

Abstract
The extremely invasive phenotypes and genotypes related to progression of gliomatosis cerebri (GC) remain unclear although GC has been removed as an independent entity from the 2016 WHO classification. Hence, categorization of GC under the current WHO molecular classification is essential, and the molecular subgroups that might contribute to GC progression should be compared with the histopathological differences between initial and new lesions identified during follow‐up. Analyses of IDH1/2 and TERTp mutations and 1p/19q co‐deletion, and immunohistochemistry of IDH1‐R132H, ATRX, p53 and galectin‐3 were performed. Anaplastic astrocytoma, IDH‐wildtype (AA‐IDHwt) was the common molecular subgroup (52.8%), followed by diffuse astrocytoma, IDH‐wildtype (DA‐IDHwt) and AA, IDH‐mutant (AA‐IDHmt) (each 16.9%), DA‐IDHmt (7.9%), glioblastoma (GBM)‐IDHwt (3.3%) and GBM‐IDHmt (2.2%). Approximately 92% of the AA‐IDHwt lesions progressed to histologically confirmed GBM in the newly enhanced lesions harboring the TERTp mutation and expressing galectin‐3. Similar to primary GBMs, GC‐related GBMs that progressed from the IDHwt subgroups showed microvascular proliferation, palisading necrosis or thrombotic occlusion, implying that a subset of IDHwt subgroups may evolve to overt GBM. Molecular subgrouping did not provide the perfect prediction for the survival of GC patients. The AA‐IDHwt group showed worse overall and progression‐free survival (PFS) than the AA‐IDHmt group. Biopsy plus radiotherapy, chemotherapy and temozolomide treatment for DA‐IDHwt, and resection plus radiotherapy and temozolomide treatment for AA‐IDHwt prolonged PFS. In conclusions, majority of GC was of the AA‐IDHwt subgroup, which progressed to GBM. Molecular subgroups may assist in the selection of treatment modalities, because “GC pattern” still remains as a special growth of gliomas in WHO 2016 classification without established treatment guideline.
Keywords: disease progression, gliomatosis cerebri, histology, IDH wildtype glioblastoma, World Health Organization 2016 classification
Introduction
Although gliomatosis cerebri (GC), a rare and extensively infiltrating glioma involving multiple contiguous lobes of the brain, has been removed as an independent tumor entity from the 2016 WHO classification, it still remains a special “GC growth pattern” that can occur in both astrocytic and oligodendroglial tumors 2, 9, 24. Till date, both the phenotype and genotype of GCs remain unexplained 31. As described previously, the clinical behavior of GC is similar to that of WHO grade III gliomas, and secondary progression to highly malignant tumor may occur 5. However, the unique extremely invasive phenotype of GC is unlike those of other ordinary high‐grade gliomas 14, as GC infiltrates the brain extensively, but usually preserves the local parenchymal architecture, which is completely different from the destructive and necrotic infiltrating pattern observed in high‐grade gliomas 5, 36. The significant neovascularization and mitotic activity that are commonly present in high‐grade gliomas are not prominent features of GC 14, 31. Rather, the majority of GC tends to present histologically low‐grade features such as rod cell‐like astrocytic tumor cells with elongated fusiform nuclei without prominent microvascular proliferation or necrosis 5, 29. Because of the discrepancy between tumor behavior and histological grades of GC, patients with GC show worse prognosis than patients with diffuse glioma of corresponding grades 5, 36.
Newly developed contrast‐enhanced lesions on follow‐up radiological images indicate the progression of GC 21. Although GC is first diagnosed using radiographic findings, which are subsequently supported by histopathological evaluation, the histological spectra of initial to newly developed lesions have been rarely specified. Therefore, histopathological review of new contrast‐enhancing lesions and the initial lesions categorized under the current WHO 2016 molecular classification via consideration of IDH mutation as well as 1p/19q copy number status is required, which will improve our understanding regarding the relationship of genotype with GC progression.
The aim of this study was to classify GC into one of the WHO 2016 diagnostic categories of infiltrating glioma and analyze whether the molecular subgroups provide specific information regarding the histological and prognostic features of GC. We also attempted to determine the molecular subgroups that might contribute to the progression of GC and compared them with the histopathological observations on initial and newly developed lesions. This demonstrates the high‐risk molecular subgroup within GCs for disease progression and clinical relevance of molecular subgrouping for selection of the best treatment in the clinic.
Patients and Methods
Patients, histological and radiological evaluations and molecular classification
We retrospectively identified 100 consecutive patients with GC who were documented in the electronic database of the Samsung Medical Center during the 20‐year period from 1995 to 2015 and whose informed consent was obtained from the patients. The diagnosis of GC was based on the following criteria 5, 29: (i) T2‐ or fluid‐attenuated inversion recovery (FLAIR)‐weighted magnetic resonance imaging (MRI) showing a diffuse process of infiltration involving more than three different lobes and relative preservation of the anatomical architecture and (ii) histological tissue analysis confirming glial cell proliferation, either diagnostic of or consistent with an infiltrative glioma. Eleven patients were excluded because of insufficient formalin‐fixed paraffin‐embedded (FFPE) tumor tissue from three patients and reclassification of six patients into glioblastoma (GBM), one patient into oligodendroglioma and one patient into “diffuse midline glioma,” among which four patients mainly affected in the thalamus or basal ganglia based on immunohistochemistry for H3.3‐K27M. Finally, 89 patients, the features of whom were radiologically and histopathologically consistent with those of GC, were enrolled in this study. Among these 89 patients, 31 patients underwent second biopsy or surgical resection for histological examination of the newly developed contrast‐enhancing lesions, which was absent in the previous studies involving follow‐up brain imaging. The medical records and results of radiological investigations were reviewed to obtain clinical data. This study was approved by the Institutional Review Board of the Samsung Medical Center (Seoul, Korea).
Neuropathologists (YLS and MJK) reviewed all the glass slides, and histological grading was based on a set of pathological factors (cellularity, nuclear atypia, mitoses, microvascular proliferation and necrosis) and was assigned to grades as follows: grade II, gliomas with mildly increased cellularity and mild nuclear atypia; grade III, gliomas with moderate hypercellularity with nuclear atypia or mildly increased cellularity with focal or dispersed anaplasia and mitotic activity (in biopsy samples); grade IV, high‐grade glioma with severe hypercellularity, significant nuclear atypia and mitotic activity, microvascular proliferation and/or necrosis 5. Rod cells with fusiform hyperchromatic naked nuclei were considered present when the longer diameter was more than thrice that of the shorter diameter 29.
All MRIs were reviewed by a neuroradiologist and were classified as type 1 or type 2 GCs, according to a previously reported criterion 5, 29. Type 1 GCs were diffuse neoplastic growths and enlargements of the involved existing structure, without the formation of a discrete tumor mass at the initial clinical presentation 12. Type 2 GCs were lesions with an obvious neoplastic mass, in addition to a diffuse infiltrative lesion involving more than three different lobes at the time of diagnosis.
The molecular classification was based on the WHO 2016 classification of brain tumors as follows 24: (i) oligodendroglioma, IDH mutant and 1p/19q co‐deleted; (ii) diffuse astrocytoma, IDH mutant (DA‐IDHmt) or IDH wildtype (DA‐IDHwt); (iii) anaplastic astrocytoma, IDH mutant (AA‐IDHmt) or IDH wildtype (AA‐IDHwt); (iv) glioblastoma, IDH mutant (GBM‐IDHmt) or IDH wildtype (GBM‐IDHwt).
