

Abstract
The proinflammatory cytokine IFN-γ, which is chronically elevated in multiple sclerosis, induces pathologic quiescence in human oligodendrocyte progenitor cells (OPCs) via upregulation of the transcription factor PRRX1. In this study using animals of both sexes, we investigated the role of heparan sulfate proteoglycans in the modulation of IFN-γ signaling following demyelination. We found that IFN-γ profoundly impaired OPC proliferation and recruitment following adult spinal cord demyelination. IFN-γ-induced quiescence was mediated by direct signaling in OPCs as conditional genetic ablation of IFNγR1 (Ifngr1) in adult NG2+ OPCs completely abrogated these inhibitory effects. Intriguingly, OPC-specific IFN-γ signaling contributed to failed oligodendrocyte differentiation, which was associated with hyperactive Wnt/Bmp target gene expression in OPCs. We found that PI-88, a heparan sulfate mimetic, directly antagonized IFN-γ to rescue human OPC proliferation and differentiation in vitro and blocked the IFN-γ-mediated inhibitory effects on OPC recruitment in vivo. Importantly, heparanase modulation by PI-88 or OGT2155 in demyelinated lesions rescued IFN-γ-mediated axonal damage and demyelination. In addition to OPC-specific effects, IFN-γ-augmented lesions were characterized by increased size, reactive astrogliosis, and proinflammatory microglial/macrophage activation along with exacerbated axonal injury and cell death. Heparanase inhibitor treatment rescued many of the negative IFN-γ-induced sequelae suggesting a profound modulation of the lesion environment. Together, these results suggest that the modulation of the heparanome represents a rational approach to mitigate the negative effects of proinflammatory signaling and rescuing pathologic quiescence in the inflamed and demyelinated human brain.
SIGNIFICANCE STATEMENT The failure of remyelination in multiple sclerosis contributes to neurologic dysfunction and neurodegeneration. The activation and proliferation of oligodendrocyte progenitor cells (OPCs) is a necessary step in the recruitment phase of remyelination. Here, we show that the proinflammatory cytokine interferon-γ directly acts on OPCs to induce pathologic quiescence and thereby limit recruitment following demyelination. Heparan sulfate is a highly structured sulfated carbohydrate polymer that is present on the cell surface and regulates several aspects of the signaling microenvironment. We find that pathologic interferon-γ can be blocked by modulation of the heparanome following demyelination using either a heparan mimetic or by treatment with heparanase inhibitor. These studies establish the potential for modulation of heparanome as a regenerative approach in demyelinating disease.
Keywords: demyelination, human, interferon, oligodendrocyte progenitor, quiescence, remyelination
Introduction
Multiple sclerosis (MS), an autoimmune disease of the CNS, is characterized by demyelination, neurologic dysfunction, and neurodegeneration (Lassmann, 2014). Oligodendrocyte progenitor cells (OPCs) are vital for the endogenous regenerative process known as remyelination. Remyelination prevents ongoing neurodegeneration that typify progressive forms of MS (Franklin and Ffrench-Constant, 2017). The failure of remyelination in MS has been largely attributed to a failure of the generation of new oligodendrocytes. This is likely because of impaired oligodendrocyte differentiation along with a combination of insufficient migration, death, or pathologic quiescence of OPCs. The inhibitory tissue environment is characterized by reactive astrogliosis and astroglial scar formation (Ponath et al., 2018), as well as proinflammatory microglial/macrophage infiltration (Lassmann et al., 2001; Wang et al., 2019). Together, the environment acts to limit the recruitment of OPCs into demyelinated lesions and may thereby prevent proliferation, differentiation, and remyelination.
Interferon-γ (IFN-γ) is a pleotropic proinflammatory cytokine implicated in MS pathogenesis (Popko et al., 1997; Steinman, 2001). IFN-γ induces microglial M1 polarization (Martinez and Gordon, 2014), upregulation of class I/II major histocompatibility complex (MHC), and increased expression of proinflammatory cytokines. In MS patients, IFN-γ levels are frequently elevated before the onset of clinical symptoms (Beck et al., 1988). Indeed, IFN-γ treatment in MS patients resulted in severe disease exacerbation (Panitch et al., 1987). However, the precise roles of IFN-γ are more complex as studies in animals have also implicated IFN-γ in neuroprotection (for review, see Arellano et al., 2015). IFN-γ signals via the ubiquitously expressed IFNγR1 that is coupled to the JAK (Janus kinase)/STAT1 (signal transducer and activator of transcription 1) pathway and results in the upregulation of interferon regulatory factor 1 (IRF1). Interestingly, single nucleotide polymorphisms in IRF1 are associated with progressive MS (Fortunato et al., 2008), and IRF1-expressing oligodendrocyte lineage cells are present in MS lesions (Ren et al., 2011; Loda and Balabanov, 2012). In vitro high doses of IFN-γ induce OPC apoptosis (Vartanian et al., 1995; Baerwald and Popko, 1998; Horiuchi et al., 2006; Wang et al., 2010). However, at lower doses and in the absence of serum, IFN-γ instead induces reversible cell-cycle quiescence in rodent OPCs (Agresti et al., 1996; Tanner et al., 2011) and human OPCs (hOPCs; Wang et al., 2018). In vivo excessive IFN-γ during CNS development induces hypomyelination (Corbin et al., 1996; LaFerla et al., 2000) and demyelination (Horwitz et al., 1997). Following demyelination, IFN-γ impairs remyelination in both experimental autoimmune encephalomyelitis (EAE) and cuprizone mice models (Lin et al., 2006). Oligodendrocyte-specific SOCS1 (suppressor of cytokine signaling 1) overexpression, a negative regulator of IFN-γ receptor, antagonized the hypomyelinating effects of IFN-γ supporting a direct cell-autonomous role following IFN-γ signaling (Balabanov et al., 2006).
Recently, we identified PRRX1 as a transcriptional regulator of pathologic quiescence in hOPCs that is upregulated by IFN-γ and Bmp signaling (Wang et al., 2018). Moreover, IFN-γ coinjection with lysolecithin results in a model of proinflammatory focal demyelination that recapitulates PRRX1 upregulation and OPC quiescence (Wang et al., 2018). In this study, we further examined the role of endogenous IFN-γ signaling on OPCs and the potential for rescuing the negative effects of IFN-γ by modulating the local OPC heparanome that regulates remyelination (Saraswat et al., 2020). We found that PRRX1 mRNA upregulation occurs in OPCs located within chronically demyelinated lesions in MS and, importantly, that IFN-γ directly influences OPC recruitment in vivo. Moreover, the modulation of the heparanome by treatment with the heparan sulfate mimetic PI-88 results in antagonism of IFN-γ-mediated signaling in OPCs both in vitro and in vivo rescuing the negative effects of IFN-γ on OPC proliferation, recruitment, differentiation, and remyelination. We previously found that PI-88 acts via inhibition of OPC-expressed sulfatases (Sulf1/2) to enhance remyelination (Saraswat et al., 2020). In contrast, the effects of PI-88 on IFN-γ signaling were not dependent on Sulf1/2 but rather were associated with inhibition of IFN-γ-induced heparanases. Intriguingly, IFN-γ signaling in OPCs resulted in activation of Wnt and Bmp target genes, suggesting a possible mechanism by which oligodendrocyte differentiation was delayed. Together, our data demonstrate that pharmacological manipulation of the heparanome using the heparan mimetic PI-88 or OGT2115 a small-molecule heparanase inhibitor may be sufficient to overcome the proinflammatory demyelinated lesion microenvironment induced by IFN-γ and rescue OPCs from pathologic quiescence.
Materials and Methods
Human tissue samples and cell culture
Human fetal brain tissue samples (gestational age, 20–22 weeks) were obtained from patients who consented to tissue use under protocols approved by the University at Buffalo Research Subjects Institutional Review Board. Forebrain tissue was minced and dissociated using papain and DNase as described previously (Conway et al., 2012). Magnetic sorting for CD140a was performed as described in the study by Conway et al. (2012). Cells were maintained on plates coated with poly-l-ornithine and laminin, in human neural differentiation (ND) media as described in the study by Abiraman et al. (2015). Cells were plated at a density of 25 k/ml and maintained as progenitors by supplementing ND media with 20 ng/ml PDGF-AA (PeproTech) and 5 ng/ml NT-3 (PeproTech).
Proliferation assay
Human CD140a+ OPCs were cultured for 24 h in serum-free media supplemented with 20 ng/ml PDGF-AA and 5 ng/ml NT-3. Twenty-four hours postseeding, cells were pretreated with PI-88 (2 µg/ml; gift of Medigen Biotechnology) for 30 min and then with IFN-γ (10 ng/ml; PeproTech) with or without PI-88 (2 µg/ml). EdU (50 μm) was then added, and cells were fixed 24 h later. Cells were immunostained for Olig2 (1:500; Millipore) and EdU using the Click-iT EdU Flow Cytometry Assay Kit (Thermo Fisher Scientific) according to the manufacturer protocol. DNA was stained with 1 µg/ml DAPI. A percentage of EdU+ Olig2+ stained nuclei was obtained from 10 fields for each fetal sample.
Luciferase assays
OPCs were seeded at a density of 25 k/ml and maintained as progenitors in ND media supplemented with PDGF-AA and NT-3. One day postseeding, cells were infected with Cignal Lenti GAS luciferase reporter (CLS-OO9L, Qiagen) for 24 h, after which media was changed to fresh ND media supplemented with growth factors and PI-88 (2 µg/ml). After 30 min of PI-88 (2 µg/ml), IFN-γ (1 and 10 ng/ml) was added with or without PI-88 (2 µg/ml). Twenty-four hours post-treatment, luminescence response was quantified using the Promega Bright-Glo reagent and a Bio-Tek plate reader, in accordance with manufacturer recommendations. Background luminescence was subtracted from all measurements, and luminescence readings were normalized to the average value of the untreated control in each experiment. Data are presented as the mean ± SEM for three technical replicates run on the same plate. For human OPCs, experiments were repeated at least three times using separate fetal samples to account for biological variation.
Oligodendrocyte differentiation and immunocytochemistry
To initiate differentiation, PDGF-AA and NT-3 growth factors were removed, and cells cultured in ND media supplemented with IFN-γ (PeproTech), OGT2155 (Tocris Bioscience), and/or PI-88 (gift from Medigen Biotechnology), as designated. Forty-eight hours following growth factor removal, cells were live stained with O4 IgM hybridoma supernatant (1:20; gift from James Goldman, Columbia University), fixed in 4% paraformaldehyde, and stained with secondary antibody. Differentiation was quantified as the proportion of O4+ cells in 10 random fields at 10× magnification (IX83 microscope, Olympus), representative of >3000 cells total in each condition.
Drug treatments in mice spinal cord demyelination model
All experiments were performed according to protocols approved by the University at Buffalo Institutional Animal Care and Use Committee. NG2creER:Rosa26YFP animals were a gift from Akiko Nishiyama (University of Connecticut, Storrs, CT; Zhu et al., 2011). NG2creER/IFNγR1fl/fl mice were derived from strain 025394 (The Jackson Laboratory). Animals were bred to generate NG2CreER offspring homozygous for IFNγR1 floxed alleles (NG2creERcre/wt:Rosa26YFPIFNγR1fl/fl). Conditional gene knockout (KO) by cre-mediated recombination in NG2+ OPCs was achieved by intraperitoneal administration of tamoxifen (200 mg/kg; Millipore Sigma) for 5 d, the last of which occurred 7 d before surgery. All experimental mice received tamoxifen in an identical manner, and littermate controls lacking cre were compared with cre-containing cKO mice. For experiments using IFN-γ and PI-88, 8-week-old female BALB/c mice were purchased from Envigo. Sulf1/2 cKO mice were used as described in the study by Saraswat et al. (2020). Recombination efficiency was determined using Olig2 and yellow fluorescent protein (YFP) immunofluorescence. We observed YFP expression in >50% of Olig2+ oligodendrocyte lineage cells, suggesting a high rate of cre-mediated recombination for both IFN-gR1 cKO mice (59.0 ± 5.2%) and Sulf1/2 cKO mice (55.2 ± 5.0%; n = 6–7 animals). Recombination efficiency was similar to previous findings using NG2creER driver line with the muscarinic M3R cKO mouse (Welliver et al., 2018).
Focal demyelination of the young adult (age, 8–11 weeks) mouse spinal cord was induced as previously described (Welliver et al., 2018). Briefly, animals were anesthetized under isoflurane and 0.5 μl of 1% lysolecithin (Lα-lysophosphatidylcholine, Millipore Sigma) was directly injected into the dorsal and ventral funiculus of the spinal cord between two thoracolumbar vertebrae. For experiments involving IFN-γ, two different doses were used (3 ng/injection and 10 ng/injection) with or without PI-88 (10 μg/ml). Drugs were first diluted in 0.9% saline and then mixed with 1% lysolecithin to get a final concentration. For the induction of demyelination drug and lysolecithin mixtures were injected into the spinal cord (Fancy et al., 2011; Lee et al., 2015). Postoperative analgesia was provided by subcutaneous injection of buprenorphine (0.05 mg/ml). Terminal BrdU/EdU was administered by intraperitoneal injection at 48 and 24 h before animals were killed (BrdU 100 mg/kg, EdU 50 mg/kg).
For heparanase inhibition, OGT2115 (Tocris Bioscience) stocks were added to 1% lysolecithin (w/v) immediately before lesion induction to a final concentration of 0.4 μm OGT2115 directly into the dorsal and ventral funiculus of the spinal cord between two thoracolumbar vertebrae at the time of lesion formation. Control animals received only lysolecithin injections.
Tissue processing
Animals were killed at 5–7 d postlesion (dpl) by transcardial perfusion of saline followed by 4% paraformaldehyde under deep anesthesia. The spinal column was extracted and postfixed for 30 min in 4% paraformaldehyde, and spinal cords were dissected and cryopreserved in sucrose gradient (7.5, 15%) for 24 h and were snap frozen in OCT (optimal cutting temperature) compound and sectioned coronally at 16 µm thickness using a Leica cryostat.
To identify lesions, one slide from each series was dehydrated in an ethanol gradient, rehydrated, and stained for 15 min in a solution containing 0.2% (w/v) Erichrome Cyanine (Millipore Sigma), 5.6% (w/v) FeCl3 · 6H2O (Thermo Fisher Scientific), and 0.5% (v/v) H2SO4. Slides were differentiated in 5.6% (w/v) FeCl3 · 6H2O until proper depth of color was achieved, dehydrated by sequential ethanol gradient followed by xylene, and mounted in Optic-Mount I (Mercedes Medical). Lesions were identified as regions of lighter Eriochrome (Solochrome) cyanine staining in the ventrolateral and/or dorsal white matter. Cross-sectional lesion areas were calculated using NIH ImageJ software, and the section with the largest area for each lesion was designated as the lesion center.
Immunohistochemistry
Slides immediately adjacent to the lesion centers, identified by Solochrome cyanine staining, were used for all immunohistochemical procedures. In general, sections were permeabilized for 30 min with 1% Triton X-100 (Alfa Aesar) and 0.25% Tween 20 (Calbiochem). Sections were blocked for 1 h with either a commercial mouse-on-mouse kit (Vector Laboratories) or a solution containing 0.5% Triton X-100 and 5% goat serum (Thermo Fisher Scientific). Primary antibodies used were rabbit β Amyloid Polyclonal (APP1; 1:250; Thermo Fisher Scientific), rabbit cleaved caspase-3 (Asp175; 1:400; Cell Signaling Technology), mouse Pan axonal Neurofilament (SMI-312R; 1:1000; BioLegend), mouse pan neuronal neurofilament (SM1-311R; 1:1000; Covance), rabbit anti-Olig2 (1:500; Millipore), mouse anti-GFAP (1:300; Millipore Sigma), rabbit anti-Iba1 (1:300; Wako Chemicals), mouse anti-INOS1 (1:100; BD Biosciences), rabbit anti-IRF1 (1:100; Santa Cruz Biotechnology), mouse anti-Stat1 (1:100; Thermo Fisher Scientific), rat BrdU (1:100; Bio-Rad), rat anti-Pdgfra (1:100; BD Biosciences), and rabbit anti-Prrx1a/b (1:1000; a gift from Dr. Basil Bakir University of Pennsylvania, PA; Takano et al., 2016). Alexa Fluor 488-, 594-, and 647-conjugated secondary antibodies (Thermo Fisher Scientific) were used at 1:500. Images of spinal cords were captured at 20× magnification using an Olympus IX51 microscope with motorized Prior XY stage and were stitched together using FIJI software. Images of immunostained sections were aligned with those taken of sections on adjacent, Solochrome cyanine-stained slides to accurately mark the lesion boundaries. For each marker, quantification was performed on at least two sections, which were averaged to generate a single data point for the comparison of treatment groups. Lesions with cross-sectional area <10,000 µm2 were excluded from the analysis, as were lesions that extended into the gray matter. All quantification was performed by an investigator blinded to the identity of the samples.
Electron microscopy
For assessment of remyelination, tissue was processed as previously described (Welliver et al., 2018; Saraswat et al., 2020). Briefly, mice were killed at 14 d postlesion by transcardial perfusion with 2% glutaraldehyde in 0.1 m phosphate buffer and spinal cords were extracted (n = 5/group), 1-mm-thick blocks surrounding the spinal cord lesion were processed through osmium tetroxide, dehydrated through ascending ethanol washes, and embedded in TAAB resin (TAAB Laboratories). One micrometer sections were cut, stained with 1% Toluidine Blue (Sigma-Aldrich), and examined by light microscopy to identify lesions. Selected blocks with lesions were trimmed, and ultrathin sections were cut and examined by electron microscopy (model H600, Hitachi). Images were acquired at 2900–6800× magnification. Analyses of remyelinated axons and g-ratios were performed blinded. For remyelination counts, a minimum of 800 axons was counted for each animal from at least six different fields, with all animals per treatment group. Analysis of the g-ratio was performed as described in the study by Saraswat et al. (2020). Briefly, axon and fiber diameter were measured using FIJI (diameter = 2 × √[area/π]). A minimum of 50 axons was analyzed per animal. The frequency distribution of axon diameter and g-ratio of remyelinated axons was calculated by binning on axon diameter and g-ratio.
