

Abstract
The histopathological spectrum of human epileptogenic brain lesions is widespread including common and rare variants of cortical malformations. However, 2–26% of epilepsy surgery specimens are histopathologically classified as nonlesional. We hypothesized that these specimens include also new diagnostic entities, in particular when presurgical magnetic resonance imaging (MRI) can identify abnormal signal intensities within the anatomical region of seizure onset. In our series of 1381 en bloc resected epilepsy surgery brain specimens, 52 cases could not be histopathologically classified and were considered nonlesional (3.7%). An increase of Olig2‐, and PDGFR‐alpha‐immunoreactive oligodendroglia was observed in white matter and deep cortical layers in 22 of these patients (42%). Increased proliferation activity as well as heterotopic neurons in white matter were additional histopathological hallmarks. All patients suffered from frontal lobe epilepsy (FLE) with a median age of epilepsy onset at 4 years and 16 years at epilepsy surgery. Presurgical MRI suggested focal cortical dysplasia (FCD) in all patients. We suggest to classify this characteristic histopathology pattern as “mild malformation of cortical development with oligodendroglial hyperplasia (MOGHE).” Further insights into pathomechanisms of MOGHE may help to bridge the diagnostic gap in children and young adults with difficult‐to‐treat FLE.
Keywords: epilepsy, frontal lobe, hyperplasia, neuropathology, oligodendrocytes
Introduction
Two—26% of epilepsy surgery specimens are histopathologically classified as nonlesional. 1, 11, 30, 34, 45 As seizure control following epilepsy surgery is less successful in magnetic resonance imaging (MRI)‐negative (nonlesional) compared with lesional focal epilepsies, MRI‐positive brain lesions in the anatomical region of seizure onset, which cannot be confirmed by histopathological examination, remain a matter of clinical and scientific debate. 4 The histopathological spectrum of epilepsy‐associated brain lesions is broad covering common and rare entities often difficult to classify due to their variable cellular composition. 5, 7, 9, 10, 31, 33 Seizures are triggered by abnormal neuronal networks and various underlying pathomechanisms, that is, either following selective neuronal cell loss, dysmorphic neuronal phenotypes or aberrant molecular expression patterns. In contrast, the contribution of glial cells to epileptogenic networks is less clear. Advanced research studies discovered pathophysiologic and molecular mechanisms in astroglia with the potential to lower seizure threshold in neuronal networks 2, 40. Only few studies and case reports describe aberrant oligodendroglial cells in focal epilepsies, ranging from oligodendroglial hamartoma 29, to microdysgenesis 25, 26, 27, oligodendroglial hyperplasia 12, 20, increased oligodendroglial cell densities in patients with TLE 24, 39 or diffuse low‐grade brain tumors with an oligodendroglial‐like phenotype. 22 Their impact on seizure generation and development of epiletogenic networks remains ambiguous, as white matter is not considered to trigger seizures.
We reviewed our series of 1381 en bloc resected epilepsy surgery brain specimens to further characterize the oligodendroglial cell population in focal epilepsies hitherto described as histopathologically nonlesional.
Material and Methods
Patient selection, clinical and imaging findings
From January 2003 to December 2013, histopathological examination was performed at our institution in 1381 brain specimens submitted from various epilepsy surgery centers. Specific diagnoses have been confirmed in 1329 cases (96.3%), including hippocampal sclerosis 9, epilepsy‐associated brain tumors 5, malformations of cortical development 10 as well as vascular malformations, glial scars and inflammatory processes. In 3.7% (n = 52), our neuropathology report remained descriptive. Forty‐one cases were obtained from frontal lobe, 8 from temporal lobe, 2 multilobar and 1 from parietal lobe. Forty one percent of these nonlesional cases presented with blurred gray‐white‐matter boundaries due to heterotopic neurons in white matter and increased numbers of subcortical oligodendroglial cells (n = 22). These 22 patients were 16.00 ± 9.72 years old at surgery (median 15.5 years, range 2–49 years), and presented with a mean duration of epilepsy prior to surgery of 11.80 ± 8.25 years, (median = 8.85 years, range 1–36 years; epilepsy onset at 4.3 ± 3.6 years). All patients suffered from intractable focal epilepsies as they failed to achieve sustained seizure freedom following adequate trials of at least two tolerated, appropriately chosen and used antiepileptic drug schedules. 28 Presurgical evaluation with various diagnostic tools including video‐Electroencephalography (EEG)‐monitoring and MR‐imaging localized the epileptogenic area to the left frontal lobe in 10 cases and to the right frontal lobe in 12 cases. High‐resolution MRI with epilepsy‐specific protocols revealed decreased cortico‐medullary differentiation and signal alterations within frontal lobe subcortical white matter in all cases (hyperintense on T2/FLAIR and hypointense on T1) leading to presurgical hypotheses of focal cortical dysplasia (FCD; Figure 1). Patients underwent surgery and we received en bloc tissue specimen originating from the frontal lobe. Post‐operative seizure control was assessed according to Engel or ILAE classification scales one year after surgery. 15, 43 Clinical data are summarized in Table 1.
Figure 1.
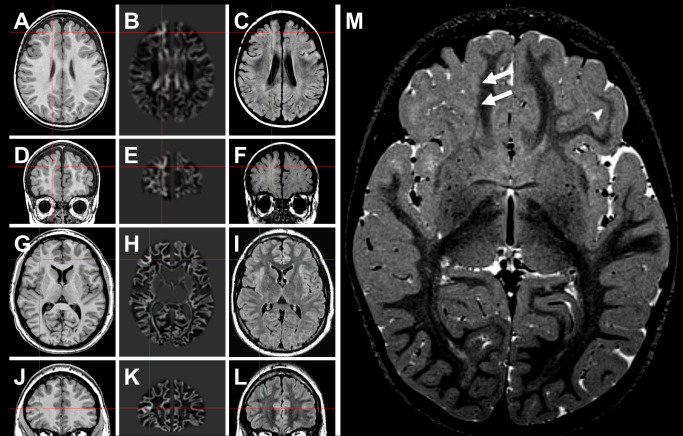
Neuroimaging findings in MOGHE. A–F. Patient #2 (Table 1). G–L. Patient #13 (Table 1). In the latter patient, the lesion became evident only after morphometric MRI analysis (junction image in 2nd column). 23 Red crosses highlight a suspected lesion in T1 (1st column), and postprocessed junction maps (2nd column). 3rd column: FLAIR sequences at same section planes. M. Three‐year old female patient with band‐like signal alteration at the gray‐white matter junction in right frontal lobe (arrows in T2 weighted turbo echo spin image) and histopathologically confirmed MOGHE.
