wide association findings show good correspondence across both studies. Overall, there emerge 32 apparently genuine AD risk loci at the day of writing of this review (August 2019), 27 (out of 32) from common variant (Table 2) and 5 (out of 5) from rare‐variant based‐results (Table 3). Note that the latter results also include two loci, that is, PLCG2 and ABI3, which recently emerged from an “exome chip” GWAS (20) on what appears to be largely the same dataset also utilized for the IGAP‐2019 GWAS. Variants in both loci showed P‐values < 0.01 in the Jansen et al (14) results (Table 3), thus, fulfilling our criteria of “replication.”
Insights into Genomic Mechanisms of AD From GWAS
The identification of complex trait genetic loci represents only the first step in what inevitably proves to be a “long and winding road” to advancing our understanding of the pathogenic mechanisms underlying the trait in question. This process amounts to “making sense” of GWAS findings and putting them into the (correct) context with other molecular data to understand why and when a specific disease has developed in cases, but not in controls. A next and probably more important aspect is to utilize these genetic insights to predict an individual's risk for the disease in question prior to the actual onset of first symptoms, with the aim to offer specific genetic counseling and/or therapeutic options (if these exist) similar to what is now possible for many monogenic disease mutations. In the beginning of this review, we summarized this procedure as the development of “early prediction—early detection—early intervention” procedures in AD. Notwithstanding its clinical importance, the translation of polygenic genomics findings into medical genetics practice is not discussed further in this review. Instead, we focus on the interpretation of the GWAS findings from a “disease mechanism” viewpoint.
In the genomics community, the set of analyses focusing on the interpretation (in terms of putative disease mechanisms and/or translational potential) of specific GWAS findings is often described as “post‐GWAS analyses.” In general terms, these approaches entail the utilization of different computational tools and analyses with the aim to integrate high‐resolution data from other genomics domains (eg, transcriptomics or epigenomics), as well as other “‐omics” data in general (eg, proteomics and metabolomics). The main aims of these efforts can perhaps be summarized (and simplified) as follows: First, delineate the functionally relevant genes located within the identified AD GWAS loci. This is important because many AD loci actually contain several plausible gene candidates in the immediate vicinity of associated lead variants. Second, characterize the functionally relevant molecular genetic mechanisms within delineated gene candidates, for example, assess whether the predominant effect on pathogenesis is the result of an exonic variant changing the function of the encoded protein or that of a regulatory variant changing the gene's expression. Third, if a non‐exonic variant is the likely culprit, assess whether it affects the expression profile of implicated genes in the tissue of interest, for example, in the case of AD the brain, for example, hippocampus or specific cortical areas. Fourth, delineate the overarching mechanistic “themes,” for example, specific pathways or gene/protein networks, with the aim to assess how the genomics findings may fit with evidence from other molecular domains, for example, neuropathological data. Almost always, these and other analyses are applied in parallel and in combination to arrive at the most likely solution. The hope behind these efforts is to arrive at a tractable mechanistic hypothesis which can then be tested by dedicated laboratory experiments to prove or disprove causality of a specific disease‐associated sequence variant.
Owing to the complexity of the matter, we can here only provide some first and early insights into potential mechanisms offered by the newest AD genomics findings. For the sake of simplicity, we only touch on the four main “mechanistic domains” outlined in the previous paragraph and summarize selected results of post‐GWAS analyses presented in the two primary GWAS publications that form the basis of this review. We refer to the other papers in this Mini‐Symposium for insights into additional potential mechanisms underlying AD pathogenesis.