Human multiple sclerosis tissue
Multiple sclerosis tissues were acquired and prepared as described in the study by Dutta et al. (2019). Briefly, brains were collected as part of the tissue procurement program approved by the Cleveland Clinic Institutional Review Board. Brains were removed according to a rapid autopsy protocol, sliced (1 cm thick), and then fixed in 4% paraformaldehyde, and 30 µm sections were cut for morphologic studies. Lesions were characterized for demyelination by immunostaining using proteolipid protein (PLP) and immune activity using MHC class II HLA-DR CR3/43 (MHCII). Chronic active demyelinated lesions were analyzed from two patients in whom secondary progressive MS (age, 53–57 years; disease duration, >10 years; postmortem interval, <8 h) was diagnosed. Control tissue was used from two non-MS patient's brains (age, 75–88 years; postmortem interval, <8 h). To control for nonspecific antibody binding, isotype control antibodies at the same concentrations were used to validate antibody staining. We did not observe any background staining.
Fluorescence in situ hybridization
Fluorescence in situ hybridization was performed with probes targeting Prrx1 (GenBank accession #NM_011127.2), Pdgfra1 (GenBank accession #NM_011058.2), Ki67 (GenBank accession #NM_001081117.2), Apcdd1 (GenBank accession #NM_133037.3), and Id4 (GenBank accession #NM_031166.2) mRNA by using the RNAscope Fluorescent Multiplex Detection Kit (Advanced Cell Diagnostics) according to the manufacturer instructions. Fixed sections were baked at 60°C for 1 h, washed with ethanol, followed by tissue pretreatment, and probe hybridization following RNAscope fluorescence multiplex assay, and sections were counterstained with DAPI to visualize nuclei. Positive signals were identified as punctate dots present in nucleus and cytoplasm. Confocal z-stack images with an optical thickness of 0.2 µm were taken, and stacked images are shown (LSM 510 Meta Confocal Microscope, Zeiss).
Quantitative RT-PCR
Total RNA was isolated from demyelinating white matter lesions area from four chronic progressive MS brain samples. Surrounding normal appearing white matter (NAWM) tissue was used as a control for the experiment. Total RNA was isolated with RNeasy Mini Kit (catalog #74104, Qiagen) and reverse transcribed to cDNA with SuperScript VILO cDNA Synthesis Kit (#11754050, Thermo Fisher Scientific) as recommended by the suppliers. The expression of reported genes (PRRX1, IRF1, STAT1, and MBP) were checked using TaqMan Gene expression assays (catalog #4453320). GAPDH was used as endogenous control in the reaction. Each sample was run in triplicate. Delta (Δ) Ct values were used to determine relative expression changes (fold change, 2-ΔΔCT). Samples in RT-PCR analysis were compared using Student's t tests, and a p value of <0.05 was considered to be a significant difference.
Statistical analyses
All quantification and data analyses were performed by an investigator blinded to the identity of the samples. All statistical analyses were performed using GraphPad Prism. Data were compared by Student's t test, one-way ANOVA, or two-way ANOVA, where appropriate significance was considered at p < 0.05. Each figure legend contains the statistical test and significance values. Data are reported as the mean ± SEM.
Results
PRRX1 mRNA is expressed by quiescent OPCs in regions of chronic demyelination in MS
IFN-γ signaling upregulates PRRX1 mRNA expression and leads to quiescence in human and mouse OPCs (Wang et al., 2018). Here, we examined the expression of PRRX1 in chronic active lesions in patients with MS and in control brains (Fig. 1). Chronic MS lesions were characterized using PLP1 and MHCII immunohistochemistry (IHC; Fig. 1A,B). Increased PRRX1 expression within the lesion correlated with STAT1, which reflects increased IFN-γ signaling (Fig. 1C,D). STAT1 immunostaining was present in the nucleus and processes of many cells within the lesion, which is consistent with previous reports (Lillemeier et al., 2001; Barcia et al., 2011). PRRX1/STAT1 upregulation in PDGFRA+ OPCs was restricted to lesion and surrounding tissue with little/no expression of PRRX1 in NAWM. Within chronic active lesions, we further confirmed upregulation of PRRX1 mRNA in PDGFRA+ OPCs by fluorescence in situ hybridization (Fig. 1E). The majority of OPCs were restricted to the lesion border, but, regardless of localization, we found that PDGFRA+ OPCs coexpressed PRRX1 (Fig. 1E,F). mRNA extracted from chronic active MS lesions (Tripathi et al., 2019) was highly enriched for PRRX1 compared with NAWM (n = 4/group; p < 0.005, unpaired t test; Fig. 1G). Similarly, IRF1 and STAT1 mRNA were both upregulated in lesions (IRF1: 1.56 ± 0.13-fold and 0.79 ± 0.19-fold; STAT1: 1.42 ± 0.14-fold and 0.9 ± 0.04-fold relative to NAWM, n = 3–4/group, p < 0.05, p < 0.05 unpaired t test). MBP mRNA was decreased, and no significant difference in PDGFRA expression was observed in MS lesions (p = 0.98). Together, these data were consistent with increased IFN-γ signaling within chronic MS lesions leading to an upregulation of PRRX1 expression by OPCs located within regions of demyelination.
Figure 1.
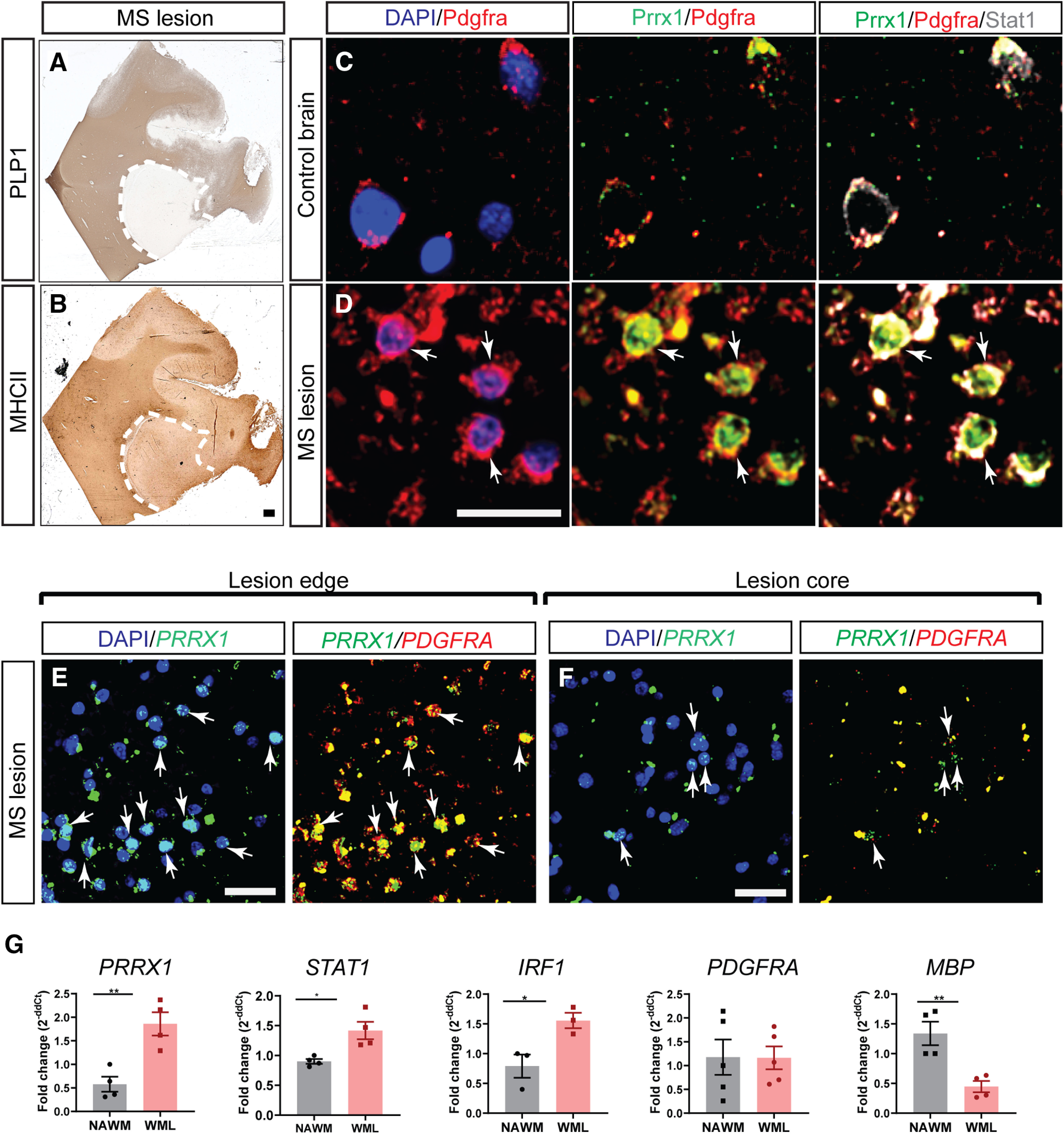
PRRX1 mRNA is enriched in OPCs located within multiple sclerosis lesions. A, B, IHC for PLP1 and MHCII for characterization of chronic lesion in the multiple sclerosis patient brain. C, D, PDGFRA, PRRX1, and STAT1 immunofluorescence in control brain and MS lesion brain (PDGFRA, red; PRRX1, green; STAT1, gray). NAWM from control brain expressed low levels of PRRX1 and STAT1. Both PRRX1 and STAT1 proteins were upregulated in OPCs within a chronic active lesion from a secondary progressive MS patient (arrows indicate colocalization of PRRX1/STAT1 with PDGFRA. E, Fluorescence RNA-scope in situ hybridization illustrates colocalization of PRRX1 mRNA with PDGFRA mRNA-expressing OPCs found at lesion edges. F, Despite being less frequent, PDGFRA+ OPCs in the lesion core also expressed PRXX1+. Arrows represents colocalization of PRRX1 with PDGFRA. G, Quantitative PCR analyses of PRRX1, IRF1, STAT1, PDGFRA, and MBP on RNA extracted from chronic active MS lesions and NAWM (mean ± SEM, n = 3–4 SPMS patients). GAPDH was used as internal control. Student's t test: *p < 0.05 and **p < 0.01, respectively. Scale bars: A, B, 100 µm; C, D, 10 µm; E, F, 20 µm.
Cell-autonomous IFNγR1 signaling mediates the inhibitory effects of IFN-γ signaling on OPC recruitment and proliferation
Treatment with the proinflammatory cytokine IFN-γ induces quiescence in purified human OPCs in vitro (Wang et al., 2018). As such, we hypothesized that IFN-γ would act directly on OPCs following demyelination and thereby reduce OPC recruitment and proliferation. To test this hypothesis, we performed conditional ablation of IFN-γ receptor 1 (IFN-γR1) in adult NG2+ OPCs by injection of tamoxifen into NG2CreERT2: IFN-γR1fl/fl: Rosa26-Yfp (IFN-γR1 cKO) mice and littermate cre-negative controls. Tamoxifen was administered for 5 d to 8- to 9-week-old mice, and lysolecithin demyelinating lesions were induced 7 d after the last injection to ensure clearance of tamoxifen (n = 3–5 animals/group). To assess the role of IFN-γR1 signaling in OPC proliferation and recruitment, mice were killed at 5 dpl (Fig. 2A). Deletion of IFN-γR1 in OPCs had no significant effect on lesion dynamics (lesion size, GFAP+ astrogliosis, and infiltration of Iba1+ microglial/macrophages; Fig. 2B,C). Next, we assessed the overall effect of IFN-γ R1 cKO on OPC recruitment and proliferation (Fig. 2D). In the absence of exogenous IFN-γ, OPC-specific IFN-γR1 ablation did not alter progenitor recruitment and proliferation relative to wild-type controls (two-way ANOVA with Holm–Sidak post-test, p > 0.4; Fig. 2F,G).
Figure 2.
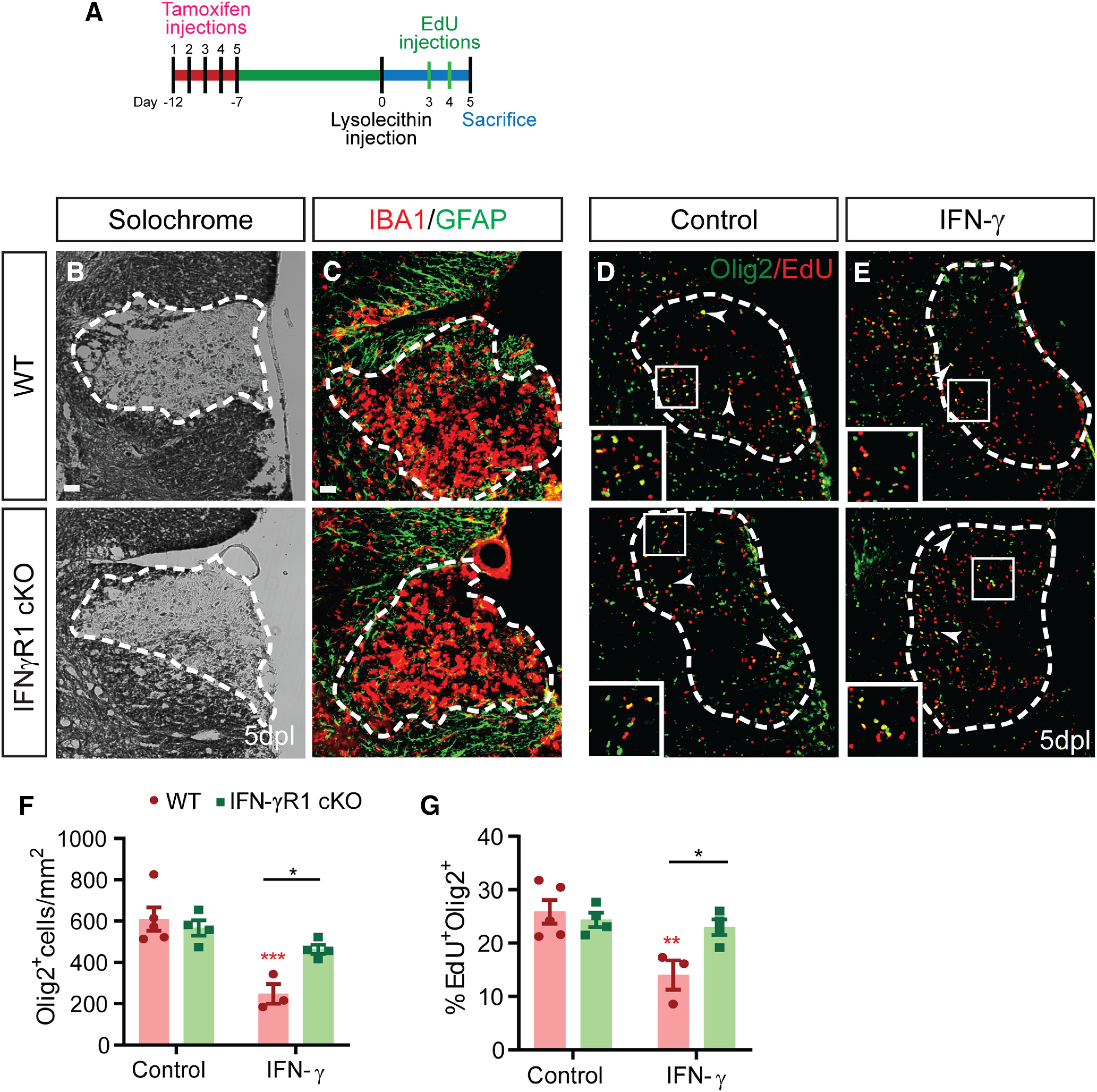
Conditional IFNγR1 knockout in OPCs enhances progenitor recruitment and proliferation following focal demyelination. A, OPC-specific IFNγR1 conditional knockout was induced in adult NG2CreERT2: Rosa-YFP: IFNγR1fl/fl mice by daily intraperitoneal administration of 200 mg/kg tamoxifen for 5 d. Animals underwent spinal cord demyelination 1 week later with (E) or without (B–D) simultaneous administration of 3 ng of IFN-γ directly into the lesion. Animals were killed at 5 dpl to assess the effects on OPC recruitment. B, Solochrome cyanine was used to identify demyelinated lesions. C, The innate inflammatory response was assessed by Gfap and Iba1 immunofluorescence. D–F, In control (D) and IFN-γ-treated (E) lesions, OPC recruitment was assessed using the oligodendrocyte lineage marker Olig2, and Olig2+ cell density quantified in F (cells/mm2, mean ± SEM). G, The percentage of EdU+ cells among Olig2+ cells was determined as a measure of OPC proliferation. Arrowhead and inserts indicate proliferating EdU+ OPCs. IFN-γ treatment significantly reduced OPC recruitment and proliferation. These effects were dependent on IFNγR1 expression in OPC, as IFNγR1 mice were unresponsive to IFN-γ treatment (n = 3–4 mice/group; two-way ANOVA interaction, p < 0.05 for Olig2+ cell density and p < 0.05 for the percentage of proliferating OPCs). *p < 0.05, Holm–Sidak multiple-comparisons post-test). Red ** and *** indicate Holm-Sidak p < 0.01 and p < 0.0001 respectively compared to matched wildtype control. Two-way ANOVA (Extended Data Fig. 2-1). Scale bar, 20 µm.
Two-way ANOVA showing effect of exogenous IFN-γ treatment in WT and IFNγR1 cKO mice and the interactions. Download Figure 2-1, DOCX file (23.7KB, docx) .