Table 1.
Patients included in this study. Abbreviations: F = female; M = male; MRI = magnetic resonance imaging; n.a. = not available; very right column indicated postoperative seizure outcome according to Engel's classification scheme. 15 Two patients (asterisk in very left column) have been re‐operated in 2014 with outcome data not yet available. Second resection showed neuropathologically remnants of oligodendroglial hyperplasia in these two cases. Antiepileptic drug (AED) medication; CBZ = Carbamazepine; CLB = Clobazam; CNZ = Clonazepam; ESM = Ethosuximide; GBP = Gabapentin; LCM = Lacosamide; LEV = Levetiracetam; LTG = Lamotrigine; MSX = Mesuximide; OXC = Oxcarbazepine; PB = Phenobarbital; PGN = Pregabalin; PHT = Phenytoin; PRM = Primidone; RUF = Rufinamide; STM = Sultiame; TPM = Topiramate; VGB = Vigabatrine; VPA = Valproic Acid; ZNS = Zonisamide.
| Patient ID | Gender | Side of resection | Localization | Onset of epilepsy (years) | Duration of epilepsy (years) | Age at surgery (years) | Diagnosis (MRI) | AED | Post‐Op outcome (Engel) |
|---|---|---|---|---|---|---|---|---|---|
| P1 | M | left | frontal | 13 | 5 | 18 | FCD | CBZ, LEV, LTG, TPM.VPA | I |
| P2 | M | right | frontal | 0,7 | 2,6 | 3 | FCD | ESM, LEV | n.a. |
| P3 | F | right | frontal | 1 | 1 | 2 | FCD | CBZ, CNZ, OXC, LEV, TPM, VGB, VPA | III |
| P4 | M | left | frontal | 1 | 3 | 4 | FCD | n.a. | n.a. |
| P5 | M | right | frontal | 2 | 13 | 15 | FCD | CLB, ESM, OXC, PB, TPM, STM, VGB, VPA | III |
| P6* | F | left | frontal | 0,3 | 9,7 | 10 | FCD | CBZ, LCM, LEV, LTG, MSX, OXC, PHT, STM, TPM, VGB, VPA, ZNS | III |
| P7 | M | left | frontal | 2 | 2 | 4 | FCD | CLB, LEV, LTG, STM, VGB, VPA | II |
| P8 | F | right | frontal | 1 | 18 | 19 | FCD | CBZ, CLB, GBP, LEV, LTG, MSX, OXC, PB, PHT, STM, TPM, VGB, VPA | III |
| P9 | F | right | frontal | 13 | 36 | 49 | FCD | n.a. | n.a. |
| P10 | M | left | frontal | 2 | 5 | 7 | FCD | CLB, LTG, OXC | Ill |
| P11 | M | left | frontal | 1,5 | 15,5 | 17 | FCD | ESM, LEV, OXC, TPM, VPA | III |
| P12 | F | right | frontal | 0,3 | 4,7 | 5 | FCD | CLB, ESM, LEV, LTG, TPM, VPA | I |
| P13* | M | right | frontal | 4 | 23 | 27 | FCD | CLB.LCM, LEV, LTG | III |
| P14 | F | left | frontal | 10 | 8 | 18 | FCD | ESM, LTG | IV |
| P15 | M | right | frontal | 11 | 35 | 46 | FCD | CBZ, LCM, LTG, PB, PHT, PGN, STM, VGB, VPA | III |
| P16 | F | right | frontal | 1 | 3 | 4 | FCD | LEV, TPM, OXC, STM, VGB, VPA | I |
| P17 | M | left | frontal | 5 | 24 | 29 | FCD | CBZ, LCM,LEV,PHT,VPA | I |
| P18 | M | left | frontal | 0,5 | 3,5 | 4 | FCD | LEV, LTG, OXC, STM, VGB, VPA | I |
| P19 | M | left | frontal | 2,5 | 13,5 | 16 | FCD | LTG, VPA | III |
| P20 | M | right | frontal | 12 | 10 | 22 | FCD | LCM, LEV, OXC, ZNS | III |
| P21 | F | right | frontal | 6 | 5 | 11 | FCD | ESM, LEV, OXC, RUF, VPA | n.a. |
| P22 | M | right | frontal | 4 | 19 | 22 | FCD | LEV, OXC, PRM, STM, VGB, VPA | I |
For histopathological group comparison, we selected a series of 10 age‐matched and location‐matched patients suffering from medically intractable FCD ILAE Type I in the frontal lobe, age‐matched temporal lobe epilepsy (TLE; temporal neocortex; n = 10), 5 cases with dysembryoplastic neuroepithelial tumors (DNTs) (WHO I°) in the temporal lobe as well as 2 oligodendrogliomas (WHO II°) in the frontal lobe. In addition, 10 age‐matched and location‐matched autopsy controls without clinical history of epilepsy or any other neurological disorder were included. Informed consent for additional scientific investigations was obtained from all patients’ or next of kin, as approved by the local ethics committee of the University of Erlangen, Bavaria, Germany. All procedures were conducted in accordance with the declaration of Helsinki.
Neuropathological evaluation
After tailored resection, surgical tissue specimens were fixed overnight in 4% formalin and routinely processed into liquid paraffin according to standardized histopathology protocols. All sections were cut at 4 μm with a microtome (Microm, Heidelberg, Germany), mounted on positively charged slides (Superfrost, Germany) and routinely stained with hematoxylin and eosin (H&E) or Nissl‐Luxol‐Fast‐Blue (Nissl‐LFB). Immunohistochemical stainings were performed on selected slides with a semiautomated staining apparatus (Ventana Benchmark, Roche Diagnostics, Mannheim, Germany) using haematoxylin counterstaining. Following antibodies were used according to manufacturer protocols: CD34 (Class II, Clone QBEnd‐10; Dako, Glostrup, Denmark), CD45 (Clones 2b11 + PD7/26; Dako), CD68 (Clone KP1; Dako), CNPase (2′,3′‐cyclic nucleotide 3′ phosphodiesterase, 1:200, clone 11‐5B, Millipore, Temecula, Canada), GFAP (Clone 6F2; Dako), IDH1 R123H (Clone H09; Dianova, Hamburg, Germany), Ki‐67 (Cell Marque, Rocklin, USA), MAP2 [microtubule‐associated protein 2, 1:100, clone c, courtesy of Dr. Riederer; 6], NeuN (Clone A60; Millipore, Temecula, USA), Neurofilament H nonphosphorylated (SMI32; Covance, Princeton, USA), Olig2 (Clone 211F1.1; Millipore), Olig2 (IBL, Gunma, Japan), p53 (clone DO‐7; Dako), PDGFR‐alpha (Cell Signaling, Danvers, USA).