Mutational analysis of IDH1, IDH2 and TERT promoters
Genomic DNA was extracted from 10 µm‐thick sections of 10% neutral FFPE blocks using the Maxwell® 16 FFPE tissue LEV DNA purification kit (Promega, USA). IDH1 (R132) and IDH2 (R140 and R172) mutations were identified using PNAClamp™ IDH1/2 mutation detection kit (PANAGENE, Daejeon, Korea) and TERT promoter (TERTp) mutations (C250 and C228) were identified using the PNAClamp™ TERT mutation detection kit (PANAGENE) according to the manufacturer's instructions. For equivocal cases, direct sequencing analyses of IDH1, IDH2 and TERTp were repeated, as described previously 22.
1p/19q Co‐deletion
While 25 cases were previously analyzed for 1p/19q status using fluorescence in situ hybridization (FISH) at the time of diagnosis, the remaining 64 cases were analyzed using PCR‐based loss of heterozygosity test for the 1p/19q deletion 17, 18. Five microsatellite markers (D1S508, D1S2734, D1S186, D19S412 and D19S219) were used. For equivocal cases, FISH was performed on the FFPE tissue sections using probes for 1p36 and 19q13 (Vysis, Downers Grove, IL, USA) 34. At least 100 non‐overlapping neoplastic nuclei containing a minimum of two control signals were evaluated. No target signal or only a single signal was interpreted as a deletion. 1p deletion was defined by a combined target‐to‐control signal ratio <0.75 or nucleus cutoff with 1 or 0 target signal >50% 34. 19q deletion was defined by a combined target‐to‐control signal ratio <0.8 and nucleus cutoff with 1 or 0 target signal >30% 34.
Immunohistochemistry
Immunohistochemical staining was performed on 4 μm‐thick FFPE sections using a Leica Microsystems Bond‐Max autostainer system (Leica Biosystems, Wetzlar, Germany), according to the manufacturer's protocols 7, 19, 34. The primary antibodies included IDH1‐R132H (clone H09, 1:10, Dianova, Hamburg, Germany), ATRX (HPA001906, 1:200, Sigma‐Aldrich, St Louis, MO, USA), galectin‐3 (9C4, 1:100, Novocastra, Newcastle, UK) and p53 (BP53‐12, 1:400, Invitrogen, USA). IDH1‐R132H expression was assessed by the presence of strong cytoplasmic staining in tumor cells 3, 22. Loss of nuclear staining in the majority of the tumor cells in the presence of an internal positive control (endothelium) was interpreted as loss of ATRX expression 34. Only nuclear staining of >5% tumor cells was considered positive for p53, which has been previously validated as the appropriate cutoff value in our institute 7, 29. Galectin‐3 was considered positive in the presence of both nuclear and cytoplasmic expression of tumor cells 23, 27.
Statistical analysis
The Chi square test, Fisher's exact test and an independent t‐test were used appropriately. Progression‐free survival (PFS) was defined as the time between the first surgical intervention and the appearance of a new lesion on the follow‐up MRI, as indicated by the treating clinician. Overall survival (OS) was defined as the period from the day of first surgery until death, or the end of follow‐up. Analysis of the survival data was performed in September 2017. Survival differences among groups were calculated using the Kaplan‐Meier method with a log‐rank test. For the subgroup survival analysis, the log‐rank test with Bonferroni's correction was used. All tests were two‐tailed, and the significance level was set at 0.05. The SPSS statistical software (version 22, Statistical Package for the Social Sciences) was used for all statistical analyses.
Results
Clinicopathological characteristics and patient management
Among the 89 patients, 52 (58.4%) were men and 37 (41.6%) were women, with median age of 49 years (range, 10–80 years) at the time of diagnosis. Based on the MRI findings, 65 tumors (73.0%) were classified as type 1 GC and 24 tumors (27.0%) as type 2 GC. In histological evaluation, histological grade III (69.7%, 62/89) was the most common, followed by grade II (24.7%, 22/89) and grade IV (5.6%, 5/89). Hyperchromatic rod cells were variably observed in all the cases. The cellularity was predominantly mild (62.9%, 56/89) [22 into grade II, 33 into grade III and 1 into grade IV], followed by moderate (36.0%, 32/89) [29 into grade III and 3 into grade IV] and severe (1.1%, 1/89) [1 into grade IV]. Pleomorphic or multinucleated giant tumor cells were identified in 21 GCs (23.6%) [20 in grade III and 1 in grade IV]. Microvascular proliferation was detected in five GCs, which were classified as grade IV. Necrosis was not identified in 89 GCs. The representative histological observations of GC are shown in Figure 1.
Figure 1.
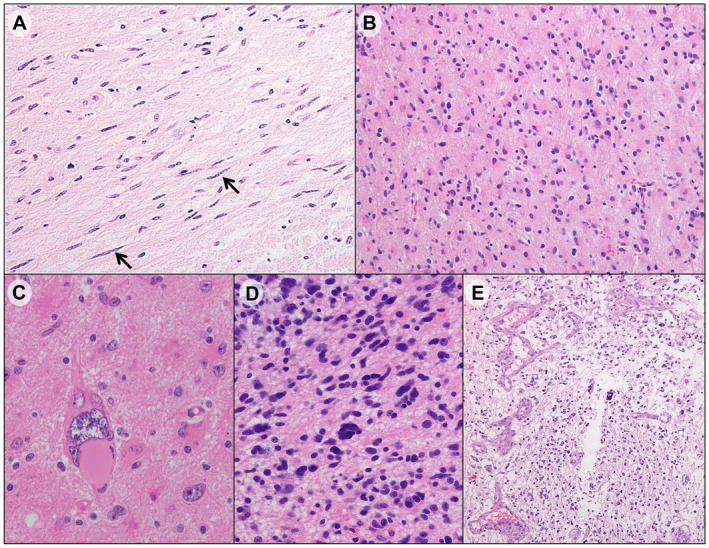
Representative histologic findings of gliomatosis cerebri. A. Infiltrating rod cells (Arrows) in anaplastic astrocytoma, IDH wildtype. B. Mildly increased cellularity and mild nuclear atypia in histological grade II (diffuse astrocytoma, IDH wildtype). C. Pleomorphic tumor cells in anaplastic astrocytoma, IDH wildtype. D. Moderate hypercellularity with nuclear atypia in histological grade III (anaplastic astrocytoma, IDH mutant). E. Microvascular proliferation in glioblastoma, IDH wildtype.
Twenty‐four patients (27.0%) underwent partial tumor resection and 65 patients (73.0%) underwent stereotactic biopsy for the purpose of histological diagnosis and decompression after the initial radiological diagnosis. Twenty‐five patients underwent either partial tumor resection or biopsy and did not receive any further adjuvant treatment. Postoperative therapy usually included radiotherapy, with or without chemotherapy. In 65 resected patients, 6 patients (6.7%) received local radiotherapy, with conventional fractionation (2 Gy/fraction) and 58 Gy median total dose (range, 54–60 Gy). The extent of the radiotherapy was determined by the area of tumor involvement. Fourteen patients (15.7%) received adjuvant chemotreatment following radiotherapy, using temozolomide (TMZ) (6.7%) or nitrosourea‐based regimen (2.3%) such as BCNU (carmustine‐[1,3‐bis (2‐chloroethyl)‐1‐nitrosourea]) and PCV (procarbazine, carmustine and vincristine), and combined TMZ and nitrosourea‐based chemotherapy (6.7%).
