

Abstract
This study evaluates newly diagnosed IgM (6%, N=75/1174) vs. non-IgM light chain amyloidosis (AL) patients. IgM AL patients had lower light chains (12.5 vs. 22.5 mg/dL; p<0.001). Heart (56% vs. 73%, p=0.002) and >1 organ involvement (31% vs. 44%, p=0.02) was less common in IgM AL, while soft tissue and peripheral nerve involvement was more common. t(11;14) was less common (27% vs. 50%, p=0.008) in IgM AL. Rates of MYD88L265P and CXCR4WHIM mutation in IgM AL were 58% (29/50) and 17% (8/46). Diagnosis after hematopathology review in IgM AL was pure plasma cell neoplasm (PPCN) in 23% (16/70), lymphoplasmacytic neoplasm (LPL) in 63% (44/70) patients and other (14%). LPL vs. PPCN groups had distinct genetic abnormalities: t(11;14): 0% (0/18) vs. 60% (9/15), p<0.001; MYD88L265P mutation: 84% (27/32) vs. 0% (0/14), p<0.001; CXCR4 mutation: 29% (8/28) vs. 0% (0/14), p=0.04. Overall survival was shorter in IgM AL when stratified by Mayo 2012 stage; stage 1/2 (59 vs. 125.9 months, p=0.003) and stage 3/4 (6.5 vs. 12.9 months, p=0.075), likely due to lower hematologic response rates (6-month: 39% vs. 59%, p=0.008). We characterized two subtypes of IgM AL (LPL/PPCN). This can aid in therapeutic decision-making, with treatment directed at the clonal disease.
Keywords: light chain amyloidosis, heavy chain amyloidosis, IgM, Waldenström’s Macroglobulinemia, lymphoplasmacytic lymphoma, fluorescence in-situ hybridization, MYD88 mutation, CXCR4 mutation
INTRODUCTION
IgM associated light chain amyloidosis (AL) accounts for 5–7% of all patients with systemic AL.(1–4) Amongst patients with Waldenström’s Macroglobulinemia (WM)/lymphoplasmacytic lymphoma (LPL), amyloidosis has been reported in 7.5% of patients, with half of the patients having a concurrent diagnosis of both WM/AL.(5) Patients with IgM AL have more soft tissue, lung and peripheral nerve involvement, while cardiac involvement is less common as compared to non-IgM AL patients.(1–4, 6–9) A recent analysis of IgM AL patients suggests that liver and nerve involvement are additional adverse prognostic factors.(9)
Prior studies have enhanced our understanding of the clinical outcomes in IgM AL. However, our knowledge of the underlying disease biology remains limited. For example, the characteristics of the underlying neoplasm as well as the prevalence of chromosomal abnormalities such as t(11;14) and mutations such as MYD88L265P and CXCR4WHIM in this entity remains largely unknown. In systemic AL, which is typically associated with an underlying plasma cell neoplasm, t(11;14) is seen in about 50% of patients, with possible prognostic and therapeutic implications.(10, 11) In contrast, MYD88L265P mutation is found in the majority of patients with WM, with one-third also having CXCR4WHIM mutation. (12) These are both prognostic and predictive of survival.(12, 13)
Our group recently reported a small series of IgM AL patients where MYD88L265P mutation was observed in 71% (10/14) of patients.(14) There have been case-reports of patients with co-existing WM/AL with CXCR4 mutation/wildtype MYD88.(15) However, this information has not yet been reported in large uniform cohorts of IgM AL patients. An enhanced understanding of the clonal cells and genetic abnormalities can support therapeutic decision making, as it remains unclear whether these patients should be treated with approaches similar to WM or AL. This study evaluated disease characteristics and outcomes of patients with IgM vs. non-IgM AL. We also conducted an in-depth assessment of the bone marrow morphology and genetic abnormalities in IgM AL.
METHODS
Patients with newly diagnosed systemic AL seen at our institution from 01/2006 to 12/2015 were included in this institutional review board (IRB) approved study. Patients provided consent for review of medical records for research purposes. Amyloid was confirmed by Congo Red staining, and subtype was assessed by mass spectrometry or immunohistochemistry of amyloid deposits. Patients were classified into the IgM cohort if they had an IgM circulating monoclonal protein. Organ involvement and response were determined by consensus criteria.(16–18) Hematologic response was assessed by consensus criteria for difference in involved and uninvolved free light chains (dFLC).(17) For patients with dFLC < 5 mg/dL, response assessment included evaluating for complete response (CR) or decrease in dFLC<1mg/dL, based on recent data.(19–21) Additionally, in IgM AL patients with a baseline monoclonal protein of at least 0.5 g/dL, response was also assessed by criteria for WM response.(22) Assessment was intention to treat, i.e. patients who had died by the landmark were considered non-responders.
Retrospective pathology review of archived bone marrow biopsies and fat aspirates was performed by an expert hematopathologist (RLK), including evaluation of disease morphology and burden, amyloid distribution, clonality and phenotype of the plasma cells, as differences have been noted in plasma cells from LPL compared with pure plasma cell neoplasms with respect to CD45 and CD19 expression.(23–25) MYD88L265P was routinely incorporated in our clinical practice in late 2014. CXCR4 testing was not done clinically in any of the patients. For patients with cryopreserved bone marrow aspirates (under an IRB approved biobanking protocol), molecular testing was performed for MYD88L265P and CXCR4 mutations. MYD88L265P mutation was tested by allele specific PCR, as previously described.(14) CXCR4 testing was done by bridged nucleic acid (BNA)-enhanced Sanger sequencing. (Supplementary data) Fluorescence in situ hybridization (FISH) testing and mass spectrometry were available historically in some patients, while in others this testing was done on cryopreserved bone marrow aspirates/archival specimens, respectively, as described before and in supplementary data.(26–28)
Analysis was carried out using JMP® 14 (SAS Institute Inc., Cary, NC) statistical software. Chi-Square and Fischer Exact tests were used to carry out univariate analysis for categorical variables and Wilcoxon Rank Sum/Kruskal Wallis for continuous variables. Survival analysis was carried out using the Kaplan-Meier method and Log-Rank test was used to compare survival curves. Overall survival (OS) was defined as the time from diagnosis of systemic AL to death and progression free survival (PFS) was defined as the time from diagnosis of systemic AL to death or progression requiring change in therapy. Cox proportional hazards model was used for multivariate survival analysis. Hazard ratios (HR) with 95% confidence interval (CI) are reported.
RESULTS
IgM AL patients comprised 6% (N=75) of 1174 newly diagnosed AL patients from 2006–15. An antecedent plasmacytic/lymphoplasmacytic disorder was diagnosed in 31% (23/75) of IgM AL patients, including IgM MGUS (70%; 16), smoldering LPL (9%; 2) and previously treated LPL (22%; 5). The median time from diagnosis of prior disorder to IgM AL was 65 months (range 8–157).
