

Abstract
Peroxisomes are organelles with diverse metabolic tasks including essential roles in lipid metabolism. They are of utmost importance for the normal functioning of the nervous system as most peroxisomal disorders are accompanied with neurological symptoms. Remarkably, the cerebellum exquisitely depends on intact peroxisomal function both during development and adulthood. In this review, we cover all aspects of cerebellar pathology that were reported in peroxisome biogenesis disorders and in diseases caused by dysfunction of the peroxisomal α‐oxidation, β‐oxidation or ether lipid synthesis pathways. We also discuss the phenotypes of mouse models in which cerebellar pathologies were recapitulated and search for connections with the metabolic abnormalities. It becomes increasingly clear that besides the most severe forms of peroxisome dysfunction that are associated with developmental cerebellar defects, milder impairments can give rise to ataxia later in life.
Keywords: ataxia, cerebellum, fatty acid, peroxisome, plasmalogen
Introduction
Peroxisomal function and dysfunction
Peroxisomes are omnipresent in mammalian cells but they have a flexible abundance and content depending on the cell type, developmental and physiological state. Beside peroxide‐generating oxidases and peroxide‐degrading enzymes such as catalase, these organelles harbor several pathways of lipid metabolism 139, 143, 144. Via α‐oxidation they chain shorten the branched‐chain fatty acid phytanic acid taken up via the diet and hydroxy‐fatty acids that are enriched in myelin. Peroxisomal β‐oxidation is not only essential for the degradation of very long‐chain fatty acids (VLCFA) and for the further breakdown of branched‐chain fatty acids, but also for the synthesis of polyunsaturated fatty acids (PUFA) such as docosahexaenoic acid (DHA, C22:6ω‐3) and the conversion of cholesterol into bile acids. Moreover, peroxisomes are required for the synthesis of ether lipids, including plasmalogens that are abundantly present in myelin. In addition, they take part in purine, polyamine, glyoxylate and amino acid metabolism. The ongoing determination of the peroxisomal proteome 58 is unveiling new peroxisomal proteins, which might lead to the identification of new peroxisomal disorders.
Peroxisomal diseases can be categorized in biogenesis disorders (PBD) and single enzyme or transporter defects. PBDs arise when one of 14 PEX genes encoding so‐called peroxins are defective 16. All these proteins are involved in a chain of events that is necessary to import cytosolically synthesized proteins into the peroxisomal membrane or lumen, or for peroxisome fission 150. Peroxisomal proteins that are mislocalized to the cytosol are mostly instable or inactive. Depending on the severity of the PEX mutation, this causes an array of disorders with developmental and/or degenerative pathology currently known as the Zellweger spectrum 16, 124.
Other peroxisomal disorders with neurological pathologies are due to mutations in peroxisomal enzymes or membrane transporters of the above mentioned lipid pathways 145. Although their metabolic deficits are less wide‐ranging, the pathologies also vary from lethality in the postnatal period to neurological demise in adulthood, depending on the affected gene and residual protein function.
We first discuss the cerebellar pathologies in single enzyme diseases as these can be better correlated with the metabolic dysfunction before elaborating on the PBDs. The findings in patients and in the corresponding mouse models are dealt with in parallel.
Cerebellar development and architecture
The cerebellum is involved in the coordination of smooth, purposeful motor activities and in learning new motor skills. Also, a growing body of evidence infers additional cerebellar functions such as cognitive processing (eg, language) and emotional reactivity (eg, fear) 114.
In humans, cerebellar development is a slow process, starting in the embryonic stage and finishing in the first postnatal years, whereas in mice, it occurs postnatally over a 3‐week period. In newborn pups, Purkinje cells (PCs) extend multiple, non‐organized dendrites evolving in a monolayer with primary dendrites branching in a single, parasagittal plane by postnatal day 21. By providing the only projections to the deep cerebellar nuclei and vestibular nuclei, PCs are the sole output neurons of the cerebellar cortex (Figure 1). During the same timeframe, granule cells migrate from the external granule cell layer to the internal granule cell layer and extend their parallel fibers (PF). This process of cell migration is carefully guided by Bergmann glia fibers projecting into the molecular layer. After arrival in the internal granule cell layer, granule cells mature and synaptic contacts are established.
Figure 1.
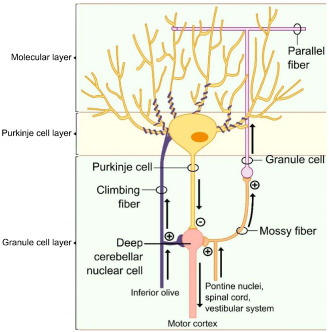
Cerebellar architecture. The cerebellar cortex harbors five cells types. The PCs are the most obtrusive, appearing as an ordered monolayer located in between the inner granular and the outer molecular cell layer. By providing the only projections to the deep cerebellar and vestibular nuclei, these cells are the sole cerebellar output neurons. Mossy fibers arising from the pontine nuclei, spinal cord and vestibular system project into the granule cell layer to establish synaptic contacts on the granule cells. In turn, PFs arising from granule cells project into the molecular layer to make excitatory synapses on the elaborated dendritic tree of the PC. CFs arising from the inferior olive provide the second PC excitatory innervation. The deep cerebellar nuclei are stimulated by mossy fibers and efferents originating from the inferior olive, while PCs provide inhibitory input.
Mature PCs are unique neurons with an elaborate dendritic tree harboring many spines, allowing a multitude of synaptic contacts. It has been shown that extrinsic signals such as normal synaptic input, growth factors, hormones and neurotrophins are necessary for dendritic outgrowth and refinement. Initially, each PC receives excitatory input from multiple climbing fibers (CF), originating from the contralateral inferior olivary nucleus of the medulla. By postnatal day 20, surplus CF are eliminated, resulting in mono innervation. This CF–PC connection is one of the strongest in the central nervous system (CNS). Indirect PC innervation originates from axons arising from the spinal cord and brainstem. These so‐called “mossy fibers” project to the granule cells, which in turn send PF making synaptic contacts with dendritic spines of multiple PCs. Excitatory inputs on PCs are counteracted by basket‐ and stellate cells, both inhibitory interneurons present in the molecular layer. The circuit is closed by inhibitory feedback connections from the deep cerebellar nuclei to the inferior olivary nucleus and an excitatory feedback projection between the cerebellar nuclei and the granule cells (Figure 1). Moreover, neuronal connections between the cerebellum and diverse brain regions such as the basal ganglia, hippocampus, thalamus and hypothalamus should not be overlooked. Given this extensive cerebellar wiring, the aforementioned additional roles in higher order brain functions are not surprising.
Pathologies causing abnormalities in PC development, cerebellar malfunctioning or PC loss generally cause a range of motor disturbances (eg, tremor, dysmetria and hypotonia) collectively referred to as “cerebellar ataxia.” Currently, more than 60 different types of cerebellum‐based ataxias have been identified. In what follows, cerebellar anomalies caused by peroxisomal dysfunction during development and adulthood will be discussed in men and mice.