IFN-γ signaling is the predominant cytokine in immune-mediated demyelination and is largely dependent on release by infiltrating CD4+ Th1 cells. While transient T-cell infiltration occurs rapidly following lysolecithin-induced demyelination, IFN-γ itself was not detected (Ghasemlou et al., 2007). As such to better recapitulate a proinflammatory environment following demyelination, we sought to drive IFN-γ signaling by coinjecting IFN-γ along with lysolecithin. IFN-γ coadministration (3 ng) had a profound negative impact on both OPC recruitment and proliferation at 5 dpl (Fig. 2E). We noted a large 2.5-fold reduction in the density of Olig2+ oligodendrocyte lineage cells (Fig. 2F) and an almost twofold reduction in the rate of OPC proliferation (percentage of Olig2+EdU+ cells among total Olig2+) in IFN-γ-treated lesions relative to control (Fig. 2G; IFN-γ: 248 ± 83 Olig2+ cells/mm2 and 14.0 ± 5% Olig2+EdU+; vs control: 610 ± 127 Olig2+ cells/mm2 and 26 ± 5% Olig2+EdU+; two-way ANOVA with Holm–Sidak post-test, p < 0.001, p < 0.01, n = 3–5). Importantly, the deletion of IFN-γR1 largely rendered OPCs unresponsive to the effects of exogenous IFN-γ treatment. In IFN-γ-augmented lesions, both OPC recruitment (Olig2+ density) and proliferation (EdU percentage) were significantly increased in IFN-γR1 cKO mice compared with wild type (Fig. 2D–G; two-way ANOVA with Holm–Sidak post-test, both p < 0.05). Importantly, two-way ANOVA revealed a highly significant interaction between genotype and IFN-γ treatment on all parameters (Extended Data Fig. 2-1). Thus, IFN-γ directly regulated OPC quiescence and recruitment following demyelination via cell-autonomous IFN-γR1 signaling.
PI-88 treatment inhibits IFN-γ signaling in hOPCs and attenuates OPC quiescence
To better understand the downstream effects of IFN-γ on hOPCs, we performed pathway analysis on the transcriptional profile of hOPCs treated with IFN-γ (Wang et al., 2018). Interestingly, IFN-γ treatment of hOPCs results in the modulation of genes involved in heparan sulfate (HS) biosynthesis, postsynthetic modifications of HS, and genes encoding core membrane-bound HS proteoglycans (HSPGs; Extended Data Fig. 3-1). As HS sulfation is critical in the regulation of OPC signaling in response to demyelination (Saraswat et al., 2020), we hypothesized that PI-88, a heparin/heparan sulfate mimetic might interfere with IFN-γ signaling in hOPCs. PI-88 blocks the interactions of heparan sulfate with various growth factors (Karoli et al., 2005), inhibits both endosulfatases (Hossain et al., 2010) and heparanases (Parish et al., 1999) acting as a noncleavable substrate at their catalytic site. To test this hypothesis, hOPCs were purified from fetal brain and cultured in PDGF-AA/NT-3-supplemented serum-free media (Fig. 3A). As previously described (Sim et al., 2011), human fetal CD140a/PDGFRA+ cells exhibited typical bipolar morphology and expressed Olig2. Treatment with IFN-γ (10 ng/ml, 24 h) significantly reduced hOPC proliferation measured as the percentage of EdU+Olig2+ cells among total Olig2+ cells (one-away ANOVA with Holm–Sidak post-test, p < 0.01, n = 4 fetal samples). The IFN-γ-mediated reduction in proliferation was not because of increased apoptosis (cleaved-caspase 3; Wang et al., 2018). Using a dose of PI-88 known to inhibit Wnt/Bmp-dependent transcriptional activation (Saraswat et al., 2020), PI-88 treatment (2 μg/ml) rescued the antiproliferative effect of IFN-γ increasing EdU to that of controls (p < 0.05; Fig. 3B,C).
Figure 3.
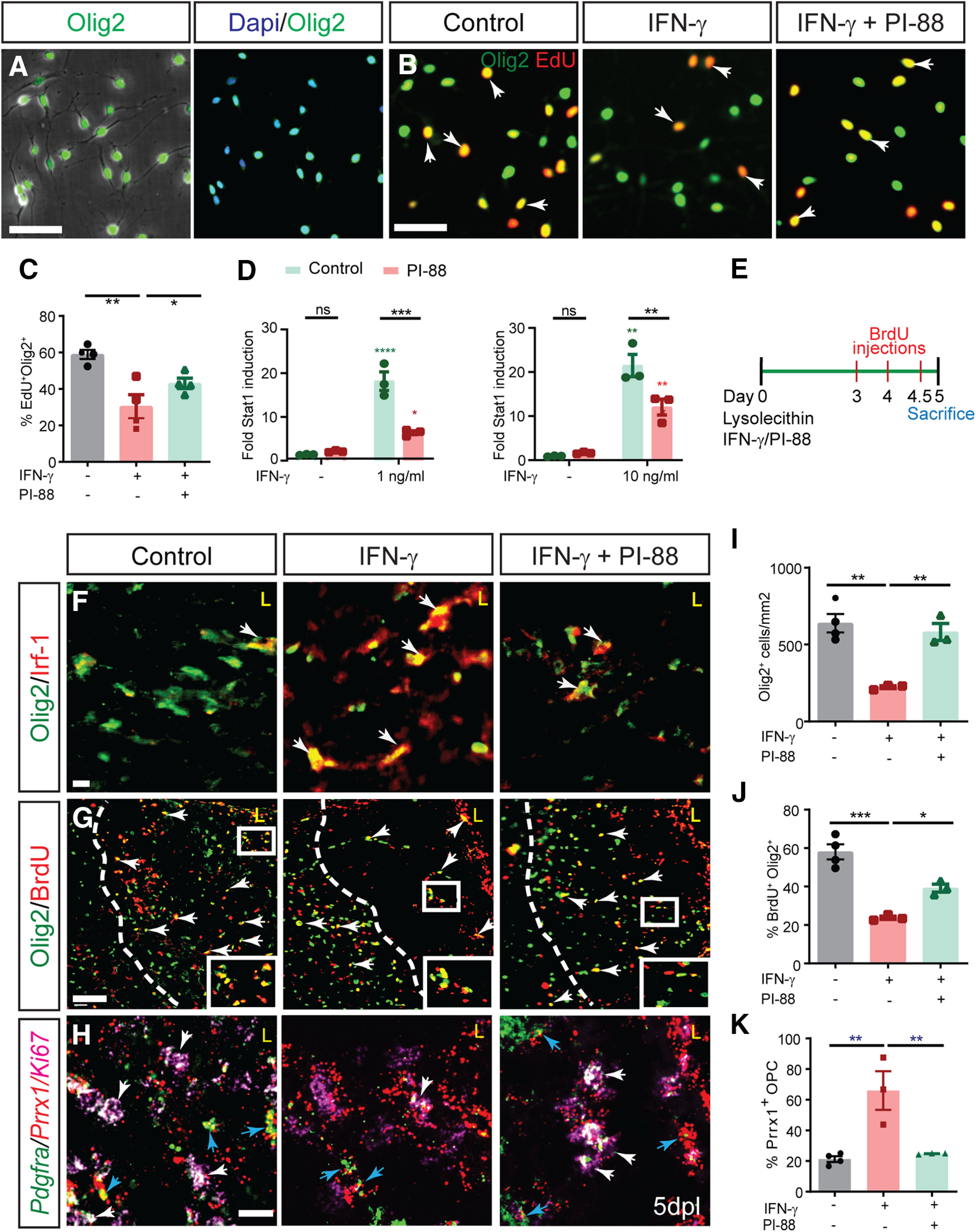
IFN-γ-mediated quiescence of human and mouse OPCs is rescued by heparan sulfate mimetic PI-88. A, Phase contrast image of CD140a/PDGFRA+ OPCs isolated from human fetal brain illustrates the typical bipolar morphology, and >95% isolated cells were positive for Olig2, an oligodendrocyte lineage marker. IFN-γ treatment of hOPCs was found to regulate a wide variety of genes associated with heparan sulfate proteoglycan synthesis and modulation (Extended Data Fig. 3-1). B, hOPCs were treated with or without heparan sulfate mimetic PI-88 (2 µg/ml) for 0.5 h and then exposed to IFN-γ (10 ng/ml). C, At 24 h, cells were pulsed with EdU for an additional 24 h and the proportion of EdU+ cells was quantified. IFN-γ treatment significantly reduced OPC proliferation (n = 4 fetal human samples, one-way ANOVA with Holm–Sidak post-test, p < 0.01). PI-88 rescued the negative effects of IFN-γ on proliferation (p < 0.05). D, The activity of a luciferase-expressing STAT1 reporter in transduced hOPCs was assessed following 24 h of IFN-γ treatment (1 and 10 ng/ml) with or without PI-88 (2 μg/ml). Luciferase activity was measured in triplicate for each human sample (n = 3 fetal human samples). Holm–Sidak test: *p < 0.05, **p < 0.01, ***p < 0.001, ****p < 0.0001 for each pairwise comparison following two-way ANOVA. Green indicates comparison with untreated cells, red indicates comparison with PI-88 control. Two-way ANOVA (Extended Data Fig. 3-2). E, Injection of 10 ng of IFN-γ with or without 10 μg/ml PI-88 directly into demyelinated lesion in 8-week-old mice. F, The effect of PI-88 on oligodendrocyte lineage cell density and IFN-γ/STAT1 pathway activity at 5 dpl was assessed by immunofluorescence for Olig2 (green) and Irf-1 (red), respectively. PI-88 treatment decreased colocalization of Irf1 in Olig2+ cells. G, I, J, OPC density and proliferation was assessed by Olig2 and by determining the proportion of BrdU+Olig2+ OPCs (G; quantified in I and J; mean ± SEM, n = 3–4). I, J, IFN-γ treatment significantly decreased OPC recruitment (I) and proliferation (J), while PI-88 injections rescued the effects of IFN-γ. Holm–Sidak test: *p < 0.05, **p < 0.01, ***p < 0.001 following one-way ANOVA. H, Fluorescence in situ hybridization at 5 dpl using Prrx1, Pdgfra, and Ki67-specific mRNA probes (red, green, and pink, respectively). Prrx1 expression was upregulated following demyelination in the presence of IFN-γ and PI-88 treatment decreased Prrx1 expression within the lesion. White arrows denote proliferating OPCs (Ki67+Pdgfra+), which were typically Prrx1 low/negative cells, while blue arrows denote nonproliferating (Ki67–) Pdgfra+ OPCs that expressed high levels of Prrx1 mRNA. A subset of astrocytes expressed Prrx1 following IFN-γ-augmented demyelination, but colocalization with Iba+ microglia was not observed (Extended Data Fig. 3-3). K, IFN-γ treatment significantly increased Prrx1 expression in Pdgfra+ OPCs, while PI-88 treatment decreased expression of Prrx1 in OPCs. Holm–Sidak test: *p < 0.05, **p < 0.01, ***p < 0.001 following one-way ANOVA. Scale bars: A, B, 50 µm; and F–H, 20 µm. ns denotes not significant.
IFN-γ treatment of human OPCs alters the transcriptional profile of genes associated with HS synthesis and presentation. Human hOPCs were treated with IFN-γ (10 ng/ml) for 24 h, and RNA-sequencing was performed as described previously (Wang et al., 2018). A heatmap demonstrating the clustering of individual HS-related genes (n = 2 human fetal samples). IFN-γ treatment had a profound effect on the hOPC heparanome. The side bar indicates statistical significance—upregulated (red) and downregulated (blue) genes (q < 0.05). Expression values for each gene were centered and scaled to facilitate visualization (z-score). Download Figure 3-1, TIF file (1.5MB, tif) .
Two-way ANOVA showing activation of STAT1 GAS luciferase by IFN-γ and PI-88 treatment and the interactions. Download Figure 3-2, file (2.5MB, tif) .
Increased Prrx1 expression in Gfap+ astrocytes following IFN-γ treatment after demyelination. RNAscope in situ hybridization (ISH) and IHC of mouse spinal cord after demyelination revealed increased Prrx1 expression by astrocytes but not Iba1+ microglial cells in lesions as assessed by immunohistochemistry of Gfap and Iba1 and Prrx1 mRNA transcripts. Arrow indicates colocalization of Prrx1 and Gfap. Scale bar, 20 μm. Download Figure 3-3, DOCX file (23.7KB, docx) .
To determine whether PI-88 directly influenced IFN-γ signaling, we transduced hOPCs with a lentiviral luciferase reporter of STAT1 activity. IFN-γ treatment resulted in robust induction of the STAT1-GAS reporter (18- and 21-fold vs control; 1 and 10 ng/ml IFN-γ. respectively; n = 3 fetal samples; Fig. 3D). PI-88 treatment had no effect on STAT1 activity in the absence of IFN-γ treatment. However, the upregulation of STAT1 reporter activity following IFN-γ treatment was significantly reduced by PI-88 treatment (two-way ANOVA with Holm–Sidak post-test, p < 0.001). Low- and high-dose IFN-γ/STAT1 signaling was similarly inhibited by PI-88 treatment (Fig. 3D). Importantly, at both doses, two-way ANOVA revealed a highly significant interaction between PI-88 and IFN-γ on proliferation of OPCs (p < 0.01 and p < 0.05; Extended Data Fig. 3-2). These results indicate that PI-88 can directly modulate IFN-γ signaling in hOPCs and thereby rescue the antiproliferative effects of IFN-γ in vitro.
PI-88 treatment attenuates IFN-γ-mediated OPCs quiescence following demyelination
We next examined whether PI-88 could attenuate OPC quiescence in inflammatory IFN-γ-augmented demyelinating lesions. To mimic the proinflammatory environment of chronic MS lesions, we coadministered high-dosage IFN-γ (10 ng/injection) with lysolecithin directly into the ventrolateral and dorsal white matter of the adult mice spinal cord at the time of lesion formation (Fig. 3E–H). In control lysolecithin lesions, IRF1 expression, a marker of active IFN-γ/STAT signaling was only found in a small subset of OPCs at 3 dpl and expression was lost by 5 dpl. In contrast, IFN-γ augmented lesions exhibited prolonged and substantially increased levels of IRF1 expression with >80% Olig2+ cells coexpressed IRF1 at 5 dpl (Fig. 3F).
In control lysolecithin lesions, a large number of Olig2+ OPCs were recruited into the lesion at 5 dpl (Fig. 3G). In contrast, IFN-γ augmented lesions exhibited a dramatic 2.9-fold reduction in Olig2+ oligodendrocyte lineage cell density (cells/mm2, n = 3–4 animals/group, one-way ANOVA with Holm–Sidak post-test, p < 0.01). PI-88 treatment rescued the effects of IFN-γ administration and significantly increased the density of Olig2+ cells compared with IFN-γ alone (p < 0.01; Fig. 3I). Interestingly, we observed an accumulation of Olig2+ cells outside the region of demyelination. Following IFN-γ treatment, this accumulation was reversed by PI-88 treatment. As described above, IFN-γ reduced the percentage of BrdU+ proliferating OPCs (p < 0.001). Importantly, PI-88 treatment significantly rescued OPC proliferation compared with IFN-γ alone (p < 0.05; Fig. 3J). These data suggest that PI-88 can reverse the antiproliferative and antimigratory effects of IFN-γ on OPCs.
As described above, Prrx1 expression in OPCs is associated with chronic demyelination and cell cycle quiescence (Wang et al., 2018). In control lysolecithin lesions, we found numerous dividing Ki67+Pdgfra+ OPCs and quiescent Ki67-Pdgfra+ OPCs that expressed Prrx1 (Fig. 3H). As expected, IFN-γ treatment increased Prrx1 expression in OPCs following demyelination (Fig. 3K). Prrx1 expression was also upregulated by small subsets of Gfap+ astrocytes around lesion edges but was not found colocalized with Iba1+ microglia (Extended Data Fig. 3-3). Intriguingly, OPCs that had accumulated outside the lesion were proliferative and lacked Prrx1 expression at 5 dpl (data not shown). In contrast, PI-88 treatment significantly reduced both the overall level of Prrx1 expression within the lesion and the proportion of Prrx1-expressing Pdgfra+ OPCs (Fig. 3H,K). Along with the reduced Prrx1 expression, we noted increased density of proliferating Ki67+Pdgfra+ proliferating OPCs. Together, these results indicate that the negative effects of IFN-γ signaling on OPC recruitment and quiescence can be antagonized by PI-88 treatment.
PI-88 treatment abrogates the proinflammatory effects of IFN-γ signaling on microglial cells
As astrogliosis can block migration and infiltration of OPCs (Wang et al., 2017) and IFN-γ induces reactive astrogliosis (Yong et al., 1991; Balasingam et al., 1994), the accumulation of OPCs outside IFN-γ-augmented demyelinating lesions suggested that glial scar formation following IFN-γ exposure may impede recruitment. We first assessed astrogliosis at 5 dpl by immunofluorescence for GFAP (n = 3–4 animals; Fig. 4A). Interestingly, following IFN-γ treatment, astrocytes exhibited a substantially altered morphology, characterized by hypertrophic cell bodies and processes (Fig. 4A), as well as extensive process interdigitation and overlap consistent with a proinflammatory state (Fig. 4A, higher-magnification image inserts; Sofroniew and Vinters, 2010). In both control and IFN-γ-augmented lysolecithin lesions, GFAP+ astrocytes did not express Irf1 protein at 5 dpl, suggesting that non-cell-autonomous effects drive astrogliosis following IFN-γ treatment (Fig. 4B). Consistent with the antagonism of IFN-γ signaling, PI-88 treatment qualitatively rescued the proinflammatory effect of IFN-γ on astrogliosis with the pattern of GFAP staining rescued to control.
Figure 4.