In 65 biopsied patients (73.0%), 14 patients (15.7%) received only radiotherapy. Twenty‐three patients (25.9%) received adjuvant chemotherapy following radiotherapy, using TMZ (7.9%) or nitrosourea‐based regimen (10.1%) and combined TMZ and nitrosourea‐based chemotherapy (7.9%).
The follow‐up period ranged from 2 to 128 months (mean ± SD, 31.1 ± 27.5 months). During the follow‐up period, 69 patients (77.5%) died and 52 patients (58.4%) showed disease progression. The median survival time of patients with GC was 23 months. The percentages of 1‐year OS, 2‐year OS and 3‐year OS for patients with GC were 78.7%, 43.8% and 25.8%, respectively.
Molecular and immunohistochemical results and their associations with growth types
The summary of patient characteristics, molecular and immunohistochemical results and correlation coefficients with GC growth types are listed in Table S1.
The IDH1 and IDH2 mutations were successfully analyzed in all 89 samples; however, the analysis of the TERTp mutation was successful in 34 samples. IDH1 mutations were detected in 24 (27.0%) out of 89 samples [23 in CGT > CAT (R132H) and 1 in CGT > AGT (R132S)]. No IDH2 mutation (0%) was detected. The 1p/19q co‐deletions were detected in three cases (3.4%), all of which were histological grade III astrocytic tumors with type 1 GC. Two of them harboring 1p/19q co‐deletions later progressed to GBMs. TERTp mutations were detected in 7 (20.6%) of 34 samples [three in C228T and four in C250T], all of which were type 1 GC of grade III (n = 6) and grade IV (n = 1) and showed intact ATRX expression. Hence, 1p/19q co‐deletions and TERTp mutations were detected exclusively in Type 1 GCs.
The loss of ATRX expression was observed in 14 of 73 cases (19.2%), in which 11 were of type 1 GC and 3 were type 2 GC. p53 positivity was observed in 12 of 27 tumors (44.4%), all of which were type 1 GCs. p53 expression correlated with loss of ATRX expression (Spearman's correlation r = 0.441; P = 0.035). Galectin‐3 was expressed in 6 of 20 tumors (30.0%), all of which were type 1 GCs and 50% (3/6) of the galectin‐3 positive GCs later progressed to higher grades.
Regarding histological features, compared to type 2 GC, type 1 GC was significantly associated with lack of pleomorphic cells (P = 0.015). The AA‐IDHwt subgroup was the most common molecular subtype in both type 1 GC and type 2 GC, which accounted for 52.3% and 54.2% cases, respectively. There was no significant correlation between GC growth types and WHO 2016 molecular subgroups (P = 0.302).
Overall, compared to type 2 GCs, the type 1 GCs appeared to show a trend toward high frequencies of 1p/19q co‐deletions, loss of ATRX expression, TERTp mutations, p53 and galectin‐3 expression and the AA‐IDHwt subgroup, whereas they lacked pleomorphic tumor cells, although most of these associations did not reach statistical significance.
Molecular Subgroupings and their correlation with survival
Based on the molecular subgroups of WHO 2016 classification (Table 1), all 89 GCs were classified into astrocytic tumors of 65 IDHwt (73.0%) [grade II (16.9%, 15/89), grade III (52.8%, 47/89) and grade IV (3.3%, 3/89)] and 24 IDHmt (27.0%) [grade II (7.9%, 7/89), grade III (16.9%, 15/89) and grade IV (2.2%, 2/89)]. No oligodendroglioma was designated to the 89 GCs, as three tumors harboring 1p/19q co‐deletions did not fit the 2016 WHO criteria (IDH mutant and complete 1p/19q co‐deleted) of oligodendrogliomas (two tumors showed AA‐IDHwt and one showed a typical histology of GBM‐IDHmt). These three cases with 1p/19q co‐deletions were interpreted to harbor partial deletions involving the 1p36 and 19q13 by FISH, and their morphologies were astrocytic, without typical histological features of oligodendroglioma.
Table 1.
Clinicopathological characteristics according to WHO 2016 molecular subgrouping of gliomatosis cerebri. Bold indicates statistically significant (P < 0.05). Abbreviations: IDHwt = IDH‐wildtype; IDHmt = IDH‐mutant; OS = overall survival.
| Total | IDHwt subgroups | IDHmt subgroups | P | |||||
|---|---|---|---|---|---|---|---|---|
| II | III | IV | II | III | IV | |||
| N = 89 (%) | n = 15 (16.9%) | n = 47 (52.8%) | n = 3 (3.3%) | n = 7 (7.9%) | n = 15 (16.9%) | n = 2 (2.2%) | ||
| Sex | 0.927 | |||||||
| Male | 52 (57.4) | 9 (60.0) | 27 (57.4) | 1 (33.3) | 5 (71.4) | 9 (60.0) | 1 (50.0) | |
| Female | 37 (41.6) | 6 (40.0) | 20 (42.6) | 2 (66.7) | 2 (28.6) | 6 (40.0) | 1 (50.0) | |
| Age (year) | 0.794 | |||||||
| <18 | 5 (5.6) | 1 (6.7) | 4 (8.5) | 0 (0.0) | 0 (0.0) | 0 (0.0) | 0 (0.0) | |
| ≥18 | 84 (94.4) | 14 (93.3) | 43 (91.5) | 3 (100) | 7 (100) | 15 (100) | 2 (100) | |
| OS† time (month) | 23 | 25 | 18 | 15 | 24 | 58 | 43 | 0.078 |
| 1 year‐OS rate | 78.7% | 73.3% | 72.3% | 100% | 85.7% | 93.3% | 100% | 0.433 |
| 2 year‐OS rate | 43.8% | 53.3% | 31.9% | 33.3% | 42.9% | 66.7% | 100% | 0.104 |
| 3 year‐OS rate | 25.8% | 20.0% | 17.0% | 33.3% | 28.6% | 46.7% | 100% | 0.044 |
| 1p/19q | 0.060 | |||||||
| Co‐deletion | 3 (3.4) | 0 (0.0) | 2 (4.3) | 0 (0.0) | 0 (0.0) | 0 (0.0) | 1 (50.0) | |
| No codeletion | 86 (96.6) | 15 (100) | 45 (95.7) | 3 (100) | 7 (100) | 15 (100) | 1 (50.0) | |
| ATRX (n = 73) | <0.001 | |||||||
| Intact | 59 (80.8) | 13 (92.9) | 38 (95.0) | 1 (100) | 2 (40.0) | 5 (41.7) | 0 (0.0) | |
| Loss | 14 (19.2) | 1 (7.1) | 2 (5.0) | 0 (0.0) | 3 (60.0) | 7 (58.3) | 1 (100) | |
| TERTp (n = 34) | 0.626 | |||||||
| Wildtype | 27 (79.4) | 5 (100) | 11 (73.3) | 1 (100) | 2 (100) | 7 (77.8) | 1 (50.0) | |
| Mutated | 7 (20.6) | 0 (0.0) | 4 (26.7) | 0 (0.0) | 0 (0.0) | 2 (22.2) | 1 (50.0) | |
| P53 (n = 27) | 0.103 | |||||||
| Positive | 12 (44.4) | 1 (20.0) | 4 (28.6) | 1 (100) | 2 (100) | 3 (75.0) | 1 (100) | |
| Negative | 15 (55.6) | 4 (80.0) | 10 (71.4) | 0 (0.0) | 0 (0.0) | 1 (25.0) | 0 (0.0) | |
| Galectin‐3 (n = 20) | 0.230 | |||||||
| Positive | 6 (30.0) | 1 (20.0) | 5 (55.6) | ‐ | 0 (0.0) | 0 (0.0) | 0 (0.0) | |
| Negative | 14 (70.0) | 4 (80.0) | 4 (44.4) | ‐ | 2 (100) | 3 (100) | 1 (100) | |
| Pleomorphic cells | 0.038 | |||||||
| Absent | 68 (76.4) | 15 (100) | 30 (63.8) | 2 (66.7) | 7 (100) | 12 (80.0) | 2 n | |
| Present | 21 (23.6) | 0 (0.0) | 17 (36.2) | 1 (33.3) | 0 (0.0) | 3 (20.0) | 0 (0.0) | |
OS time indicates median survival time.