Defects in α‐Oxidation of Phytanic Acid
Patients lacking the first enzyme of the peroxisomal α‐oxidation pathway, phytanoyl‐CoA hydroxylase (PHYH), suffer from Refsum disease, an inherited disorder resulting in the accumulation of phytanic acid in several tissues and body fluids 148, 152. Although the majority of Refsum patients carry PHYH gene mutations, the disease can also be caused by PEX7 mutations, impairing the import of PHYH into the peroxisome 63, 66, 67, 134 (Figure 2). Patients lacking other enzymes of the peroxisomal α‐oxidation pathway have not been identified. Phytanic acid is derived from chlorophyll by bacterial metabolism in the gut of ruminants and is primarily taken up through ruminant meat and dairy products. Other peroxisome‐related disorders characterized by increased phytanic acid levels are Zellweger spectrum disorders, multifunctional protein 2 (MFP2) deficiency, and rhizomelic chondrodysplasia punctata (RCDP) type 1.
Figure 2.
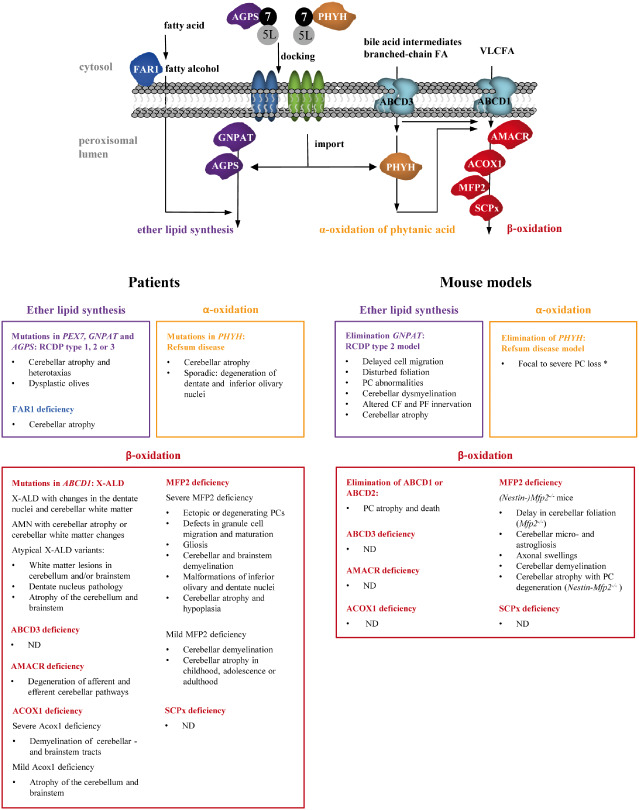
Schematic overview of cerebellar and brainstem pathologies in peroxisomal single enzyme and transporter defects in men and mice. Defects in either of the three major lipid metabolic pathways can cause developmental and/or degenerative pathologies. Ataxia is a common but not obligate clinical presentation. Defects in ether lipid synthesis occur through mutations in peroxisomal enzymes AGPS or GNPAT, or in FAR1 located at the outer peroxisomal membrane that produces fatty alcohols. AGPS needs to be imported through PEX7 (7) that binds the long form of PEX5 (5L) before docking at the peroxisomal membrane. A similar mechanism is involved in the import of PHYH, the first enzyme of peroxisomal α‐oxidation, which functions in the breakdown of the branched‐chain FA phytanic acid reaching the peroxisomal lumen via import through ABCD3. Peroxisomal β‐oxidation defects related to cerebellar dysfunction are caused by mutations in the ABCD1 transporter or in any of the enzymes shown involved in the degradation pathway. Import of these enzymes occurs through PEX5 (see Figure 3). ABCD2 (not depicted), which transports VLCFA and other substrates, was only inactivated in mice. Figure adapted from (150). ND = not documented; * after phytol treatment.
Refsum disease
Cerebellar and brainstem pathologies in Refsum patients
Refsum was the first to describe cerebellar signs as one of the main manifestations of the disease that was named after him, although it is now clear that cerebellar problems can be late in onset compared with retino‐ or neuropathy 147. The clinical presentation of Refsum disease is variable, and cerebellar signs do not always occur 3, 120. Surprisingly, only few post‐mortem histopathological investigations on Refsum patients were performed and findings in the cerebellum and brainstem are scarce, revealing atrophy of the cerebellar vermis and patchy cell degeneration in the dentate and inferior olivary nuclei 22, 55, 106. It should be noted that cerebellar pathology may not be the sole origin of ataxia as gait disturbances without cerebellar pathological changes have been reported 46, 112. The loss of proprioception caused by the demyelinating neuropathy will cause a sensory ataxia on top of the cerebellar ataxia 46, 88. As their gait disturbances ameliorated upon dietary phytanic acid restrictions, this strongly indicated that phytanic acid accumulation induces toxicity 33. Interestingly, phytanic acid concentrations are 3 to 4 times higher in the peripheral nerves of Refsum patients as compared with the brain, caused by impaired access through the blood‐brain barrier 128. Phytanic acid accumulations in the peripheral nervous system slow down nerve conduction velocity, impair reflexes and perturb sensation. The demyelinating polyneuropathy generally starts before the cerebellar dysfunction, making it sometimes clinically difficult to detect the cerebellar ataxia. However, signs like nystagmus cannot be explained by the polyneuropathy, and other signs of ataxia are out of proportion with the degree of sensory involvement 133.
The Refsum mouse model
The in vivo short‐term pathological processes of Refsum disease were studied by Ferdinandusse et al 42, using PHYH‐deficient mutant mice on chow supplemented with phytol. Phytanic acid is derived from dietary sources only and the concentration of branched‐chain fatty acids or their precursors in standard rodent food is low. A moderate increase in the plasma concentration of phytanic acid was obtained when Phyh −/− mice were supplemented with 0.1% phytol for 6 weeks, whereas 3 weeks of 0.25% phytol‐supplementation yielded plasma levels similar to those reported in Refsum disease patients (>1 mmol/L). Phytanic acid markedly accumulated in the cerebellum, although levels were 3–10‐fold lower compared with testis, kidney and liver, in line with the relative impermeability of the blood–brain barrier 128. The phytol diet induced prominent gait disturbances in the mutant mice. Immunohistochemistry revealed cerebellar modifications ranging from focal PC loss in Phyh−/− mice fed 0.1% phytol to severe cell loss and morphological changes of the remaining cells in Phyh−/− mutants on the 0.25% phytol‐supplemented diet. As in patients, also in the phytol‐supplemented Phyh−/− mice, the peripheral nervous system contributed to the symptoms as motor nerve conduction velocity was decreased, indicating peripheral neuropathy.
Molecular aspects of phytanic acid accumulation
The improvement of ataxia following reduced phytanic acid intake in Refsum patients, as well as the dosage‐dependent severity of PC degeneration in the phytol‐treated Refsum mouse model, strongly suggest a detrimental role of high phytanic acid concentrations on the cerebellum.