Delineating Functionally Relevant Genes Within the AD Associated Genetic Loci
In the context of GWAS, the term “genetic locus” describes a specific stretch of genomic DNA collectively showing association evidence with the outcome trait in question, here the onset of AD. The physical dimensions, that is, length, of each locus can vary quite substantially between chromosomal regions and mainly depend on the extent of correlation between DNA sequence variants located within the locus, a situation reffered to as “linkage disequilibrium”. A genomic region not much affected by chromosomal recombination at meiosis will be larger (and possibly contain more genes) than recombination “hotspots” which are smaller and typically contain fewer genes. In the GWAS by Jansen et al, the average AD locus encompassed ~138 000 base pairs (bp) in length (range 1–823 000 bp; calculated based on information from table S2 of (14)). Using three different mapping strategies the authors of that study concluded that up to 192 genes may be linked to the 29 AD loci identified by genome‐wide screening. Using equivalent mapping strategies, the authors of the IGAP‐2019 study delineated up to 400 gene candidates underlying the GWAS signals in their analyses (16). In theory, all of these could be involved in AD pathogenesis and hence represent culprits for eliciting the observed GWAS signal within each locus. In some instances, it is even possible that several DNA sequence variants within the same locus are independently associated with disease risk and these do not necessarily have to be located in the same gene. The APOE locus on chromosome 19q13.32 shall serve as a illustrative example of the problem faced when attempting to translate “GWAS loci” to “disease genes” (Figure S2): here, the entire AD‐associated GWAS locus extends over ~823 000 bp. Near the main association signal, it contains several highly correlated variants mapping into different genes located in close proximity. Most of the other non‐APOE AD GWAS loci highlighted in the GWAS by Jansen et al (14) and Kunkle et al (16) contain several, albeit less than in the APOE region, such gene candidates. Knowledge of the other “mechanistic domains” (see below) can help to further pinpoint the pathophysiological culprits, that is, genes eliciting the observed GWAS signals.
Characterization of the Underlying Molecular Genetic Mechanisms
An integral part of mapping relevant disease genes within GWAS loci is characterizing the assumed molecular genetic mechanisms of the associated DNA sequence variants. For instance, an exonic loss‐of‐function variant, for example, introducing a premature stop‐codon severely affecting the function of the encoded protein, may elicit stronger and more significant disease‐effects than an intronic variant without any obvious molecular consequences. However, exonic variants (and others near the coding sequence) are quite rare and rarely serve as plausible mechanistic explanation for the disease association. Figure 2 depicts the distribution of variant locations for all genome‐wide significant loci in the Jansen et al (14) GWAS (Figure S3 for both GWAS, side‐by‐side). It shows that the two most frequent disease‐associated variant categories are “intergenic” (ie, outside the coding sequence, between genes) and “intronic.” While these categories may at first appear less compelling than “exonic” or “splicing” or “UTR” it is becoming increasingly clear from other work that much of the intergenic space is actively involved in the regulation of gene expression (15) and may, therefore, be of relevance in the onset and progression of disease. Systematic functional mapping of variants in the 29 loci highlighted in the Jansen et al (14) GWAS revealed that in addition to APOE a total of only four loci (ie, CR1 [chr. 1], PILRA [chr. 7], APH1B [chr. 15], and CD33 [chr. 19]) contained non‐synonymous exonic variants deemed to be “credible causal” for the observed association signals (see table S9 in (14)); conversely, the association signals in the other loci are, therefore, likely elicited by non‐exonic variants. Interestingly, within the APOE locus on chromosome 19 this approach identified a total of 16 non‐synonymous exonic variants across nine different genes: the one deemed as “credible causal” using the same framework was the well‐known epsilon4‐allele (at variant rs429358) in the APOE gene itself.
Figure 2.
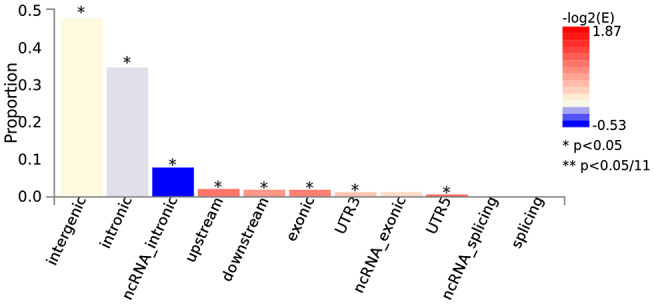
Functional consequences of SNPs on genes for the GWAS by Jansen et al (14). The histogram displays the proportion of SNPs (all SNPs in LD with independent significant SNPs) which have corresponding functional annotation assigned by ANNOVAR. Bars are colored by log2 (enrichment) relative to all SNPs in the selected reference panel. Plots are made with FUMA (25) using data described in “Data Availability Statement” section. For equivalent results of the GWAS by Kunkle et al (16) see Figure S3.