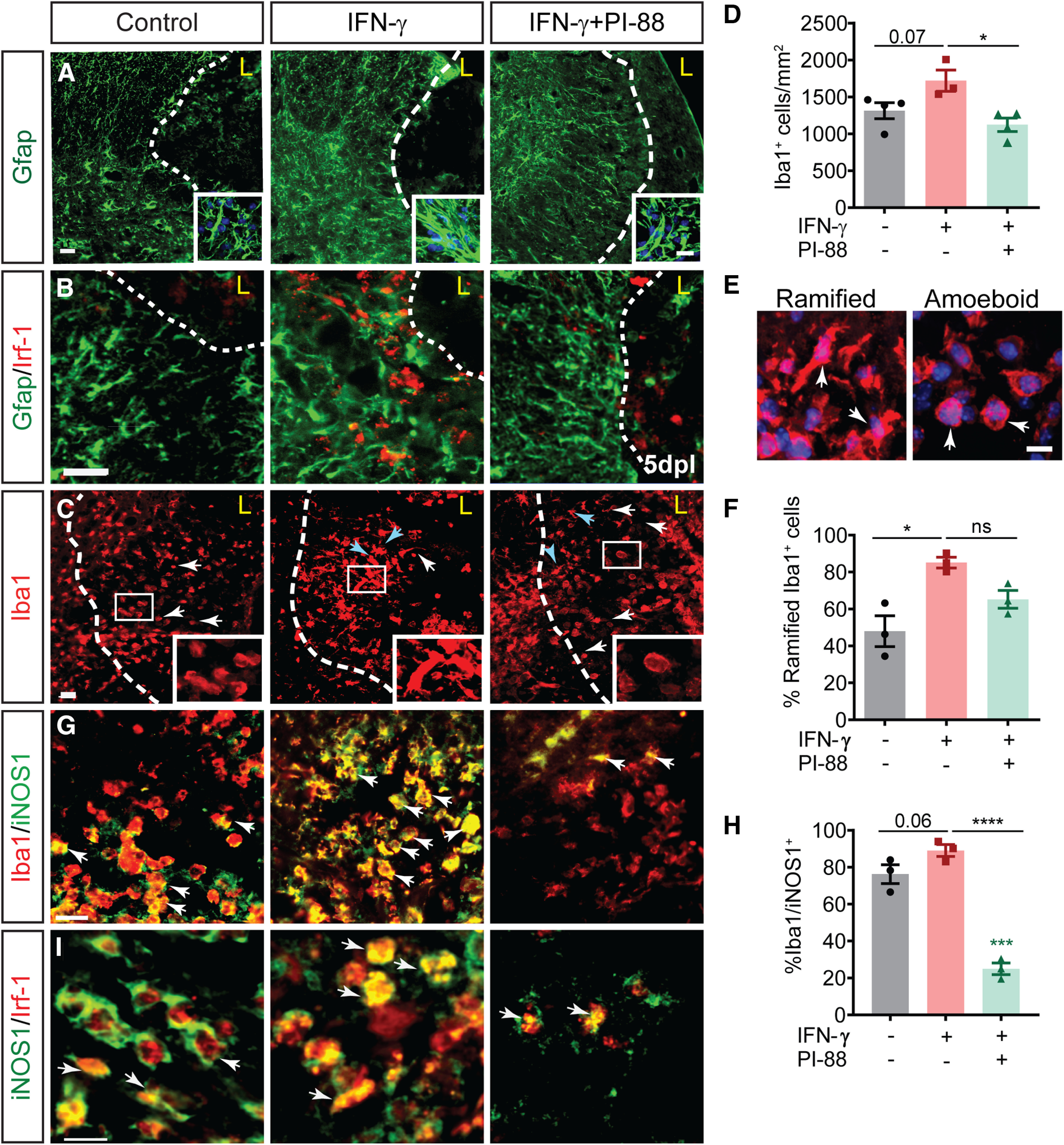
PI-88 rescues IFN-γ-mediated proinflammatory effects on glial cells following demyelination. A, Following demyelination, astrogliosis was assessed by Gfap immunofluorescence at 5 dpl (inserts shows higher-magnification confocal image). Gfap was substantially increased by IFN-γ, while PI-88 treatment decreased reactive astrogliosis (n = 3–4/group). B, IFN-γ-dependent signaling in Gfap+ astrocytes was not apparent as Irf1 immunoreactivity was not colocalized with Gfap. C, D, Microglial infiltration was assessed by Iba1 immunofluorescence in lesion (C), quantified in D. Density of Iba1+ microglial cells increased following IFN-γ treatment, whereas PI-88 treatment decreased microglial infiltration (mean ± SEM, n = 3–4). Holm–Sidak test: *p < 0.05, ***p < 0.001, ****p < 0.0001 following one-way ANOVA, respectively. White arrows denote amoeboid shaped microglial cells, while blue arrows indicate reactive ramified. E, F, Confocal images showing examples of ramified and amoeboid shape microglia, quantified in F. G, The presence of proinflammatory M1 microglia was assessed by iNOS1 immunofluorescence. White arrows indicate double-positive cells. H, Percentage of iNOS+Iba1+ microglial cells among total Iba1+ cells (Holm–Sidak post-test shown). I, IFN-γ signaling was assessed by colocalization of Irf-1 and iNOS+ proinflammatory microglia. Scale bar, 20 µm. ns denotes not significant.
Microglial cells are exquisitely sensitive to interferon signaling (Martinez and Gordon, 2014; Ottum et al., 2015; Vergara et al., 2019) and microglial polarization is known to regulate positive and negative aspects of remyelination (Miron et al., 2013). IFN-γ administration increased Iba1+ microglial cell infiltration by 25% relative to the control group (n = 3–4 animals/group; one-way ANOVA with Holm–Sidak post-test, p = 0.07; Fig. 4C,D). Within the lesion, we observed distinct Iba1+ microglial morphologies that are known to correlate with activation state, phagocytic capacity, and MHC class II expression (Mathieu et al., 2010). Microglial cells were classified as activated ramified microglial (elongated cell bodies with long thick processes, rounded cell bodies with small thick processes) or phagocytic amoeboid microglial (rounded shape with no processes; Fig. 4E). We observed a significant 1.4-fold increase in ramified microglia in the IFN-γ group compared with control (n = 3 −4 animals/group; one-way ANOVA, Holm–Sidak post-test, p < 0.01; Fig. 4F) and as a consequence the percentage of amoeboid microglial was reduced by 2.9-fold. Consistent with IFN-γ-induced M1 polarization (Martinez and Gordon, 2014; Tang and Le, 2016), we observed that ∼90% of Iba1+ cells coexpressed inducible nitric oxide synthase (iNOS) following IFN-γ treatment (Fig. 4G,H). This represented a small increase from control lysolecithin lesions (n = 3–4 animals/group; one-way ANOVA, Holm–Sidak post-test, p = 0.06). Likewise, Irf1 expression in microglial cells was similarly enhanced in lesions with IFN-γ compared with control lesions (Fig. 4I).
Importantly, PI-88 treatment essentially rescued the effects of IFN-γ administration on the microglial response. We observed significant reduction in the density of infiltrating Iba1+ cells following PI-88 treatment (p < 0.05; Fig. 4C,D). Furthermore, PI-88 treatment restored microglial morphology to control levels, with the frequency of amoeboid microglia increased by 2.4-fold relative to IFN-γ alone (Fig. 4C–F). Importantly, this corresponded with altered M1/M2 polarization with PI-88 treatment reducing the percentage of iNOS-expressing microglia by 3.6-fold (p < 0.0001; Fig. 4G,H). Surprisingly, PI-88 administration reduced iNOS expression twofold below that of control lesions, indicating a likely modulation of microglial responses regardless of IFN-γ treatment (p < 0.001). Following PI-88 administration, Irf1 expression was restricted to INOS1+ M1 microglia while INOS1– M2 microglia did not express Irf1 (Fig. 4I). Together, these findings suggest that PI-88 treatment modulates the demyelinated lesion microenvironment by directly inhibiting IFN-γ signaling on astrocytic and microglial cells.
IFN-γ-mediated exacerbation of lesion dynamics is rescued by PI-88
As IFN-γ has multiple proinflammatory functions in the CNS, we next assessed the effect of IFN-γ administration on lesion size, cell death, and axonal injury using a higher dose of 10 ng/injection. At 5 dpl, we observed that high-dose IFN-γ lesions were significantly larger than control lesions being increased by 2.7-fold relative to control group (n = 3–4; one-way ANOVA with Holm–Sidak post-test, p < 0.05; Fig. 5A,B). IFN-γ induces necrosis and cell death in oligodendrocytes in MS and EAE, and following toxin-induced demyelination (Agresti et al., 1996; Horiuchi et al., 2006). To assess cell death, we examined the expression of cleaved caspase 3 (Fig. 5C,D). We noted a fivefold increase in the density of cleaved caspase3+ cells and debris following IFN-γ (n = 3–4; Holm–Sidak post-test, p < 0.001). In MS, the accumulation of App1 occurs in injured axons located with regions of active demyelination (Gehrmann et al., 1995). We assessed axonal injury by App1 immunofluorescence (Fig. 5E). We noted that IFN-γ treatment significantly increased the density of App1+ fibers (1.7-fold, Holm–Sidak post-test, p < 0.01; Fig. 5F). We further confirmed that IFN-γ-induced APP was largely localized to neurofilament-positive axons (heavy and light chain) by confocal microscopy (Fig. 5G).
Figure 5.
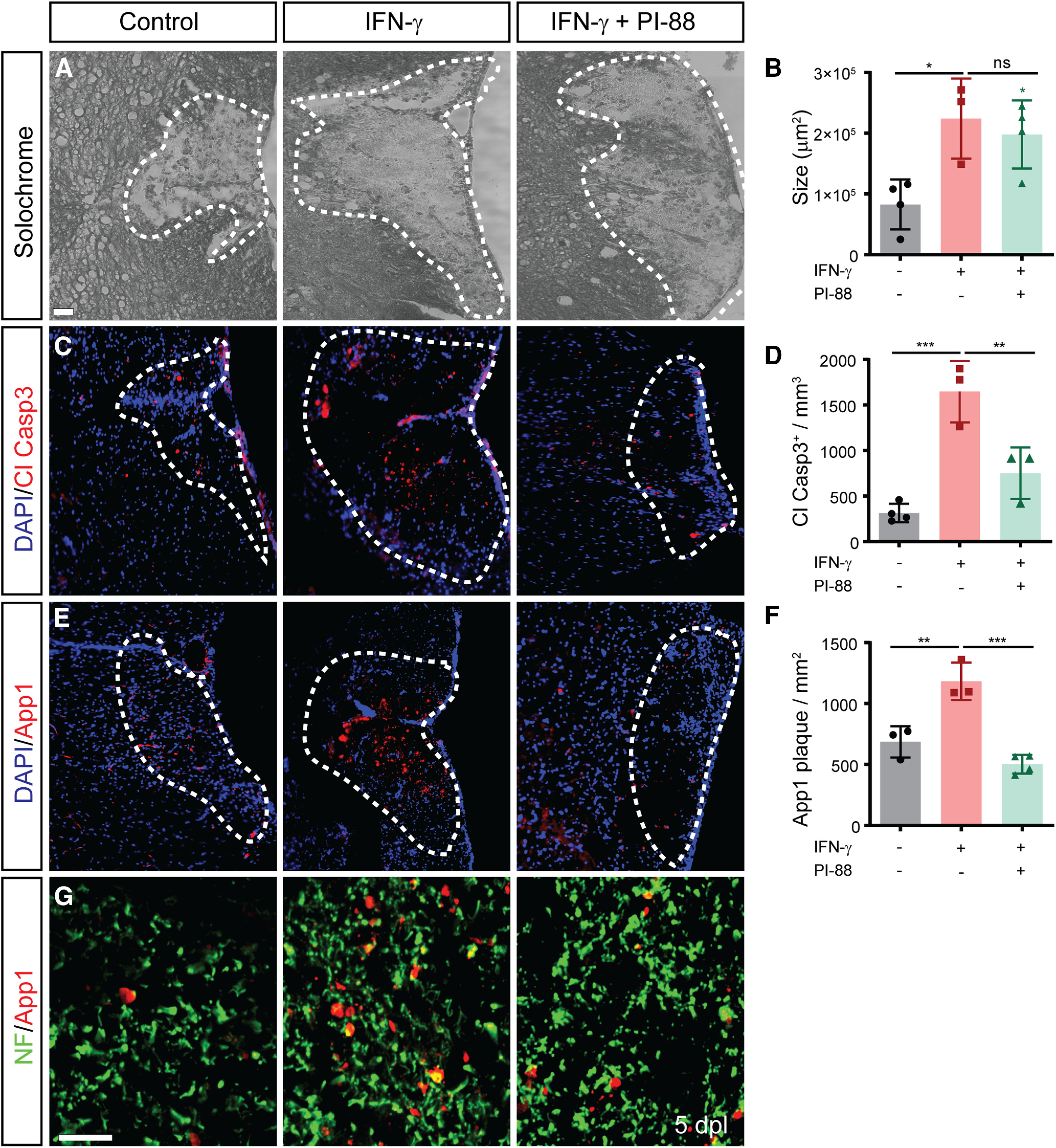
IFN-γ exacerbates cell and axonal injury following demyelination which was rescued by PI-88 treatment. A, Lesion size was assessed by Solochrome cyanine lipid stain at 5 dpl. B, IFN-γ treatment increased lesion size, which was not significantly affected by PI-88 treatment (area: mean ± SEM, n = 3–4/group). C, D, Cell death was assessed by analysis of cleaved caspase 3-stained particles (C) and quantified in D. E, F, Axon damage was assessed by the accumulation of App1 following demyelination (E) and quantified in F. Holm–Sidak test: *p < 0.05, **p < 0.01, ***p < 0.001 following one-way ANOVA. Cell death and axon injury was exacerbated by IFN-γ, and both of these measures of tissue injury were rescued by concurrent PI-88 treatment. G, App1 was restricted to neurofilament-positive axons (SMI31/32). Scale bars: A–C, 50 µm; D, 20 µm. ns denotes not significant.
We previously found that PI-88 did not alter lesion size or the extent of astrogliosis and microglial response (Saraswat et al., 2020). As PI-88 treatment was capable of moderating IFN-γ-induced effects on OPC recruitment and microglial polarization, we next asked whether PI-88 could rescue the effects of IFN-γ on lesion size, cell death, and axonal injury. PI-88 treatment had no effect on IFN-γ-induced lesion size (p = 0.54). Importantly, PI-88 treatment attenuated IFN-γ-induced caspase 3 activity (p < 0.01; Fig. 5D), as well as reducing the accumulation of axonal neurofilament-positive APP1 plaques relative to IFN-γ alone (2.3-fold, p < 0.001; Fig. 5F,G). Together, these data demonstrate that PI-88 treatment rescues the degenerative effects of IFN-γ signaling in demyelinating lesions in vivo.
Sulfatase modulation does not alter IFN-γ signaling following demyelination
PI-88 possesses a high level of 6-O sulfation (Yu et al., 2002) and acts as a noncleavable substrate for sulfatases (SULF1/2; Hossain et al., 2010). We have previously shown that PI-88 acts via sulfatase inhibition to modulate Wnt and Bmp signaling following demyelination (Saraswat et al., 2020). Therefore, we investigated whether PI-88 acts to modulate IFN-γ signaling in hOPC via the inhibition of sulfatases in vitro. As SULF2 is the principal sulfatase expressed in hOPCs, hOPCs were infected with scrambled or SULF2-targeted knock-down (KD) lentivirus before transduction with luciferase STAT1 reporter (Fig. 6A). Strikingly, luciferase activity inducted by IFN-γ signaling was unaffected by SULF2 KD (n = 2 human samples; two-way ANOVA with Holm–Sidak post-test, p > 0.99). As shown above, PI-88 treatment significantly attenuated the IFN-γ-mediated induction of STAT1 in both scrambled control and SULF2 KO (p < 0.01). Importantly, and unlike Wnt and Bmp signaling (Saraswat et al., 2020), there was no significant interaction between SULF2 KD and PI-88 (two-way ANOVA, p = 0.99; Extended Data Fig. 6-1). These findings suggests that SULF2 does not regulate IFN-γ signaling in human OPCs.
Figure 6.

IFN-γ signaling in OPCs following demyelination was not influenced by sulfatases (Sulf1/2). A, The activity of a STAT1-dependent luciferase reporter in human OPCs following 24 h of IFN-γ treatment (10 ng/ml) with or without PI-88 (2 μg/ml) and following infection with lentiviral SULF2 or control scrambled shRNAi was assessed. Luciferase activity was measured in triplicate for each human sample (n = 2 human fetal samples). Holm–Sidak test: *p < 0.05, **p < 0.01, ***p < 0.001 for each pairwise comparison following two-way ANOVA (mean ± SEM normalized to maximal response). SULF2 KD did not influence IFN-γ-dependent STAT activity, two-way ANOVA (Extended Data Fig. 6-1). B, OPC-specific Sulf1/2 knockout was induced in adult NG2: CreER; Rosa26IsIYFP; Sulf1fl/fl-Sulf2fl/fl (Sulf1/2 cKO) mice by daily intraperitoneal administration of 200 mg/kg tamoxifen for 5 d. Littermate cre-negative mice were similarly injected with tamoxifen as controls. Animals were lesioned 1 week after the last day of injection with or without simultaneous administration of 3 ng of IFN-γ directly into the lesion site. Animals were killed at 5 dpl, and spinal cord tissue was processed. C, D, Olig2/EdU immunofluorescence of wild-type control and Sulf1/2 cKO mice with or without IFN-γ treatment (n = 4–5 mice/group). Insert shows colocalization of EdU (red) in a subset of Olig2+ cells (green). E, F, Sulf1/2 cKO did not influence the effects of IFN-γ on OPC recruitment (Olig2+ cells/mm2; E) and proliferation (percentage EdU among Olig2+ cells; F). Examination of the interaction of these variables by two-way ANOVA did not reveal a significant effect, suggesting independent modes of action for Sulf1/2 and IFN-γ (p = 0.17, p = 0.31). Two-way ANOVA (Extended Data Fig. 6-2). Scale bar, 20 µm. ns denotes not significant.
Two-way ANOVA showing activation of STAT1 GAS luciferase in control and SULF2 KO by IFN-γ and PI-88 treatment and the interactions. Download Figure 6-1, DOCX file (23.7KB, docx) .
Two-way ANOVA showing effect of exogenous IFN-γ treatment in WT and Sulf1/2 cKO mice and the interactions. Download Figure 6-2, DOCX file (23.7KB, docx) .