Fluorescence‐immunohistochemistry and in situ hybridization analysis
Double immuno‐fluorescence labeling of Olig2 and Ki‐67 was detected with the above mentioned primary antibodies. Used secondary antibodies were Cy3 (goat anti‐mouse, 1:100; Dianova, Hamburg, Germany) and Alexa Fluor 488 (goat antirabbit; 1:100, Invitrogen, Karlsruhe, Germany). Cellular nuclei have been counterstained with Hoechst 33342 (Sigma‐Aldrich, Schnelldorf, Germany). We used fluorescence‐labeled polynucleotide probes recognizing human specific chromosomes 19q13,19p13 and 1p36,1q25 for in situ hybridization to detect tumor‐specific co‐deletions of chromosomes 1p/19q 13, as supplied by and according to the manufacturer's protocol (ZytoVision GmbH, Bremerhaven, Germany).
Semiquantitative histopathological assessment of oligodendroglial cell densities and proliferation activity
Semiquantitative cell density measurements were performed on 4 μm thin sections using Olig2 and Ki‐67 immunohistochemistry and evaluated with a microcomputer imaging system (ColorView II CCD Camera, Soft Imaging Systems SIS, Stuttgart, Germany) attached to a BX51 Microscope (Olympus, Tokyo, Japan). Olig2‐ and Ki‐67 immunoreactive cells within the subcortical white matter were counted at 20× objective magnification in 16 regions of interest (250 × 250 μm) for Olig2, and in 32 fields for Ki‐67 and expressed as mean numbers of Olig2‐positive cells and Ki‐67‐positive cells per mm2 using AnalySIS imaging software (SIS). PDGFR‐alpha‐immunoreactive cells were counted at 40× object magnification in 10 high power fields and expressed as immunoreactive cells per mm2. Co‐expression of Olig2/Ki‐67 immunoreactivity was quantified with a microcomputer imaging system (ColorView II CCD Camera) attached to an IX70 Microscope using Cell^F imaging software (Olympus). Proliferating (Ki‐67‐positive) Olig2‐immunoreactive cells have been counted at 40× objective magnification in 20 high power fields and expressed as double‐stained cells per mm2.
Statistical analysis
Statistical analyses were performed using SPSS 22 (IBM, IBM SPSS statistics 22, USA). Values of P < 0.05 were considered significant unless otherwise stated. We tested for the presence of significant differences between the cohort of patients with MOGHE and each control group regarding densities of Olig2‐immunoreactive cells/mm2 and Ki‐67‐immunoreactive cells/mm2. A Shapiro‐Wilk test decided on the presence of normal distribution in each group, the null hypothesis was rejected at P < 0.1; the equality of variances was examined using Levene's test and was rejected at P < 0.05. Because of these preliminary tests we chose ANOVA to study the distribution of Olig2‐immunoreactive cells/mm2, contrast analyzes compared MOGHE with each control group; Mann‐Whitney test was considered as a suitable test for the comparison of Ki‐67‐immunoreactive cells/mm2, and again MOGHE was correlated with each control group. Finally, taking into account the problem of multiple comparisons, P‐values were adjusted using the Bonferroni correction. Within the cohort of MOGHE we described relationships between Olig2‐positive cells/mm2, Ki‐67‐positive cells/mm2 and co‐expressing cells/mm2 against the age at surgery and duration of epilepsy with bivariate correlation using Pearson's and Spearman's correlation coefficients.
Results
Neuropathological evaluation
All 22 cases with “blurred gray‐white‐matter boundaries due to heterotopic neurons in white matter and increased numbers of subcortical oligodendroglial cells” described in our neuropathology report (see above) were systematically reviewed. Available surgical tissue specimens were anatomically complete including large areas of cortical gray and subcortical white matter. NeuN immunohistochemistry confirmed a six‐layered architecture of the cortical ribbon without evidence for radial microcolumns or horizontal dyslamination, when analyzed in areas cut perpendicular to the pial surface. Dysmorphic neurons or balloon cells were neither detectable on H&E stains nor when using immunohistochemistry for nonphosphorylated neurofilament proteins (SMI32). Thus, none of these specimens met the criteria for a diagnosis of FCD according to the ILAE classification scheme. 8 Instead, all cases showed foci of gray‐white matter blurring with increased densities of heterotopic neurons subjacent to white matter (Supporting Information Figure S1) but not increased in remote (deep) areas of white matter, defined as 500 μm distant from the cortical junction, as previously reported in several epilepsy conditions. 14, 21, 32, 36, 42 The lack of heterotopic neurons in deep white matter regions excluded also the histopathological diagnosis of mild malformations of cortical development Palmini Type II. 33 We did not observe signs for chronic or acute inflammation (negative for CD45 and CD68 immunolabeling) nor diffuse neuroepithelial tumor infiltration (negative for p53, IDH1, Supporting Information Figure S2). 5 Fluorescence in situ hybridization for the detection of chromosomal 1p/19q losses did not reveal any co‐deletion in the oligodendroglial cell component (Supporting Information Figure S2).