As a result, AA‐IDHwt was the most common molecular subgroup in GC. AA‐IDHwt harbored the most common TERTp mutation (4/7) and pleomorphic or multinucleated tumor giant cells (81.0%, 17/21), which was statistically higher than the number of pleomorphic cells of AA‐IDHmt (14.3%, 3/21) (P = 0.038).
Overall, molecular subgrouping did not provide statistically significant differences in OS and PFS in patients with GC (P = 0.078 and P = 0.435, respectively; Figure 2A‐B). The shortest median survival was 15 months for the GBM‐IDHwt group, followed by 18 months for the AA‐IDHwt, 24 months for the DA‐IDHmt, 25 months for the DA‐IDHwt, 43 months for the GBM‐IDHmt and 58 months for the AA‐IDHmt groups, which showed a trend toward shorter survival along with increase in histological grades in the IDHwt subgroups.
Figure 2.
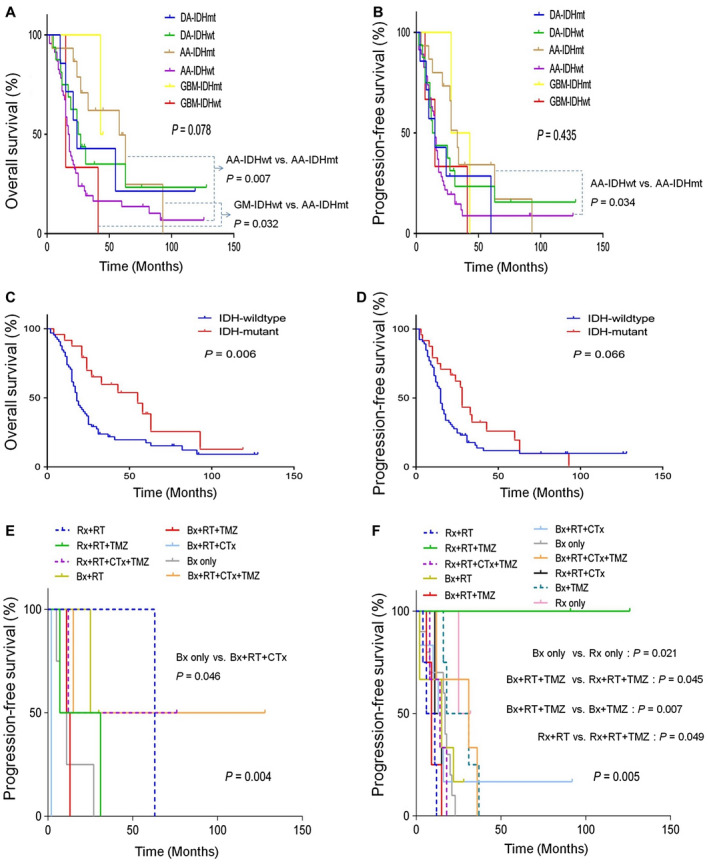
Overall survival (OS) (A) and progression‐free survival (PFS) (B) in gliomatosis cerebri (GC) according to WHO 2016 molecular subgroups. OS (C) and PFS (D) according to IDH mutational status. PFS according to treatment modalities in DA‐IDHwt subgroup (E) and AA‐IDHwt subgroup (F).
Comparison between subgroups revealed that the AA‐IDHwt (31.5 ± 5.3 months) and GBM‐IDHwt groups (23.7 ± 8.7 months) showed statistically worse OS than the AA‐IDHmt subgroup (54.5 ± 9.7 months) (P = 0.007 and P = 0.032, respectively). The AA‐IDHwt subgroup (27.4 ± 5.4 months) also showed statistically poorer PFS than the AA‐IDHmt subgroup (41.7 ± 8.7 months) (P = 0.034). There were no statistically significant prognostic differences between the other molecular subgroups.
Survival and prognosis
We also evaluated the prognostic impact of the histological grades, GC growth types and IDH or TERTp mutational status on survival of patients with GC. The presence of IDH1 mutation (median 55 months) was associated with better OS than that of wild‐type IDH1 (median 18 months) in all patients (P = 0.006), whereas the IDH1 mutation showed a trend of borderline association with favorable PFS (P = 0.066) (Figure 2C,D). However, OS or PFS did not vary significantly among GCs with respect to GC growth types, TERTp mutational status, or histological grades (P = 0.625 and P = 0.321; P = 0.455 and P = 0.098; P = 0.805 and P = 0.858, respectively).
In multivariate analysis (Table 2), the presence of IDH1 mutation was the only independent favorable prognostic factor predicting increased OS [P = 0.009, HR 0.201, 95% CI 0.060–0.671]. In addition, the IDH1 mutation, partial resection, TERTp wildtype and absence of newly developed enhancing lesion were independent favorable prognostic factors associated with increased PFS in patients [P = 0.010, HR 0.247, 95% CI 0.086–0.712; P = 0.021, HR 0.204, 95% CI 0.053–0.785; P = 0.009, HR 5.334, 95% CI 1.519–18.729; P = 0.004, HR 6.099, 95% CI, 1.754–21.204, respectively].
Table 2.
Multivariate analyses for overall survival and progression‐free survival using the Cox proportional hazard model. Abbreviations: HR = hazard ratio; CI = confidential interval; TMZ = temozolomide.
| Overall survival | Progression‐free survival | |||
|---|---|---|---|---|
| HR (95% CI) | P | HR (95% CI) | P | |
| Age (year) (>18 vs. ≤18) | 0.232 (0.023–2.352) | 0.216 | 0.697 (0.091–5.305) | 0.727 |
| Sex (male vs. female) | 1.518 (0.441–5.232) | 0.508 | 1.171 (0.375–3.662) | 0.786 |
| IDH1 (Wildtype vs. Mutated) | 0.201 (0.060–0.671) | 0.009 | 0.247 (0.086–0.712) | 0.021 |
| TERTp (Wildtype vs. Mutated) | 2.581 (0.650–10.255) | 0.178 | 5.334 (1.519–18.729) | 0.009 |
| Growth type (type 1 vs. type 2) | 0.206 (0.024–1.765) | 0.149 | 0.983 (0.187–5.170) | 0.984 |
| Histological grade (II‐III vs. IV) | 1.591 (0.411–6.160) | 0.501 | 0.763 (0.215–2.705) | 0.676 |
| Newly enhancing lesion (yes vs. no) | 1.483 (0.462–4.757) | 0.507 | 6.099 (1.754–21.204) | 0.004 |
| Partial resection (yes vs. no) | 0.294 (0.071–1.226) | 0.093 | 0.204 (0.053–0.785) | 0.021 |
| Radiotherapy (yes vs. no) | 0.736 (0.204–2.660) | 0.641 | 0.757 (0.228–2.513) | 0.649 |
| Chemotherapy† (yes vs. no) | 2.039 (0.484–8.581) | 0.331 | 2.590 (0.632–10.616) | 0.186 |
| TMZ (yes vs. no) | 2.787 (0.767–10.128) | 0.119 | 2.774 (0.825–9.327) | 0.099 |
Nitrosourea‐based chemotherapy.