During the past decade, several in vitro studies were performed to elucidate the mechanisms underlying the toxicity caused by phytanic acid. These include experiments with isolated cerebellar mitochondria, cerebellar homogenates, cultured neural cells or fibroblasts incubated with phytanic acid concentrations in the range of those found in plasma of Refsum disease patients (1–500 μM). Impairments of mitochondrial function and morphology were commonly found, including a reduced matrix NAD(P)H content, alterations in energy production, loss of membrane potential and opening of the transition pore 19, 68, 74, 108, 110, 115. Phytanic acid also induced oxidative stress in some in vitro preparations 108.
As to the precise molecular impact, opposing mechanisms were proposed, probably because of the varying experimental setups. In view of the low permeability of the blood–brain barrier to phytanic acid 128, it can be questioned whether exposure of neural cells, and in particular isolated mitochondria, to phytanic acid concentrations occurring in Refsum plasma reflects the in vivo situation. In some experiments, 80% of cultured glial cells died after a 4‐h incubation with 100 μM phytanic acid 110 whereas, with exception of PCs, no overt cell death was reported in the brain of Refsum mice treated with a high dose of phytol 42. In addition, it is unfortunate that in several studies control incubations with straight‐chain fatty acids were not included. In a well‐controlled study using permeabilized and intact human fibroblasts 74, it was shown that phytanic acid acts as a protonophore leading to mitochondrial membrane depolarization and reduced ATP production. It was further proven that this activity depended on the branched‐chain and carboxyl function as it was mimicked by pristanic acid, the α‐oxidized shortened product of phytanic acid, but not by phytol or the straight‐chain palmitic acid with the same number of carbons in the backbone. In addition to mitochondrial dysfunction, it was reported that phytanic acid impairs Ca2+ homeostasis 68, 110 and deregulates synaptic Na+, K+‐ATPase in the cerebellum 20.
Taken together, phytanic acid seems to exert adverse effects on cerebellar PCs, which contribute to an unstable gait in men and mice. Nevertheless, the potential mechanisms deduced from in vitro experiments need to be confirmed in the in vivo situation, which is possible using the Refsum mouse model.
Peroxisomal β‐Oxidation Disorders
Currently, six divergent genetic diseases are known in which peroxisomal fatty acid β‐oxidation is deficient. In order of decreasing incidence the following pathologies are distinguished: (i) X‐linked adrenoleukodystrophy (X‐ALD), (ii) multifunctional protein‐2 (MFP2) deficiency, (iii) acyl‐CoA oxidase 1 (ACOX1) deficiency, (iv) 2‐methylacyl‐CoA racemase (AMACR) deficiency, (v) sterol carrier protein X (SCPx) deficiency and (vi) ABCD3 deficiency (Figure 2).
X‐linked adrenoleukodystrophy
Adrenoleukodystrophy protein, the peroxisomal ABC transporter encoded by the ATP binding cassette D1 (ABCD1) gene, transports VLCFA either as free acids or as CoA esters into peroxisomes prior to β‐oxidation 27, 153. As a consequence, adrenoleukodystrophy protein deficiency is biochemically characterized by C24:0 and C26:0 accumulations in plasma and tissues such as the CNS, adrenal cortex and gonads.
Cerebellar and brainstem pathologies in X‐ALD patients
In X‐ALD patients, the clinical spectrum ranges from asymptomatic and Addison‐only (isolated adrenocortical insufficiency) to progressive neurological dysfunction caused by cerebral demyelination in childhood, adolescence or adulthood 70.
The majority of X‐ALD males present either with childhood cerebral ALD (CCALD), characterized by major inflammatory demyelination of the cerebral white matter, or with adrenomyeloneuropathy (AMN), a progressive disorder involving the spinal cord and peripheral nerves (with more frequent axonal than demyelinating polyneuropathy) in adulthood. In both patient groups, adrenocortical failure, which also occurs as the sole clinical involvement, is frequent. About half of AMN patients develop cerebral white matter lesions, typically with a less aggressive evolution than the lesions in CCALD. These different presentations can occur in the same family (with the same ABCD1 mutation), and the factors determining who will develop which clinical presentation remain largely unknown. Moreover, recent evidence indicates that bone marrow transplantation, the therapy for CCALD, is not effective to prevent AMN, again indicating the differences in pathogenesis of these presentations 136. In about 1% of the patients 85, white matter damage in CCALD and AMN starts in the cerebellar white matter, resulting in cerebellar ataxia as a neurological presentation. Also, a form of X‐ALD presenting with primary cerebellar (neuronal) degeneration has been described 79. Almost all of these latter cases were reported before genetic confirmation of X‐ALD was part of clinical routine, and before the mild presentations of PBDs 136 (among which ataxia) were known. As the finding of elevated VLCFA was the only biochemical proof of the diagnosis, several of these cases may in fact have been patients with a mild PBD instead of X‐ALD. At least one report describes profound vermian atrophy in a genetically confirmed X‐ALD patient, even without white matter changes. The family history was suggestive of a cerebellar presentation in other affected family members, and the authors proposed that this presentation was linked to this particular mutation, delC1321in exon 2 31. Reports of more confirmed patients with this presentation would help to establish its real frequency.
Insights from mouse models
Three Abcd1 knockout mouse models were generated independently 48, 70, 72, 87, all showing VLCFA accumulation in the brain. A late‐onset phenotype affecting motor performance developed in 20‐month‐old Abcd1−/− mice and was accompanied by axonal degeneration in the spinal cord, resembling AMN pathology. Remarkably, inflammatory demyelination as seen in CCALD patients did not develop in the CNS 104. However, PC death was reported in the cerebellum of aged ABCD1‐deficient mice 45, corresponding with the rare X‐ALD patients with primary cerebellar atrophy.
ABCD2 deficiency, so far not described in men, was created in mice. This transporter partially overlaps in function with ABCD1 92. Similar to Abcd1 mutants, ABCD2‐deficient mice developed cerebellar problems including PC atrophy and loss, concomitant with whole‐body tremor and cerebellar ataxia. Not surprisingly, the pathological signs occurred earlier and were more severe in Abcd1/Abcd2 double mutants 45.
The toxicity of VLCFA
The only metabolic abnormality in X‐ALD is the accumulation of VLCFA. Because of their exceptional length, increased levels of VLCFA are thought to be detrimental when incorporated into membrane phospholipids 61. The toxicity of C26:0 was shown in several in vitro studies using diverse cell types including neural cells. In some experimental designs, tissues or cells with Abcd1 gene knockout or knockdown were used giving rise to elevated VLCFA levels from endogenous origin 4, 53. In other studies, cells were incubated with exogenously added C26:0 up to 50 μM 60, 86, 154. It should however be noted that this exceeds the pathological concentrations of C26:0 in X‐ALD patients which were estimated to be 1–5 μM 5, 28, 129, 137. Oxidative stress and mitochondrial impairments were recurrent cellular dysfunctions triggered by the VLCFA and were assumed to cause axonal degeneration in the spinal cord 52 and, together with an inflammatory component, the cerebral phenotype 119. Whether these mechanisms also underlie the demise of PCs is however unresolved.