Increased proliferation and oligodendroglial cell densities
A previously reported semiquantitative approach was used to measure Olig2‐immunoreactive cell densities in subcortical white matter. 32 The most striking finding in MOGHE was the increase in Olig2‐immunoreactive oligodendroglial‐like cells in regions with blurred gray‐white‐matter junctions (P < 0.001), compared with our control series of frontal lobe FCD ILAE Type I, temporal neocortex obtained from patients with hippocampal sclerosis (TLE), age‐matched and location‐matched postmortems without any neurological disorder and temporal lobe DNTs (Figures 2 and 3). The increase of oligodendroglial‐like cells in MOGHE was multifocal with patchy areas of diffuse infiltration, thereby distinguishable from previously reported perivascular clustering in TLE specimens. 25, 39 Interestingly, Olig2‐positive cell densities were similar to those observed in low‐grade oligodendrogliomas (WHO II°). We also studied PDGFR‐alpha, a marker for putative oligodendroglial precursor cells to confirm the oligodendroglial lineage of Olig2‐positive cells in MOGHE (Supporting Information Figure S3). Proliferation activity in mature human brain was low with only sporadic nuclear Ki‐67 labeling in surgically resected FCD, TLE or postmortem controls (Figure 3). Proliferation levels were significantly higher in MOGHE (P < 0.001) and reached levels similar to those observed in DNTs. In contrast, proliferation in oligodendrogliomas showed a multifold increase. Double‐immunofluorescence staining identified Olig2‐positive cells as proliferation active in MOGHE (Supporting Information Figure S2). Proliferation activity could not be detected in GFAP‐postive astroglial cells (not shown). We also performed qualitative analysis of the myeloarchitecture using Nissl‐LFB histochemistry as well as CNPase and myelin basic protein immunoreactivity, but were not able to detect a specific pattern indicative of hyper‐ or dysmyelination in MOGHE specimens. Some areas showed rather less myelination and axon fiber densities (Figure 4), but these findings will require systematic electron microscopy for validation.
Figure 2.
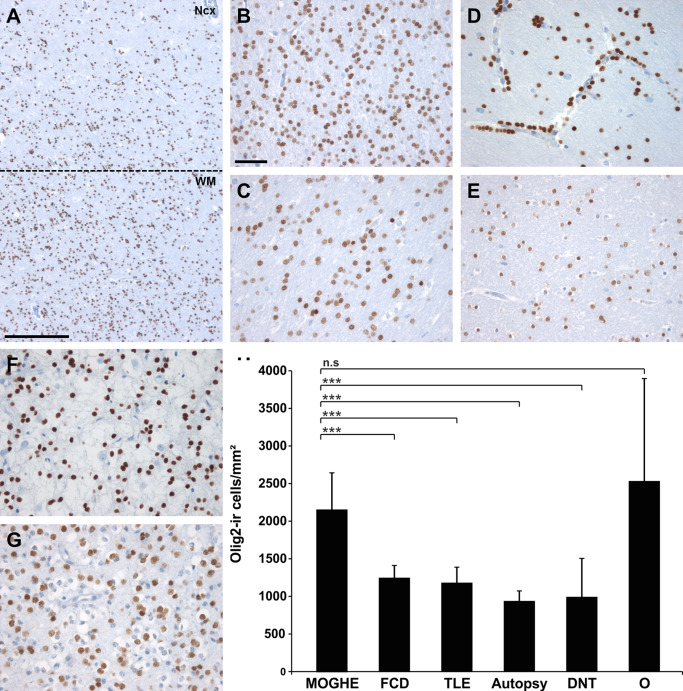
Oligodendroglial cell densities in epilepsy specimens and controls. A. Olig2 immunohistochemistry at the gray (NCx) ‐white matter (WM) junction in MOGHE (dotted line). Scale bar = 500 μm. B–G. higher magnification from white matter regions in different pathology samples and post‐mortem controls (autopsy; E). B. MOGHE; (C) FCD ILAE Type I, (D) temporal lobe epilepsy (TLE) with perivascular clustering of oligodendroglia‐like cells 25, 39; (F) DNT (WHO I°), (G) oligodendroglioma (O; WHO II°). Scale bar in B = 20 μm, applies also to C–G. H. a statistically significant increase was detected for Olig2‐positive cells in MOGHE and oligodendrogliomas (O). *** = P < 0.001, n.s. = not significant.
Figure 3.
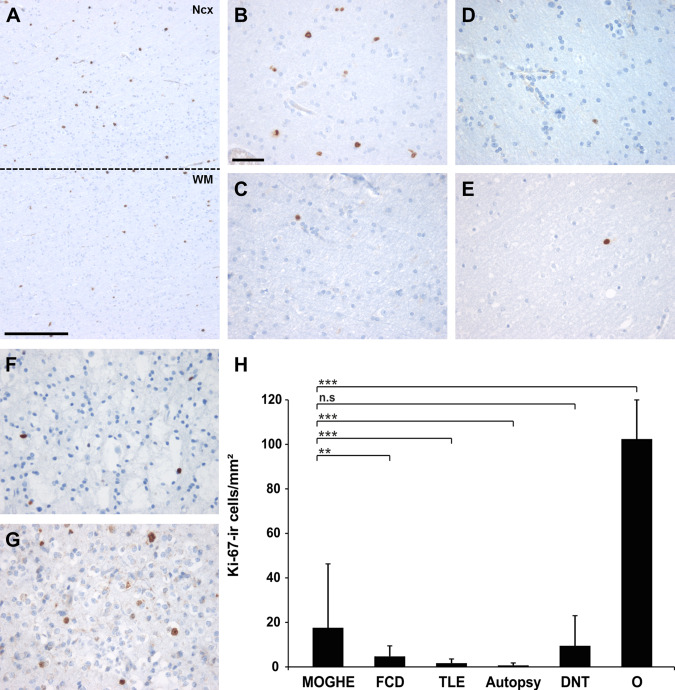
Proliferation activity in epilepsy specimens and controls. A. Ki‐67 immunohistochemistry at gray (NCx) ‐ white matter (WM) junction (dotted line) in MOGHE. Scale bar = 500 μm. B–G. higher magnification from white matter regions in different pathology samples and post‐mortem controls (autopsy; E). B. MOGHE; C. FCD ILAE Type I, D. temporal lobe epilepsy (TLE) with perivascular clustering of oligodendroglia‐like cells; F. DNT (WHO I°), G. oligodendroglioma (WHO II°). Scale bar in B = 20 μm, applies also to C–G. H. A statistically significant increase of Ki‐67 positive cells was detected in MOGHE compared with FCD I, TLE and autopsy controls (** = P < 0.01; *** = P < 0.001). A similar proliferation activity was encountered in DNTs (WHO I°, n.s. = not significant), whereas oligodendrogliomas (O) showed significantly higher proliferation (P < 0.001).