Prognostic correlations depending on the treatment modalities including biopsy (Bx) or partial tumor resection (Rx) with or without radiotherapy (RT), nitrosourea‐based chemotherapy (CTx) and TMZ were further investigated. The most favorable median OS periods were observed in Rx + RT + TMZ (91 months), followed by Bx + TMZ (37 months), Bx + RT + CTx + TMZ (31 months), Bx + RT + CTx (25 months), Bx only (21 months), Bx + RT + TMZ (17 months), Rx + RT + CTx (15 months) and Bx + RT (15 months), Rx + RT + CTx + TMZ (14 months) and Rx + RT (12 months). The most favorable median PFS was detected in Rx only (34 months), followed by Bx + TMZ (31 months), Bx + RT + CTx + TMZ (27 months), Bx only (18 months), Bx + RT + CTx (15 months) and Bx + RT (15 months), Rx + RT + CTx + TMZ (13 months), Rx + RT (11 months), Rx + RT + CTx (11 months) and Bx + RT + TMZ (11 months).
Among the WHO 2016 molecular subgroups, the DA‐IDHwt and AA‐IDHwt subgroups showed statistically significant differences in PFS depending on treatment modalities (P = 0.004 and P = 0.005, respectively) (Figure 2E‐F). However, no other molecular subgroup showed statistically significant differences in OS or PFS with respect to treatments.
In the DA‐IDHwt subgroup, Bx + RT + CTx + TMZ (median 71.5 months) showed the best outcomes, followed by Rx + RT (63 months), Bx + RT (27.5 months), Rx + RT + TMZ (19 months), Rx + RT + CTx + TMZ (12 months), Bx + RT + TMZ (12 months), Bx only (9 months) and Bx + RT + CTx (2 months). As a result, Bx + RT + CTx + TMZ led to the most favorable PFS, whereas Bx + RT + CTx resulted in a PFS worse than those observed with other treatment modalities in the DA‐IDHwt subgroups.
In the AA‐IDHwt subgroup, Rx + RT + TMZ (108.5 months) showed the best outcomes of PFS, followed by Bx + RT + CTx + TMZ (31 months), Rx only (28.5 months), Bx + TMZ (24.5 months), Bx only (17 months), Rx + RT + CTx + TMZ (16 months), Bx + RT (15 months), Bx + RT + CTx (14 months), Rx + RT + CTx (13 months), Bx + RT + TMZ (9 months) and Rx + RT (median 8.5 months). As a result, Rx + RT + TMZ showed the most favorable PFS, whereas Rx + RT tended to show the worst PFS compared to other treatments in the AA‐IDHwt subgroups. Taken together, Bx + RT + CTx + TMZ for the DA‐IDHwt subgroup and Rx + RT + TMZ for the AA‐IDHwt subgroup increased PFS rates in GCs.
In particular, several therapeutic options were statistically better for PFS of the AA‐IDHwt subgroup; for example, Rx only was better than Bx only (P = 0.021), Rx + RT + TMZ was better than Bx + RT + TMZ (P = 0.045), Bx + TMZ was better than Bx + RT + TMZ (P = 0.007) and Rx + RT + TMZ was better than Rx + RT (P = 0.049).
For 12 patients, there was an unusually long OS of >5 years after diagnosis of GC (range 61–128 months) (Table 3). Prominent therapeutic modalities were not identified in these patients. However, the effects of Bx + RT + CTx + TMZ for the DA‐IDHwt subgroup and Rx + RT + TMZ for the AA‐IDHwt subgroup were also well appreciated in two patients who survived >10 years. These patients were female with type 2 GC of the IDHwt subgroups (DA‐IDHwt and AA‐IDHwt) who underwent treatment, including radiotherapy and TMZ.
Table 3.
Clinicopathological characteristics from shorter survival to longer survival time of 12 patients who lived more than 5 years after diagnosis of gliomatosis cerebri. Abbreviations: OS = overall survival; DA‐IDHwt = Diffuse astrocytoma, IDH‐wildtype; DA‐IDHmt = Diffuse astrocytoma, IDH‐mutant; AA‐IDHwt = Anaplastic astrocytoma, IDH‐wildtype; AA‐IDHmt = Anaplastic astrocytoma, IDH‐mutant; F = female; M = male; Rx = resection; RT = radiotherapy; TMZ = temozolomide; Bx = biopsy; CTx = nitrosourea‐based chemotherapy.
| No. | WHO2016 | OS (month) | Survival | Sex | Age (year) | Growth type | Treatment† |
|---|---|---|---|---|---|---|---|
| 1 | AA‐IDHmt | 61 | Alive | M | 42 | Type 2 | Rx |
| 2 | AA‐IDHmt | 63 | Died | M | 28 | Type 2 | Bx RT |
| 3 | DA‐IDHwt | 63 | Died | M | 52 | Type 1 | Rx RT |
| 4 | DA‐IDHwt | 76 | Alive | F | 48 | Type 2 | Rx RT CTx TMZ |
| 5 | AA‐IDHwt | 77 | Alive | M | 40 | Type 1 | Bx TMZ |
| 6 | AA‐IDHwt | 82 | Died | F | 49 | Type 1 | Bx RT CTx TMZ |
| 7 | AA‐IDHwt | 91 | Died | M | 55 | Type 2 | Rx RT TMZ |
| 8 | AA‐IDHwt | 92 | Alive | M | 15 | Type 1 | Bx RT CTx |
| 9 | AA‐IDHmt | 93 | Died | F | 28 | Type 2 | Rx RT TMZ |
| 10 | DA‐IDHmt | 119 | Alive | M | 68 | Type 1 | Bx RT TMZ |
| 11 | AA‐IDHwt | 126 | Alive | F | 49 | Type 2 | Rx RT GKS TMZ |
| 12 | DA‐IDHwt | 128 | Alive | F | 51 | Type 2 | Bx Bx RT CTx TMZ |
Treatment includes the overall treatment after initial biopsy or resection before follow‐up biopsy.
Histopathological comparison between initial lesion at first diagnosis and newly developed contrast‐enhancing lesion during follow‐up
The median interval time to new appearance of contrast‐enhancing lesions in follow‐up MRI was 13 months, ranging from 1 to 89 months. Thirty‐one patients were available for histological review of both initial and later newly‐developed lesions. However, two cases were excluded because of complete necrosis in the follow‐up biopsied specimens, and the remaining 29 specimens were only evaluated for histological comparisons (Table S2). The schematic diagnostic spectra from initial lesion to disease progressed lesion have been summarized in Figure 3.
Figure 3.
