
Figure 3.
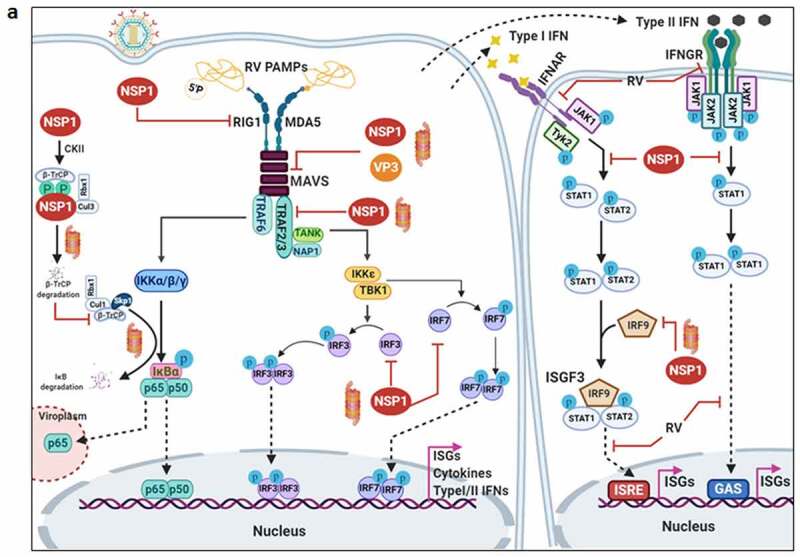
Evasion of host cellular antiviral responses by RV. (a) Evasion of host cellular IFN response. Within RV infected cells, RV RNA species with exposed 5ʹ-phosphate groups or with incomplete 5ʹ-O-methylated “cap” structures act as PAMPs and are recognized by host cellular PRRs RIG1 and MDA5. Subsequent oligomerization of the mitochondrial adaptor MAVS forms a platform for recruitment of TRAFs and two kinase complexes IKK-α/β/γ and TBK1-IKKε leading to the activation of IRF3/7 and NF-κB-dependent transcriptional programme (IFNs, ISGs, cytokines). NF-κB contains two subunits p50 and p65. IKK-α/β/γ phosphorylates NF-κB inhibitory protein IκB at the α subunit resulting in its proteasomal degradation by SCFβ-TrCP (Skp1-Cullin1-F-box containing protein β-TrCP) E3 ubiquitin ligase complex. Being freed of IκB, p50-p65 heterodimeric NF-κB translocates to nucleus and causes transcriptional activation. RV-NSP1 targets many host proteins of this pathway proteasomally (such as MAVS, β-TrCP, IRF3/7, TRAF2) or non-proteasomally (RIG1) and causes their degradation in a RV strain-dependent manner. RV-VP3 has also been shown to target MAVS for proteasomal degradation. Moreover, at least for β-TrCP degradation, a fostering role of hijacked host cellular Cul3-Rbx1 E3 ubiquitin ligase machinery has been implicated. NSP1-β-TrCP interaction also requires CK-II-directed NSP1 phosphorylation reactions. Some RV strains block NF-κB function through sequestration of NF-κB p65 away from nucleus into viroplasmic puncta. The amplification step of IFN response includes binding of IFNs (type I and type II) to cognate receptors (type I IFN receptor IFNAR and type II IFN receptor IFNGR) to activate JAK-STAT signaling. The phosphorylated forms of STAT1 and STAT2 form a complex called ISGF3 by associating with IRF9. This heterotrimeric complex translocates to the nucleus, binds to IFN-stimulated response elements (ISREs) and trans-activates a series of ISGs of cyto-protective and antiviral nature. Homodimeric phosphorylated STAT1 downstream of IFNGR signaling also trans-activates ISGs by nuclear translocation and binding to Gamma interferon activation site (GAS). RV infection curtails IFN amplification pathway by promoting degradation of IRF9, IFN receptors, and by preventing STAT1 phosphorylation as well as STAT1-STAT2 nuclear translocation. RV-NSP1 is responsible for degradation of IRF9 proteasomally and for inhibition of STAT1 phosphorylation. (b) Evasion of host cellular OAS/RNase L pathway by RV. Within virus infected cells, viral dsRNA population can induce oligomerization-dependent activation of the enzyme OAS which further catalyzes the formation of 2ʹ-5ʹAs. Upon interacting with 2ʹ-5ʹAs, RNase L gets activated through dimerization and triggers cleavage of RNA including the viral RNA species. RV evades the deleterious effects of OAS/RNase L pathway by its structural protein VP3. RV-VP3 has intrinsic 2ʹ, 5ʹ-phosphodiesterase (2ʹ-5ʹPDE) motif through which it disintegrates 2ʹ-5ʹA structures, thereby preventing RNase L activation and viral RNA cleavage. (c) Evasion of innate antiviral impacts of host RNAi machinery by RV. Double stranded RV replication intermediates can potentially be subjected to trimming by host cellular DICER resulting in production of virus‐derived small interfering RNAs (viRNAs). Incorporation of viRNAs into the RISC containing the catalytic effector AGO2 can subsequently target viral RNA population. During early hours of RV-SA11 and RV-A5-13 infection, RV-NSP1 interacts with and ubiquitylates AGO2 leading to proteasomal demise