Figure 4.
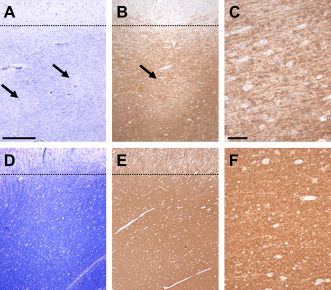
Decreased myelin staining densities in MOGHE. A. Nissl‐LFB staining in MOGHE compared with nonepileptic controls (D) revealed decreased staining intensities in juxtacortical areas (arrows). Dotted lines indicate gray‐white matter boundary. B. CNPase immunoreactivity was also decreased, often occurring with a pachty pattern (arrow). C. higher magnification of area indicated by arrow in B. E/F. a homogeneous distribution of CNPase immunoreactivity was observed in our control series. Scale bar in A: 400 μm, applies also for B,E; Scale bar in C: 50 μm, applies also for F.
Clinico‐pathological correlation
All 22 patients of our series with mMCD and oligodendroglial hyperplasia (MOGHE) suffered from frontal lobe epilepsy (FLE). Epilepsy onset varied from 0.3 to 13 years with a median onset at 2 years. Despite early onset of drug‐resistant seizures, epilepsy surgery was performed after variable time periods of 1–36 years (mean 11.8, median 8.8 years). Although there was no significant correlation between oligodendroglial cell densities and the duration of epilepsy prior to surgery, the proliferation activity was higher in younger patients (Spearman rank test, P < 0.01). This correlation became more evident when calculating the coefficient of total Olig2‐positive cells divided by Olig2 proliferating cells (Spearman rank test, P < 0.01; Figure 5).
Figure 5.
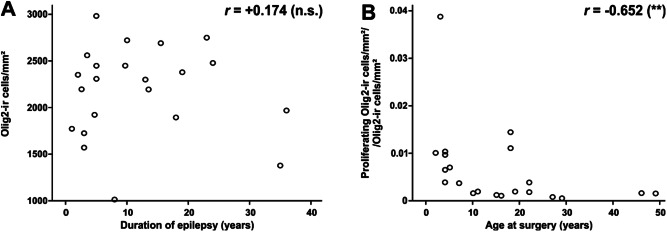
Clinico‐pathological correlation in MOGHE. A. increased oligodendroglial cell densities did not change significantly with duration of epilepsy (Spearman rank test, not significant n.s.). B. significantly higher proliferation activity of Olig2‐positive cells was identified at younger age of operation (Spearman rank test, ** P < 0.01).
Postoperative seizure outcome was available in 18/22 patients. Six patients were free of disabling seizures (33%). One patient presented with rare disabling seizures, whereas the majority (11/18; 61%) of patients presented with ongoing seizures (class III: 10/18 and class IV: 1/18). Due to seizure relapse two patients were reoperated (P6 and P13, asterisk in Table 1). Second resection revealed histopathologically remnants of mMCD with oligodendroglial hyperplasia in both patients.
Discussion
We identified a distinct group of patients suffering from early onset FLE and presenting with a variety of MRI hyperintense brain lesions (presurgically classified as FCD) within the electrophysiologically identified seizure onset zone. All patients were, therefore, considered surgical candidates, but primary histopathology reports classified the specimen as nonlesional. Clustered oligodendroglial hyperplasia with increased proliferative activity in white matter involving the gray‐white matter junction was a common finding when re‐evaluating these surgical specimens.
The diagnostic term of oligodendroglial hyperplasia was first mentioned by Burger in 2002 to define previously described increased oligodendroglial cell densities in brain specimens obtained from epilepsy surgery. 1, 12, 26 The term oligodendroglial hyperplasia was also used by Hamilton and Nesbit to describe increased cell numbers of oligodendrocytes in juxta‐cortical white matter in a patient with FLE and focal cortical thickening in high‐field MRI. 20 Oligodendroglial abnormalities were recognized for many years as perineuronal and perivascular clustering in surgical specimen from patients with temporal lobe epilepsy 1, 26, 37, which were also identified in our series of TLE specimens. These abnormalities were histopathologically distinct from MOGHE (Figure 2) and previously also described as oligodendrogliosis 37, oligodendroglial hamartoma 29 or microdysgenesis, 25, 26, 27, 39, but not included into Palmini's international classification scheme of FCD 33 nor into the ILAE classification of FCDs from 2011. 10 None of the original reports observed or examined proliferation activity of the oligodendroglial cell population in the temporal lobe, and these perivascular oligodendroglial clusters were shown none‐proliferative in our cohort of control samples (Figure 3). Interestingly, the degree of proliferation of oligodendrocytes in the frontal lobe varied in different age groups of our cohort of 22 patients. We detected highest proliferation indices only at young age of operation compared with older patients of our series (Figure 5). This finding suggests either a maldevelopmental or secondary regenerative component 18 in the pathogenesis of these lesions rather than indicating neoplastic transformation.
MOGHE appears as a distinct clinico‐pathologic entity in difficult‐to‐treat FLE, presenting with MRI visible changes. Presurgical evaluation led, therefore, to the hypothesis of an underlying and surgically treatable FCD in all 22 patients. However, seizure outcome was less favorable than expected in these patients. Careful re‐examination of MRI signal changes identified one or more of the following features: (a) frontal lobe localization; (b) involvement of gray‐white matter boundaries, (c) increased signal intensity in FLAIR protocols and (d) presence of multifocal lesions. The latter suggest also a large anatomical extent of MOGHE, and surgical failure may be due to subtotal resections in our patient series.
The interpretation of epileptogenic potential of MOGHE remains challenging, as neither glia cells elicit directly any action potential nor has intracerebral EEG recording so far been able to provide evidence for seizure onset in white matter. In contrast, white matter abnormalities are frequent findings in focal epilepsies including heterotopic neurons, blurred gray‐white matter junctions, myelin and axonal alterations as well as gliosis 17, 32, 35, 38. Glial cells have a huge impact on neuronal network organization and contribute either directly or indirectly to neuronal membrane excitability. This applies most prominently to astroglia 40, which can degrade adenosine, an antiepileptogenic master regulator molecule. 2, 16, 19, 44 In contrast, oligodendroglial cells are a key target in multiple sclerosis and viral CNS infections not associated with chronic seizure disorders. The involvement of this glial cell population in epilepsy awaits, therefore, further clarification.