Characterization of Potential Variant Effects on Gene Expression
Non‐exonic (and even some exonic) variants are hypothesized to exert their molecular effects by influencing the regulation of gene expression (10). To this end, the recent advent of high‐dimensional reference datasets for tissue‐specific gene expression profiles based on whole transcriptome RNA sequencing data, has vastly facilitated the interpretation of putative regulatory effects of DNA sequence variants (26). Such variants are often called “expression quantitative trait loci” (eQTL), meaning that they elicit quantitative (and statistically significant) effects on the expression of genes. In essence, these studies are nothing but GWAS using quantitative gene expression data as outcome variables. The field was revolutionized by the launch of the “GTEx” (Genotype‐Tissue Expression) project, whose overarching aim is to characterize variation in gene expression across individuals and diverse tissues in humans (1). Owing to the availability of gene expression data in 13 different brain regions, data accumulated by the GTEx consortium are highly relevant to the neuroscience community (26). Importantly for the efforts described here, all GTEx data and results are made freely available via a dedicated website (www.gtexportal.org) and numerous variant‐mapping algorithms have integrated these data into their “variant effect prediction.” Among these is the “Functional Mapping and Annotation” (FUMA) of GWAS findings tool (25), developed by the same group that also spearheaded the Jansen et al (14) AD GWAS.
There are multiple different ways of connecting DNA sequence variant to gene expression data. One way routinely applied by FUMA are “gene property analyses for tissue specificity.” Essentially, these analyses probe for tissue specificity of the phenotype by testing for potential relationships between tissue specific gene expression profiles (based on GTEx data) and disease‐gene associations (based on genome‐wide gene‐based association statistics of the underlying single‐variant results). Applying these analyses to the discovery phase gene‐based GWAS results from the Jansen et al (14) study revealed study‐wide significant association between AD risk genes and tissue‐specific gene expression in spleen, whole blood, and lung (Figures 3 and S4A). Interestingly, no significant association was observed between AD risk genes and gene expression in the various different brain regions analyzed by GTEx. A very similar pattern was observed when analyzing the discovery‐phase AD GWAS results from the Kunkle et al study (16) which also showed significant tissue‐specific expression in non‐brain regions such as whole blood, liver, and spleen (Figure S4B). These and other related sets of gene expression results converged on the notion that AD risk genes overlap more with immune‐system related tissues than with brain‐ or neuron‐specific datasets. For instance, the brain cell types with the highest expression of AD risk genes were microglia, a cell type involved in the brain's immune system response, rather than neuronal cell types (see figure 4 of (14)).
Figure 3.
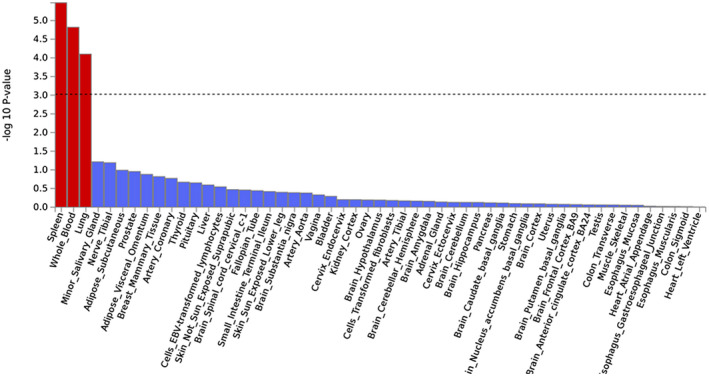
Results of tissue expression analysis for the GWAS by Jansen et al (14). Results are based on MAGMA gene‐property analyses as implemented in FUMA (25) using GTEx (1) v6 data on 53 tissue types. Input GWAS data as described in “Data Availability Statement” section. Red bars represent study‐wide significant results correcting for the 53 tissue types.