of this catalytic effector. In this respect, RV-NSP1 acts as a putative viral‐suppressor‐of‐RNAi (VSR). In absence of AGO2, siRNA/shRNA-guided RNAi and also potential viRNA-directed RNAi are rendered nonfunctional during early hours of RV infection. Clonal overexpression of AGO2 shows anti-RV effects. (d) Attenuation of host cellular anti-oxidant defense system by RV. Nrf2 is the master transcription factor which deals with cellular redox stress by transcribing anti-oxidant and cyto-protective effectors such as HO-1, NQO1, SOD1. Under unstressed condition, Nrf2 is constantly turned over in a ubiquitin-proteasome-dependent way by cellular Keap1-Rbx1-Cul3 machinery. (Left panel) In RV infected cells, reactive oxygen species (ROS) is induced during early hours leading to Keap1 inhibition and Nrf2 upregulation. Elevated Nrf2, further primed by PKC-mediated phosphorylation, translocates to nucleus and trans-activate stress responsive genes which contain Nrf2-binding motif [anti-oxidant response element (ARE)] in their promoter regions. Quenching ROS by NAC has an antagonizing effect on RV infection. (Right panel) During later hours of RV infection, Nrf2 is expelled out of the nucleus, ubiquitylated by a non-canonical E3 ubiquitin ligase (other than the canonical Keap1-Rbx1-Cul3 machinery) and degraded proteasomally. Levels of HO-1, NQO1, and SOD1 were also reduced. Agonists of Nrf2/ARE pathway, such as Keap1 inhibitors CDDO-Me and RA-839, show potent anti-RV effects. (e) Time-dependent regulation of host cellular apoptotic cell death by RV. (Left panel) During early hours of infection, anti-apoptotic pathways are activated and pro-apoptotic pathways are inhibited for ensuring viral replication. The prime most survival pathway includes activation of PI3K-Akt signaling as a result of interaction of RV-NSP1 with PI3K. Interaction of the chaperone Hsp90 with Akt has an agonistic effect on this pathway. Inhibition of survival pathways through targeting PI3K, phospho-Akt and Hsp90 by LY, 294–002, triciribine and 17-AAG, respectively, sensitized RV replication. Inhibition of pro-apoptotic pathways is multifaceted. One of them is the upregulation of the miRNA population hsa-miR-142-5p by RV-NSP5. Elevated hsa-miR-142-5p sensitizes its targets TGFβR II and SMAD3 leading to attenuation of p38MAPK-ERK1/2-JNK-dependent apoptotic signaling in HT29 cell line. Another strategy is the ubiquitylation and proteasomal degradation of p53 during early infection period. RV-NSP1 plays a pivotal role in this regulation. In absence of p53, transcription of p53-dependent apoptotic genes is prevented. Yet another anti-apoptotic modality in early hours of RV infected cells is the prevention of RV-NSP4 translocation to mitochondria. This is enabled by the ubiquitin-proteasome-dependent demise of the mitochondrial chaperonin Hsp60 which facilitates NSP4 mitochondrial import. A phosphorylation event of Hsp60 carried out by the autophosphorylated and activated form of Src kinase (SrcY416) imparts proteasomal sensitivity to Hsp60. Targeting hsa-miR-142-5p by its anti-miR and Src kinase by a small molecule SKI-I exert anti-RV activity. (Right panel) During late phase of infection, apoptotic pathways are activated and/or de-repressed and outweigh the survival pathways. Intrinsic pathway of apoptosis observed in late hours of RV infected cells is partially dependent on Bax. Subsequent release of cytochrome c into cytosol results in apoptosome formation and activation of executioner caspases. Reduced level of hsa-miR-142-5p results in de-repression of TGFβR II-SMAD3-p38MAPK-ERK1/2-JNK-dependent apoptotic