To further examine the potential for sulfatase-dependent modulation of IFN-γ signaling following demyelination, we investigated whether conditional Sulf1/2 deletion in adult NG2+ OPCs could influence exogenous IFN-γ signaling and proinflammatory lesion development. Following tamoxifen injection, adult Sulf1/2 cKO and littermate control mice were coinjected with IFN-γ and lysolecithin at the time of lesion formation as described previously (Saraswat et al., 2020). We assessed OPC proliferation at 5 dpl (Fig. 6B) as well as the density of Olig2+ and EdU+Olig+ proliferating progenitors (n = 4–5/group; Fig. 6C–F). As shown previously (Saraswat et al., 2020), Sulf1/2 cKO resulted in a significant increase in Olig2+ density at 5 dpl (two-way ANOVA: Sulf1/2 cKO, p <0.001) but did not influence OPC proliferation (EdU percentage; two-way ANOVA p = 0.80). Following IFN-γ treatment, we again observed a significant reduction in both the Olig2+ cell density and the proportion of Olig2+EdU+ proliferating OPCs. Importantly, these effects occurred regardless of Sulf1/2 genotype (effect of IFN-γ on Olig2 density, p < 0.01; EdU percentage, p < 0.01; Fig. 6E,F). Two-way ANOVA did not reveal a significant interaction between Sulf1/2 genotype and IFN-γ treatment on either parameter (p = 0.17, p = 0.31; Extended Data Fig. 6-2). Thus, the effects of IFN-γ augmentation on OPC recruitment were not influenced by sulfatases expressed by OPCs and, together, these findings suggest that sulfatases do not regulate IFN-γ signaling in human and mouse OPCs.
Inhibition of heparanase by PI-88 moderates IFN-γ signaling following inflammatory demyelination
We further investigated the possible mechanism by which PI-88 influences IFN-γ signaling and its effects on the lesion microenvironment. PI-88 (Muparfostat) is currently in clinical trials as a heparanase and sulfatase inhibitor for treatment of malignant cancer. Heparanase modulates the tumor microenvironment (Vlodavsky et al., 2011), and is upregulated in Alzheimer's disease (García et al., 2017) and following cerebral ischemia (Li et al., 2012). As sulfatase inhibition was not responsible for the actions of PI-88 on OPC proliferation, we hypothesized that PI-88 may act via inhibition of heparanase. Therefore, we compared the effects of PI-88 and OGT2115, a small-molecule heparanase inhibitor (Cheng et al., 2014; Hammond et al., 2014) with or without IFN-γ at the time of spinal cord lysolecithin injection (Fig. 7). Similar to our previous results, PI-88 treatment induced a 2.2-fold increase in OPC recruitment in IFN-γ augmented lesions (n = 3–4 animals/group, one-way ANOVA with Holm–Sidak, p < 0.001; Fig. 7C). PI-88 treatment in control lysolecithin lesions did not significantly increase the percentage of proliferating OPCs (Olig2+EdU+) among total Olig2+ cells (p = 0.95; Fig. 7D). However, PI-88 treatment again rescued the IFN-γ-mediated inhibition of OPC proliferation by 1.5-fold (p < 0.05).
Figure 7.
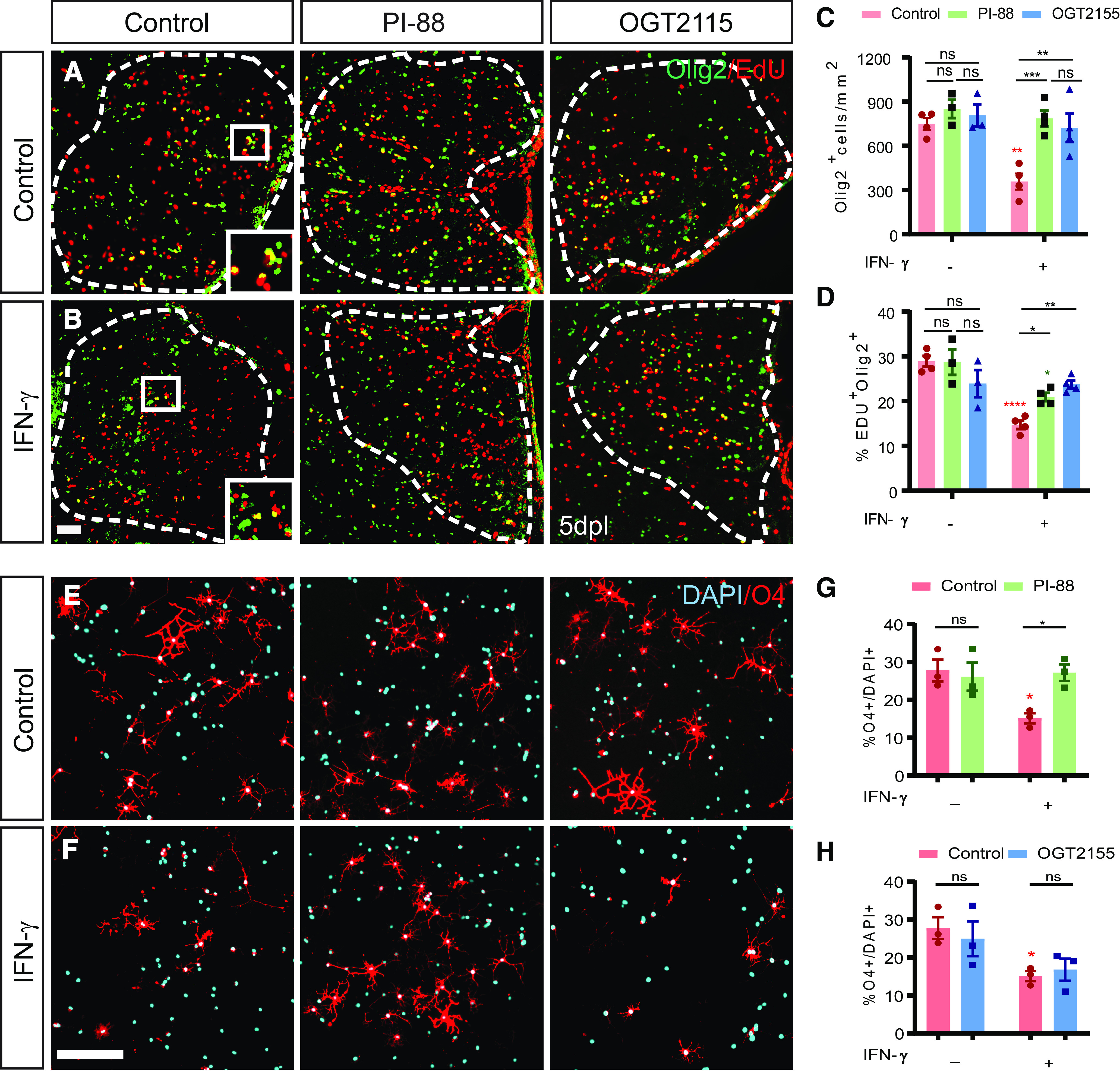
PI-88 acts via heparanase inhibition to rescue IFN-γ-mediated quiescence of OPCs. A, B, Control (A) and IFN-γ (B) mouse groups underwent focal spinal cord demyelination with or without PI-88 or OGT2115 injection and were killed at 5 dpl. Immunofluorescence for Olig2 (green) and EdU (red) was performed. C, D, OPC recruitment (Olig2+ cells/mm2; C) and proliferation (EdU percentage among Olig2+ cells; D) were quantified. Both PI-88 and OGT2115 treatment rescued the negative effects of IFN-γ on OPC recruitment (G) and proliferation (H) following demyelination (mean ± SEM; n = 3–4 mice/group). Two-way ANOVA interaction for Olig2+ cell density and percentage EdU OPCs were Holm–Sidak test: *p < 0.05, **p < 0.01 (Extended Data Fig. 7-1). Red and green denotes p value compared to matched untreated controls. E, F, at 0.5 h following treatment with or without PI-88 (2 μg/ml) or OGT2155 (0.4 μM) hOPCs were treated with vehicle (E) or IFN-γ (F) (10 ng/ml). O4+ oligodendrocyte (red) differentiation was assessed at 48 h and the proportion of O4+ cells quantified (mean ± SEM, n = 3 fetal samples) following PI-88 (G) or OGT2155 (H). IFN-γ has a significant inhibitory effect of differentiation (red, *), which is rescued by PI-88 (two-way ANOVA; Extended Data Fig. 7-2) but not OGT2115 treatment (two-way ANOVA; Extended Data Fig. 7-3). Holm–Sidak test: *p < 0.05 following two-way ANOVA. Scale bars: A, B, 20 µm; E, F, 50 µm. ns denotes not significant. ***p < 0.001, ****p < 0.0001.
Two-way ANOVA showing effects of heparanase inhibitor (PI-88 and OGT2115)-mediated modulation of IFN-γ signaling effects and the interaction. Download Figure 7-1, DOCX file (23.7KB, docx) .
Two-way ANOVA showing effects of heparanase inhibitor (PI-88)-mediated modulation of IFN-γ signaling effects on hOPC differentiation and the interaction. Download Figure 7-2, DOCX file (23.7KB, docx) .
Two-way ANOVA showing effects of heparanase inhibitor (OGT2155)-mediated modulation of IFN-γ signaling effects on hOPC differentiation and the interaction. Download Figure 7-3, DOCX file (23.7KB, docx) .
Similar to PI-88, heparanase inhibitor OGT2115 treatment did not significantly influence Olig2+ cell density (p = 0.80) or percentage of proliferating cells (Olig2+EdU+) among total Olig2+ cells (p = 0.17) in control lesions (Fig. 7C,D). Recruitment into proinflammatory IFN-γ-treated lesions was significantly rescued by OGT2115 treatment, being increased by twofold relative to IFN-γ lesions (Olig2+ cells/mm2, p < 0.05), and, importantly, OGT2115 treatment increased OPCs proliferation by 1.7-fold (p < 0.01; Fig. 7C,D). Two-way ANOVA revealed a significant interaction of both drugs with IFN-γ, suggesting that they act with a similar mechanism (two-way ANOVA, p < 0.05 and p < 0.01; Extended Data Fig. 7-1). Together, this suggests that PI-88 may act via inhibition of heparanase to counteract the proinflammatory lesion microenvironment.
Next, to address whether heparanase inhibitors act in a cell or non-cell-autonomous manner on OPC differentiation, we treated hOPCs with either PI-88 or OGT2155 and exposed them to IFN-γ (10 ng/ml). hOPCs were treated with IFN-γ with or without PI-88 (2 µg/ml) following mitogen withdrawal and immature oligodendrocyte differentiation assessed at 48 h (Fig. 7E–I). As shown in rodent OPCs (Tanner et al., 2011), IFN-γ significantly blocked human O4+ oligodendrocyte differentiation (n = 3 fetal human samples; two-way ANOVA with Holm–Sidak post-test, p < 0.01; Extended Data Fig. 7-2). PI-88 treatment rescued IFN-γ induced inhibition of oligodendrocyte differentiation (Fig. 7K, Extended Data Fig. 7-2; p < 0.05). In contrast, the heparanase inhibitor OGT2155 (0.4 μm/ml) did not influence the inhibitory effects of IFN-γ on differentiation (n = 3 fetal samples; two-way ANOVA with Holm–Sidak post-test, p = 0.79; Fig. 7L, Extended Data Fig. 7-3). The lack of effect by OGT2115 is consistent with the absence of heparanase expression by hOPCs (Saraswat et al., 2020; Extended Data Fig. 3-1) and suggests a non-cell-autonomous effect of OGT2115 in vivo. In contrast, as PI-88 was able to restore differentiation in the presence of IFN-γ, this suggests that additional mechanisms act to block IFN-γ signaling such as by directly blocking IFN-γ ligand/receptor binding.
IFN-γ signaling in OPCs potentiates Wnt/Bmp signaling during oligodendrocyte differentiation
To determine whether OPC-specific IFN-γ signaling contributes to impaired oligodendrocyte differentiation in vivo and, whether PI-88 treatment could rescue this, we examined oligodendrocyte differentiation in lysolecithin lesions in littermate controls and OPC-specific IFN-γR1 cKO mice with or without concurrent PI-88 treatment. Interestingly, the deletion of IFN-γR1 resulted in a 2.5-fold increase in the density of Olig2+CC1+ postmitotic oligodendrocytes and a 1.8-fold increase in the percentage of Olig2+CC1+ oligodendrocytes relative to wild-type control animals (n = 4–5 animals/group; two-way ANOVA with Holm–Sidak post-test, p < 0.01, p < 0.001, respectively; Fig. 8A–D). Although IFN-γR1 cKO had no effect on Olig2+ cell density at 5 dpl, robust oligodendrogenesis in IFN-γR1 cKO animals was accompanied by a small but significant increase in the total density of Olig2+ oligodendrocyte lineage cells relative to wild-type controls (Holm–Sidak post-test, p < 0.05; Fig. 8B). As shown previously (Saraswat et al., 2020), PI-88 treatment of wild-type animals significantly increased both Olig2+CC1+ oligodendrocyte density and CC1+ oligodendrocyte percentage (p < 0.05 and p < 0.001, respectively; Fig. 8C,D). Interestingly, while the effect of PI-88 treatment and IFN-γR1 cKO on the percentage of CC1+ oligodendrocytes was additive (p < 0.05), there were no differences observed in the densities of either Olig2+ oligodendrocytes lineage cells or Olig2+CC1+ oligodendrocytes between PI-88-treated groups (p > 0.05). Importantly, two-way ANOVA did not reveal a significant interaction between genotype and PI-88 treatment on all parameters (p = 0.28, p = 0.17, p = 0.09; Extended Data Fig. 8-1). Thus, the effects of OPC IFN-γ signaling and PI-88 treatment on oligodendrocyte differentiation at 7 dpl are likely mediated via distinct pathways.
Figure 8.

Excessive IFN-γ signaling blocks OPC differentiation following demyelination in a cell-autonomous manner and is associated with the activation of Wnt/Bmp target genes in OPCs. A, Wild-type and IFNγR1 cKO were subjected to lysolecithin-mediated focal demyelination of the spinal cord with or without coadministration of 10 μg/ml PI-88. To assess the formation of newly generated oligodendrocytes, animals were killed at 7 d post-lesion (7 dpl). Olig2 (Red) and CC1 (green) colocalization was used to assess OL differentiation. B–D, The densities of Olig2+ oligodendrocytes lineage cells (cells/mm2; B) and Olig2+CC1+ postmitotic oligodendrocytes (C) was quantified along with the percentage of CC1+ oligodendrocytes (D; n = 3–5 mice/group; mean ± SEM). Holm–Sidak test: *p < 0.05, **p < 0.01, ***p < 0.001 for each pairwise comparison following two-way ANOVA. Red denotes p values compared with control wild-type, while green denotes p values compared with IFNγR1 cKO group. E, F, Two-way ANOVA did not reveal a significant interaction between genotype and PI-88 treatment on all parameters (p = 0.28, p = 0.17, p = 0.09; Extended Data Fig. 8-1). G, H, Quantification of Wnt target gene expression Apcdd1 (G) and BMP target gene Id4 (H) was determined in Pdgfra+ OPCs at 7 dpl (n = 4–5 mice/group, mean ± SEM) and at 5 dpl (Extended Data Fig. 8-2). t test: *p < 0.05. Both Wnt and Bmp target genes were regulated in an IFNγR1-dependent manner in OPCs. Scale bar, 20 μm. ns denotes not significant.
Two-way ANOVA showing effects of PI88 treatment in WT and IFNγR1 cKO and the interaction. Download Figure 8-1, TIF file (623.1KB, tif) .
Temporal regulation Wnt/Bmp signaling by IFN-γ signaling in OPCs following demyelination. A, RNAscope in situ hybridization for Wnt and Bmp target genes (Apcdd1/Pdgfra and ID4/Pdgfra, respectively), at 5 and 7 dpl (mean ± SEM, n = 3–6 mice/group). Two-way ANOVA revealed that there was no interaction between IFN-γR1 genotype and the time for Wnt target expression, suggesting that IFN-γ does not influence Wnt signaling via a direct effect on OPCs (p = 0.19). In contrast, we observed a significant interaction of time and IFN-γR1 genotype with regard to Bmp target gene expression, suggesting that IFN-γR1 signaling potentiates Bmp activation in a time-dependent manner (p < 0.01). Holm–Sidak multiple-comparisons post-test revealed a significant effect of IFN-γR1 signaling on both target genes at 7 dpl but no significant effect at 5 dpl (*p < 0.05 and **p < 0.01, respectively). Download Figure 8-2, DOCX file (23.7KB, docx) .
PI-88 treatment antagonizes Wnt and Bmp signaling following demyelination leading to accelerated oligodendrocyte differentiation (Saraswat et al., 2020). As the effect of PI-88 treatment was similar in magnitude to IFN-γR1 cKO, we next asked whether OPC IFN-γ signaling could modulate Wnt/Bmp signaling in OPCs. We examined expression of Wnt and Bmp target gene expression in Pdgfra+ OPCs using multicolor fluorescence in situ hybridization (Fig. 8E,G). In IFN-γR1 cKO mice, we observed a significant 1.6-fold decrease in expression of Wnt target gene Apcdd1+ and a corresponding 1.9-fold decrease in Bmp target gene Id4 within Pdgfra+ OPCs at 7 dpl relative to littermate controls (n = 4–5 animals/group, unpaired t test, both p < 0.05; Fig. 8F,H). This suggested that Wnt/Bmp signaling at 7 dpl was dependent on OPC-specific IFN-γ signaling and that IFN-γ signaling via Wnt/Bmp contributes to the inhibitory microenvironment that prevents efficient oligodendrocyte differentiation. Next, to determine whether Wnt/Bmp target gene expression in OPCs during recruitment was dependent on IFN-γR1, we examined target gene expression at 5 dpl in cKO and control mice (Extended Data Fig. 8-2). Intriguingly, at 5 dpl there was no significant difference in Apcdd1+ and Id4+ OPC target gene expression between IFN-γR1 cKO and wild-type groups. As such, IFN-γ-mediated signaling in OPCs that results in impaired OPC recruitment and proliferation is likely not mediated by pathologic Wnt/Bmp signaling.