Our findings promote different scenarios for underlying pathogenic mechanisms. Subgranular cortical layers may be directly compromized by mMCD and oligodendroglial hyperplasia, thereby contributing to aberrant neuronal network activity. Vice versa, sustained (epileptic) network activity may directly trigger oligodendrogliogenesis. Oligodendrogliogenesis and differentiation is indeed promoted by action potential firing via adenosine as axonal signal for differentiation of oligodendrocyte progenitor cells. 41 Implication of neuronal activity could be recently shown also in an experimental animal model using an optogenetic approach to activate neuronal populations. 18 MOGHE areas might then develop secondary to epileptogenic frontal lobe networks when physiologically advancing myelination is compromized by early onset frontal lobe seizures. Indeed, our histochemical and immunohistochemical analysis reveal decreased myelin densities in some affected white matter regions (Figure 4A–C), suggesting that regenerative mechanisms during a compromised myelination period could also play a role.
It remains challenging to include MOGHE into existing clinico‐pathologic or genetic classification schemes. 3 We provisionally classify MOGHE as mild malformation of cortical development due to increased numbers of heterotopic neurons in white matter observed in all cases. However, we are aware of difficulties and controversies adherent with our proposal to assign glial hyperplasia in white matter into the spectrum of cortical dysplasias. 10, 33 Further discussion and research will have to justify terminology use and to clarify the biological nature of this intriguing new disease entity manifesting in FLE.
Supporting information
Additional Supporting Information may be found in the online version of this article at the publisher's web‐site:
Figure S1. Histopathological findings in MOGHE A. In all MOGHE cases, NeuN immunoreactivity showed no evidence for architectural abnormalities of the cortical ribbon. Arrow points to area with close up shown in (B). B. many heterotopic NeuN‐positive neurons can be detected at the gray‐white matter junction of MOGHE regions, and were described as “blurring”. C. numerous oligodendroglial cells at the gray‐white matter junction (H&E staining). Boxed areas magnified in (D) and (E). D. close up of increased oligodendroglial clusters along small blood vessels and neurons of deep cortical layers. E. MOGHE in white matter adjacent to neocortex. F,G. Double‐ fluorescence microcopy for NeuN (in green) and Olig2 (in red). Compared with normal controls (G), the MOGHE patient in (F) showed increased oligodendroglial cell densities as well as more heterotopic white matter neurons with blurring of the gray – white matter junction (dotted line). Scale bar in A = 500 μm. Scale bar in B = 200 μm, applies also to C. Scale bar in D = 50 μm, applies also to E. Scale bar in F = 500 μm, applies also to G.
Figure S2. Proliferative oligodendroglial cells in MOGHE did not share features of oligodendroglial tumors (A) Olig2 immunofluorescence in white matter of MOGHE. B. same Olig2 positive cells express also the nuclear Ki‐67 proliferation epitope (arrows). C. triple channel co‐registration with Hoechst counterstaining of all cellular nuclei. Scale bar in A = 100 μm, applies also to B,C. Immunohistochemical studies for p53 in (D) and IDH‐1 in (E) did not reveal any staining in MOGHE areas. Scale bar in D: 50 μm applies also for E. FISH analysis for chromosomal losses of 1p (F) and 19q (G). Scale bar in F: 5 μm, applies also for G.
Figure S3. Oligodendroglial lineage marker PDGFRα in MOGHE PDGFR‐alpha labeled increased numbers of oligodendrocytic precursor cells in MOGHE (A,C,D), compared with controls (B), Autopsy case shown here). Scale bar in A: 50 μm, applies also for B; Scale bar in C: 20 μm, applies also for (D). E. a statistically significant increase of PDGFRα positive cells was detected in MOGHE compared with FCD I, TLE and autopsy controls (*** = P < 0.001).
Figure S4. GFAP—immunoreactivities in MOGHE compared with other focal epilepsies GFAP—immunoreactivity in MOGHE (A: P1, B: P3, C: P5) showed no distinct presentation compared with autopsy controls (D), TLE (E) or FCD I (F). Scale bar in F: 50 μm, applies also for A–E.
Acknowledgment
This work was supported by the European Union FP7 DESIRE consortium (GA No. 644125). The present work was performed in fulfillment of the requirements for obtaining the doctoral title ‘‘Dr. med.’’ (School of Medicine, Friedrich‐Alexander University Erlangen‐Nuremberg).
The authors declare that they have no conflicts of interest to disclose.