Baseline Characteristics:
Table 1 shows the baseline characteristics of patients with IgM vs non-IgM AL. Patients with IgM AL were more likely to be older and less likely to have lambda as involved LC. Median dFLC (12.5 vs. 22.5 mg/dL; p<0.001) and proportion of patients with evaluable dFLC of ≥ 5 mg/dL (69% vs. 87, p<0.001) was lower in the IgM AL cohort. IgM AL patients were less likely to have t(11;14) (27% vs. 50%, p=0.008). Trisomies were also less common, though the difference was not statistically significant (11% vs. 24%, p=0.08). There were significant differences in organ involvement as described in Table 1, including a lower cardiac involvement (56% vs. 73%, p=0.002),fewer patients with Mayo 2012 stage 3 or 4 disease (40% vs. 55%, p=0.03) and less frequent multi-organ involvement (heart, kidney liver; 31% vs. 44%, p=0.02 ) in the IgM AL group.
Table 1:
Baseline characteristics of patients with IgM vs. non-IgM associated AL amyloidosis
| Characteristic | IgM (N=75) N (%) or median (IQR) |
Non-IgM (N=1099) N (%) or median (IQR) |
P-value |
|---|---|---|---|
| Males | 57 (76) | 710 (65) | 0.04 |
| Age at diagnosis, years | 69 (63–74) | 64 (57–71) | <0.001 |
| iFLC- Lambda | 49 (65) | 820 (76) | 0.048 |
| BM clonal cells % | 10 (5–27) | 10 (5–15) | 0.07 |
| Serum M-protein, g/dL | 1 (0.5–1.5) | 0 (0 – 0.6) | <0.001 |
| dFLC, mg/dL | 12.5 (3.2 – 36.2) | 22.5 (8.6–59.1) | <0.001 |
| dFLC ≥ 5 mg/dL | 48 (69), N=70 | 923 (87), N=1067 | <0.001 |
| Troponin-T, ng/mL | 0.01 (0.01–0.06) | 0.03 (0.01–0.08) | 0.08 |
| NTProBNP, pg/mL | 1576 (223–3907) | 2486 (490–7446) | 0.006 |
| GFR, ml/min/m2 | 69 (40–84) | 62 (43–78) | 0.34 |
| 24 hour urine protein, mg | 690 (162–5099) | 1142 (185–5256) | 0.65 |
| Alkaline phosphatase, IU/mL | 86 (69–113) | 90 (70–134) | 0.20 |
| Mayo 2012 stage (%) | 22/13/13/13, N=61 (36/21/21/21) | 217/217/248/291, N=973 (22/22/26/30) | 0.10 |
| Mayo 2004 Stage (%) | 18/22/21, N=61 (26/25/19) | 184/370/431, N=985 (19/38/44) | 0.12 |
| Renal stage | 36/16/12, N=64 (56/25/19) | 548/392/105, N=1045 (52/38/10) | 0.04 |
| FISH, trisomy/tetrasomy | 4/35 (11) | 152/648 (24) | 0.08 |
| FISH, t(11;14) | 9/34 (27) | 320/647 (50) | 0.008 |
| Organ involvement | |||
| Heart | 42 (56) | 806 (73) | 0.002 |
| Renal | 39 (52) | 630 (57) | 0.37 |
| Liver | 8 (11) | 182 (17) | 0.16 |
| Gastrointestinal | 15 (20) | 267 (24) | 0.39 |
| Autonomic nervous system | 9 (12) | 136 (12) | 0.92 |
| Peripheral nerve | 24 (32) | 169 (15) | <0.001 |
| Soft tissue | 30 (40) | 222 (20) | <0.001 |
| Lung | 5 (7) | 31 (3) | 0.07 |
| >1 major organ (heart/kidney/liver) | 23 (31) | 488 (44) | 0.02 |
abbreviations: BM: bone marrow: dFLC: difference in involved and uninvolved free light chains; iFLC: involved free light chain; FISH: fluorescence in-situ hybridization; GFR: glomerular filtration rate
Bone Marrow Morphology:
Archival bone marrow biopsy slides were available for review in 70/75 (93%) of IgM AL patients. The final morphologic diagnosis was a pure plasma cell neoplasm (PPCN type IgM AL) in 23% (16/70) and a lymphoplasmacytic neoplasm (LPL type IgM AL) in 63% (44/70) of patients, which included characteristic LPL in 39% (27/70) of patients and a low grade B-cell lymphoma with plasmacytic differentiation, not further classifiable in 24% (17/70) of patients and other diagnoses in the remaining patients. (Table 2) By flow cytometry, CD19 was positive in 68% (19/25) and CD45 in 58% (14/24) of patients in whom it was tested, which is similar to prior observations in LPL.(23) Congo red staining revealed amyloid in 76% (45/59) of patients in bone marrow biopsy and in 84% (38/45) of patients on fat aspirate. Amyloid typing as was confirmed by mass spectrometry in 79% (59/75) of patients and by immunohistochemistry in 16 patients.
Table 2:
Bone marrow morphology and MYD88 L265P and CXCR4WHIM mutations in IgM AL amyloidosis patients
| n/N (%) | Additional details | |
|---|---|---|
| Morphology | ||
| Pure plasma cell neoplasm | 16/70 (23) | |
| Lymphoplasmacytic lymphoma | 27/70 (39) | LPL type, 44/70 (63%) |
| Low grade BCL with plasmacytic differentiation | 17/70 (24) | |
| Low grade B-cell lymphoma, other | 4/70 (6) | |
| Other | 3/70 (4) | |
| Too few clonal cells to classify | 3/70 (4) | |
| Flow Cytometry | ||
| CD19 (plasma cells), N=25 | 17/25 (68) | |
| CD45 (plasma cells), N=24 | 14/24 (58) | |
| CD5 (B cells), N=31 | 8/31 (26) | |
| CD10 (B cells), N=31 | 1/31 (3) | |
| MYD88 and CXCR4 mutations | ||
| MYD88 L265P mutation | 29/50 (58) | |
| 8/46 (17) | c.1013 C>A p.S338 non-sense mutation, n=2; c.1013 C>G p.S338 | |
| CXCR4 WHIM mutation | non-sense mutation, n=4; c.982dup, p.Ile328Asnfs*16, n=1 c.972dup, p.Ser325Glnfs*19, n=1 | |
| MYD88 L265P mutated/ CXCR4 WHIM wild type | 18/46 (39) | |
| Both MYD88 L265P /CXCR4 WHIM mutated | 7/46 (15) | |
| MYD88 L265P wildtype/CXCR4 WHIM mutated | 1/46 (2) | |
| Both wildtype | 20/46 (44) | |
abbreviations, LPL: lymphoplasmacytic lymphoma
Genomic features:
Overall, abnormal FISH results were seen in 35% (13/37) of IgM AL patients, including CCND1/IGH associated with t(11;14) in 27% (9/34) and trisomies in 11% (4/35), including trisomy 3 (n=2), 9 (n=1) and 17 (n=1). FISH testing was insufficient in eight patients and not done in 30 patients. MYD88L265P mutation was observed in 58% (29/50) and CXCR4WHIM mutation in 17% (8/46) of IgM AL patients. (Table 2) The CXCR4 mutation was an S338 non-sense mutation in 6/8 patients. Data on both MYD88 and CXCR4 mutation was available in 46 patients. In this group, 39% of patients were MYD88 mutated/ CXCR4 wildtype, 15% had both mutations, 2% had only CXCR4 mutation and 44% were wildtype for both. Conventional chromosome analyses were available in 48 IgM patients, including a normal karyotype or isolated Y chromosome deletion in 94% (45/48) of patients. In the remaining three, an abnormal karyotype was seen that included t(6;11) and deletion 9q; trisomy 12 and trisomy 8 in one patient each.