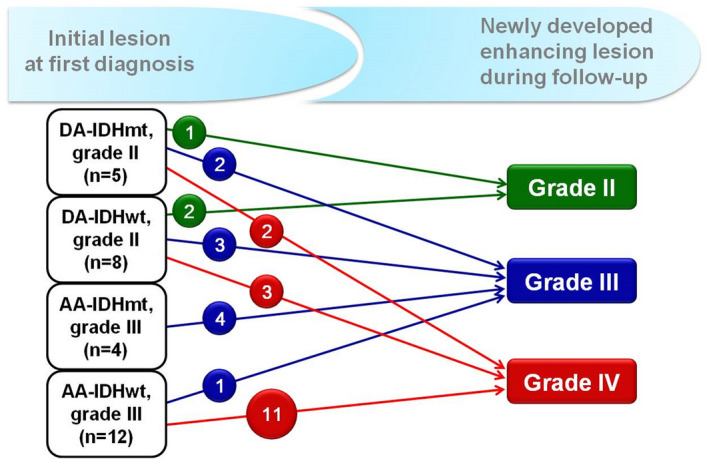
Diagnostic spectrums from initial lesion to newly developed contrast‐enhanced lesions during the follow‐up.
The newly developed contrast‐enhancing lesions initially originated from 20 IDHwt subgroups (69.0%) [7 grade II (24.2%), 12 grade III (41.4%), and 1 grade IV (3.4%)] and 9 IDHmt subgroups (31.0%) [5 grade II (17.2%) and 4 grade III (13.8%)]. Of those 29 cases, 20 (69.0%) [16 IDHwt (55.2%) and 4 IDHmt (13.8%)] progressed to grades higher than the initial grades, in which 15 cases (51.7%), including 13 IDHwt (44.8%) [2 DA‐IDHwt and 11 AA‐IDHwt] and 2 DA‐IDHmt (6.9%), developed to GBM. The remaining five grade II cases [three DA‐IDHwt (60.0%) and two DA‐IDHmt (40.0%)] cases became grade III lesions. The median interval times to GBM progression were 13 months for the IDHwt subgroups (range 1–34 months) and 7.5 months for the IDHmt subgroups (range 2–13 months). However, none of the AA‐IDHmt subgroups progressed to GBM. In contrast, the histological grades of the remaining nine cases of contrast‐enhancing new lesions (31.0%), including four IDHwt [two grade II, one grade III, one grade IV] and five IDHmt [one grade II and four grade III], were identical to the initial grades.
Discussion
In the present study, we have shown that AA‐IDHwt is the most common molecular subgroup (52.8%) in GC, with approximately 92% (11/12) of AA‐IDHwt exhibiting progression to GBM in the newly enhanced lesions. Molecular subgrouping, according to the 2016 WHO classification, did not accurately predict the survival of patients with GC. However, the AA‐IDHwt subgroup showed worse prognoses in terms of both OS and PFS than the AA‐IDHmt subgroup. We also observed that Bx + RT + CTx + TMZ treatment for the DA‐IDHwt subgroup and Rx + RT + TMZ treatment for the AA‐IDHwt subgroup prolonged the PFS rates. To the best of our knowledge, this is the first study demonstrating the differences between the histological spectra of initial and progressed lesions, and the clinical outcomes of the treatment modalities depend on specific molecular subgroups in GC.
Fifty percent GCs was subcategorized into AA‐IDHwt, followed by DA‐IDHwt (16.9%), AA‐IDHmt (16.9%), DA‐IDHmt (7.9%), GBM‐IDHwt (3.3%) and GBM‐IDHmt (2.2%). No oligodendroglioma was identified. The majority of GCs were IDHwt subgroups (73.0%), with the most common being AA‐IDHwt. A prominent histological feature of the AA‐IDHwt subgroup was the high frequency of occurrence (81.0%) of pleomorphic cells, which distinguished it from the AA‐IDHmt subgroup. In the newly developed enhancing lesions observed during follow‐up, AA‐IDHwt and DA‐IDHwt comprised the two top‐ranking molecular subgroups progressing to GBM. Interestingly, the TERTp mutation was only identified in the AA‐IDHwt subgroup, which later progressed to GBM. Galectin‐3 expression was also positive in the DA‐IDHwt and AA‐IDHwt subgroups, which later progressed to GBM. The TERTp mutation is related to the transformation of glial progenitor cells to primary GBM and its associated aggressive phenotype 15, 24, 28. Galectin‐3 expression in glial tumor cells has been reported to be related to infiltrative cell motility away from hypoxic regions 27. Our molecular and immunohistochemical observations are interesting in the context of previously published data, as GC has been considered a lesion of intermediate malignancy in the progression of diffuse glioma to GBMs, and the progression of GC is mediated by tumor stem cells 20. As GC shares characteristics with neural stem cells, GC may show migratory behavior, diversity of progeny and proliferative potential 20. Herrlinger et al 9 have shown that 52.2% GCs show GBM‐IDHwt‐associated methylation profile, suggesting a close relationship between GC and GBM at the molecular level. Recently, IDHwt TERTp‐mutated astrocytomas were found to be typically present as GC in one series 11, suggesting that IDHwt GCs indeed possessed the potential of malignant transformation to GBMs. In fact, rapid transformations from initially IDH1‐wildtype, low‐grade GC without contrast‐enhancement on MRI to massive GBM or higher‐grade glioma have been rarely reported within the short interval of 3–7 months after initial presentation 13, 16, 26, 32, 35.
In addition, the newly appeared GBM originating from the IDHwt subgroups showed microvascular proliferation (n = 3), palisading necrosis (n = 2) or thrombotic occlusion (n = 1), which are not related to therapeutic effects, but rather corresponded to typical histological features of primary GBM. These differences in the histological spectra between initial and progressed lesions may contradict the well‐known mechanism that only IDHmt low‐grade gliomas secondarily succumb to GBM, as well as the fact that GBM‐IDHwt arise de novo with no precursor lesions 24. Nevertheless, we observed that a considerable number of IDHwt GCs truly progressed to GBM‐IDHwt in our study, implying a subset of IDHwt subgroups may be precursors or early‐stage GBMs that ultimately evolve to overt GBM.
Molecular subgrouping, according to the WHO 2016 classification, did not provide detailed prognostic stratification for patients with GC. Rather, the presence of the IDH1 mutation in GC patients demonstrated independent favorable prognostic factor for both OS and PFS. The favorable prognostic value of IDH1 mutations has been consistently reported in patients with GC 4, 6, 22. The absence of reliable prognostic and predictive markers for GCs despite application of the WHO 2016 molecular subgrouping guidelines, highlights the importance of IDH1 mutation status in patients with GCs.