References
- 1. Armstrong DD (1993) The neuropathology of temporal lobe epilepsy. J Neuropathol Exp Neurol 52:433–443. [DOI] [PubMed] [Google Scholar]
- 2. Aronica E, Sandau US, Iyer A, Boison D (2013) Glial adenosine kinase ‐ a neuropathological marker of the epileptic brain. Neurochem Int 63:688–695. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Barkovich AJ, Guerrini R, Kuzniecky RI, Jackson GD, Dobyns WB (2012) A developmental and genetic classification for malformations of cortical development: update 2012. Brain 135(Pt 5):1348–1369. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Bien CG, Raabe AL, Schramm J, Becker A, Urbach H, Elger CE (2013) Trends in presurgical evaluation and surgical treatment of epilepsy at one centre from 1988‐2009. J Neurol Neurosurg Psychiatry 84:54–61. [DOI] [PubMed] [Google Scholar]
- 5. Blumcke I, Aronica E, Urbach H, Alexopoulos A, Gonzalez‐Martinez JA (2014) A neuropathology‐based approach to epilepsy surgery in brain tumors and proposal for a new terminology use for long‐term epilepsy‐associated brain tumors. Acta Neuropathol 128:39–54. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Blumcke I, Becker AJ, Normann S, Hans V, Riederer BM, Krajewski S, Wiestler OD, Reifenberger G (2001) Distinct expression pattern of microtubule‐associated protein‐2 in human oligodendrogliomas and glial precursor cells. J Neuropathol Exp Neurol 60:984–993. [DOI] [PubMed] [Google Scholar]
- 7. Blumcke I, Coras R, Miyata H, Ozkara C (2012) Defining clinico‐neuropathological subtypes of mesial temporal lobe epilepsy with hippocampal sclerosis. Brain Pathol 22:402–411. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Blumcke I, Muhlebner A (2011) Neuropathological work‐up of focal cortical dysplasias using the new ILAE consensus classification system ‐ practical guideline article invited by the Euro‐CNS Research Committee. Clin Neuropathol 30:164–177. [DOI] [PubMed] [Google Scholar]
- 9. Blumcke I, Thom M, Aronica E, Armstrong DD, Bartolomei F, Bernasconi A et al (2013) International consensus classification of hippocampal sclerosis in temporal lobe epilepsy: a Task Force report from the ILAE Commission on Diagnostic Methods. Epilepsia 54:1315–1329. [DOI] [PubMed] [Google Scholar]
- 10. Blumcke I, Thom M, Aronica E, Armstrong DD, Vinters HV, Palmini A et al (2011) The clinico‐pathological spectrum of Focal Cortical Dysplasias: a consensus classification proposed by an ad hoc Task Force of the ILAE Diagnostic Methods Commission. Epilepsia 52:158–174. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Bruton CJ (1988) The neuropathology of temporal lobe epilepsy. In: Maudsley Monographs, Russel G, Marley E, Williams P (eds), pp. 1–158. Oxford University Press: London. [Google Scholar]
- 12. Burger PC, Scheithauer BW, Vogel FS (2002) Surgical Pathology of the Nervous System and its Coverings, 4th edn. Churchill Livingstone: New York, Edinburgh, London, Philadelphia. [Google Scholar]
- 13. Cairncross JG, Ueki K, Zlatescu MC, Lisle DK, Finkelstein DM, Hammond RR et al (1998) Specific genetic predictors of chemotherapeutic response and survival in patients with anaplastic oligodendrogliomas. J Natl Cancer Inst 90:1473–1479. [DOI] [PubMed] [Google Scholar]
- 14. Emery JA, Roper SN, Rojiani AM (1997) White matter neuronal heterotopia in temporal lobe epilepsy: a morphometric and immunohistochemical study. J Neuropathol Exp Neurol 56:1276–1282. [DOI] [PubMed] [Google Scholar]
- 15. Engel J (1987) Outcome with respect to epileptic seizures. In: Surgical Treatment of the Epilepsies, Engel JJ (ed), pp. 553–571. Raven: New York. [Google Scholar]
- 16. Fedele DE, Gouder N, Guttinger M, Gabernet L, Scheurer L, Rulicke T et al (2005) Astrogliosis in epilepsy leads to overexpression of adenosine kinase, resulting in seizure aggravation. Brain 128(Pt 10):2383–2395. [DOI] [PubMed] [Google Scholar]
- 17. Garbelli R, Milesi G, Medici V, Villani F, Didato G, Deleo F et al (2012) Blurring in patients with temporal lobe epilepsy: clinical, high‐field imaging and ultrastructural study. Brain 135(Pt 8):2337–2349. [DOI] [PubMed] [Google Scholar]
- 18. Gibson EM, Purger D, Mount CW, Goldstein AK, Lin GL, Wood LS et al (2014) Neuronal activity promotes oligodendrogenesis and adaptive myelination in the mammalian brain. Science 344:1252304. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Guttinger M, Fedele D, Koch P, Padrun V, Pralong WF, Brustle O, Boison D (2005) Suppression of kindled seizures by paracrine adenosine release from stem cell‐derived brain implants. Epilepsia 46:1162–1169. [DOI] [PubMed] [Google Scholar]
- 20. Hamilton BE, Nesbit GM (2009) MR imaging identification of oligodendroglial hyperplasia. AJNR Am J Neuroradiol 30:1412–1413. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Hildebrandt M, Pieper T, Winkler P, Kolodziejczyk D, Holthausen H, Blumcke I (2005) Neuropathological spectrum of cortical dysplasia in children with severe focal epilepsies. Acta Neuropathol 110:1–11. [DOI] [PubMed] [Google Scholar]
- 22. Honavar M, Janota I, Polkey CE (1999) Histological heterogeneity of dysembryoplastic neuroepithelial tumour: identification and differential diagnosis in a series of 74 cases. Histopathology 34:342–356. [DOI] [PubMed] [Google Scholar]
- 23. Huppertz HJ, Grimm C, Fauser S, Kassubek J, Mader I, Hochmuth A et al (2005) Enhanced visualization of blurred gray‐white matter junctions in focal cortical dysplasia by voxel‐based 3D MRI analysis. Epilepsy Res 67:35–50. [DOI] [PubMed] [Google Scholar]
- 24. Iyer PM, Moroney J, Mullins G, Farrell M, Delanty N (2012) Status of memory loss. BMJ Case Rep. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Kasper BS, Paulus W (2004) Perivascular clustering in temporal lobe epilepsy: oligodendroglial cells of unknown function. Acta Neuropathol (Berl) 108:471–475. [DOI] [PubMed] [Google Scholar]
- 26. Kasper BS, Stefan H, Buchfelder M, Paulus W (1999) Temporal lobe microdysgenesis in epilepsy versus control brains. J Neuropathol Exp Neurol 58:22–28. [DOI] [PubMed] [Google Scholar]
- 27. Kasper BS, Stefan H, Paulus W (2003) Microdysgenesis in mesial temporal lobe epilepsy: a clinicopathological study. Ann Neurol 54:501–506. [DOI] [PubMed] [Google Scholar]
- 28. Kwan P, Arzimanoglou A, Berg AT, Brodie MJ, Allen Hauser W, Mathern G et al (2010) Definition of drug resistant epilepsy: consensus proposal by the ad hoc Task Force of the ILAE Commission on Therapeutic Strategies. Epilepsia 51:1069–1077. [DOI] [PubMed] [Google Scholar]