Sub-classification of IgM amyloidosis:
Based on bone marrow morphology, patients with IgM AL were further classified as LPL type (N=44) and PPCN type (N=16), as described above. Table 3 shows the differences between these two groups of patients. Patients with LPL type IgM AL had a higher tumor mass. A majority of the patients in both groups had clonal plasma cells on flow cytometry (87% vs.94%), while clonal B cells were observed predominantly in patients with LPL morphology (90% vs. 14%, p<0.001). Only one patient with PPCN had monoclonal B cells and there were two distinct, unrelated disease processes (plasma cell and lymphoid) in the bone marrow of this patient. This patient was also positive for t(11;14) in plasma cells and did not have MYD88 or CXCR4 mutation. None of the patients in the LPL group (0/18) harbored t(11;14) compared with 60% (9/15) of patients in the PPCN group (p<0.001). Trisomies were seen in 15% vs. 8% of patients respectively, p>0.99. MYD88L265P mutation was observed in 84% (27/32) of patients in the LPL group compared with only 0% (0/14) in the PPCN group. CXCR4 mutation was observed in 29% (8/28) vs. 0% (0/14) of the patients in the LPL vs. PPCN group, p<0.001. Patients in the LPL group were more likely to have cardiac involvement (68% vs. 35%, p=0.02).
Table 3:
Pathologic and clinical features based on morphologic subtypes of IgM AL amyloidosis
| Lymphoplasmacytic lymphoma (LPL) morphology N=44 | Pure plasma cell disorder (PPCD) morphology N=16 | P-value | |
|---|---|---|---|
| BM disease burden at AL diagnosis: <5%/5–10%/>10% | 7/11/26 (16/25/59) | 7/7/3 (41/41/18) | 0.009 |
| Clonal plasma cells by flow cytometry | 32/37 (87) | 15/16 (94) | >0.99 |
| Clonal B cells by flow cytometry | 28/31 (90) | 1/7 (14) | <0.001 |
| FISH | |||
| t(11;14) | 0/18 (0) | 9/15 (60) | <0.001 |
| Trisomies | 3/20 (15) | 1/13 (8) | >0.99 |
| MYD88 and CXCR testing | |||
| MYD88L265P mutation | 27/32 (84) | 0/14 (0) | <0.001 |
| CXCR4 mutation | 8/28 (29) | 0/14 (0) | 0.04 |
| MYD88 L265P mutated/CXCR4 wild type | 16/28 (57) | 0/14 (0) | |
| Both MYD88 L265P /CXCR4 mutated | 7/28 (25) | - | |
| MYD88 L265P wildtype/CXCR4 mutated | 31/28 (4) | - | |
| Both wildtype | 4/28 (14) | 14/14 (100) | |
| Organ involvement | |||
| Cardiac | 30 (68) | 6 (35) | 0.02 |
| Renal | 21 (44) | 11 (65) | 0.23 |
| Liver | 4 (9) | 2 (12) | 0.76 |
| Gastrointestinal | 9 (21) | 3 (18) | 0.80 |
| Autonomic nervous system | 7 (16) | 0 (0) | - |
| Peripheral nerve | 16 (36) | 16 (35) | 0.94 |
| Soft tissue | 20 (46) | 4 (24) | 0.15 |
| Lung | 5 (11) | 0 (0) | - |
abbreviations, BM: bone marrow
Treatment and Response:
Treatment details were known in 64 patients in the IgM amyloid cohort. Significant heterogeneity was observed in the treatment regimens, which are described in Supplementary Table 1. Therapy was autologous stem cell transplant (ASCT) based in 25% (16/64) of patients, with or without induction therapy. The conditioning regimens for transplant were melphalan based (200 mg/m2:10, 140 mg/m2: 3) in the majority (81%, 13) and three patients received BEAM (carmustine, etoposide, cytarabine and melphalan) conditioning. For those not undergoing transplant, rituximab based combinations were used in 39% (25/64) of patients, including bortezomib in six patients. Non-rituximab based treatment was used in 25% (16/64) of patients, including bortezomib in four patients, while 11% of patients (7/64) did not receive any treatment. Overall, 16% of treated patients received bortezomib. Comparatively, the treatment regimens in the non-IgM cohort (N=980) were ASCT based in 29% (280), bortezomib based in 23% (224), alkylator based in 34% (330), while 4% (43) received other regimens and 10% (102) of patients did not receive any treatment. There was no difference in the rates of ASCT (28%, 15/67 and 32%, 281/878) amongst treated patients in the two groups (p=0.53)
In the IgM cohort, hematologic response by dFLC criteria, including new criteria for patients with dFLC < 5 mg/dL were 39% (18/46) at six months and 52% (25/48) as best response. Rates of deep response (at least very good partial response or response in dFLC < 5 mg/dL) were 24% (11/46) at six months and 44% (21/48) as best response. (Supplementary Table 2) Monoclonal protein response was seen in 85% of patients, (28/33; CR:1, VGPR:16; PR: 7, minor response: 4). Thirty-two patients were evaluable by both criteria. All patients (100%, 19/19) who achieved a dFLC response also achieved a monoclonal protein response. Of the 13 patients who did not achieve a dFLC response, 62% (8/13) achieved a monoclonal protein response. Patients who achieved a dFLC response had longer OS (83.4 vs. 8.9 months, p=0.006), including in the LPL morphology sub-cohort (83.4 vs. 6.6 months, p=0.01). Similarly, achievement of monoclonal protein response was associated with longer OS (83.4 months vs. 6.5 months, p <0.001). Although patient numbers were small, we compared outcomes of patients who achieved monoclonal protein response, but not dFLC response (n=8) to those not achieving either response (n=5); median OS was not reached vs. 6.5 months, p=0.004. Median dFLC at start of therapy in these two groups was 7.0 mg/dL and 9.6 mg/dL respectively. (Supplementary Table 3) Hematologic response rates were lower in the IgM cohort compared with non-IgM cohort, both for overall response (39% vs. 59%, p=0.008) and deep response (24% vs. 41%, p=0.02) at six months. Best organ response rates for heart, kidney and liver in the IgM cohort were 35% (6/17), 40% (10/25) and 0% (0/5), respectively. Organ response rates were lower than that observed in the non-IgM amyloidosis cohort (heart: 47%, kidney: 48%, liver: 41%), though the differences were not statistically significant. (Supplementary Table 4)
Treatment in the LPL IgM AL group was ASCT based in 27% (10/37), rituximab based combinations in 47% (18/37, bortezomib, n=5), non-rituximab based in 14% (5/37, bortezomib=2), while 11% (4/37) of patients did not receive any treatment. Treatment received by the PPCN group was ASCT based in 33% (5/15), rituximab based in 20% (3/15, bortezomib=2) and non-rituximab based in 47% (7/15, bortezomib=1). There was no difference in hematologic response rates in the two groups at six months (41%, 11/27 in LPL and 42%, 5/12 in PPCN, p=0.96).