IFN-γ-mediated axonal damage and hypomyelination is rescued by heparanase modulation
As PI-88 was able to effectively antagonize the negative effects of IFN-γ on OPC recruitment and differentiation, we asked whether heparanase inhibition by PI-88 or OGT2155 could modulate inflammatory remyelination. As such, we treated IFN-γ-treated lesions (3 ng) with either PI-88 or OGT2155 at the time of lesion induction and analyzed remyelination by electron microscopy at 14 dpl, during the onset of remyelination (Fig. 9A–E). Consistent with the increased App1 immunofluorescence at 5 dpl, IFN-γ induced a diverse pattern of axonal degeneration at 14 dpl (Fig. 9F,G). To assess axonal degeneration, we calculated the proportion of degenerated axons among total axons (Fig. 9I). We noted a 4.8-fold increase in axonal degeneration following IFN-γ (n = 3–6, Holm–Sidak post-test, p < 0.0001). PI-88 and OGT2155 treatment was capable of rescuing the IFN-γ-induced effects on axonal injury. Both PI-88 and OGT2155 treatment substantially attenuated IFN-γ induced axonal damage (n = 3-6; Holm–Sidak post-test, p < 0.0001, p < 0.001). To assess remyelination, we focused on the healthy-appearing axons in each group and degenerating axons were excluded (Fig. 9A–E,H). We found that the proportion of remyelinated axons was significantly reduced following IFN-γ treatment consistent with impaired spontaneous remyelination (Fig. 9J). Both heparanase inhibitors PI-88 and OGT2155 rescued the negative effect of IFN-γ and significantly increased the proportion of remyelinated axons at 14 dpl (Holm–Sidak post-test, p < 0.01, p < 0.0001). The g-ratio analysis revealed that remyelinated axons in the IFN-γ group exhibited thinner myelin (higher g-ratio) compared with mice in other groups (Fig. 9K). Linear regression analysis of axon diameter versus g-ratio demonstrated a significant increase in g-ratio following IFN-γ treatment (Fig. 9L), which was in turn rescued by PI-88 (Fig. 9M) and OGT2155 (Fig. 9O) treatment. The distribution of axonal diameter within the lesion suggested that axonal swelling among healthy-appearing axons was not a significant feature (Fig. 9P). Interestingly, the rescue of hypomyelination by PI-88 and OGT2155 was more efficient among small-diameter axons (<2 μm). The distribution of g-ratios demonstrated a significant increase in thinly remyelinated axons following IFN-γ treatment, which was rescued by PI-88 and OGT2155 (Fig. 9Q). Importantly, neither heparanase inhibitor induced axonal damage following demyelination. We have previously shown that PI-88 does not induce axonal injury (Saraswat et al., 2020). Here, OGT2155 similarly did not increase axonal damage compared with control (p = 0.26; Fig. 9I). Unlike PI-88 (Saraswat et al., 2020), the heparanase inhibitor OGT2155 did not substantially improve spontaneous remyelination compared with control lesion (percentage of remyelinated axons, p = 0.12; Fig. 9J,P). However, we did observe a small but significant increase in myelin sheath thickness of remyelinated axons following OGT2115 treatment (Fig. 9K,N,Q). Together, these data demonstrate that heparanase inhibition in demyelinating lesion rescues the degenerative effects of IFN-γ and improves remyelination of inflammatory demyelinating lesions.
Figure 9.
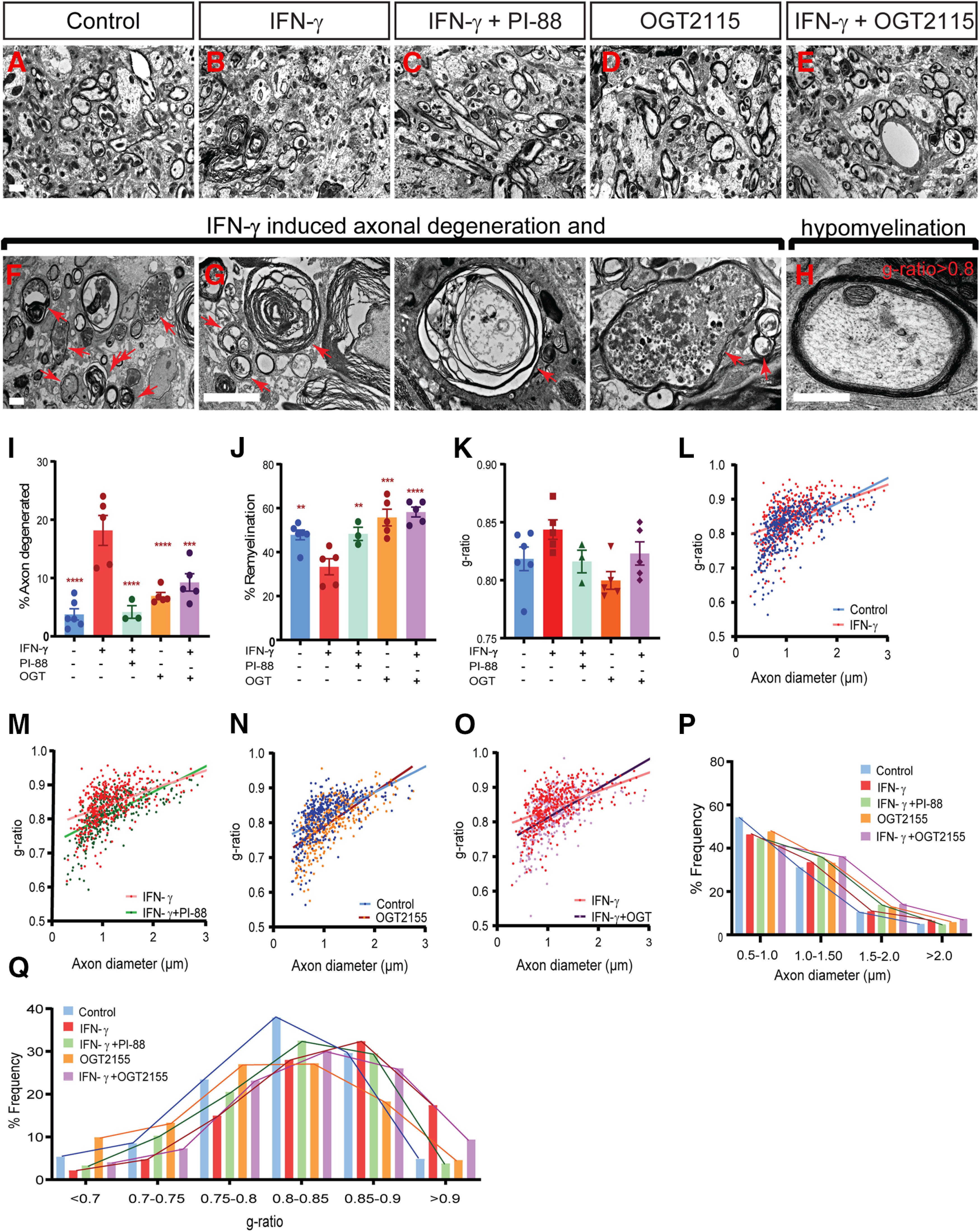
Heparanase inhibitors PI-88 and OGT2115 rescue remyelination and reduce axonal injury induced by IFN-γ treatment. A–E, Injection of 3 ng of IFN-γ with or without PI-88 (10 μg/ml) or OGT2155 (0.4 μm) directly into demyelinated lesions in mice (8–10 weeks of age). Animals were killed at 14 dpl, and spinal cord tissue was processed for electron microscopy. Representative fields shown within the lesion periphery. F, G, IFN-γ treatment induces various forms of axonal injury and degeneration. H, Several remyelinated axons also exhibit very thin remyelination following IFN-γ treatment. Arrows represent degenerating axons. I–K, The proportions of degenerating axons among total axons (I), the proportion of remyelinated axons among healthy-appearing axons (J), and the g-ratio of normal-appearing remyelinating axons (K) were calculated in each animal (mean ± SEM; n = 3–6 animals/group). I, J, Both PI-88 and OGT2115 treatment rescued the negative effects of IFN-γ on axonal damage (I) and the percentage of remyelination (J) following demyelination. L–O, Relationship between axon diameter and g-ratio (linear regression shown) between groups (L; control vs IFN-γ; M, IFN-γ vs IFN-γ + PI-88; N, control vs OGT2155; and O, IFN-γ vs IFN-γ + OGT2155). P, Q, Frequency distribution of axonal diameter (P) and g-ratio (Q) in lesion (n = 3–6 animals/group, ≥400 axons). Holm–Sidak test: **p < 0.01, ***p < 0.001, ****p < 0.0001 for each pairwise comparison following one-way ANOVA. Scale bar, 2 μm.
Discussion
Multiple sclerosis is a devastating autoimmune disease characterized by chronic demyelination. While the causes for failed remyelination in human disease are unknown, the presence of nondividing OPCs in chronic MS lesions (Wolswijk, 1998; Kuhlmann et al., 2008) suggests that pathologic quiescence may prevent OPC activation and thereby impair oligodendrocyte recruitment and differentiation following demyelination (Dietz et al., 2016). IFN-γ acts on human OPCs to induce PRRX1 expression and pathologic quiescence in vitro, and PRRX1 prevents successful engraftment and remyelination following transplantation (Wang et al., 2018). As such, we hypothesized that IFN-γ may directly contribute to pathologic quiescence in OPCs by upregulating PRRX1 following demyelination. In this study, we found that PRRX1 was indeed expressed in OPCs present within chronic MS lesions and that IFN-γ signaling critically influenced the lesion environment and OPC quiescence following experimental demyelination in mice. Importantly, IFN-γ acted directly on OPCs recruited into the region of demyelination as OPC-specific ablation of IFN-γR1 rescued the IFN-γ-mediated effects on quiescence. In addition to other ECM components (Lau et al., 2013), we recently showed that the heparanome has a profound effect on OPC recruitment and differentiation following demyelination and that sulfatases act to promote inhibitory Wnt and Bmp signaling (Saraswat et al., 2020). In the current study, we have extended these observations to show that modification of the heparanome by the inhibition of heparanase, via either PI-88 or OGT2115, rescues the proinflammatory effects of IFN-γ on OPC recruitment and lesion environment.
Unlike EAE and multiple sclerosis itself, IFN-γ production by CD4+ Th1 cells is not thought to be a key hallmark of toxin-based experimental models of demyelination such as lysolecithin and cuprizone (Ghasemlou et al., 2007; Remington et al., 2007). As such, we first characterized the effect of exogenous IFN-γ on lesion formation and focused on the effects on OPC recruitment. IFN-γ exacerbated the lysolecithin-induced demyelination, leading to increased lesion volume and cell death following demyelination with attendant axonal injury consistent with its well described role as a proinflammatory cytokine. This was likely mediated by direct IFN-γ signaling on microglial/macrophage cells as we noted substantially increased Irf1 within Iba1+ cells. Indeed, we observed increased iNOS expression among macrophages coincident with an activated ramified morphology consistent with polarization toward the proinflammatory phenotype M1, as observed previously (Miron et al., 2013). In addition, IFN-γ-augmented lesions displayed pronounced astrogliosis surrounding the demyelinated region that was substantially greater than matched control lesions. IFN-γ along with numerous other cytokines induce reactive astrogliosis (Balasingam et al., 1994). While we cannot exclude that IFN-γ directly induced astrogliosis following demyelination, we did not observe Irf1 expression in astrocytes at 5 d postlesion, suggesting that if direct IFN-γ signaling occurred in astrocytes, it was transient in nature. Astrogliosis following IFN-γ treatment would be also expected as a result of microglial-mediated release of TNF-α and other proinflammatory mediators. Similar to our results, in both MS tissue and EAE, astrocytes are less responsive to IFN-γ compared with both microglia and oligodendrocyte lineage cells (Ren et al., 2011).
Paracrine influences of IFN-γ modulate many aspects of the OPC response to demyelination. Genomic IFN-γ receptor deletion accelerates OPC recruitment proliferation following cuprizone-mediated demyelination (Maña et al., 2006), but it is not known whether IFN-γ signaling on OPCs directly influences recruitment or contributes to pathologic quiescence following demyelination. Unlike previous studies, which have largely focused on the role of IFN-γ signaling in postmitotic oligodendrocytes (for review, see Lin and Popko, 2009), to investigate the role of OPC autonomous IFN-γ signaling, we used conditional deletion of IFN-γR1 in NG2-expressing OPCs. Interestingly, while endogenous IFN-γ signaling did not alter OPC proliferation or recruitment, conditional deletion of IFN-γR1 resulted in accelerated differentiation at 7 dpl. Furthermore, we found that conditional IFN-γR1 deletion was sufficient to overcome the negative effects of exogenous IFN-γ treatment while not influencing the increased astrogliosis and microglial cell infiltration (data not shown). Together, this suggests that cell-autonomous IFN-γ signaling in OPCs is critically important in the regulation of recruitment and differentiation following inflammatory demyelination. Human OPC-autonomous IFN-γ signaling acts via STAT1 activation and upregulates PRRX1 to cause cell cycle arrest (Wang et al., 2018). Herein, we show that STAT1 and PRRX1 upregulation is a common hallmark in chronic active MS lesions with numerous PRRX1-expressing quiescent OPCs present in the lesion border and within the lesion core. This is consistent with a key role of IFN-γ and participation of PRRX1 in the failure of remyelination in MS.
Previous studies have suggested that low-level or prior exposure to IFN-γ exerts a protective role in MS and EAE (Ottum et al., 2015). A low dose of IFN-γ induces moderate endoplasmic reticulum stress in mature oligodendrocytes and inhibits the unfolded protein response (UPR) and apoptosis (Lin and Popko, 2009; Ottum et al., 2015). Indeed, IFN-γ exposure before the induction of EAE is protective and prevents demyelination (Lin et al., 2007). We found that coadministration of IFN-γ at the time of demyelination resulted in impaired OPC–oligodendrocyte differentiation. This effect may be a result of the failed recruitment of OPCs or the activation of the UPR. Intriguingly, both Wnt and Bmp target gene expression in OPCs was dependent on IFN-γ signaling, and the deletion of IFN-γ receptors in OPCs resulted in the downregulation of both target genes during the period of maximal oligodendrocyte generation. Wnt and Bmp signaling are key pathways that inhibit OPC differentiation and remyelination (Mabie et al., 1997; Grinspan et al., 2000; Fancy et al., 2009; Feigenson et al., 2009; Sabo et al., 2011). There is accumulating evidence that link IFN-γ signaling with Wnt activation (Nava et al., 2010) and, via inhibition of TGF-β1 (Ishida et al., 2004), the activation of Bmp signaling (Yuan et al., 2018; Ning et al., 2019). We previously found that IFN-γ treatment of human OPCs induces a gene expression signature that is consistent with the activation of Wnt signaling (REACTOME_SIGNALING_BY_WNT, GSEA q = 2.6 × 10−3; Wang et al., 2018). Furthermore, IFN-γ treatment upregulated glypican-2 (GPC2) in hOPCs. GPC2, a cell surface heparan sulfate proteoglycan, is a known regulator of Wnt signaling (Li et al., 2017). These data suggest that the IFN-γ pathway may intimately regulate Wnt and Bmp signaling following demyelination. In contrast, we did not observe that the regulation of Wnt and Bmp targets genes at an earlier time point during which time OPCs are actively recruited into the region of demyelination and are undergoing rapid proliferation. This suggests that the mechanisms by which IFN-γ influences OPC recruitment and differentiation are distinct and provide novel insight into the mechanisms by which Wnt/Bmp pathways become dysregulated in inflammatory demyelinating disease.
The local heparanome surrounding OPCs is critical in the regulation of OPC recruitment, differentiation, and the overall success of remyelination (Saraswat et al., 2020). Our previous RNA-sequencing analysis of human OPCs exposed to IFN-γ suggested that the heparanome might be directly regulated by IFN-γ. Intriguingly, IFN-γ dimer binding to HSPG and to IFN-γ receptors are mutually exclusive events (Sadir et al., 1998). As such, we investigated the potential for PI-88, a heparin mimetic known to block sulfatases (Hossain et al., 2010) and heparanases (Parish et al., 1999), and thereby to rescue the negative effects of IFN-γ following demyelination. PI-88 treatment was able to oppose the effects of IFN-γ on human OPC proliferation, differentiation, and STAT signaling in vitro, and, following IFN-γ-augmented demyelination, to improve OPC recruitment and remyelination as well as to reduce cell death, axonal injury, reactive gliosis, and the macrophage bias toward an iNOS-expressing M1 phenotype. As PI-88 blocks endosulfatases (Sulf1/2) to improve OPC recruitment and differentiation following demyelination (Saraswat et al., 2020), we first tested whether PI-88 blocks IFN-γ signaling in a sulfatase-dependent manner. In cultured hOPCs, which do not express heparanase (HPSE1 and HPSE2), SULF2 knockdown had no effect of IFN-γ signaling and did not influence PI-88-mediated antagonism of IFN-γ. Consistent with previous reports (Sarrazin et al., 2005), this suggested that PI-88, acting as a heparan sulfate mimetic, could directly bind to and block IFN-γ. Similarly, conditional deletion of Sulf1/2 in NG2-expressing OPCs increased OPC density, as shown previously (Saraswat et al., 2020), but sulfatase deletion did not influence the effects of IFN-γ on reduced OPC proliferation or recruitment. Together, this suggests distinct nonoverlapping mechanisms of sulfatase deletion and PI-88 treatment following IFN-γ treatment.