- 29. Marucci G, Giulioni M, Martinoni M, Volpi L, Michelucci R (2011) Oligodendroglial hamartoma: a potential source of misdiagnosis for oligodendroglioma. J Neurooncol 101:325–328. [DOI] [PubMed] [Google Scholar]
- 30. Mathieson G (1975) Pathologic aspects of epilepsy with special reference to the surgical pathology of focal cerebral seizures. Adv Neurol 8:107–138. [PubMed] [Google Scholar]
- 31. Mischel PS, Nguyen LP, Vinters HV (1995) Cerebral cortical dysplasia associated with pediatric epilepsy. Review of neuropathologic features and proposal for a grading system. J Neuropathol Exp Neurol 54:137–153. [DOI] [PubMed] [Google Scholar]
- 32. Muhlebner A, Coras R, Kobow K, Feucht M, Czech T, Stefan H et al (2012) Neuropathologic measurements in focal cortical dysplasias: validation of the ILAE 2011 classification system and diagnostic implications for MRI. Acta Neuropathol 123:259–272. [DOI] [PubMed] [Google Scholar]
- 33. Palmini A, Najm I, Avanzini G, Babb T, Guerrini R, Foldvary‐Schaefer N et al (2004) Terminology and classification of the cortical dysplasias. Neurology 62(6 Suppl. 3):S2–8. [DOI] [PubMed] [Google Scholar]
- 34. Plate KH, Wieser HG, Yasargil MG, Wiestler OD (1993) Neuropathological findings in 224 patients with temporal lobe epilepsy. Acta Neuropathol 86:433–438. [DOI] [PubMed] [Google Scholar]
- 35. Rodriguez‐Cruces R, Concha L (2015) White matter in temporal lobe epilepsy: clinico‐pathological correlates of water diffusion abnormalities. Quant Imaging Med Surg 5:264–278. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Rojiani AM, Emery JA, Anderson KJ, Massey JK (1996) Distribution of heterotopic neurons in normal hemispheric white matter: a morphometric analysis. J Neuropathol Exp Neurol 55:178–183. [DOI] [PubMed] [Google Scholar]
- 37. Sakuma S, Halliday WC, Nomura R, Ochi A, Otsubo H (2014) Increased population of oligodendroglia‐like cells in pediatric intractable epilepsy. Neurosci Lett 566:188–193. [DOI] [PubMed] [Google Scholar]
- 38. Shepherd C, Liu J, Goc J, Martinian L, Jacques TS, Sisodiya SM, Thom M (2013) A quantitative study of white matter hypomyelination and oligodendroglial maturation in focal cortical dysplasia type II. Epilepsia 54:898–908. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Stefanits H, Czech T, Pataraia E, Baumgartner C, Derhaschnig N, Slana A, Kovacs GG (2012) Prominent oligodendroglial response in surgical specimens of patients with temporal lobe epilepsy. Clin Neuropathol 31:409–417. [DOI] [PubMed] [Google Scholar]
- 40. Steinhauser C, Boison D (2012) Epilepsy: crucial role for astrocytes. Glia 60:1191. [DOI] [PubMed] [Google Scholar]
- 41. Stevens B, Porta S, Haak LL, Gallo V, Fields RD (2002) Adenosine: a neuron‐glial transmitter promoting myelination in the CNS in response to action potentials. Neuron 36:855–868. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Thom M, Sisodiya S, Harkness W, Scaravilli F (2001) Microdysgenesis in temporal lobe epilepsy. A quantitative and immunohistochemical study of white matter neurones. Brain 124(Pt 11):2299–2309. [DOI] [PubMed] [Google Scholar]
- 43. Wieser HG, Blume WT, Fish D, Goldensohn E, Hufnagel A, King D, Sperling MR et al (2001) ILAE Commission Report. Proposal for a new classification of outcome with respect to epileptic seizures following epilepsy surgery. Epilepsia 42:282–286. [PubMed] [Google Scholar]
- 44. Williams‐Karnesky RL, Sandau US, Lusardi TA, Lytle NK, Farrell JM, Pritchard EM et al (2013) Epigenetic changes induced by adenosine augmentation therapy prevent epileptogenesis. J Clin Invest 123:3552–3563. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Wolf HK, Wiestler OD (1993) Surgical pathology of chronic epileptic seizure disorders. Brain Pathol 3:371–380. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Additional Supporting Information may be found in the online version of this article at the publisher's web‐site:
Figure S1. Histopathological findings in MOGHE A. In all MOGHE cases, NeuN immunoreactivity showed no evidence for architectural abnormalities of the cortical ribbon. Arrow points to area with close up shown in (B). B. many heterotopic NeuN‐positive neurons can be detected at the gray‐white matter junction of MOGHE regions, and were described as “blurring”. C. numerous oligodendroglial cells at the gray‐white matter junction (H&E staining). Boxed areas magnified in (D) and (E). D. close up of increased oligodendroglial clusters along small blood vessels and neurons of deep cortical layers. E. MOGHE in white matter adjacent to neocortex. F,G. Double‐ fluorescence microcopy for NeuN (in green) and Olig2 (in red). Compared with normal controls (G), the MOGHE patient in (F) showed increased oligodendroglial cell densities as well as more heterotopic white matter neurons with blurring of the gray – white matter junction (dotted line). Scale bar in A = 500 μm. Scale bar in B = 200 μm, applies also to C. Scale bar in D = 50 μm, applies also to E. Scale bar in F = 500 μm, applies also to G.
Figure S2. Proliferative oligodendroglial cells in MOGHE did not share features of oligodendroglial tumors (A) Olig2 immunofluorescence in white matter of MOGHE. B. same Olig2 positive cells express also the nuclear Ki‐67 proliferation epitope (arrows). C. triple channel co‐registration with Hoechst counterstaining of all cellular nuclei. Scale bar in A = 100 μm, applies also to B,C. Immunohistochemical studies for p53 in (D) and IDH‐1 in (E) did not reveal any staining in MOGHE areas. Scale bar in D: 50 μm applies also for E. FISH analysis for chromosomal losses of 1p (F) and 19q (G). Scale bar in F: 5 μm, applies also for G.
Figure S3. Oligodendroglial lineage marker PDGFRα in MOGHE PDGFR‐alpha labeled increased numbers of oligodendrocytic precursor cells in MOGHE (A,C,D), compared with controls (B), Autopsy case shown here). Scale bar in A: 50 μm, applies also for B; Scale bar in C: 20 μm, applies also for (D). E. a statistically significant increase of PDGFRα positive cells was detected in MOGHE compared with FCD I, TLE and autopsy controls (*** = P < 0.001).
Figure S4. GFAP—immunoreactivities in MOGHE compared with other focal epilepsies GFAP—immunoreactivity in MOGHE (A: P1, B: P3, C: P5) showed no distinct presentation compared with autopsy controls (D), TLE (E) or FCD I (F). Scale bar in F: 50 μm, applies also for A–E.