While palliative surgery, radiotherapy and chemotherapy have been used to treat patients with GCs, the optimal therapeutic strategy for GCs remains unclear. There is no standard guideline for surgery and the extent of surgical resection in GC. A previous study has reported that any survival benefit is not provided between partial resection and biopsy (median survival, 18 vs. 21 months) in 30 GC patients 30. Radiotherapy has been reported to be effective in approximately 50% patients with GCs and to increase OS 10. However, large field radiotherapy is associated with the risk of severe toxicity. Chemotherapy is often administered to patients with GC, and upfront chemotherapy produces an objective radiological response rate in 25%–45% patients and a median PFS of 13–17 months 6, 14. However, its efficacy still remains controversial. TMZ is currently widely used as a first‐line treatment for GCs because it is more tolerable and less toxic than nitrosourea‐based chemotherapy 14, and may be a cost‐effective treatment for GC 33. In these circumstances, the molecular subgrouping within GC may be of considerable clinical significance for guided decision‐making regarding the appropriate therapeutic strategy. Especially, we have shown that Bx + RT + CTx + TMZ therapy for the DA‐IDHwt subgroup and Rx + RT + TMZ therapy for the AA‐IDHwt subgroup increased PFS rates in GCs. In particular, in the AA‐IDHwt subgroup, RT + TMZ may be recommended post‐resection but not post‐biopsy. After tumor resection, RT + TMZ may be related to more favorable PFS than RT. However, after biopsy, initial TMZ may be recommended instead of RT + TMZ. In the context, the effects of Bx + RT + CTx + TMZ for the DA‐IDHwt subgroup and Rx + RT + TMZ for the AA‐IDHwt subgroup were also well appreciated in two patients who unusually survived >10 years. Although a long indolent course and prolonged survival are rarely observed in GC patients 8, the reasons for long survivors with GC should be further more clearly elucidated in the future study. Therefore, our study suggests that molecular subgrouping may allow prediction of clinical outcomes regarding treatments and enable selection of patients who might benefit from treatment.
In our GCs, four cases were located in the midline, in addition to more than three cerebral lobes, which were used for immunohistochemistry for H3.3‐K27M. Among them, only one case of a 31‐year‐old male was confirmed via H3.3‐K27M positivity as diffuse midline glioma and was excluded from the current study. The characteristics of adult H3‐K27M mutant gliomas in association with GC are yet to be completely elucidated 25, 31. Whether GC with H3‐K27M mutation may represent a rare molecular pattern of GCs or not requires further investigation.
Based on our results, it could not be conclusive that GC is a specific subtype of diffuse gliomas pathologically and prognostically. The current WHO 2016 classification describes “GC pattern” only in IDH‐wildtype gliomas 24. However, the present study has shown that GC consists of both IDH‐wildtype and IDH‐mutant subtypes, although the majority of GCs are AA‐IDHwt that would progress to GBM. The prognosis of GC is importantly related to IDH mutational status. The favorable prognosis would be anticipated when the therapeutic strategies such as concurrent chemoradiation indicative of GBM are applied to GC patients, regardless the histological grades.
The limitations of the current study are a lack of entire GC lesions caused by resection or biopsy samples for possible regional heterogeneity. Unfortunately, because of sample limitations we could not fully evaluate all cases for genetic alterations associated with IDH‐wildtype astrocytic tumors that are proposed by the Consortium to Inform Molecular and Practical Approaches to CNS Tumor Taxonomy (cIMPACT‐NOW) recently issued update 3 1, because of insufficient sample quantity mostly obtained from streotactic biopsy for additional molecular study. The current study also took long time until accomplishing both molecular and immunohistochemical results, their validations and repeated experiments for confirmation before recent cIMPACT‐NOW recommendation. Nevertheless, we were able to interpret several trends from these results. AA‐IDHwt was the most common molecular subgroup of GC and was also the most remarkable subgroup potentially progressing to GBM. Molecular subgrouping within GC appears to be insufficient to completely explain the survival outcomes of GC, highlighting the importance of IDH1 mutation predicting favorable survival in GC. Because the term of “GC pattern” still remains as a special growth of gliomas in WHO 2016 classification with neither established prognostic and predictive markers nor treatment guideline in GC, molecular subgroups may indicate the best treatment options for GC patients, who might benefit from various treatment modalities.
Author Contributions
Yeon‐Lim Suh and Mi Jung Kwon designed the study, interpreted the data, wrote the manuscript and drafted of the manuscript; So Young Kang carried out the experiments and data acquisition; Haeyon Cho, Jung Il Lee and Sung Tae Kim were involved in the acquisition, analysis or interpretation of clinical data.
Conflicts of Interest
All authors have no conflicts of interests to disclose.
Supporting information
Table S1 . Patient characteristics of 89 patients with gliomatosis cerebri and correlations between growth type and clinicopathological characteristics.
Table S2 . Clinicopathological features of the 29 patients with second biopsy at disease progression site during follow‐up.
Acknowledgments
This work was supported by the National Research Foundation of Korea (NRF) grant funded by the Korea government (Ministry of Science and ICT) (No. NRF‐2019R1C1C1004463).
Data Availability Statement: Data available on request because of privacy/ethical restrictions. The data that support the findings of this study are available on request from the corresponding author.
Data Availability Statement
Data available on request because of privacy/ethical restrictions. The data that support the findings of this study are available on request from the corresponding author.
References
- 1. Brat DJ, Aldape K, Colman H, Holland EC, Louis DN, Jenkins RB et al (2018) cIMPACT‐NOW update 3: recommended diagnostic criteria for “Diffuse astrocytic glioma, IDH‐wildtype, with molecular features of glioblastoma, WHO grade IV”. Acta Neuropathol 136:805–810. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Broniscer A, Chamdine O, Hwang S, Lin T, Pounds S, Onar‐Thomas A et al (2016) Gliomatosis cerebri in children shares molecular characteristics with other pediatric gliomas. Acta Neuropathol 131:299–307. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Capper D, Weissert S, Balss J, Habel A, Meyer J, Jager D et al (2009) Characterization of R132H mutation‐specific IDH1 antibody binding in brain tumors. Brain Pathol 20:245–254. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Desestret V, Ciccarino P, Ducray F, Criniere E, Boisselier B, Labussiere M et al (2011) Prognostic stratification of gliomatosis cerebri by IDH1(R132H) and INA expression. J Neurooncol 105:219–224. [DOI] [PubMed] [Google Scholar]
- 5. Fuller GN, Kros JM (2007) Gliomatosis cerebri. In: WHO Classification of Tumours of the Central Nervous System, Louis DN, Ohgaki H, Wiestler OD, Cavenee WK, Ellison DW, Figarella‐Branger D et al (eds), pp. 50–52. IARC Press: Lyon. [Google Scholar]