Delineating Overarching Mechanistic “Themes” by Means of Pathway Analyses
The final set of “functional characterization” analyses discussed in this section relate to various types of “pathway analyses” to elucidate common mechanistic themes combining genetics with protein function. There are a multitude of different approaches to link disease genes to pathways, but probably the most widely used test in this context is to probe for an enrichments of associated genes in certain predefined gene sets (eg, converging on certain biological mechanisms defined by gene ontology [GO] classification). Applying such GO‐based enrichment analyses for the two GWAS discussed in this review highlighted two biological mechanisms in both studies, that is, processes involved in “lipid metabolism” and “APP metabolism/ Aβ formation.” In addition, the GWAS by Kunkle et al (16) also identified “immune system response” and “tau protein binding” as significantly enriched among their set of AD‐associated GWAS genes. While the connection to lipid metabolism and immune system response pathways had already been described in similar enrichment analyses of the IGAP‐2013 data (13), the connection to APP metabolism represents a new outcome of the refined AD association results of the two largest and most recent GWAS. As such it provides further support for the notion that the “amyloid hypothesis” of AD—which posits that Aβ mismetabolism is the primary driver of AD‐related pathogenesis (for reviews see refs (22) and (19))—may also be at play in late‐onset, polygenic AD, in addition to its well‐known role in monogenic AD.
As for all other “functional mapping” domains highlighted in this section, there is a vast array of additional types of analyses allowing to integrate the evidence from genetics studies (such as GWAS) to those from other “‐omics” domains with the aim to derive common pathways relevant for pathogenesis and/ or therapeutic interventions. A substantial number of additional results from these analyses are highlighted in the primary publications by Jansen et al (14) and Kunkle et al (16). To learn more about these findings and their implications we encourage the interested reader to scrutinize the very detailed Supporting Information provided with both publications on the Nature Genetics website.
Concluding Remarks and Outlook
At the day of writing (August of 2019), AD genomics can be summarized succinctly with the results from two GWAS investigating a large number of independent case control datasets of European descent using state‐of‐the‐art methodology. From these studies, a total of 32 independent genomic loci showing compelling association with AD emerge in analyses of both common (Table 2) and rare (Table 3) genomic variants. While the exact nature of the underlying genes biologically responsible for the observed genetic associations remains elusive for most of the implied loci, a number of overlapping mechanistic themes emerge. First, both GWAS converge on the notion that most AD associated DNA variants are located in non‐coding portions of the genome, especially in regions with effects on gene transcription. This is in line with GWAS results from other complex phenotypes (8, 24) and has important bearings on the design of future genomic studies: if most of the functionally relevant variation occurs outside genes, technologies focusing on coding regions only (such as exon variant genotyping or whole‐exome sequencing) will likely not be suitable to decipher the genetic basis of AD and other conditions. Instead, more emphasis should be laid on the regions between genes (eg, using whole‐genome sequencing) and their functional implications and interactions (eg, using epigenomic and transcriptomic profiling, covered elsewhere in this Mini‐Symposium). Second, and in line with previous work in the AD field, the computational modeling performed by both GWAS emphasizes lipid metabolism and immune system response as crucial components in the pathogenesis of AD. For the latter, this is corroborated by gene‐set enrichment results showing expression in immune system‐related tissues (ie, whole blood, spleen, and liver) and, perhaps more importantly, in a key population of immune cells in the brain (microglia). Third, while no direct association signals were observed in the genes causing early‐onset monogenic AD (ie, APP, PSEN1, and PSEN2) the GWAS variants by both teams show a highly significant enrichment in other genes involved in the regulation of “APP metabolism/ Aβ formation.” This is the first time that APP metabolism emerged as a main functional category in genetic analyses of polygenic AD, providing further support for the amyloid hypothesis in this form of the disease. Last but not least, the results of both GWAS were published in “pre‐print” form prior to entering the peer‐review process (https://www.biorxiv.org/). The authors should be commended for this decision, as it has effectively allowed the community to work with their findings for almost a year before formal publication.
Despite their seminal scopes and unique analytic angles extending our understanding of genomic mechanisms underlying AD pathogenesis, the current GWAS results still leave some important questions unanswered. For instance, the new data in both studies did not markedly increase the proportion of phenotypic variance explained by genetics, a situation often described as the “missing heritability problem” in complex traits (27). Hence, if the phenotypic variance cannot be sufficiently explained by “simple” DNA variants of the type typically assayed by GWAS (eg, single base‐changes and small insertion‐deletions), the elusive heritability must be “hidden” elsewhere, for instance in other types of genomic variants (necessitating other genotyping/sequencing methods) and/or in genetic interactions among loci (necessitating novel analytic approaches highlighted elsewhere in this Mini‐Symposium). Third, both GWAS discussed here (as most GWAS published for AD to date), focus on datasets of European descent. The reason for this apparent selectivity is often simply convenience: European‐descent populations are typically those with the most readily available phenotype and genotype data. It will be exciting to observe how the utilization of datasets from other descent‐groups will (re‐)shape our knowledge and understanding of genomics mechanisms underlying AD (12). Finally, delineating the precise molecular mechanisms linking “genomic dysfunction” to “cognitive dysfunction,” for example, via “immune system dysfunction,” are still, works‐in‐progress and will require the development and application of novel methods effectively linking readouts from “‐omics”‐based studies to cellular function in vivo to establish causality of the observed statistical associations.