Survival Outcomes:
Median follow up of the entire cohort was 91 months. Median OS in IgM vs. non-IgM amyloidosis was 20.6 months vs. 38.7 months, p=0.11. (Figure 1a) Amongst patients with Mayo 2012 stage 1 and 2, median OS was significantly inferior in the IgM cohort (59 vs. 125.9 months, p=0.003). Amongst patients with Mayo 2012 stage 3 and 4, median OS was 6.5 vs. 12.9 months, p=0.075. Median OS by Mayo 2004 stage stratification in the IgM vs. non-IgM cohort was as follows, stage 1: 83.4 vs. 149.4 months, p=0.02; stage 2: 30.9 vs 68.9 months, p=0.14; stage 3a: 5.5 vs. 20.1 months, p <0.001 and stage 3b: 2.4 vs. 5.2 months, p=0.049. (Supplementary Figure 1) PFS was not significantly different for all patients with IgM vs. non-IgM amyloidosis. However, in the Mayo stage 3 and 4 subgroup, patients with IgM AL had inferior PFS, 4.6 vs. 7.8 months, p=0.047. (Supplementary Figure 2) OS was inferior in patients with liver involvement for the entire AL cohort (9.8 vs. 47.4 months, p<0.001), as well as in patients with IgM AL (2.9 vs. 22.8 months, p<0.001). (Supplementary Figure 3) Not surprisingly, the median OS from amyloid diagnosis in IgM AL patients with previously treated WM (n=5) was lower compared with the remaining 70 patients (6.6 vs. 20.9 months, p=0.15)
Figure 1:
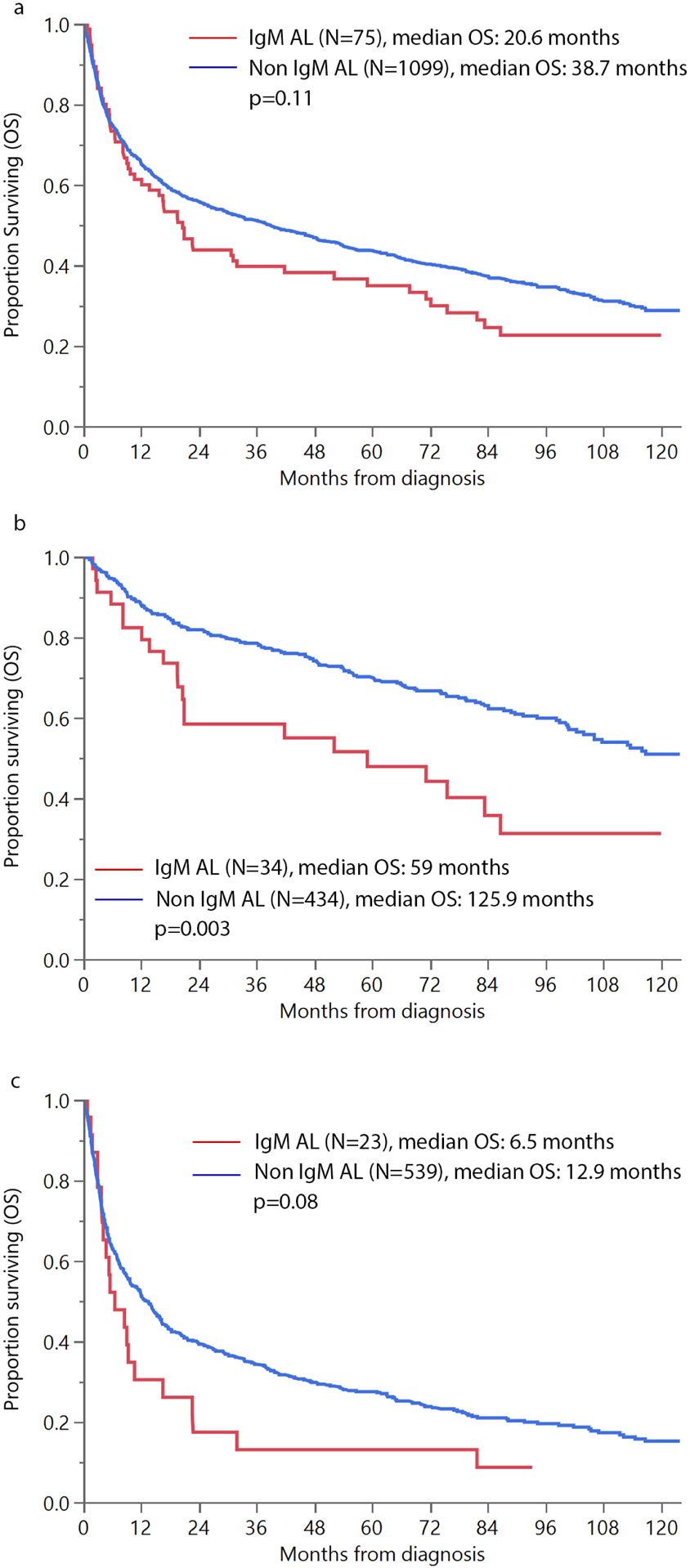
Overall survival (OS) in patients with IgM vs. non IgM light chain amyloidosis (a) all patients (b) Mayo 2012 stage 1 and 2 (c) Mayo 2012 Stage 3 and 4
Figure 2 depicts OS outcomes by morphology and mutation status in the IgM AL. OS in LPL vs. PPCN IgM AL was 20.3 vs. 37.4 months, p=0.27. OS in patients with MYD88 mutation present vs. not was 20.9 vs 52.1 months, p=0.51. Patients with CXCR4 mutation had a trend towards inferior OS, with median OS being 14.5 vs. 59 months, p=0.07. Median OS in patients who harbored either or both mutations vs. those with both wildtype was 32.3 vs. 62.2 months, p=0.73. (Figure 2) Amongst the LPL subtype, median OS in those with MYD88 mutation present vs. not was 19.6 vs. 30.7 months, p=0.96 and CXCR4 mutation present vs. not was 14.5 vs 41.8 months, p=0.22, while either mutation vs. both wild type was 21.9 vs. 53.2 months, p=0.90.