Acyl‐CoA oxidase‐1 deficiency
ACOX1 catalyzes the first step in peroxisomal β‐oxidation. Its enzymatic function is restricted to the oxidation of VLCFA, long‐chain dicarboxylic acids and PUFA. ACOX1 deficiency is often referred to as pseudo‐neonatal ALD because of the similarities in clinical presentation with the PBD neonatal adrenoleukodystrophy (see below) 100. Patients have increased levels of VLCFA 41 although some ACOX1‐deficient patients with normal plasma VLCFA concentrations have also been described 111.
Currently, the pathogenic knowledge of ACOX1 deficiency is restricted to what has been observed in a limited number of patients worldwide. Delays in motor development such as head control, crawling and non‐assisted walking were described. In addition to a delay in attaining early developmental milestones, almost all ACOX1‐deficient children aged 24 to 48 months showed regression of motor achievements. Brain MRI studies demonstrated myelin loss starting in the white matter of the cerebellum, cerebellar peduncles and brainstem tracts, progressively extending to the midbrain and the cortical white matter 21, 80, 100, 111, 125, 146, 149.
In two ACOX1‐deficient siblings with a mild disease course, developmental milestones were normal but clumsiness appeared at an age of 8–10 years evolving in progressive unsteadiness and severe ataxia. Interestingly, the cerebellum, middle cerebellar peduncles and brainstem showed marked atrophy whereas the cerebrum was only mildly affected 43. These patients survived into their fifties. This moderate clinical phenotype was associated with only borderline increased levels of VLCFA, likely caused by a mutation outside of the catalytically active parts of the enzyme 51, 125.
Acox1−/− mice present with a severe hepatic phenotype and, to our knowledge, the neuropathology has not been thoroughly investigated. Given that VLCFA accumulate in blood and ACOX1 expression was confirmed in neural and glial cells of the cerebellum and other motor‐related regions such as the pontine nuclei and inferior olive 35, 49 it is surprising that no neurological signs were reported.
Multifunctional protein‐2 deficiency
MFP2 deficiency occurs with an incidence of approximately 1:100 000 39. MFP2, alternately known as D‐bifunctional protein, is encoded by the hydroxy‐steroid dehydrogenase type 4 (HSD17B4) gene. MFP2 catalyzes the oxidation of a broad spectrum of substrates including VLCFA, 2‐methyl‐branched‐chain fatty acids (eg, pristanic acid), bile acid intermediates [dihydroxycholestanoic acid (DHCA) and trihydroxycholestanoic acid (THCA)], leukotrienes and long‐chain dicarboxylic acids. In addition, MFP2 is involved in the biosynthesis of DHA, the most common PUFA in the mammalian brain and retina 39.
Cerebellar and brainstem pathologies in MFP2‐deficient patients
The importance of MFP2 in the developing CNS is highlighted by the fact that the majority of MFP2‐deficient patients display severe abnormalities at birth, presenting with hypotonia and brain malformations, and die within the first years of life 39, 145. This phenotype is indistinguishable from Zellweger disease (see below). Concerning the cerebellum, imaging experiments uncovered hypoplasia, demyelination and atrophy. Brain autopsy studies revealed ectopic or degenerating PCs, gliosis and defects in the migration and maturation of granule neurons. Brainstem pathologies often include demyelination and malformations of the inferior olivary and dentate nuclei 39, 69, 71, 95, 98, 138.
Depending on the nature of the HSD17B4 mutation, MFP2‐deficient patients may achieve some developmental milestones followed by regression. Similar to mild PBD patients (see below), central cerebellar demyelination is then a common feature 39, 122. Importantly, several patients who presented with progressive cerebellar ataxia during childhood, adolescence or adulthood combined with peripheral neuropathy and hearing loss were identified in recent years as HSD17B4 mutants by next generation sequencing 82, 84, 89. The residual enzyme activity led to normal plasma levels of VLCFA and branched‐chain fatty acids and they were proposed to represent a novel subtype of MFP2 deficiency based on their clinical, genetic and biochemical features.
Of interest, several syndromes encompassing cerebellar manifestations could also be ascribed to MFP2 deficiency. For example, HSD17B4 mutations are the first recognized genetic cause of Perrault syndrome 99, characterized by ovarian dysgenesis and sensorineural hearing loss but often also associated with neurological problems. Shrinkage of the cerebellum and motor problems in Perrault patients are similar to the clinical profile of mild MFP2‐deficient patients 47, 57, 96, 99. However, mutations in several other genes have been identified to cause Perrault syndrome. Furthermore, the importance of MFP2 in ataxic phenotypes can be deduced from patients with Stiff‐man syndrome, a rare disorder of the CNS characterized by the production of auto‐antibodies against glutamic acid decarboxylase, resulting in muscle rigidity and spasms. MFP2 is an autoimmune target in a subset of Stiff‐man patients 26, 30, 62 and gait ataxia is a facultative neurological symptom 75. A correlation between the concentration of MFP2 auto‐antibodies and the severity of cerebellar ataxia has not been established so far, but the expression of both GAD and MFP2 97 in cerebellar PCs is in favor of PC dysfunction underlying the motor problems in these patients. Another post‐developmental disease linked to MFP2 deficiency is optico‐cochleo‐dentate degeneration characterized by cerebellar symptoms, regression of motor performance and degeneration of the optic nerve, the dentate nucleus and the cochlear nerve 116. Clinically, the distinction between mild MFP2 deficiency, Perrault syndrome caused by MFP2 deficiency, and optico‐cochleo‐dentate degeneration appears artificial, and all these syndromes could be gathered under “MFP2 ataxia syndrome.”
Cerebellar and brainstem abnormalities in mouse models with MFP2 deficiency
In contrast to patients with a total ablation of MFP2 function, cerebral development was normal and cerebellar formation was only mildly affected in Mfp2−/− mice 6, 65. A delay in cerebellar foliation was observed in 7 days old Mfp2−/− mice, but cerebellar architecture appeared normal in the post weaning period 77. However, progressive motor problems such as clasping of hind legs and poor performance on the rotarod already presented in four weeks old Mfp2−/− mice and evolved in immobility and death by the age of 6 months 65, 140. Peripheral sensorimotor abnormalities were absent in Mfp2−/− mice, pointing to MFP2 deficiency in the CNS as the origin of the motor problems 65. Studies in Nestin‐Mfp2−/− mice, a model in which MFP2 is deleted from neural cells, showed a similar motor phenotype as the Mfp2−/− mice 140.