Supporting information
Figure S1. Manhattan plots of “discovery phase” GWAS findings from the two GWAS discussed in this review. A. Jansen et al (14); B. Kunkle et al (16). Equivalent to Figure 1 except that P‐values were not truncated.
{kind=link}
Figure S2. Regional association plot for the APOE region on chromosome 19q13.32 in the GWAS by Jansen et al (14). Plots were constructed with FUMA (25) on data described in “Data Availability Statement” section. Color code for SNPs: Each SNP is color‐coded based on the highest r2 to one of the independent significant SNPs, if that is greater or equal to r2 = 0.6. Other SNPs (ie, r2 < 0.6) are colored in grey. The top lead SNPs in genomic risk loci, lead SNPs and ind. sig. SNPs are circled in black and colored in dark‐purple, purple and red, respectively. Color code for genes: Red: Mapped genes. Blue: Non‐mapped protein‐coding genes. Dark grey: Non‐mapped non‐coding genes.
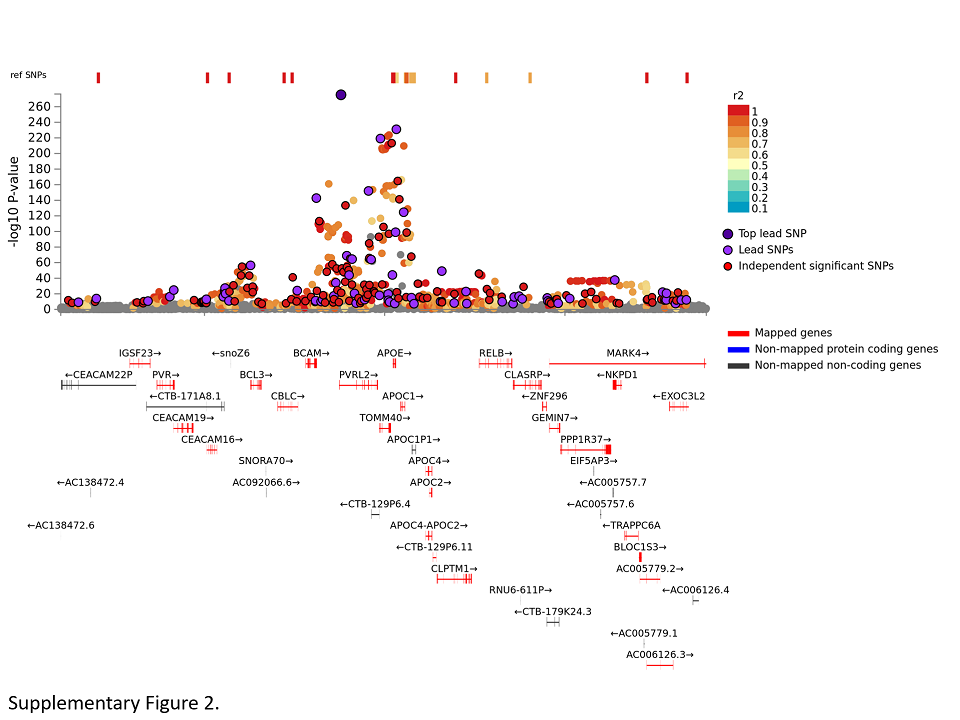{kind=link}
Figure S3. Functional consequences of SNPs on genes for the GWAS by Jansen et al 14 and Kunkle et al (16). See legend to Figure 2 for more details.