Figure 2:
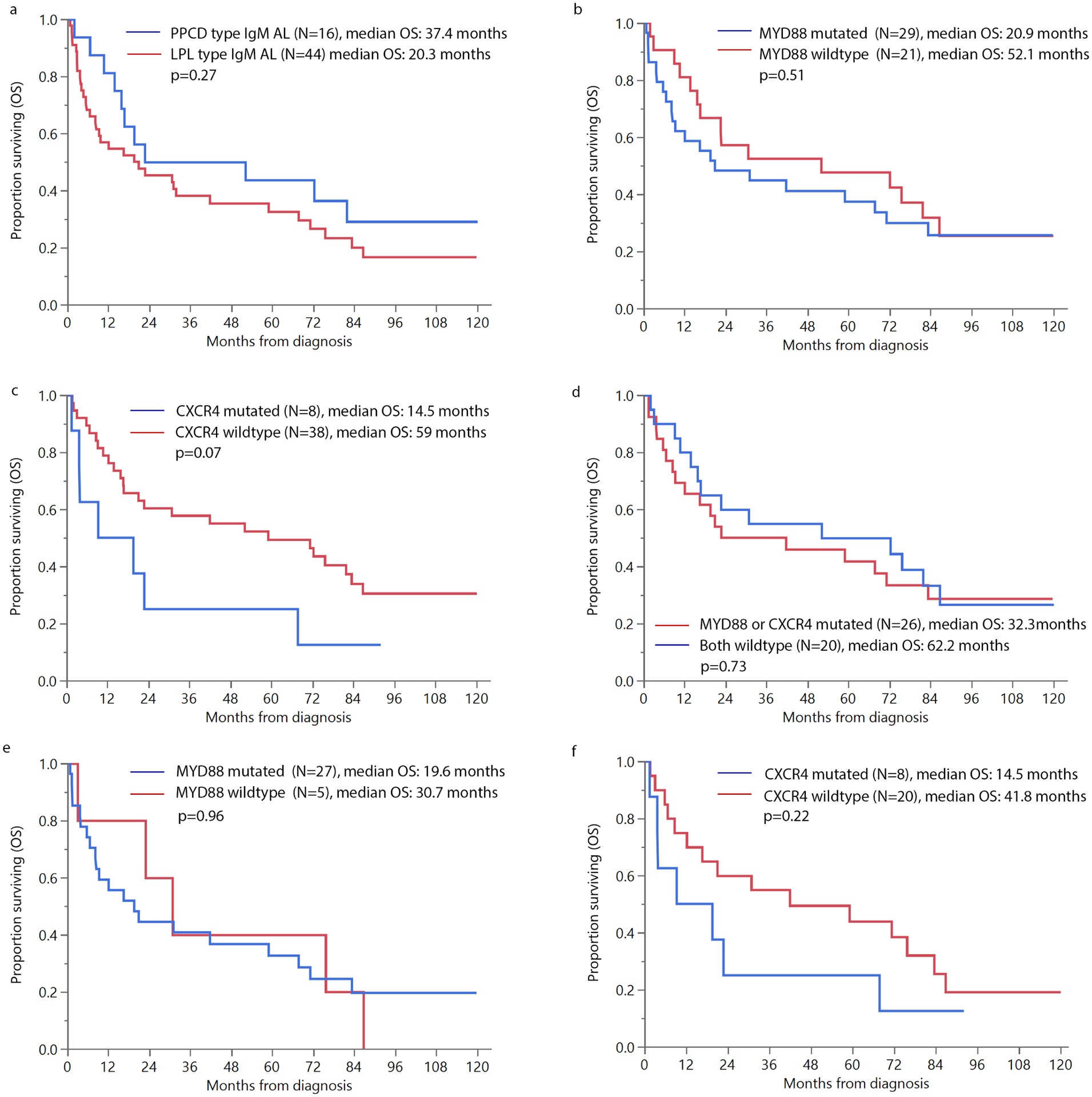
Overall survival (OS) in patients with IgM amyloidosis (a) pure plasma cell disorder (PPCD) vs. lymphoplasmacytic lymphoma (LPL) morphology; (b) MYD88 mutation vs. not; (c) CXCR4 mutation vs. not; (d) CXCR4 and/or MYD88 mutation vs. both wildtype; (e) MYD88 mutation vs. not in LPL subtype IgM AL and (f) CXCR4 mutation vs. not in LPL subtype IgM AL
Table 4 describes multivariate analysis for OS for all AL patients and the IgM AL cohort. Patients with IgM AL had inferior OS (HR: 1.49, 95% CI: 1.04–2.14), even after accounting for age, ASCT and Mayo 2012 stage. This risk remained elevated (HR: 1.55, 95% CI: 1.08 – 2.22, p=0.02) even when accounting for liver involvement, which was an adverse prognostic factor in the entire cohort and sub-cohort of IgM AL patients. However, adjusting for deep hematologic response appeared to mitigate the adverse prognosis seen in IgM AL (HR: 1.32, 95% CI: 0.89–1.97, p=0.17), suggesting that the difference in survival may be explained by the lack of deep hematologic responses in the IgM cohort. Amongst the IgM cohort, liver involvement was a significant adverse prognostic factor (HR 5.89, 95% CI: 2.36–14.7, p <0.001), even after adjusting for Mayo 2012 stage and underlying morphology (PPCN vs. not). The adverse prognosis conferred by liver involvement (HR: 9.93, 95% CI: 2.80–35.1, p <0.001) was also independent of presence of CXCR4 mutation and Mayo stage in another model. We did not observe any difference in survival based on peripheral or autonomic nerve involvement. Hence these variables were not included in multivariate analysis.