signaling in HT29 cell line. Weakened interaction of p53 with RV-NSP1 stabilizes p53 and causes p53-dependent transcription of pro-apoptotic genes (PUMA, Bax. Bak). In absence of SrcY416, Hsp60 is no longer phosphorylated and therefore escorts NSP4 across mitochondria. NSP4 also positively regulates apoptotic mitochondrial fragmentation by promoting Cdk1-dependent phosphorylation of Drp1 at Serine 616 residue and further recruiting them to mitochondria. NSP4 also promotes mitochondrial translocation of Parkin which reduces mitochondrial fusion by degrading Mfn1. Targeting Drp1 and Cdk1 by respective small molecule inhibitors Mdivi-1 and RO-3306 prevented apoptotic mitochondrial fragmentation and viral progeny release. (f) Subversion of host UPR by RV. Accumulation of misfolded proteins in the ER leads to uncoupling of GRP78 from UPR sensors, resulting in activation of the three branches of UPR-ATF6 pathway, PERK-dependent pathway, and IRE1-based signaling. RV activates two (ATF6 and IRE1) of the three branches of UPR, but limits maturation of the activated UPR pathways. Following RV infection, dissociation of GRP78 from ATF6 (ATF6p90; the transcriptionally inactive fragment) triggers translocation of ATF6 to the Golgi apparatus where it is cleaved and the transcriptionally active fragment ATF6p50 is transported to nucleus to trans-activate UPR elements (CHOP, GADD34, GRP78 and GRP94). Despite the initial activation of ATF6 arm of UPR, RV inhibits further transcription of UPR elements by immobilization of the ATF6p50 fragment into viroplasms. UPR element proteins are also sequestered within viroplasms and further synthesis of them is inhibited by NSP3-induced host translational stasis. Release of PERK from GRP78 leads to homo-dimerization and phosphorylation of PERK; however, RV sequesters p-PERK in the viroplasms inhibiting further activation. Uncoupling of GRP78 from IRE1 leads to homo-dimerization and autophosphorylation of IRE1. Phosphorylated IRE1 (p-IRE1) triggers splicing of xbp1 mRNA (xbp1u) to form a spliced variant (xbp1s). However, further translation of the xbp1s is prevented as a result of general host translational inhibition mediated by RV-NSP3. RV also induces IRE1-independent alternative splicing of xbp1 leading to generation of an exon-skipped splice variant (xbp1es). This event of xbp1 alternative splicing was found to concur with NSP3-mediated PABPC1 nuclear translocation. (g) Evasion of antiviral impacts of SGs and PBs and viroplasmic sequestration of host cellular RBPs. Translational shut off because of phosphorylation of eIF2α is a classical trigger for formation of SGs which accumulate many cellular proteins (see figure). PBs and GW bodies also contain protein conglomerates (see figure). In RV infected cells, eIF2α becomes phosphorylated by PKR, but SG formation is prevented. Punctate PB structures are also absent in infected cells. PB component Pan3 and GW body component AGO2 are degraded proteasomally by RV-NSP1. At later hours, AGO2 relocalizes to viroplasmic niches. Other PB/SG/GW body components are also relocated to different subcellular niches such as nuclear compartment (XRN1, hDcp1, DDX6) or viroplasms (ADAR1, Caprin1, CPEB, eIF2α, PKR, Staufen1, PPM1A, LSM1, PARN, GW182, Caf1) or remained dispersed in cytosol (G3BP1, ZBP1). Moreover, many PB/SG components interact with viroplasmic RV-NSP2 and RV-NSP5. Additionally, many hnRNPs and ARE-BPs are re-located from nucleus to cytosol, get sequestered to viroplasms, and interact with RV-NSP2 and RV-NSP5 within RV infected cells. Many relocated RBPs are also absorbed by the copious viral transcripts