- 6. Glas M, Bahr O, Felsberg J, Rasch K, Wiewrodt D, Schabet M et al (2011) NOA‐05 phase 2 trial of procarbazine and lomustine therapy in gliomatosis cerebri. Ann Neurol 70:445–453. [DOI] [PubMed] [Google Scholar]
- 7. Ha SY, Kang SY, Do IG, Suh YL (2013) Glioblastoma with oligodendroglial component represents a subgroup of glioblastoma with high prevalence of IDH1 mutation and association with younger age. J Neurooncol 112:439–448. [DOI] [PubMed] [Google Scholar]
- 8. Herrlinger U, Felsberg J, Kuker W, Bornemann A, Plasswilm L, Knobbe CB et al (2002) Gliomatosis cerebri: molecular pathology and clinical course. Ann Neurol 52:390–399. [DOI] [PubMed] [Google Scholar]
- 9. Herrlinger U, Jones DTW, Glas M, Hattingen E, Gramatzki D, Stuplich M et al (2016) Gliomatosis cerebri: no evidence for a separate brain tumor entity. Acta Neuropathol 131:309–319. [DOI] [PubMed] [Google Scholar]
- 10. Inoue T, Kumabe T, Kanamori M, Sonoda Y, Watanabe M, Tominaga T (2011) Prognostic factors for patients with gliomatosis cerebri: retrospective analysis of 17 consecutive cases. Neurosurg Rev 34:197–208. [DOI] [PubMed] [Google Scholar]
- 11. Izquierdo C, Barritault M, Poncet D, Cartalat S, Joubert B, Bruna J et al (2018) Radiological Characteristics and Natural History of Adult IDH‐Wildtype Astrocytomas with TERT Promoter Mutations. Neurosurgery 85:E448–E456. [DOI] [PubMed] [Google Scholar]
- 12. Jennings MT, Frenchman M, Shehab T, Johnson MD, Creasy J, LaPorte K, Dettbarn WD (1995) Gliomatosis cerebri presenting as intractable epilepsy during early childhood. J Child Neurol 10:37–45. [DOI] [PubMed] [Google Scholar]
- 13. Jimenez Caballero PE, Mollejo Villanueva M, Marsal Alonso C (2007) Gliomatosis cerebri: evolution to glioblastoma multiforme. Neurologia 22:395–398. [PubMed] [Google Scholar]
- 14. Kaloshi G, Everhard S, Laigle‐Donadey F, Marie Y, Navarro S, Mokhtari K et al (2008) Genetic markers predictive of chemosensitivity and outcome in gliomatosis cerebri. Neurology 70:590–595. [DOI] [PubMed] [Google Scholar]
- 15. Killela PJ, Reitman ZJ, Jiao Y, Bettegowda C, Agrawal N, Diaz LA Jr et al (2013) TERT promoter mutations occur frequently in gliomas and a subset of tumors derived from cells with low rates of self‐renewal. Proc Natl Acad Sci USA 110:6021–6026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Kim Y, Hong M, Do IG, Ha SY, Lee D, Suh YL (2015) Wnt5a, Ryk and Ror2 expression in glioblastoma subgroups. Pathol Res Pract 211:963–972. [DOI] [PubMed] [Google Scholar]
- 17. Kim SH, Kim H, Kim TS (2005) Adequate microsatellite markers for 1p/19q loss of heterozygosity of oligodendroglial tumors in Korean patients. Korean J Pathol 39:22–33. [Google Scholar]
- 18. Kim SH, Kim H, Kim TS (2005) Clinical, histological, and immunohistochemical features predicting 1p/19q loss of heterozygosity in oligodendroglial tumors. Acta Neuropathol 110:27–38. [DOI] [PubMed] [Google Scholar]
- 19. Kim HK, Yu IK, Kim SM, Kim JH, Lee SH, Lee SY (2017) Rapid progression of gliomatosis cerebri to secondary glioblastoma, factors that affect the progression rate: a case report. J Korean Soc Radiol 76:221–228. [Google Scholar]
- 20. Kong DS, Kim ST, Lee JI, Suh YL, Lim DH, Kim WS et al (2010) Impact of adjuvant chemotherapy for gliomatosis cerebri. BMC Cancer 10:424. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Kong DS, Kim MH, Park WY, Suh YL, Lee JI, Park K et al (2008) The progression of gliomas is associated with cancer stem cell phenotype. Oncol Rep 19:639–643. [PubMed] [Google Scholar]
- 22. Kwon MJ, Kim ST, Kwon MJ, Kong DS, Lee D, Park S et al (2012) Mutated IDH1 is a favorable prognostic factor for type 2 gliomatosis cerebri. Brain Pathol 22:307–317. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Kwon MJ, Sung CO, Kang SY, Do IG, Suh YL (2013) Differential expression of extracellular matrix‐related genes in rare variants of meningioma. Hum Pathol 44:260–268. [DOI] [PubMed] [Google Scholar]
- 24. Louis DN, von Deimling A, Cavenee WK (2016) Diffuse astrocytic and oligodendroglioal tumours. In: WHO Classification of Tumours of the Central Nervous System, Louis DN, Ohgaki H, Wiestler OD, Cavenee WK, Ellison DW, Figarella‐Branger D et al (eds), Chapter 1, pp. 15–77. IARC Press: Lyon. [Google Scholar]
- 25. Meyronet D, Esteban‐Mader M, Bonnet C, Joly MO, Uro‐Coste E, Amiel‐Benouaich A et al (2017) Characteristics of H3 K27M‐mutant gliomas in adults. Neuro Oncol 19:1127–1134. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Nagata R, Ikeda K, Nakamura Y, Ishikawa Y, Miura K, Sato R et al (2010) A case of gliomatosis cerebri mimicking limbic encephalitis: malignant transformation to glioblastoma. Intern Med 49:1307–1310. [DOI] [PubMed] [Google Scholar]
- 27. Neder L, Marie SK, Carlotti CG Jr, Gabbai AA, Rosemberg S, Malheiros SM et al (2004) Galectin‐3 as an immunohistochemical tool to distinguish pilocytic astrocytomas from diffuse astrocytomas, and glioblastomas from anaplastic oligodendrogliomas. Brain Pathol 14:399–405. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Ohgaki H, Kleihues P (2013) The definition of primary and secondary glioblastoma. Clin Cancer Res 19:764–772. [DOI] [PubMed] [Google Scholar]
- 29. Park S, Suh YL, Nam DH, Kim ST (2009) Gliomatosis cerebri: clinicopathologic study of 33 cases and comparison of mass forming and diffuse types. Clin Neuropathol 28:73–82. [DOI] [PubMed] [Google Scholar]
- 30. Perkins GH, Schomer DF, Fuller GN, Allen PK, Maor MH (2003) Gliomatosis cerebri: improved outcome with radiotherapy. Int J Radiat Oncol Biol Phys 56:1137–1146. [DOI] [PubMed] [Google Scholar]
- 31. Ranjan S, Warren KE (2017) Gliomatosis cerebri: current understanding and controversies. Front Oncol 7:165. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. San Millan B, Kaci R, Polivka M, Robert G, Heran F, Gueguen A et al (2010) Gliomatosis cerebri: a biopsy and autopsy case report. Ann Pathol 30:25–29. [DOI] [PubMed] [Google Scholar]
- 33. Sanson M, Cartalat‐Carel S, Taillibert S, Napolitano M, Djafari L, Cougnard J et al (2004) Initial chemotherapy in gliomatosis cerebri. Neurology 63:270–275. [DOI] [PubMed] [Google Scholar]
- 34. Sim J, Nam DH, Kim Y, Lee IH, Choi JW, Sa JK, Suh YL (2018) Comparison of 1p and 19q status of glioblastoma by whole exome sequencing, array‐comparative genomic hybridization, and fluorescence in situ hybridization. Med Oncol 35:60. [DOI] [PubMed] [Google Scholar]
- 35. Sun P, Piao H, Guo X, Wang Z, Sui R, Zhang Y et al (2014) Gliomatosis cerebri mimicking acute viral encephalitis and with malignant transformation of partial lesions: a case report. Exp Ther Med 8:925–928. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Vates GE, Chang S, Lamborn KR, Prados M, Berger MS (2003) Gliomatosis cerebri: a review of 22 cases. Neurosurgery. 53:261–271; discussion 271. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Table S1 . Patient characteristics of 89 patients with gliomatosis cerebri and correlations between growth type and clinicopathological characteristics.
Table S2 . Clinicopathological features of the 29 patients with second biopsy at disease progression site during follow‐up.
Data Availability Statement
Data available on request because of privacy/ethical restrictions. The data that support the findings of this study are available on request from the corresponding author.