Table 4:
Multivariate analysis for overall survival (a) all patients and (b) patients with IgM amyloidosis
| (a) All patients | Univariate HR with 95% CI | Multivariate HR with 95% CI | Multivariate HR with 95% CI | Multivariate HR with 95% CI |
|---|---|---|---|---|
| Model 1 | Model 2 | Model 3 | ||
| IgM vs. Non IgM AL | 1.25 (0.95–1.65), p=0.11 | 1.49 (1.04 – 2.14), p=0.03 | 1.55 (1.08–2.22), p=0.02 | 1.33 (0.89–1.97), p=0.17 |
| Age at diagnosis ≥ 65 years | 1.80 (1.55–2.08), P<0.001 | 1.38 (1.15–1.67), p<0.001 | 1.42 (1.17–1.71), p<0.001 | 1.33 (1.08–1.62), p=0.007 |
| Mayo 2012 Stage: advanced (3/4) vs. early (1/2) | 3.38 (2.85–4.02), P<0.001 | 2.27 (1.85–2.77), p<0.001 | 2.24 (1.82–2.74), p<0.001 | 2.36(1.89–2.96), p<0.001 |
| ASCT as part of therapy | 0.23 (0.18–0.29), P<0.001 | 0.36 (0.28–0.47), P<0.001 | 0.38 (0.29–0.49), p<0.001 | 0.42 (0.32–0.56), p<0.001 |
| Liver involvement | 1.75 (1.46–2.10), p<0.001 | - | 1.42 (1.11–1.78), p=0.005 | - |
| Deep hematologic response at 6 months | 0.24 (0.19–0.30), P<0.001 | - | - | 0.31 (0.24–0.38), p<0.001 |
| (b) IgM AL patients | Univariate HR with 95% CI | Multivariate HR with 95% CI | Multivariate HR with 95% CI | Multivariate HR with 95% CI |
| Model 1 | Model 2 | Model 3 | ||
| Mayo 2012 Stage: advanced (3/4) vs. early (1/2) | 2.86 (1.54–5.30), p<0.001 | 2.93 (1.53–5.61), p=0.001 | 3.19 (1.70–6.00), P<0.001 | 2.68 (1.06–6.80), p=0.04 |
| Liver involvement vs. not | 4.51 (2.04–9.98), p<0.001 | 5.89 (2.36 –14.70), p<0.001 | 5.41 (2.19 – 13.35), p<0.001 | 9.93 (2.80 –35.1), p<0.001 |
| PPCD vs. not | 0.72 (0.37–1.39), p=0.32 | 0.71 (0.34–1.51), p=0.37 | - | - |
| CXCR4 mutation vs. wildtype | 2.14 (0.91–4.97), p=0.08 | - | - | 1.36 (0.46–4.04), p=0.58 |
| MYD88 mutated vs. wildtype | 1.25 (0.64 – 2.42), 0.51 | - | - | - |
| Peripheral nerve involvement vs. not | 0.76 (0.43–1.35) P=0.36 | - | - | - |
| Autonomic nerve involvement vs. not | 1.27 (0.60–2.69) | - | - | - |
DISCUSSION
This study provides comprehensive data on patients with IgM amyloidosis, including disease characteristics, outcomes and genomic features. We identified two main subtypes of IgM amyloidosis (LPL-type vs. PPCN-type) with distinct genetic and morphologic features. Patients might be best managed with therapies directed at their underlying disease process.
Concordant with prior reports,(1–4, 6–9) we observed a difference in the pattern of organ involvement in patients with IgM vs. non-IgM AL, with lower rates of heart involvement, but higher likelihood of peripheral nerve and soft tissue involvement, as well as a trend towards more lung involvement. However, despite having lower cardiac involvement and less frequent multi-organ involvement, survival outcomes were not better in IgM AL patients. In fact, after adjusting for cardiac biomarkers and dFLC, patients with IgM AL had inferior OS. This difference is likely attributable to the lower hematologic response rates seen in IgM AL patients, especially deep hematologic responses. In a small proportion (7%, n=5) of patients, a prior diagnosis of treated WM may have contributed to the poor OS outcomes. The median OS for all IgM AL patients in our study was 20.6 months, which is lower than the median OS of 47–76 months observed in other series.(3, 4, 7, 9) Cardiac involvement was more common in our series (56%) compared with most prior reports (35% – 45%), which might explain the differences in survival.(3, 4, 7, 9) In contrast, cardiac involvement was similar (53%) in the series published by Palladini et al.(3) However, the severity of cardiac involvement cannot be compared as data on Mayo stage is not available from that study. Similar to prior reports, we observed that IgM AL patients are less likely to have lambda LC involvement and only two-thirds are evaluable by the current dFLC response criteria. However, the majority of patients could be evaluated by additional criteria proposed for patients with low dFLC disease.(19–21)
Our study confirms that liver involvement confers an adverse prognosis in all AL patients, including IgM AL patients as reported by the European collaborative study.(9) However, unlike the European cohort, autonomic or peripheral nerve involvement was not observed to be an adverse prognostic factor. Organ response rates in our IgM cohort were low, which is similar to prior reports(9) and likely secondary to low rates of deep hematologic response.
A novel feature of our study is the comprehensive review of archival bone marrow in a large uniform cohort of consecutive IgM AL patients. Additionally, this study provides data on BM plasma cell FISH abnormalities and prevalence of MYD88 and CXCR4 mutations. Prior to this, data on MYD88 mutation in IgM AL is available from a case series of 14 patients (71% mutated) from our group and data on MYD88 (5/7 mutated) and CXCR4 (4/4 wildtype) mutations in a few previously treated patients with WM/AL receiving ibrutinib.(29) Based on our findings, we classified patients with IgM AL into two main groups, PPCN and LPL type. Patients in the PPCN group exhibited genetic changes comparable to those with non-IgM amyloidosis, with prevalence of t(11;14) being 60% vs. 50%, respectively and none of these patients had MYD88 or CXCR4 mutations. MYD88 mutation was seen in the majority of patients with LPL morphology and CXCR4 mutation was seen in a third of such patients, which is similar to observations in WM. (30)(12)
Treatment in our cohort was heterogeneous and this is similar to prior reports.(1, 3, 4, 8, 9) We suggest that the underlying disease morphology and genetic changes should be carefully reviewed at diagnosis and treatment should be directed at the underlying clonal disease. Unlike WM, where even modest reductions in monoclonal protein can provide benefit, patients with AL amyloidosis require a deep response to eliminate amyloidogenic light chains, which is the best predictor of organ improvement.(17, 18, 31, 32) Therefore, combination regimens to achieve a deep response are needed in patients with IgM AL. For patients with PPCN morphology, treatment approaches similar to non-IgM AL can be undertaken. For LPL morphology patients, we suggest that treatment should include a combination of rituximab, an alkylating agent and bortezomib to target both the lymphoid and plasmacytic components of the disease. Response rates of 78% have been observed with rituximab, bortezomib and dexamethasone and 59% with rituximab and bendamustine in IgM AL. In a recent report, ibrutinib was associated with low response rates in IgM AL, with response seen in 2 of the 8 patients.(29) Phase II studies with rituximab, bortezomib and bendamustine in low grade lymphomas (including LPL) and mantle cell lymphoma have showed high response rates of 88–94%, including CR rates of 53– 64%.(33, 34) No data is available with this approach in IgM amyloidosis and this regimen should be investigated in LPL type IgM AL. One challenge with the use of bortezomib is that a third of IgM AL patients have peripheral neuropathy. ASCT has led to excellent and durable responses in IgM AL, with a recent report from our group demonstrating a response rate of 92% and data from Boston University showing a 100% response rate.(8, 35, 36) However, only a fraction of patients with amyloidosis are transplant eligible. For transplant eligible patients, conditioning regimens in patients with LPL morphology should be similar to those used for WM, such as BEAM.(37, 38) Treatment duration should be guided by achievement of a deep hematologic response. As shown by our data and data from the European collaborative study, change in both FLC and monoclonal protein can be used to assess response in these patients. We suggest using both monoclonal protein and FLC level, especially in those with an LPL morphology, recognizing that some patients may only be evaluable by one of the parameters.