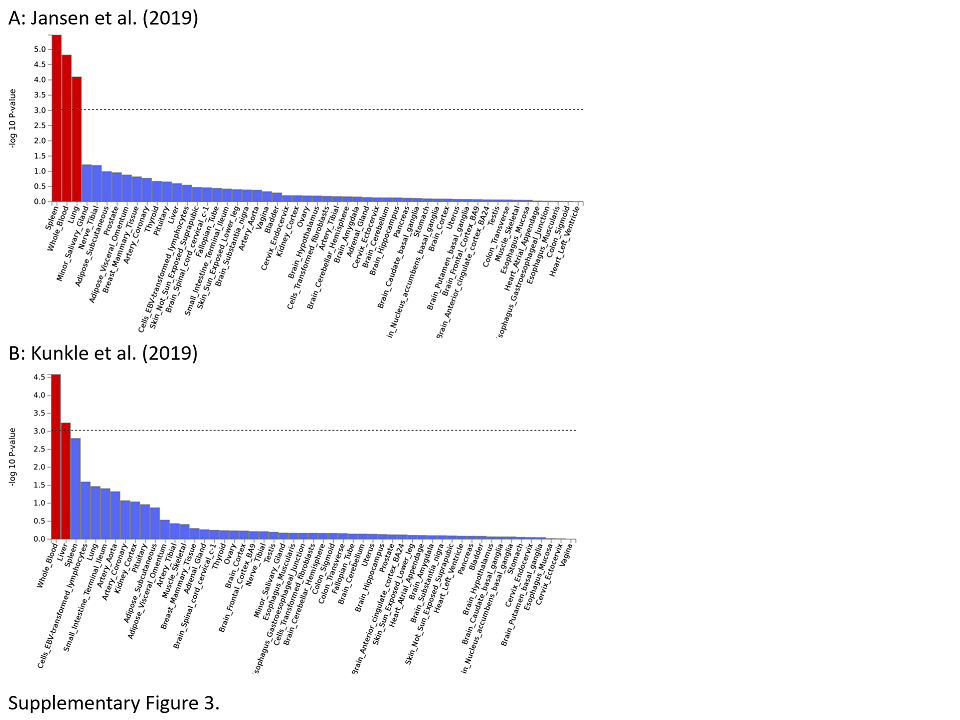{kind=link}
Figure S4. Results of tissue expression analysis for the GWAS by Jansen et al 14 and Kunkle et al (16). See legend to Figure 3 for more details.
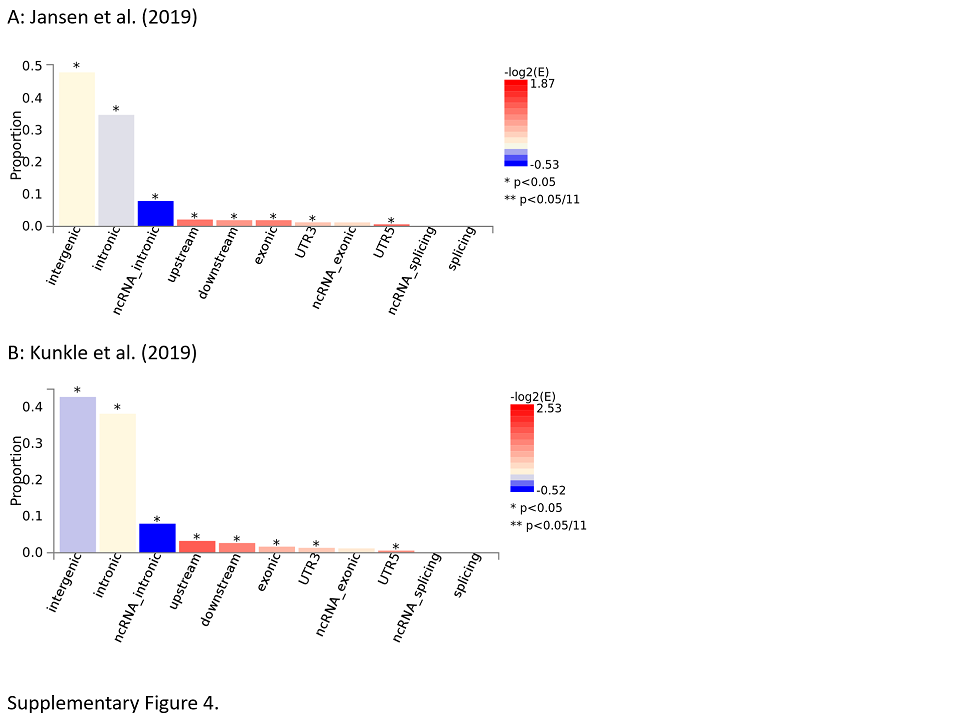{kind=link}
Acknowledgements
This work was supported by grants from the Deutsche Forschungsgemeinschaft (DFG), the European Research Council (ERC), and the Cure Alzheimer's Fund (CAF).