While heparanases (HPSE1/2) are not expressed by mouse OPCs (Zhang et al., 2014) or human OPCs (Wang et al., 2018), heparanase expression has been widely reported in microglial/macrophage and endothelial cells. HPSE expression is induced in microglial cells following proinflammatory LPS stimulation (Lively and Schlichter, 2013), and directly contributes to their activation (Gutter-Kapon et al., 2016) and migration (Takahashi et al., 2008). Heparanase can be induced in neurons and astrocytes following vascular damage (Zhang et al., 2006; Changyaleket et al., 2017). Likewise, endothelial heparanase can be induced following stimulation with proinflammatory cytokines such as IFN-γ (Edovitsky et al., 2006). Thus, while a role for heparanase in MS has not been described, it is highly likely that heparanases are present and active in demyelinating lesions (Changyaleket et al., 2017). In support of this, we also noted increased presence of Hpse1 and Hpse2 in IFN-γ augmented demyelinated lesions (data not shown). As PI-88 is also a potent inhibitor of heparanase (Parish et al., 1999), we sought to further test whether the inhibition of heparanase could rescue the effects of IFN-γ on OPC recruitment and proliferation. Treatment with OGT2115, a small-molecule nonheparan mimetic heparanase inhibitor (McKenzie, 2007), was equally effective as PI-88 in the rescue of both OPC recruitment and proliferation to control levels. This indicates that heparanase itself likely contributes to IFN-γ-mediated effects on OPC dynamics, and, therefore, it is probable that the degradation of heparan sulfate on the HSPG core by endogenous heparanases facilitates IFN-γ signaling following demyelination. While the clinical landscape for heparanase inhibitors is yet to be fully explored, several lead compounds have been identified. The most studied PI-88 is being investigated for the treatment of various cancer indications such as hepatocellular carcinoma (Liu et al., 2014), melanoma (Lewis et al., 2008), and prostate cancer (Khasraw et al., 2010). Apart from PI-88, three heparan sulfatase mimetics have entered clinical trials but small-molecule inhibitors have yet to be studied (Sanderson et al., 2017). However, given the broad interest in heparanase inhibitors in cancer, these molecules will likely be forthcoming and present an attractive target to overcome IFN-γ-mediated remyelination failure.
While the deposition of CSPGs and their negative roles following demyelination have received much attention in the past, much less research has focused on the roles of HSPG following demyelination. The role of HSPG expression on OPCs has not been widely studied. In cultured OPCs, sulfation of newly synthesized HSPG is required for the proliferative and antidifferentiation effects of FGF-2 (Bansal and Pfeiffer, 1994). HSPG deposition has been shown in MS lesions (van Horssen et al., 2006), and accumulates around small callosal lysolecithin lesions (Macchi et al., 2020). Ndst1, a key enzyme in the biosynthesis of heparan sulfate, is enriched among OPCs and OLs in this zone. Intriguingly, preventing HSPG deposition by deletion of OPCs and oligodendrocyte-expressed Ndst1 promotes OPC proliferation but does not influence resultant oligodendrocyte density. HSPGs (Syndecan 3 and Glypican 1) are expressed by human and rodent OPCs (Winkler et al., 2002; Sim et al., 2006) and likely provide a substrate for IFN-γ binding in OPC culture. Additional studies will be needed to delimit which HSPGs are influential in terms of IFN-γ binding and the induction of OPC quiescence.
In conclusion, we show that OPC autonomous IFN-γ signaling acts to limit recruitment following demyelination and may result in pathologic quiescence leading to chronic demyelination. The heparanome regulates numerous aspects of OPC signaling following demyelination, and, herein, we show that pathologic IFN-γ signaling affecting OPCs is dependent on the heparanome. As such, we show that heparanase inhibition acts to prevent IFN-γ signaling and rescue the negative sequelae associated with IFN-γ. In addition to the roles of sulfatase-mediated modulation of the heparanome on OPC dynamics following demyelination (Saraswat et al., 2020), a therapeutic strategy focused on the heparanome represents a promising approach to promoting myelin repair. Together, our findings define a novel therapeutic target for reversing pathologic quiescence in OPCs and altering the pathologic lesion microenvironment for the treatment of demyelinating disease.
Footnotes
This work was supported by National Institute of Neurological Disorders and Stroke Grant R01-NS-104021, National Center for Advancing Translational Sciences (NCATS) Grant UL1-TR-001412, National Multiple Sclerosis Society Grant RG-1701-26750, the Kalec Multiple Sclerosis Foundation, the Change MS Foundation, and the Skarlow Memorial Trust. J.J.P. received additional support from National Institute of General Medical Sciences Grant R25-GM-09545902, NCATS Grant UL1TR001412-S1, and the New York State Department of Health (Empire State Stem Cell Fund) through the Stem Cells in Regenerative Medicine Fellowship (NYSTEM Grant C30290GG). We thank the Confocal Microscope and Flow Cytometry Facility in the School of Medicine and Biomedical Sciences, University at Buffalo, for assistance.
The authors declare no competing financial interests.
References
- Abiraman K, Pol SU, O'Bara MA, Chen GD, Khaku ZM, Wang J, Thorn D, Vedia BH, Ekwegbalu EC, Li JX, Salvi RJ, Sim FJ (2015) Anti-muscarinic adjunct therapy accelerates functional human oligodendrocyte repair. J Neurosci 35:3676–3688. 10.1523/JNEUROSCI.3510-14.2015 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Agresti C, D'Urso D, Levi G (1996) Reversible inhibitory effects of interferon-gamma and tumour necrosis factor-alpha on oligodendroglial lineage cell proliferation and differentiation in vitro. Eur J Neurosci 8:1106–1116. 10.1111/j.1460-9568.1996.tb01278.x [DOI] [PubMed] [Google Scholar]
- Arellano G, Ottum PA, Reyes LI, Burgos PI, Naves R (2015) Stage-specific role of interferon-gamma in experimental autoimmune encephalomyelitis and multiple sclerosis. Front Immunol 6:492. 10.3389/fimmu.2015.00492 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baerwald KD, Popko B (1998) Developing and mature oligodendrocytes respond differently to the immune cytokine interferon-gamma. J Neurosci Res 52:230–239. [DOI] [PubMed] [Google Scholar]
- Balabanov R, Strand K, Kemper A, Lee JY, Popko B (2006) Suppressor of cytokine signaling 1 expression protects oligodendrocytes from the deleterious effects of interferon-gamma. J Neurosci 26:5143–5152. 10.1523/JNEUROSCI.0737-06.2006 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Balasingam V, Tejada-Berges T, Wright E, Bouckova R, Yong VW (1994) Reactive astrogliosis in the neonatal mouse brain and its modulation by cytokines. J Neurosci 14:846–856. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bansal R, Pfeiffer SE (1994) Inhibition of protein and lipid sulfation in oligodendrocytes blocks biological responses to FGF-2 and retards cytoarchitectural maturation, but not developmental lineage progression. Developmental biology 162:511–524. [DOI] [PubMed] [Google Scholar]
- Barcia C, Ros CM, Annese V, Gómez A, Ros-Bernal F, Aguado-Yera D, Martínez-Pagán ME, de Pablos V, Fernandez-Villalba E, Herrero MT (2011) IFN-γ signaling, with the synergistic contribution of TNF-α, mediates cell specific microglial and astroglial activation in experimental models of Parkinson's disease. Cell Death Dis 2:e142. 10.1038/cddis.2011.17 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Beck J, Rondot P, Catinot L, Falcoff E, Kirchner H, Wietzerbin J (1988) Increased production of interferon gamma and tumor necrosis factor precedes clinical manifestation in multiple sclerosis: do cytokines trigger off exacerbations? Acta Neurol Scand 78:318–323. 10.1111/j.1600-0404.1988.tb03663.x [DOI] [PubMed] [Google Scholar]
- Changyaleket B, Deliu Z, Chignalia AZ, Feinstein DL (2017) Heparanase: potential roles in multiple sclerosis. J Neuroimmunol 310:72–81. 10.1016/j.jneuroim.2017.07.001 [DOI] [PubMed] [Google Scholar]
- Cheng CC, Lee YH, Lin SP, Huangfu WC, Liu IH (2014) Cell-autonomous heparanase modulates self-renewal and migration in bone marrow-derived mesenchymal stem cells. J Biomed Sci 21:21. 10.1186/1423-0127-21-21 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Conway GD, O'Bara MA, Vedia BH, Pol SU, Sim FJ (2012) Histone deacetylase activity is required for human oligodendrocyte progenitor differentiation. Glia 60:1944–1953. 10.1002/glia.22410 [DOI] [PubMed] [Google Scholar]
- Corbin JG, Kelly D, Rath EM, Baerwald KD, Suzuki K, Popko B (1996) Targeted CNS expression of interferon-gamma in transgenic mice leads to hypomyelination, reactive gliosis, and abnormal cerebellar development. Mol Cell Neurosci 7:354–370. 10.1006/mcne.1996.0026 [DOI] [PubMed] [Google Scholar]
- Dietz KC, Polanco JJ, Pol SU, Sim FJ (2016) Targeting human oligodendrocyte progenitors for myelin repair. Exp Neurol 283:489–500. 10.1016/j.expneurol.2016.03.017 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dutta R, Mahajan KR, Nakamura K, Ontaneda D, Chen J, Volsko C, Dudman J, Christie E, Dunham J, Fox RJ, Trapp BD (2019) Comprehensive autopsy program for individuals with multiple sclerosis. J Vis Exp. Advance online publication. Retrieved July 19, 2019. doi: 10.3791/59511. [DOI] [PubMed] [Google Scholar]
- Edovitsky E, Lerner I, Zcharia E, Peretz T, Vlodavsky I, Elkin M (2006) Role of endothelial heparanase in delayed-type hypersensitivity. Blood 107:3609–3616. 10.1182/blood-2005-08-3301 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fancy SP, Baranzini SE, Zhao C, Yuk DI, Irvine KA, Kaing S, Sanai N, Franklin RJ, Rowitch DH (2009) Dysregulation of the Wnt pathway inhibits timely myelination and remyelination in the mammalian CNS. Genes Dev 23:1571–1585. 10.1101/gad.1806309 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fancy SPJ, Harrington EP, Yuen TJ, Silbereis JC, Zhao C, Baranzini SE, Bruce CC, Otero JJ, Huang EJ, Nusse R, Franklin RJM, Rowitch DH (2011) Axin2 as regulatory and therapeutic target in newborn brain injury and remyelination. Nat Neurosci 14:1009–U1099. 10.1038/nn.2855 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Feigenson K, Reid M, See J, Crenshaw EB 3rd, Grinspan JB (2009) Wnt signaling is sufficient to perturb oligodendrocyte maturation. Mol Cell Neurosci 42:255–265. 10.1016/j.mcn.2009.07.010 [DOI] [PubMed] [Google Scholar]
- Fortunato G, Calcagno G, Bresciamorra V, Salvatore E, Filla A, Capone S, Liguori R, Borelli S, Gentile I, Borrelli F, Borgia G, Sacchetti L (2008) Multiple sclerosis and hepatitis C virus infection are associated with single nucleotide polymorphisms in interferon pathway genes. J Interferon Cytokine Res 28:141–152. 10.1089/jir.2007.0049 [DOI] [PubMed] [Google Scholar]
- Franklin RJM, Ffrench-Constant C (2017) Regenerating CNS myelin - from mechanisms to experimental medicines. Nat Rev Neurosci 18:753–769. 10.1038/nrn.2017.136 [DOI] [PubMed] [Google Scholar]
- García B, Martín C, García-Suárez O, Muñiz-Alonso B, Ordiales H, Fernández-Menéndez S, Santos-Juanes J, Lorente-Gea L, Castañón S, Vicente-Etxenausia I, Piña Batista KM, Ruiz-Díaz I, Caballero-Martínez MC, Merayo-Lloves J, Guerra-Merino I, Quirós LM, Fernández-Vega I (2017) Upregulated expression of heparanase and heparanase 2 in the brains of Alzheimer's disease. J Alzheimers Dis 58:185–192. 10.3233/JAD-161298 [DOI] [PubMed] [Google Scholar]
- Gehrmann J, Banati RB, Cuzner ML, Kreutzberg GW, Newcombe J (1995) Amyloid precursor protein (APP) expression in multiple sclerosis lesions. Glia 15:141–151. 10.1002/glia.440150206 [DOI] [PubMed] [Google Scholar]
- Ghasemlou N, Jeong SY, Lacroix S, David S (2007) T cells contribute to lysophosphatidylcholine-induced macrophage activation and demyelination in the CNS. Glia 55:294–302. 10.1002/glia.20449 [DOI] [PubMed] [Google Scholar]
- Grinspan JB, Edell E, Carpio DF, Beesley JS, Lavy L, Pleasure D, Golden JA (2000) Stage-specific effects of bone morphogenetic proteins on the oligodendrocyte lineage. J Neurobiol 43:1–17. [DOI] [PubMed] [Google Scholar]
- Gutter-Kapon L, Alishekevitz D, Shaked Y, Li JP, Aronheim A, Ilan N, Vlodavsky I (2016) Heparanase is required for activation and function of macrophages. Proc Natl Acad Sci U S A 113:E7808–E7817. 10.1073/pnas.1611380113 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hammond E, Khurana A, Shridhar V, Dredge K (2014) The role of heparanase and sulfatases in the modification of heparan sulfate proteoglycans within the tumor microenvironment and opportunities for novel cancer therapeutics. Front Oncol 4:195. 10.3389/fonc.2014.00195 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Horiuchi M, Itoh A, Pleasure D, Itoh T (2006) MEK-ERK signaling is involved in interferon-gamma-induced death of oligodendroglial progenitor cells. J Biol Chem 281:20095–20106. 10.1074/jbc.M603179200 [DOI] [PubMed] [Google Scholar]
- Horwitz MS, Evans CF, McGavern DB, Rodriguez M, Oldstone MBA (1997) Primary demyelination in transgenic mice expressing interferon-gamma. Nat Med 3:1037–1041. 10.1038/nm0997-1037 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hossain MM, Hosono-Fukao T, Tang R, Sugaya N, van Kuppevelt TH, Jenniskens GJ, Kimata K, Rosen SD, Uchimura K (2010) Direct detection of HSulf-1 and HSulf-2 activities on extracellular heparan sulfate and their inhibition by PI-88. Glycobiology 20:175–186. 10.1093/glycob/cwp159 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ishida Y, Kondo T, Takayasu T, Iwakura Y, Mukaida N (2004) The essential involvement of cross-talk between IFN-gamma and TGF-beta in the skin wound-healing process. J Immunol 172:1848–1855. 10.4049/jimmunol.172.3.1848 [DOI] [PubMed] [Google Scholar]
- Karoli T, Liu L, Fairweather JK, Hammond E, Li CP, Cochran S, Bergefall K, Trybala E, Addison RS, Ferro V (2005) Synthesis, biological activity, and preliminary pharmacokinetic evaluation of analogues of a phosphosulfomannan angiogenesis inhibitor (PI-88). J Med Chem 48:8229–8236. 10.1021/jm050618p [DOI] [PubMed] [Google Scholar]
- Khasraw M, Pavlakis N, McCowatt S, Underhill C, Begbie S, de Souza P, Boyce A, Parnis F, Lim V, Harvie R, Marx G (2010) Multicentre phase I/II study of PI-88, a heparanase inhibitor in combination with docetaxel in patients with metastatic castrate-resistant prostate cancer. Ann Oncol 21:1302–1307. 10.1093/annonc/mdp524 [DOI] [PubMed] [Google Scholar]
- Kuhlmann T, Miron V, Cui Q, Cuo Q, Wegner C, Antel J, Brück W (2008) Differentiation block of oligodendroglial progenitor cells as a cause for remyelination failure in chronic multiple sclerosis. Brain 131:1749–1758. 10.1093/brain/awn096 [DOI] [PubMed] [Google Scholar]
- LaFerla FM, Sugarman MC, Lane TE, Leissring MA (2000) Regional hypomyelination and dysplasia in transgenic mice with astrocyte-directed expression of interferon-gamma. J Mol Neurosci 15:45–59. 10.1385/JMN:15:1:45 [DOI] [PubMed] [Google Scholar]
- Lassmann H (2014) Mechanisms of white matter damage in multiple sclerosis. Glia 62:1816–1830. 10.1002/glia.22597 [DOI] [PubMed] [Google Scholar]
- Lassmann H, Brück W, Lucchinetti C (2001) Heterogeneity of multiple sclerosis pathogenesis: implications for diagnosis and therapy. Trends Mol Med 7:115–121. 10.1016/s1471-4914(00)01909-2 [DOI] [PubMed] [Google Scholar]
- Lau LW, Cua R, Keough MB, Haylock-Jacobs S, Yong VW (2013) Pathophysiology of the brain extracellular matrix: a new target for remyelination. Nat Rev Neurosci 14:722–729. 10.1038/nrn3550 [DOI] [PubMed] [Google Scholar]
- Lee HK, Chaboub LS, Zhu W, Zollinger D, Rasband MN, Fancy SP, Deneen B (2015) Daam2-PIP5K is a regulatory pathway for Wnt signaling and therapeutic target for remyelination in the CNS. Neuron 85:1227–1243. 10.1016/j.neuron.2015.02.024 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lewis KD, Robinson WA, Millward MJ, Powell A, Price TJ, Thomson DB, Walpole ET, Haydon AM, Creese BR, Roberts KL, Zalcberg JR, Gonzalez R (2008) A phase II study of the heparanase inhibitor PI-88 in patients with advanced melanoma. Invest New Drugs 26:89–94. 10.1007/s10637-007-9080-5 [DOI] [PubMed] [Google Scholar]
- Li J, Li JP, Zhang X, Lu Z, Yu SP, Wei L (2012) Expression of heparanase in vascular cells and astrocytes of the mouse brain after focal cerebral ischemia. Brain Res 1433:137–144. 10.1016/j.brainres.2011.11.032 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li N, Fu H, Hewitt SM, Dimitrov DS, Ho M (2017) Therapeutically targeting glypican-2 via single-domain antibody-based chimeric antigen receptors and immunotoxins in neuroblastoma. Proc Natl Acad Sci U S A 114:E6623–E6631. 10.1073/pnas.1706055114 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lillemeier BF, Köster M, Kerr IM (2001) STAT1 from the cell membrane to the DNA. EMBO J 20:2508–2517. 10.1093/emboj/20.10.2508 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lin W, Popko B (2009) Endoplasmic reticulum stress in disorders of myelinating cells. Nat Neurosci 12:379–385. 10.1038/nn.2273 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lin W, Kemper A, Dupree JL, Harding HP, Ron D, Popko B (2006) Interferon-gamma inhibits central nervous system remyelination through a process modulated by endoplasmic reticulum stress. Brain 129:1306–1318. 10.1093/brain/awl044 [DOI] [PubMed] [Google Scholar]