Our study has some limitations. It is a retrospective study and data on treatment outcomes is not available for some patients. However, given the rarity of this disease, it is difficult to conduct a prospective study in this cohort. The advantage of our study is the long-term follow up of a uniform cohort of patients, including a contemporaneous control group of non-IgM AL patients and review of bone marrow morphology to characterize the underlying disease and genetic testing on cryopreserved biospecimens.
In conclusion, patients with IgM AL have inferior outcomes compared with non-IgM AL, which may be attributed to lower hematologic response rates. We have characterized two different subtypes in this disease based on morphology (LPL and PPCN type), with distinct genetic features. Treatment should be targeted towards the underlying clone to achieve a deep response, which may help improve outcomes in these patients in the future.
Supplementary Material
ACKNOWLEDGEMENTS:
We would like to acknowledge Ms. Kim Henderson and Ms. Katie Halverson for their assistance in testing stored biospecimens.
Research support/Grant Funding:
Katherine McCleary Research Fund and K. Edward Jacobi Research Partners Fund of the International Waldenström’s Macroglobulinemia Foundation; Amyloidosis Foundation
Footnotes
CONFLICT OF INTEREST:
AD: Research Funding from Celgene, Takeda, Prothena, Jannsen, Pfizer, Alnylam, GSK; PK: Research Funding from Celgene, Takeda. MQL: Research Funding from Celgene; SKK: Research Funding and membership on an entity’s Board of Directors or advisory committees: AbbVie, Celgene, Janssen, KITE, Merck. Membership on an entity’s Board of Directors or advisory committees: Oncopeptides, Takeda. Research funding from Novartis and Roche.; MAG: Honoraria/consultancy from Ionis, Alnylam, Prothena, Celgene, Janssen, Specytrum, Annexon, Apellis, Amgen, Medscape, Abbvie, Research to Practice, Physcians Education Resource and Teva. Remaining authors: None
REFERENCES:
- 1.Gertz MA, Kyle RA, Noel P. Primary systemic amyloidosis: a rare complication of immunoglobulin M monoclonal gammopathies and Waldenstrom’s macroglobulinemia. J Clin Oncol. 1993;11(5):914–20. [DOI] [PubMed] [Google Scholar]
- 2.Gertz MA, Buadi FK, Hayman SR. IgM amyloidosis: clinical features in therapeutic outcomes. Clin Lymphoma Myeloma Leuk. 2011;11(1):146–8. [DOI] [PubMed] [Google Scholar]
- 3.Palladini G, Russo P, Bosoni T, Sarais G, Lavatelli F, Foli A, et al. AL amyloidosis associated with IgM monoclonal protein: a distinct clinical entity. Clinical lymphoma & myeloma. 2009;9(1):80–3. [DOI] [PubMed] [Google Scholar]
- 4.Wechalekar AD, Lachmann HJ, Goodman HJ, Bradwell A, Hawkins PN, Gillmore JD. AL amyloidosis associated with IgM paraproteinemia: clinical profile and treatment outcome. Blood. 2008;112(10):4009–16. [DOI] [PubMed] [Google Scholar]
- 5.Zanwar S, Abeykoon JP, Ansell SM, Gertz MA, Dispenzieri A, Muchtar E, et al. Primary systemic amyloidosis in patients with Waldenstrom macroglobulinemia. Leukemia. 2019;33(3):790–4. [DOI] [PubMed] [Google Scholar]
- 6.Milani P, Merlini G. Monoclonal IgM-related AL amyloidosis. Best practice & research Clinical haematology. 2016;29(2):241–8. [DOI] [PubMed] [Google Scholar]
- 7.Terrier B, Jaccard A, Harousseau JL, Delarue R, Tournilhac O, Hunault-Berger M, et al. The clinical spectrum of IgM-related amyloidosis: a French nationwide retrospective study of 72 patients. Medicine (Baltimore). 2008;87(2):99–109. [DOI] [PubMed] [Google Scholar]
- 8.Sissoko M, Sanchorawala V, Seldin D, Sworder B, Angelino K, Broce M, et al. Clinical presentation and treatment responses in IgM-related AL amyloidosis. Amyloid. 2015;22(4):229–35. [DOI] [PubMed] [Google Scholar]
- 9.Sachchithanantham S, Roussel M, Palladini G, Klersy C, Mahmood S, Venner CP, et al. European Collaborative Study Defining Clinical Profile Outcomes and Novel Prognostic Criteria in Monoclonal Immunoglobulin M-Related Light Chain Amyloidosis. J Clin Oncol. 2016;34(17):2037–45. [DOI] [PubMed] [Google Scholar]
- 10.Muchtar E, Dispenzieri A, Kumar SK, Ketterling RP, Dingli D, Lacy MQ, et al. Interphase fluorescence in situ hybridization in untreated AL amyloidosis has an independent prognostic impact by abnormality type and treatment category. Leukemia. 2017;31(7):1562–9. [DOI] [PubMed] [Google Scholar]
- 11.Bochtler T, Hegenbart U, Kunz C, Granzow M, Benner A, Seckinger A, et al. Translocation t(11;14) is associated with adverse outcome in patients with newly diagnosed AL amyloidosis when treated with bortezomib-based regimens. J Clin Oncol. 2015;33(12):1371–8. [DOI] [PubMed] [Google Scholar]
- 12.Treon SP, Cao Y, Xu L, Yang G, Liu X, Hunter ZR. Somatic mutations in MYD88 and CXCR4 are determinants of clinical presentation and overall survival in Waldenstrom macroglobulinemia. Blood. 2014;123(18):2791–6. [DOI] [PubMed] [Google Scholar]
- 13.Treon SP, Tripsas CK, Meid K, Warren D, Varma G, Green R, et al. Ibrutinib in previously treated Waldenstrom’s macroglobulinemia. N Engl J Med. 2015;372(15):1430–40. [DOI] [PubMed] [Google Scholar]
- 14.Chakraborty R, Novak AJ, Ansell SM, Muchtar E, Kapoor P, Hayman SR, et al. First report of MYD88 L265P somatic mutation in IgM-associated light-chain amyloidosis. Blood. 2016;127(23):2936–8. [DOI] [PubMed] [Google Scholar]
- 15.Thakral B, Kanagal-Shamanna R. Systemic AL amyloidosis associated with Waldenstrom macroglobulinemia: an unusual presenting complication. Blood. 2016;127(1):168. [PubMed] [Google Scholar]
- 16.Gertz MA, Comenzo R, Falk RH, Fermand JP, Hazenberg BP, Hawkins PN, et al. Definition of organ involvement and treatment response in immunoglobulin light chain amyloidosis (AL): a consensus opinion from the 10th International Symposium on Amyloid and Amyloidosis, Tours, France, 18–22 April 2004. American journal of hematology. 2005;79(4):319–28. [DOI] [PubMed] [Google Scholar]