Data Availability Statement
All figures shown in this review were based on the re‐analysis and plotting of summary statistics made available alongside the original GWAS using FUMA (25). Specifically, for the Jansen et al (14) study we utilized the discovery phase association results available at https://ctg.cncr.nl/software/summary_statistics. For the Kunkle et al (16) study, we used the Stage 1 summary data available at https://www.niagads.org/igap‐rv‐summary‐stats‐kunkle‐p‐value‐data.
References
- 1. Aguet F, Ardlie KG, Cummings BB, Gelfand ET, Getz G, Hadley K et al (2017) Genetic effects on gene expression across human tissues. Nature 550:204–213. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Alzheimer A (1907) Über eine eigenartige Erkrankung der Hirnrinde. Allg Zeitschr Psychiatr Psychiatr‐Gerichtl Med 109:146–148. [Google Scholar]
- 3. Bertram L, Lange C, Mullin K, Parkinson M, Hsiao M, Hogan MF et al (2008) Genome‐wide association analysis reveals putative Alzheimer's disease susceptibility loci in addition to APOE. Am J Hum Genet 83:623–632. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Bertram L, McQueen MB, Mullin K, Blacker D, Tanzi RE (2007) Systematic meta‐analyses of Alzheimer disease genetic association studies: the AlzGene database. Nat Genet 39:17–23. [DOI] [PubMed] [Google Scholar]
- 5. Bertram L, Tanzi RE (2019) Alzheimer disease risk genes: 29 and counting. Nat Rev Neurol. 15:191–192. [DOI] [PubMed] [Google Scholar]
- 6. Buniello A, MacArthur JAL, Cerezo M, Harris LW, Hayhurst J, Malangone C et al (2019) The NHGRI‐EBI GWAS catalog of published genome‐wide association studies, targeted arrays and summary statistics 2019. Nucleic Acids Res 47:D1005–D1012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Bycroft C, Freeman C, Petkova D, Band G, Elliott LT, Sharp K et al (2018) The UK Biobank resource with deep phenotyping and genomic data. Nature 562:203–209. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Canela‐Xandri O, Rawlik K, Tenesa A (2018) An atlas of genetic associations in UK Biobank. Nat Genet 50:1593–1599. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Cruts M, Van Broeckhoven C (2018) Data mining: applying the AD&FTD mutation database to progranulin. Methods Mol Biol 1806:81–92. [DOI] [PubMed] [Google Scholar]
- 10. Hannon E, Marzi SJ, Schalkwyk LS, Mill J (2019) Genetic risk variants for brain disorders are enriched in cortical H3K27ac domains. Mol Brain 12:7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Heston LL, Mastri AR, Anderson VE, White J (1981) Dementia of the Alzheimer type. Arch Gen Psychiatry 38:1085–1090. [DOI] [PubMed] [Google Scholar]
- 12. Hindorff LA, Bonham VL, Brody LC, Ginoza MEC, Hutter CM, Manolio TA, Green ED (2018) Prioritizing diversity in human genomics research. Nat Rev Genet 19:175–185. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. International Genomics of Alzheimer's Disease Consortium (IGAP) ,Jones L, Lambert JC, Wang LS, Choi SH, Harold D et al (2015) Convergent genetic and expression data implicate immunity in Alzheimer's disease. Alzheimers Dement 11:658–671. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Jansen IE, Savage JE, Watanabe K, Bryois J, Williams DM, Steinberg S et al (2019) Genome‐wide meta‐analysis identifies new loci and functional pathways influencing Alzheimer's disease risk. Nat Genet 51:404–413. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Kundaje A, Meuleman W, Ernst J, Bilenky M, Yen A, Heravi‐Moussavi A et al (2015) Integrative analysis of 111 reference human epigenomes. Nature 518:317–330. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Kunkle BW, Grenier‐Boley B, Sims R, Bis JC, Damotte V, Naj AC et al (2019) Genetic meta‐analysis of diagnosed Alzheimer's disease identifies new risk loci and implicates Aβ, tau, immunity and lipid processing. Nat Genet 51:414–430. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Lambert JC, Ibrahim‐Verbaas CA, Harold D, Naj AC, Sims R, Bellenguez C et al (2013) Meta‐analysis of 74,046 individuals identifies 11 new susceptibility loci for Alzheimer's disease. Nat Genet 45:1452–1458. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Liu JZ, Erlich Y, Pickrell JK (2017) Case‐control association mapping by proxy using family history of disease. Nat Genet 49:325–331. [DOI] [PubMed] [Google Scholar]