- Lin W, Bailey SL, Ho H, Harding HP, Ron D, Miller SD, Popko B (2007) The integrated stress response prevents demyelination by protecting oligodendrocytes against immune-mediated damage. J Clin Invest 117:448–456. 10.1172/JCI29571 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu CJ, Chang J, Lee PH, Lin DY, Wu CC, Jeng LB, Lin YJ, Mok KT, Lee WC, Yeh HZ, Ho MC, Yang SS, Yang MD, Yu MC, Hu RH, Peng CY, Lai KL, Chang SS, Chen PJ (2014) Adjuvant heparanase inhibitor PI-88 therapy for hepatocellular carcinoma recurrence. World J Gastroenterol 20:11384–11393. 10.3748/wjg.v20.i32.11384 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lively S, Schlichter LC (2013) The microglial activation state regulates migration and roles of matrix-dissolving enzymes for invasion. J Neuroinflammation 10:75. 10.1186/1742-2094-10-75 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Loda E, Balabanov R (2012) Interferon regulatory factor 1 regulation of oligodendrocyte injury and inflammatory demyelination. Rev Neurosci 23:145–152. 10.1515/revneuro-2011-068 [DOI] [PubMed] [Google Scholar]
- Mabie PC, Mehler MF, Marmur R, Papavasiliou A, Song Q, Kessler J (1997) Bone morphogenetic proteins induce astroglial differentiation of oligodendroglial–astroglial progenitor cells. J Neurosci 17:4112–4120. 10.1523/JNEUROSCI.17-11-04112.1997 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Macchi M, Magalon K, Zimmer C, Peeva E, El Waly B, Brousse B, Jaekel S, Grobe K, Kiefer F, Williams A, Cayre M, Cayre P (2020) Mature oligodendrocytes bordering lesions limit demyelination and favor myelin repair via heparan sulfate production. Elife 9:e51735. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maña P, Liñares D, Fordham S, Staykova M, Willenborg D (2006) Deleterious role of IFNgamma in a toxic model of central nervous system demyelination. Am J Pathol 168:1464–1473. 10.2353/ajpath.2006.050799 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martinez FO, Gordon S (2014) The M1 and M2 paradigm of macrophage activation: time for reassessment. F1000Prime Rep 6:13. 10.12703/P6-13 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mathieu P, Battista D, Depino A, Roca V, Graciarena M, Pitossi F (2010) The more you have, the less you get: the functional role of inflammation on neuronal differentiation of endogenous and transplanted neural stem cells in the adult brain. J Neurochem 112:1368–1385. 10.1111/j.1471-4159.2009.06548.x [DOI] [PubMed] [Google Scholar]
- McKenzie EA (2007) Heparanase: a target for drug discovery in cancer and inflammation. Br J Pharmacol 151:1–14. 10.1038/sj.bjp.0707182 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miron VE, Boyd A, Zhao JW, Yuen TJ, Ruckh JM, Shadrach JL, van Wijngaarden P, Wagers AJ, Williams A, Franklin RJ, ffrench-Constant C (2013) M2 microglia and macrophages drive oligodendrocyte differentiation during CNS remyelination. Nat Neurosci 16:1211–1218. 10.1038/nn.3469 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nava P, Koch S, Laukoetter MG, Lee WY, Kolegraff K, Capaldo CT, Beeman N, Addis C, Gerner-Smidt K, Neumaier I, Skerra A, Li L, Parkos CA, Nusrat A (2010) Interferon-gamma regulates intestinal epithelial homeostasis through converging beta-catenin signaling pathways. Immunity 32:392–402. 10.1016/j.immuni.2010.03.001 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ning J, Zhao Y, Ye Y, Yu J (2019) Opposing roles and potential antagonistic mechanism between TGF-β and BMP pathways: implications for cancer progression. EBioMedicine 41:702–710. 10.1016/j.ebiom.2019.02.033 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ottum PA, Arellano G, Reyes LI, Iruretagoyena M, Naves R (2015) Opposing roles of interferon-gamma on cells of the central nervous system in autoimmune neuroinflammation. Front Immunol 6:539. 10.3389/fimmu.2015.00539 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Panitch HS, Hirsch RL, Haley AS, Johnson KP (1987) Exacerbations of multiple sclerosis in patients treated with gamma interferon. Lancet 1:893–895. 10.1016/s0140-6736(87)92863-7 [DOI] [PubMed] [Google Scholar]
- Parish CR, Freeman C, Brown KJ, Francis DJ, Cowden WB (1999) Identification of sulfated oligosaccharide-based inhibitors of tumor growth and metastasis using novel in vitro assays for angiogenesis and heparanase activity. Cancer Res 59:3433–3441. [PubMed] [Google Scholar]
- Ponath G, Park C, Pitt D (2018) The role of astrocytes in multiple sclerosis. Front Immunol 9:217. 10.3389/fimmu.2018.00217 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Popko B, Corbin JG, Baerwald KD, Dupree J, Garcia AM (1997) The effects of interferon-gamma on the central nervous system. Mol Neurobiol 14:19–35. 10.1007/BF02740619 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Remington LT, Babcock AA, Zehntner SP, Owens T (2007) Microglial recruitment, activation, and proliferation in response to primary demyelination. Am J Pathol 170:1713–1724. 10.2353/ajpath.2007.060783 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ren Z, Wang Y, Liebenson D, Liggett T, Goswami R, Stefoski D, Balabanov R (2011) IRF-1 signaling in central nervous system glial cells regulates inflammatory demyelination. J Neuroimmunol 233:147–159. 10.1016/j.jneuroim.2011.01.001 [DOI] [PubMed] [Google Scholar]
- Sabo JK, Aumann TD, Merlo D, Kilpatrick TJ, Cate HS (2011) Remyelination is altered by bone morphogenic protein signaling in demyelinated lesions. J Neurosci 31:4504–4510. 10.1523/JNEUROSCI.5859-10.2011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sadir R, Forest E, Lortat-Jacob H (1998) The heparan sulfate binding sequence of interferon-gamma increased the on rate of the interferon-gamma-interferon-gamma receptor complex formation. J Biol Chem 273:10919–10925. 10.1074/jbc.273.18.10919 [DOI] [PubMed] [Google Scholar]
- Sanderson RD, Elkin M, Rapraeger AC, Ilan N, Vlodavsky I (2017) Heparanase regulation of cancer, autophagy and inflammation: new mechanisms and targets for therapy. FEBS J 284:42–55. 10.1111/febs.13932 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Saraswat D, Shayya HJ, Polanco JJ, Tripathi A, Welliver RR, Pol SU, Seidman RA, Broome JE, O'Bara MA, van Kuppervelt TH, Phillips JJ, Dutta R, Sim FJ (2020) Overcoming the inhibitory microenvironment surrounding oligodendrocyte progenitor cells following demyelination. bioRxiv. doi: 10.1101/2020.01.21.906073. 10.1101/2020.01.21.906073 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sarrazin S, Bonnaffé D, Lubineau A, Lortat-Jacob H (2005) Heparan sulfate mimicry: a synthetic glycoconjugate that recognizes the heparin binding domain of interferon-gamma inhibits the cytokine activity. J Biol Chem 280:37558–37564. 10.1074/jbc.M507729200 [DOI] [PubMed] [Google Scholar]
- Sim FJ, Lang JK, Waldau B, Roy NS, Schwartz TE, Pilcher WH, Chandross KJ, Natesan S, Merrill JE, Goldman SA (2006) Complementary patterns of gene expression by human oligodendrocyte progenitors and their environment predict determinants of progenitor maintenance and differentiation. Ann Neurol 59:763–779. [DOI] [PubMed] [Google Scholar]
- Sim FJ, McClain CR, Schanz SJ, Protack TL, Windrem MS, Goldman SA (2011) CD140a identifies a population of highly myelinogenic, migration-competent and efficiently engrafting human oligodendrocyte progenitor cells. Nat Biotechnol 29:934–941. 10.1038/nbt.1972 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sofroniew MV, Vinters HV (2010) Astrocytes: biology and pathology. Acta Neuropathol 119:7–35. 10.1007/s00401-009-0619-8 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Steinman L (2001) Blockade of gamma interferon might be beneficial in MS. Mult Scler 7:275–276. 10.1177/135245850100700501 [DOI] [PubMed] [Google Scholar]
- Takahashi H, Matsumoto H, Smirkin A, Itai T, Nishimura Y, Tanaka J (2008) Involvement of heparanase in migration of microglial cells. Biochim Biophys Acta 1780:709–715. 10.1016/j.bbagen.2007.12.014 [DOI] [PubMed] [Google Scholar]
- Takano S, Reichert M, Bakir B, Das KK, Nishida T, Miyazaki M, Heeg S, Collins MA, Marchand B, Hicks PD, Maitra A, Rustgi AK (2016) Prrx1 isoform switching regulates pancreatic cancer invasion and metastatic colonization. Genes Dev 30:233–247. 10.1101/gad.263327.115 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tang Y, Le W (2016) Differential Roles of M1 and M2 Microglia in Neurodegenerative Diseases. Mol Neurobiol 53:1181–1194. 10.1007/s12035-014-9070-5 [DOI] [PubMed] [Google Scholar]
- Tanner DC, Cherry JD, Mayer-Pröschel M (2011) Oligodendrocyte progenitors reversibly exit the cell cycle and give rise to astrocytes in response to interferon-γ. J Neurosci 31:6235–6246. 10.1523/JNEUROSCI.5905-10.2011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tripathi A, Volsko C, Datta U, Regev K, Dutta R (2019) Expression of disease-related miRNAs in white-matter lesions of progressive multiple sclerosis brains. Ann Clin Transl Neurol 6:854–862. 10.1002/acn3.750 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vartanian T, Li Y, Zhao M, Stefansson K (1995) Interferon-gamma-induced oligodendrocyte cell death: implications for the pathogenesis of multiple sclerosis. Mol Med 1:732–743. [PMC free article] [PubMed] [Google Scholar]
- van Horssen J, Bo L, Dijkstra CD, de Vries HE (2006) Extensive extracellular matrix depositions in active multiple sclerosis lesions. Neurobiol Dis 24:484–491. [DOI] [PubMed] [Google Scholar]
- Vergara D, Nigro A, Romano A, De Domenico S, Damato M, Franck J, Coricciati C, Wistorski M, Cardon T, Fournier I, Quattrini A, Salzet M, Furlan R, Maffia M (2019) Distinct Protein Expression Networks are Activated in Microglia Cells after Stimulation with IFN-gamma and IL-4. Cells 8:580. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vlodavsky I, Elkin M, Ilan N (2011) Impact of heparanase and the tumor microenvironment on cancer metastasis and angiogenesis: basic aspects and clinical applications. Rambam Maimonides Med J 2:e0019. 10.5041/RMMJ.10019 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang H-F, Liu X-K, Li R, Zhang P, Chu Z, Wang C-L, Liu H-R, Qi J, Lv G-Y, Wang G-Y, Liu B, Li Y, Wang Y-Y (2017) Effect of glial cells on remyelination after spinal cord injury. Neural Regen Res 12:1724–1732. 10.4103/1673-5374.217354 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang J, Saraswat D, Sinha AK, Polanco J, Dietz K, O'Bara MA, Pol SU, Shayya HJ, Sim FJ (2018) Paired related homeobox protein 1 regulates quiescence in human oligodendrocyte progenitors. Cell Rep 25:3435–3450.e6. 10.1016/j.celrep.2018.11.068 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang J, Wang J, Wang J, Yang B, Weng Q, He Q (2019) Targeting microglia and macrophages: a potential treatment strategy for multiple sclerosis. Front Pharmacol 10:286–286. 10.3389/fphar.2019.00286 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang Y, Ren Z, Tao D, Tilwalli S, Goswami R, Balabanov R (2010) STAT1/IRF-1 signaling pathway mediates the injurious effect of interferon-gamma on oligodendrocyte progenitor cells. Glia 58:195–208. 10.1002/glia.20912 [DOI] [PubMed] [Google Scholar]
- Welliver RR, Polanco JJ, Seidman RA, Sinha AK, O'Bara MA, Khaku ZM, S González DA, Nishiyama A, Wess J, Feltri ML, Paez PM, Sim FJ (2018) Muscarinic receptor M3R signaling prevents efficient remyelination by human and mouse oligodendrocyte progenitor cells. J Neurosci 38:6921–6932. 10.1523/JNEUROSCI.1862-17.2018 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Winkler S, Stahl RC, Carey DJ, Bansal R (2002) Syndecan-3 and perlecan are differentially expressed by progenitors and mature oligodendrocytes and accumulate in the extracellular matrix. J Neurosci Res 69:477–487. [DOI] [PubMed] [Google Scholar]
- Wolswijk G (1998) Chronic stage multiple sclerosis lesions contain a relatively quiescent population of oligodendrocyte precursor cells. J Neurosci 18:601–609. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yong VW, Moumdjian R, Yong FP, Ruijs TC, Freedman MS, Cashman N, Antel JP (1991) Gamma-interferon promotes proliferation of adult human astrocytes in vitro and reactive gliosis in the adult mouse brain in vivo. Proc Natl Acad Sci U S A 88:7016–7020. 10.1073/pnas.88.16.7016 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yu G, Gunay NS, Linhardt RJ, Toida T, Fareed J, Hoppensteadt DA, Shadid H, Ferro V, Li C, Fewings K, Palermo MC, Podger D (2002) Preparation and anticoagulant activity of the phosphosulfomannan PI-88. Eur J Med Chem 37:783–791. 10.1016/s0223-5234(02)01347-8 [DOI] [PubMed] [Google Scholar]
- Yuan G, Zhan Y, Gou X, Chen Y, Yang G (2018) TGF-β signaling inhibits canonical BMP signaling pathway during palate development. Cell Tissue Res 371:283–291. 10.1007/s00441-017-2757-y [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang Y, Yeung MN, Liu J, Chau CH, Chan YS, Shum DK (2006) Mapping heparanase expression in the spinal cord of adult rats. J Comp Neurol 494:345–357. 10.1002/cne.20811 [DOI] [PubMed] [Google Scholar]
- Zhang Y, Chen K, Sloan SA, Bennett ML, Scholze AR, O'Keeffe S, Phatnani HP, Guarnieri P, Caneda C, Ruderisch N, Deng S, Liddelow SA, Zhang C, Daneman R, Maniatis T, Barres BA, Wu JQ (2014) An RNA-sequencing transcriptome and splicing database of glia, neurons, and vascular cells of the cerebral cortex. J Neurosci 34:11929–11947. 10.1523/JNEUROSCI.1860-14.2014 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhu X, Hill RA, Dietrich D, Komitova M, Suzuki R, Nishiyama A (2011) Age-dependent fate and lineage restriction of single NG2 cells. Development 138:745–753. 10.1242/dev.047951 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Two-way ANOVA showing effect of exogenous IFN-γ treatment in WT and IFNγR1 cKO mice and the interactions. Download Figure 2-1, DOCX file (23.7KB, docx) .
IFN-γ treatment of human OPCs alters the transcriptional profile of genes associated with HS synthesis and presentation. Human hOPCs were treated with IFN-γ (10 ng/ml) for 24 h, and RNA-sequencing was performed as described previously (Wang et al., 2018). A heatmap demonstrating the clustering of individual HS-related genes (n = 2 human fetal samples). IFN-γ treatment had a profound effect on the hOPC heparanome. The side bar indicates statistical significance—upregulated (red) and downregulated (blue) genes (q < 0.05). Expression values for each gene were centered and scaled to facilitate visualization (z-score). Download Figure 3-1, TIF file (1.5MB, tif) .
Two-way ANOVA showing activation of STAT1 GAS luciferase by IFN-γ and PI-88 treatment and the interactions. Download Figure 3-2, file (2.5MB, tif) .
Increased Prrx1 expression in Gfap+ astrocytes following IFN-γ treatment after demyelination. RNAscope in situ hybridization (ISH) and IHC of mouse spinal cord after demyelination revealed increased Prrx1 expression by astrocytes but not Iba1+ microglial cells in lesions as assessed by immunohistochemistry of Gfap and Iba1 and Prrx1 mRNA transcripts. Arrow indicates colocalization of Prrx1 and Gfap. Scale bar, 20 μm. Download Figure 3-3, DOCX file (23.7KB, docx) .
Two-way ANOVA showing activation of STAT1 GAS luciferase in control and SULF2 KO by IFN-γ and PI-88 treatment and the interactions. Download Figure 6-1, DOCX file (23.7KB, docx) .
Two-way ANOVA showing effect of exogenous IFN-γ treatment in WT and Sulf1/2 cKO mice and the interactions. Download Figure 6-2, DOCX file (23.7KB, docx) .
Two-way ANOVA showing effects of heparanase inhibitor (PI-88 and OGT2115)-mediated modulation of IFN-γ signaling effects and the interaction. Download Figure 7-1, DOCX file (23.7KB, docx) .
Two-way ANOVA showing effects of heparanase inhibitor (PI-88)-mediated modulation of IFN-γ signaling effects on hOPC differentiation and the interaction. Download Figure 7-2, DOCX file (23.7KB, docx) .
Two-way ANOVA showing effects of heparanase inhibitor (OGT2155)-mediated modulation of IFN-γ signaling effects on hOPC differentiation and the interaction. Download Figure 7-3, DOCX file (23.7KB, docx) .
Two-way ANOVA showing effects of PI88 treatment in WT and IFNγR1 cKO and the interaction. Download Figure 8-1, TIF file (623.1KB, tif) .
Temporal regulation Wnt/Bmp signaling by IFN-γ signaling in OPCs following demyelination. A, RNAscope in situ hybridization for Wnt and Bmp target genes (Apcdd1/Pdgfra and ID4/Pdgfra, respectively), at 5 and 7 dpl (mean ± SEM, n = 3–6 mice/group). Two-way ANOVA revealed that there was no interaction between IFN-γR1 genotype and the time for Wnt target expression, suggesting that IFN-γ does not influence Wnt signaling via a direct effect on OPCs (p = 0.19). In contrast, we observed a significant interaction of time and IFN-γR1 genotype with regard to Bmp target gene expression, suggesting that IFN-γR1 signaling potentiates Bmp activation in a time-dependent manner (p < 0.01). Holm–Sidak multiple-comparisons post-test revealed a significant effect of IFN-γR1 signaling on both target genes at 7 dpl but no significant effect at 5 dpl (*p < 0.05 and **p < 0.01, respectively). Download Figure 8-2, DOCX file (23.7KB, docx) .