- 17.Palladini G, Dispenzieri A, Gertz MA, Kumar S, Wechalekar A, Hawkins PN, et al. New criteria for response to treatment in immunoglobulin light chain amyloidosis based on free light chain measurement and cardiac biomarkers: impact on survival outcomes. Journal of clinical oncology. 2012;30(36):4541–9. [DOI] [PubMed] [Google Scholar]
- 18.Palladini G, Hegenbart U, Milani P, Kimmich C, Foli A, Ho AD, et al. A staging system for renal outcome and early markers of renal response to chemotherapy in AL amyloidosis. Blood. 2014;124(15):2325–32. [DOI] [PubMed] [Google Scholar]
- 19.Milani P, Basset M, Russo F, Foli A, Merlini G, Palladini G. Patients with light-chain amyloidosis and low free light-chain burden have distinct clinical features and outcome. Blood. 2017;130(5):625–31. [DOI] [PubMed] [Google Scholar]
- 20.Dittrich T, Bochtler T, Kimmich C, Becker N, Jauch A, Goldschmidt H, et al. AL amyloidosis patients with low amyloidogenic free light chain levels at first diagnosis have an excellent prognosis. Blood. 2017;130(5):632–42. [DOI] [PubMed] [Google Scholar]
- 21.Sidana S, Tandon N, Dispenzieri A, Gertz MA, Buadi FK, Lacy MQ, et al. Clinical presentation and outcomes in light chain amyloidosis patients with non-evaluable serum free light chains. Leukemia. 2018;32(3):729–35. [DOI] [PubMed] [Google Scholar]
- 22.Owen RG, Kyle RA, Stone MJ, Rawstron AC, Leblond V, Merlini G, et al. Response assessment in Waldenstrom macroglobulinaemia: update from the VIth International Workshop. British journal of haematology. 2013;160(2):171–6. [DOI] [PubMed] [Google Scholar]
- 23.Rosado FG, Morice WG, He R, Howard MT, Timm M, McPhail ED. Immunophenotypic features by multiparameter flow cytometry can help distinguish low grade B-cell lymphomas with plasmacytic differentiation from plasma cell proliferative disorders with an unrelated clonal B-cell process. British journal of haematology. 2015;169(3):368–76. [DOI] [PubMed] [Google Scholar]
- 24.Morice WG, Chen D, Kurtin PJ, Hanson CA, McPhail ED. Novel immunophenotypic features of marrow lymphoplasmacytic lymphoma and correlation with Waldenstrom’s macroglobulinemia. Modern pathology : an official journal of the United States and Canadian Academy of Pathology, Inc. 2009;22(6):807–16. [DOI] [PubMed] [Google Scholar]
- 25.Shi M, Ternus JA, Ketterling RP, Jevremovic D, McPhail ED. Immunophenotypic and laboratory features of t(11;14)(q13;q32)-positive plasma cell neoplasms. Leuk Lymphoma. 2018;59(8):1913–9. [DOI] [PubMed] [Google Scholar]
- 26.Fonseca R, Blood E, Rue M, Harrington D, Oken MM, Kyle RA, et al. Clinical and biologic implications of recurrent genomic aberrations in myeloma. Blood. 2003;101(11):4569–75. [DOI] [PubMed] [Google Scholar]
- 27.Binder M, Rajkumar SV, Ketterling RP, Dispenzieri A, Lacy MQ, Gertz MA, et al. Occurrence and prognostic significance of cytogenetic evolution in patients with multiple myeloma. Blood Cancer J. 2016;6:e401. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Vrana JA, Gamez JD, Madden BJ, Theis JD, Bergen HR 3rd, Dogan A. Classification of amyloidosis by laser microdissection and mass spectrometry-based proteomic analysis in clinical biopsy specimens. Blood. 2009;114(24):4957–9. [DOI] [PubMed] [Google Scholar]
- 29.Pika T, Hegenbart U, Flodrova P, Maier B, Kimmich C, Schonland SO. First report of ibrutinib in IgM-related amyloidosis: few responses, poor tolerability, and short survival. Blood. 2018;131(3):368–71. [DOI] [PubMed] [Google Scholar]
- 30.Treon SP, Xu L, Yang G, Zhou Y, Liu X, Cao Y, et al. MYD88 L265P somatic mutation in Waldenstrom’s macroglobulinemia. N Engl J Med. 2012;367(9):826–33. [DOI] [PubMed] [Google Scholar]
- 31.Sidana S, Tandon N, Dispenzieri A, Gertz MA, Buadi F, Lacy M, et al. Factors predicting organ response in light chain amyloidosis (AL). Journal of Clinical Oncology. 2017;35(15_suppl):8048-. [Google Scholar]
- 32.Muchtar E, Dispenzieri A, Leung N, Lacy MQ, Buadi FK, Dingli D, et al. Optimizing deep response assessment for AL amyloidosis using involved free light chain level at end of therapy: failure of the serum free light chain ratio. Leukemia. 2019;33(2):527–31. [DOI] [PubMed] [Google Scholar]
- 33.Fowler N, Kahl BS, Lee P, Matous JV, Cashen AF, Jacobs SA, et al. Bortezomib, bendamustine, and rituximab in patients with relapsed or refractory follicular lymphoma: the phase II VERTICAL study. J Clin Oncol. 2011;29(25):3389–95. [DOI] [PubMed] [Google Scholar]
- 34.Flinn IW, Thompson DS, Boccia RV, Miletello G, Lipman A, Flora D, et al. Bendamustine, bortezomib and rituximab produces durable complete remissions in patients with previously untreated, low grade lymphoma. British journal of haematology. 2018;180(3):365–73. [DOI] [PubMed] [Google Scholar]
- 35.Sidiqi MH, Buadi FK, Dispenzieri A, Warsame R, Lacy MQ, Dingli D, et al. Autologous Stem Cell Transplant for IgM-Associated Amyloid Light-Chain Amyloidosis. Biology of blood and marrow transplantation : journal of the American Society for Blood and Marrow Transplantation. 2019;25(3):e108–e11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Gertz MA, Hayman SR, Buadi FK. Transplantation for IgM amyloidosis and IgM myeloma. Clinical lymphoma & myeloma. 2009;9(1):77–9. [DOI] [PubMed] [Google Scholar]
- 37.Marzolini MA, Thomson KJ, Dorman J, D’Sa S. BEAM-conditioned autologous SCT improves the quality of response in Waldenstrom’s macroglobulinaemia and lymphoplasmacytic lymphoma: a single centre’s 10-year experience. Bone marrow transplantation. 2014;49(9):1231–2. [DOI] [PubMed] [Google Scholar]
- 38.Kyriakou C, Canals C, Sibon D, Cahn JY, Kazmi M, Arcese W, et al. High-dose therapy and autologous stem-cell transplantation in Waldenstrom macroglobulinemia: the Lymphoma Working Party of the European Group for Blood and Marrow Transplantation. J Clin Oncol. 2010;28(13):2227–32. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.