- 19. Selkoe DJ, Hardy J (2016) The amyloid hypothesis of Alzheimer's disease at 25 years. EMBO Mol Med 8:595–608. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Sims R, van der Lee SJ, Naj AC, Bellenguez C, Badarinarayan N, Jakobsdottir J et al (2017) Rare coding variants in PLCG2, ABI3, and TREM2 implicate microglial‐mediated innate immunity in Alzheimer's disease. Nat Genet 49:1373–1384. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Suh J, Choi SH, Romano DM, Gannon MA, Lesinski AN, Kim DY, Tanzi RE (2013) ADAM10 missense mutations potentiate β‐amyloid accumulation by impairing prodomain chaperone function. Neuron 80:385–401. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Tanzi RE, Bertram L (2005) Twenty years of the Alzheimer's disease amyloid hypothesis: a genetic perspective. Cell 120:545–555. [DOI] [PubMed] [Google Scholar]
- 23. Veitch DP, Weiner MW, Aisen PS, Beckett LA, Cairns NJ, Green RC et al (2019) Understanding disease progression and improving Alzheimer's disease clinical trials: recent highlights from the Alzheimer's Disease Neuroimaging Initiative. Alzheimer's Dement. 15:106–152. [DOI] [PubMed] [Google Scholar]
- 24. Visscher PM, Wray NR, Zhang Q, Sklar P, McCarthy MI, Brown MA, Yang J (2017) 10 years of GWAS discovery: biology, function, and translation. Am J Hum Genet 101:5–22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Watanabe K, Taskesen E, van Bochoven A, Posthuma D (2017) Functional mapping and annotation of genetic associations with FUMA. Nat Commun 8:1826. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Wohlers I, Bertram L (2018) Taking genomics research to the next level: the Genotype‐Tissue expression project. Mov Disord 33:1097. [DOI] [PubMed] [Google Scholar]
- 27. Zuk O, Hechter E, Sunyaev SR, Lander ES (2012) The mystery of missing heritability: genetic interactions create phantom heritability. Proc Natl Acad Sci U S A 109:1193–1198. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Figure S1. Manhattan plots of “discovery phase” GWAS findings from the two GWAS discussed in this review. A. Jansen et al (14); B. Kunkle et al (16). Equivalent to Figure 1 except that P‐values were not truncated.
Figure S2. Regional association plot for the APOE region on chromosome 19q13.32 in the GWAS by Jansen et al (14). Plots were constructed with FUMA (25) on data described in “Data Availability Statement” section. Color code for SNPs: Each SNP is color‐coded based on the highest r2 to one of the independent significant SNPs, if that is greater or equal to r2 = 0.6. Other SNPs (ie, r2 < 0.6) are colored in grey. The top lead SNPs in genomic risk loci, lead SNPs and ind. sig. SNPs are circled in black and colored in dark‐purple, purple and red, respectively. Color code for genes: Red: Mapped genes. Blue: Non‐mapped protein‐coding genes. Dark grey: Non‐mapped non‐coding genes.
Figure S3. Functional consequences of SNPs on genes for the GWAS by Jansen et al 14 and Kunkle et al (16). See legend to Figure 2 for more details.
Figure S4. Results of tissue expression analysis for the GWAS by Jansen et al 14 and Kunkle et al (16). See legend to Figure 3 for more details.
Data Availability Statement
All figures shown in this review were based on the re‐analysis and plotting of summary statistics made available alongside the original GWAS using FUMA (25). Specifically, for the Jansen et al (14) study we utilized the discovery phase association results available at https://ctg.cncr.nl/software/summary_statistics. For the Kunkle et al (16) study, we used the Stage 1 summary data available at https://www.niagads.org/igap‐rv‐summary‐stats‐kunkle‐p‐value‐data.