Histologically, several lesions were found in the cerebellum of both mouse models. Prominent lipid droplets containing neutral lipids progressively accumulated in Bergmann glial cells, whereas smaller inclusions were occasionally observed in cerebellar PCs. Interestingly, astrogliosis and up‐regulation of the antioxidant enzyme catalase were also found in the molecular layer of the Mfp2−/− cerebellum 65, 140. These observations raised the question whether astroglial lesions underlie the neuromotor problems in MFP2 deficiency. It is however unlikely that the lipid droplets and catalase overexpression are detrimental as they also occurred in the brain of Gfap‐Pex5−/− mice (see also below) that do not develop neurological problems and have a normal life span. Additional microscopic analyses in both Mfp2−/− and Nestin‐Mfp2−/− mice uncovered swellings on myelinated PC axons, followed by cerebellar microgliosis and profound astrogliosis in the deep cerebellar nuclei. Myelin loss started in the cerebellar lobules and expanded to the central white matter. In 12 months old Nestin‐Mfp2−/− mice, a time point at which Mfp2−/− mice already passed away, degeneration of the PC axon and dendritic tree culminated in PC loss and cerebellar atrophy. The fact that axonal abnormalities preceded neurodegeneration was compatible with a dying back neuropathy 140. It was investigated whether the absence of oligodendroglial MFP2 plays a causal role in the cerebellar demyelination by using Cnp‐Mfp2−/− mice, an oligodendrocyte selective knockout model. However, these mice showed normal motor skills and their cerebellum was unaffected until the age of 12 months ruling out a primary role for peroxisomal β‐oxidation in oligodendrocytes for the maintenance of the cerebellum 140.
The early‐onset ataxic phenotype of both Mfp2−/− and Nestin‐Mfp2−/− mice clearly preceded cerebellar histopathological changes 91, 140. As in other neurodegenerative diseases such as Huntington's disease 132 and spinocerebellar ataxia type 1 64 early cell dysfunction rather than cell death might be involved in the initial disease stages. Currently, the cellular mechanisms underlying loss of PC integrity and cerebellar demise in MFP2 knockout mice are still unsolved. One of the outstanding questions is whether the PC degeneration is inherent to peroxisomal β‐oxidation loss within these cells, which is supported by their elevated MFP2 expression as compared with other neural cells 91, or by non‐cell autonomous mechanisms.
Candidate metabolites causing cerebellar defects in MFP2 deficiency
Given the broad substrate spectrum of MFP2, several metabolic disturbances could account for cerebellar degeneration in MFP2‐deficient patients and mice. However, it remains unresolved how defective peroxisomal β‐oxidation impacts on cerebellar integrity. First, as in ABCD1 deficiency, VLCFA accumulate in the nervous system after loss of MFP2. However, when comparing the mouse models, it is striking that cerebellar degeneration occurs much earlier and is more drastic in Mfp2−/− than in double Abcd1/Abcd2−/− mutants. Furthermore, the absence of a clinical phenotype in Cnp‐Mfp2−/− mice, despite elevated C26:0 levels in the cerebellum, does not support a crucial role for VLCFA in cerebellar degeneration 140.
MFP2 is also necessary for the breakdown of pristanic acid, the branched‐chain fatty acid formed by α‐oxidation of phytanic acid, both presumably having a similar toxicity profile when accumulating. As already mentioned, mice are exposed to very low levels of these fatty acids when fed a normal diet. Whereas in the Refsum mouse model, the cerebellum only degenerated after supplementation of phytol, this occurred in MFP2‐deficient mice on a standard diet. In addition, the cerebellum of Mfp2−/− and Nestin‐Mfp2−/− mice is free of phytanic or pristanic acid accumulation and the ataxic behavior of Mfp2−/− mice fed with pellets enriched in the branched‐chain fatty acid precursor phytol does not differ from MFP2 knockout mice on a normal diet 65. As already mentioned, it is striking that plasma levels of VLCFA and the branched‐chain fatty acids are normal or near‐normal in MFP2 patients with post‐developmental cerebellar degeneration (MFP2 ataxia syndrome) 43, 82, 84, 89, 99, 122.
Another potential metabolic candidate is a lack of DHA. It is well established that this PUFA requires one peroxisomal β‐oxidation cycle for its synthesis and in MFP2‐deficient patients, a shortage of this important PUFA was shown 38. Quite surprisingly, and for unclear reasons, DHA levels are normal in cerebellum and other brain regions of MFP2 knockout mice 65. Unless cell type or regional differences in DHA concentration would exist, a lack of PUFA cannot be claimed as the origin of cerebellar pathology.
In summary, the cerebellar pathology in Mfp2−/− and Nestin‐Mfp2−/− mice cannot be correlated with levels of known MFP2 substrates nor by myelin loss. It appears therefore that MFP2 could have other, unknown functions.
Alpha‐methylacyl‐CoA racemase, sterol carrier protein X and ABCD3 gene mutations
AMACR, SCPX and PMP70 are involved in the metabolism of branched‐chain substrates by peroxisomal β‐oxidation. AMACR converts 2R‐pristanic acid, 25R‐DHCA and 25R‐THCA to their S‐configuration, prior to their passage through the peroxisomal β‐oxidation pathway. Most AMACR‐deficient patients are asymptomatic until adolescence where after they start showing symptoms similar to those observed in Refsum disease patients 29, 59, 131. Cerebellar dysfunction was only seen in two out of ten patients 24, 29, 59. An MRI study on these two siblings revealed chronic degenerative lesions of efferent cerebellar pathways 59. Altered signal intensities in the dentate nucleus, superior cerebellar peduncle, thalamus and red nucleus indicated degeneration of the cerebellothalamic, dentatothalamic and dentatorubral tracts. In addition, pontine atrophy and signal changes of the transverse pontine fibers also revealed involvement of cerebellar afferents. No inferior olivary lesions were observed 59. The sensory (‐motor) neuropathy observed in most patients likely contributes to their motor abnormalities 59.
Pristanic acid also accumulates in patients with mutations in the SCPx gene. Once SCPx is imported into the peroxisome, it is split into two functional domains: a thiolase domain that catalyzes the final step of the peroxisomal β‐oxidation, and a sterol carrier protein 2 (SCP2) domain. Gait disturbances, tremor and impairment of balance in an adult SCPx‐deficient patient was consistent with cerebellar ataxia and slowed saccades indicated brainstem malfunction 40. As in other peroxisomal patients with defective branched‐chain fatty acid catabolism, the motor and sensory neuropathy should not be overlooked as an additional source of motor problems. Similar to Refsum disease patients, restriction of phytanic acid intake halted symptom progression in the SCPx patient 40, 43, while in AMACR‐deficient patients, the benefit of a phytanic acid restricted diet is inconclusive 121. Scpx knockout mice challenged with a phytol diet developed ataxia and peripheral neuropathy with an unsteady gait 117 but this was not the case in Amacr knockouts 113. Unfortunately, the cerebellum was not investigated in these mouse models.
Finally, the transport of the C27‐bile acid intermediates and branched‐chain fatty acids across the peroxisomal membrane requires ABCD3 also denoted as PMP70. In a single recently identified ABCD3‐deficient patient and in the mouse model, no cerebellar defects were observed 44. However, the patient died at young age from liver disease and the mice were only supplemented with phytol for a short period, precluding potential effects of branched‐chain fatty acids on the central or peripheral nervous system.
Ether Phospholipid Deficiency
Ether phospholipids are a subclass of glycerophospholipids with a long‐chain fatty alcohol at the sn‐1 position. Of all ether lipid species, plasmalogens that have a vinyl‐ether bond are the most abundant in mammalian tissues 15, 81, 94. The synthesis of this ether bond requires the enzymatic machinery in peroxisomes, namely glyceronephosphate O‐acyltransferase (GNPAT) and alkylglycerone phosphate synthase (AGPS) 56 (Figure 2). Being an essential constituent of myelin and neurological membranes, plasmalogens are thought to have a pivotal role in signal transduction both in the central and peripheral nervous system. At the molecular level, they were suggested to act as antioxidants, as a reservoir of PUFA and as mediators of membrane dynamics 15, 83, 95.
Rhizomelic chondrodysplasia punctata
Rhizomelic Chondrodysplasia Punctata (RCDP) has an overall incidence of 1:100 000 10. RCDP type 1 is caused by mutations in the PTS2 receptor PEX7, impairing the import of three peroxisomal enzymes, that is, AGPS, phytanoyl‐CoA hydroxylase and 3‐ketoacyl thiolase. The majority of PEX7 deficient patients show a very severe to moderate depletion in plasmalogen levels together with an increase in plasma phytanic acid levels 10. In a few PEX7 patients 63, 134 mutations are so mild that plasmalogen levels are close to normal and, as already mentioned, their phenotype is similar to Refsum disease patients, caused by impaired activity of phytanoyl‐CoA hydroxylase 67. In all other cases, the phenotype is fully determined by the impaired ether‐phospholipid synthesis. This is deduced from the fact that the clinical signs are indistinguishable from patients with RCDP type 2 or 3 with an isolated defect in ether‐phospholipid synthesis caused by mutations in respectively GNPAT and AGPS.
Cerebellar and brainstem pathologies in RCDP patients
The clinical image of RCDP types 1, 2 and 3 is generally severe but varies according to the residual capacity in plasmalogen biosynthesis 9. The most severely affected patients with undetectable plasmalogens in red blood cells showed progressive cerebellar atrophy caused by PC and granule cell degeneration in the neonatal period. The onset of cerebellar deterioration was delayed by 1 year in patients with residual albeit very low plasmalogen levels 9, 10. Non‐inflammatory myelin abnormalities were frequently observed 2, 102, 126, 127 and dysplastic olives and cerebellar heterotaxias noted in post‐mortem studies may refer to developmental defects 101, 102, 123. The achievement of motor skills in patients with the severe phenotype is poor 9. Other typical features include skeletal anomalies, audiovisual problems and psychomotor retardation with a considerably reduced life expectancy.
Less is known about the disease process of chronic RCDP patients with a milder clinical and biochemical course. The majority shows a delay in motor development such as the inability to sit or walk independently 9, 10, 12. Furthermore, patients with mild ataxia, nystagmus and cerebellar atrophy with PC and granule cell depletion have also been reported 103. As in the severely affected children, abnormalities in cerebellar architecture or myelination were not always present in these mild RCDP patients 9, 12.
Insights from mouse models
Knowledge about the consequences of deficient ether lipid synthesis has greatly increased by the analysis of Pex7−/− and Gnpat−/− (also called Dhapat−/−) mouse models 14, 17, 83, 109. Cerebellar development was mainly studied in Gnpat knockout mice revealing foliation abnormalities, a delay in granule cell migration and atrophy during the first postnatal weeks 77, 130. The PC showed multiple aberrations including axonal spheroids, defects in paranode organization and a hyperspiny appearance accompanied by altered CF and PF innervations 130. Dysmyelination occurred both in the cerebellum and the cerebrum 130 but cerebellar myelin loss was not progressive 13. Decreased rotarod and vertical pole performances in Gnpat−/− mice suggested cerebellar ataxia 130. However, a recent study on both Pex7 and Gnpat knockout mice has shown that plasmalogen deficiency in the peripheral nervous system affects axonal sorting and the myelination process by influencing Schwann cell differentiation and maturation, thereby causing peripheral neuropathy. Impairment of the AKT–GSK3β pathway induced by plasmalogen deficiency in Schwann cell membranes was proposed as the underlying mechanism 25. Therefore, the ataxia of Gnpat−/− mice may have a dual origin.
Fatty acyl‐CoA reductase 1 deficiency
The importance of plasmalogens for the maintenance of cerebellar integrity is underscored by the recent identification of a few patients with Fatty Acyl‐CoA Reductase 1 (FAR1) deficiency. FAR1 plays a critical role in the plasmalogen biosynthesis pathway by reducing saturated and unsaturated fatty acyl‐CoAs to their respective fatty alcohols. The enzyme is located at the cytosolic side of the peroxisomal membrane. Remarkably, a 19‐year‐old FAR1‐deficient patient presented with atrophy of the cerebellar vermis and hemispheres accompanied by cortical white matter lesions 18.
Peroxisome Biogenesis Defects
The import of peroxisomal matrix and membrane proteins requires the concerted action of at least 14 peroxins that have diverse functions including cytosolic receptors (PEX5 and PEX7) for enzymes carrying the peroxisome targeting signal (PTS) 1 or 2, docking proteins in the peroxisomal membrane and proteins that allow the membrane translocation and the recycling of receptors to the cytosol 150 (Figure 3). PEX7 is only needed for the import of a minority of proteins harboring a PTS2 signal (AGPS, phytanoyl‐CoA hydroxylase and 3‐ketoacyl thiolase). As already mentioned, severe PEX7 mutations cause RCDP, whereas very mild mutations cause Refsum pathologies because of, respectively, plasmalogen synthesis and α‐oxidation defects 134. Defects in all the other peroxins will impede all peroxisomal functions leading to an array of clinical presentations now designated as Zellweger spectrum disorders 16, 124. The phenotypes range from severe developmental disorders (Zellweger syndrome), that are fatal in the first year of life, to milder syndromes previously named neonatal adrenoleukodystrophy and Infantile Refsum disease that sometimes allow survival into adulthood.
Figure 3.
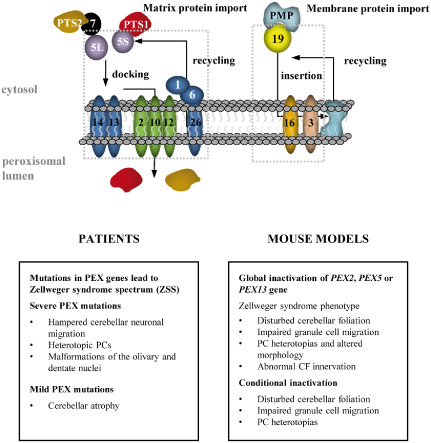
Schematic overview of cerebellar and brainstem pathologies in PBD in men and mice. PBD are caused by mutations in proteins involved in peroxisomal matrix protein import and peroxisomal membrane protein (PMP) import. PMPs contain a C‐terminal (PTS1) or a N‐terminal (PTS2) peroxisomal targeting signal. PTS1 matrix proteins are recognized by a shorter variant of the peroxisomal import receptor PEX5 (5S). The PTS1/PEX5 complex binds to the docking complex at the peroxisomal membrane, which leads to import of the protein in the peroxisomal lumen. PTS2 proteins are imported in a similar way, but are first recognized by PEX7 that binds to the longer variant of PEX5. Peroxisomal membrane proteins are incorporated via a mechanism involving PEX19, PEX16 and PEX3.
Developmental cerebellar pathologies
Newborns with Zellweger syndrome, also known as the cerebro‐hepato‐renal syndrome, present with hypotonia and craniofacial dysmorphisms 16. MRI of the brain shows gyric abnormalities including polymicrogyria in the Sylvian fissure, frontoparietal pachygyria and heterotopias 11, 151, indicative of a neuronal migration defect. Brain atrophy is another common feature. The landmark histological studies by Volpe 141 and Evrard 34 showed that in Zellweger syndrome, besides the cytoarchitectonic abnormalities of the cortex, malformations of the cerebellum and the inferior olivary nucleus occur. In the postnatal cerebellum, a subset of PCs are heterotopically located in the white matter, or abnormally positioned relative to the granule cells as a result of hampered neuronal migration. The dysplasia of the olivary and dentate nuclei also encompass laminar discontinuities. By MRI, no abnormalities were however observed in the cerebellum within the age of 2 months 11.
Cerebellar malformation in PBDs was studied in detail using mouse models lacking PEX2, PEX5 or PEX13. Depending on the genetic background, the global knockout mice die in the perinatal period or survive only for a few weeks 7. Only in the longer surviving Pex2−/− knockouts, cerebellar development could be studied as it occurs postnatally in mice. Alternatively, neural selective knockouts were generated for PEX5 and PEX13 circumventing severe liver pathology in the postnatal period and enabling to study cerebellar histogenesis 76, 93.
A thorough investigation in Pex2−/− mice revealed multiple anomalies in the developing cerebellum in which granule cells, PC and CF were affected leading to persistent abnormalities in cerebellar foliation 36. The migration of granule cells from the external granule cell layer to the internal granule cell layer was delayed. As the Bergmann glia scaffold appeared normal, the migration impairment was due to an intrinsic problem of the granule cells, which also showed increased apoptotic death. The impaired granule cell migratory capacity was confirmed using in vitro setups 37. The heterotopia of PCs was less pronounced in the mice as compared with Zellweger patients as only a minor fraction of PCs were slightly delocalized to the internal granule cell layer. However, although the initial development of PC was indistinguishable from wild type until postnatal day 3, the subsequent maturation was impaired with less complex branching of the dendritic tree and aberrant formation and orientation of dendritic spines. The latter coincided with the abnormal development of CFs. Although these innervated the PC somata at P3 in a normal fashion, the translocation of the CF dendritic tree to the PC dendrite compartment was delayed 36. In addition, the axonal compartment of PCs was affected with the occurrence of spheroids, altogether indicative of ongoing degenerative processes.
These data were confirmed in mice with inactivation of Pex13 93 or Pex5 76 restricted to neural cells by using Nestin‐Cre and respective floxed mice. Their cerebellar development was abnormal including granule cell migration impairment, increased cell death and defects in PC positioning and arborization. In cultured cerebellar Pex13−/− neurons, oxidative stress mediated by mitochondrial dysfunction was demonstrated 93. Surprisingly, no elevation of VLCFA was found in Nestin‐Pex13−/− brains eliminating this as a potential mechanism.
Remarkably, also in mice with a liver selective depletion of functional peroxisomes, major abnormalities in cerebellar morphogenesis leading to impaired foliation were observed 76, indicating that brain extrinsic mechanisms also play a role. The mechanisms were not resolved but given that bile acid treatment can partially restore the cerebellar anomalies in Pex2 knockout mice, accumulation of immature bile acids may be involved 37.
Taken together, the conservation of cerebellar dysgenesis in peroxisome‐deficient mice and men, underscores the necessity of intact peroxisomal metabolism for the normal formation of the cerebellum. Both nervous system intrinsic and extrinsic factors may impact on the intricate interplay between granule cells, CFs and PCs.
Milder peroxisome biogenesis defects and their cerebellar pathologies
The milder Zellweger spectrum disorders have a variable onset and presentation. The age at diagnosis ranges from the neonatal period up to adulthood and survival varies from a few years to several decades. Impairment of vision and hearing, psychomotor retardation and liver disease are among the most frequent clinical signs. Other symptoms include hypotonia, leukodystrophy and ataxia. A major obstacle for the diagnosis is that the typical peroxisome‐related biochemical anomalies (increased levels of VLCFA, branched‐chain fatty acids, bile acid intermediates and plasmalogens) are often not or only borderline affected. Frequently, the genetic cause is a point mutation in PEX1, the gene accounting for more than 50% of Zellweger spectrum cases 150.
Leukodystrophies initiate in the hindbrain
Infants with the adrenoleukodystrophy phenotype of PBD reach some developmental milestones but deteriorate after the age of 1 year 135. The leukodystrophy affects both the cerebrum and the cerebellum and can be stable or progressive. In the absence of clear‐cut biochemical peroxisomal hallmarks, the differential diagnosis with other leukodystrophies is challenging. By performing sequential MRI, a typical pattern was revealed for the ZSS, whereby abnormalities start in the hilus of the dentate nucleus and the superior cerebellar peduncles before affecting the cerebellar white matter and the brainstem. The white matter degeneration in the forebrain appears thereafter 135. Other unique presentations were reported such as an infant with acute neurological deterioration accompanied by inflammatory demyelination in the brainstem 78. Remarkably, in the Nestin‐Pex5 mouse model lacking functional peroxisomes in neural cells, extensive inflammatory demyelination develops. This also initiated in the cerebellum and progressed to brainstem, cortex and corpus callosum mimicking the sequence in patients 13.
PEX16 mutations as a cause of cerebellar ataxia
The PEX16 protein is essential for peroxisomal membrane assembly, which precedes the import of matrix proteins. In six PEX16 mutant patients, the most pronounced neurological defects were related to cerebellar dysfunction 32. They developed spastic paraplegia and ataxia in preschool years, whereas cognitive function was unaffected. Nevertheless, besides cerebellar atrophy, widespread changes in white matter in both sub‐ and supratentorial regions were detected and several patients were diagnosed with a demyelinating motor and sensory neuropathy. Plasma VLCFA were mildly increased in all cases, whereas branched‐chain fatty acids only in half of them.
The ring finger PEX disorders: PEX12, PEX10 and PEX2
An emerging subgroup of mild Zellweger spectrum patients present with ataxia as a primary clinical sign. Gootjes et al 54 reinvestigated a patient first diagnosed with trihydroxycholestanoic acidemia and found that PEX12 deficiency was the cause of the clinical and biochemical abnormalities. The patient presented around age 5 with mild intellectual disability, cerebellar ataxia, hypotonia and absent reflexes. Later, she developed retinopathy. Cerebellar imaging was not reported. Two patients with compound heterozygosity for PEX10 showing an analogous clinical presentation were reported by Régal et al 107. After a normal early development, they were referred to the clinic between 6 and 8 years because of worsening gait disturbances. Cerebellar abnormalities such as gait ataxia and dysarthria were confirmed by MRI, showing cerebellar atrophy. Both patients also showed an axonal motor neuropathy and posterior column dysfunction. Cognition was normal in both patients, and there was no retinopathy. Although VLCFA were not increased, a peroxisomal disorder was suspected because of increased phytanic and pristanic acid levels.
A pure cerebellar syndrome was seen in two brothers with a mutation in PEX2 118. Although both patients had the same mutation and showed similar clinical features, there was a striking difference in the onset of symptoms. One patient started to develop gait disturbances around 3.5 years and was diagnosed at 14 years with isolated progressive cerebellar ataxia. The other patient showed cerebellar signs such as impaired gait and dysmetria around the age of 18 years. Both patients showed pronounced cerebellar atrophy but no signs of myelin abnormalities or neuronal migration defects on MRI. Also in these patients, only pristanic acid and phytanic acid were moderately elevated, whereas VLCFA levels were normal. In a third PEX2 patient with a very similar clinical phenotype, biochemical analysis revealed a slight increase in VLCFA levels 90.
Although their numbers are still low, it is intriguing that these patients carry a mutation in PEX12, PEX10 or PEX2. These peroxins act as ubiquitin ligase (E3)‐like proteins and contain a RING finger domain. Mono‐ubiquitination allows PEX5 to be recycled, whereas poly‐ubiquitination targets PEX5 to the proteasome for degradation 50.
At present, it is unclear why the mutations in these genes with a common task in the peroxisome biogenesis process give rise to the cerebellar ataxic phenotype. Metabolically, these patients share mildly increased levels of branched‐chain fatty acids but minor alterations in VLCFA. Interestingly, not all mild PBDs cause ataxia. Indeed, the phenotype of mild PEX6 deficiency is characterized by retinopathy and deafness (105 and pers. comm.), signs absent in mild PEX10 and mild PEX2 deficiency, but no cerebellar degeneration. Our current knowledge of the function of these genes as necessary for global peroxisomal function does not explain why mild defects result in gene‐specific clinical syndromes. At least two possible explanations deserve further investigation. The reserve for normal peroxisomal function may be different for different PEX genes in different tissues. Another explanation would be unknown additional functions beside peroxisomal biogenesis.
Conclusion
It is striking that dysfunction of each of the three major peroxisomal lipid pathways gives rise to cerebellar defects. α‐Oxidation impairment causes a pure degenerative process that can be halted by dietary intervention. The diversity in β‐oxidation and ether lipid synthesis diseases is much wider, ranging from developmental malformations of the cerebellum and/or brainstem to degeneration later in life. It is remarkable that, during the last decade, besides the prototypical severe peroxisomal diseases such as Zellweger syndrome, MFP2 deficiency and CCALD, a number of patients have been diagnosed with mild deficiencies. Their identification is hampered by their apparent rarity, clinical signs that are not pathognomonic such as ataxia, neuropathy and psychomotor retardation in combination with (near) normal levels of peroxisomal metabolites in blood. There is no doubt that with the increasing application of genetic techniques such as exome sequencing, many more of these patients will be identified.
The mechanisms linking metabolic abnormalities to cerebellar pathology in peroxisomal disorders remain largely unresolved. Phytanic acid levels correlate with ataxia in Refsum disease, but the precise molecular impact (in PCs) needs to be further elucidated. Furthermore, it remains unclear whether and how elevated levels of VLCFA affect the cerebellum. It is most intriguing that in recently diagnosed adolescent or adult peroxisomal patients with progressive ataxia, most peroxisomal metabolites are (near) normal, contrasting with the high concentrations necessary to cause acute neurotoxicity in vitro. This raises the question whether unknown metabolic factors may be at play. To sort this out, unbiased approaches should be used to unravel whether mild peroxisome dysfunction may deregulate other metabolites. With regard to the cerebellar pathologies induced by the lack of ether lipids, the recent demonstration that AKT‐GSK3β signaling is impaired in the peripheral nervous system of an RCDP mouse model might shed new light on the pathogenesis in the brain 25.
Cerebellar PCs are among the most metabolically active of all neurons, thereby making them more vulnerable for derangement of cellular homeostasis. This not only relates to peroxisomal deficiencies but also to lysosomal 8, 142, mitochondrial 23 and autophagy 1, 73 processes. In this view, it cannot be excluded that a primary peroxisomal metabolic defect impairs mitochondrial function or autophagy processes. Whereas some peroxisomal pathologies may be PC autonomous, others are likely caused by aberrations in the circuitry innervating these pivotal cells. This can be addressed by creating mouse models with PC selective inactivation of peroxisomal proteins. It remains to be elucidated whether neurons in other brain areas develop similar pathologies at later time points as a consequence of peroxisome dysfunction.
Acknowledgments
Work by M.B. was supported by Fonds Wetenschappelijk Onderzoek Vlaanderen (G.0675.12N and G.0A15.13) and KULeuven (OT12/79). The authors thank Dr. Reyes Lόpez for contributing to the figures.
References
- 1. Alirezaei M, Kemball CC, Flynn CT, Wood MR, Whitton JL, Kiosses WB (2010) Short‐term fasting induces profound neuronal autophagy. Autophagy 6:702–710. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Alkan A, Kutlu R, Yakinci C, Sigirci A, Aslan M, Sarac K (2003) Delayed myelination in a rhizomelic chondrodysplasia punctata case: MR spectroscopy findings. Magn Reson Imaging 21:77–80. [DOI] [PubMed] [Google Scholar]
- 3. Aubourg P, Wanders R (2013) Peroxisomal disorders. Handb Clin Neurol 113:1593–1609. [DOI] [PubMed] [Google Scholar]
- 4. Baarine M, Beeson C, Singh A, Singh I (2015) ABCD1 deletion‐induced mitochondrial dysfunction is corrected by SAHA: implication for adrenoleukodystrophy. J Neurochem 133:380–396. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Baarine M, Ragot K, Athias A, Nury T, Kattan Z, Genin EC et al (2012) Incidence of Abcd1 level on the induction of cell death and organelle dysfunctions triggered by very long chain fatty acids and TNF‐alpha on oligodendrocytes and astrocytes. Neurotoxicology 33:212–228. [DOI] [PubMed] [Google Scholar]
- 6. Baes M, Huyghe S, Carmeliet P, Declercq PE, Collen D, Mannaerts GP, Van Veldhoven PP (2000) Inactivation of the peroxisomal multifunctional protein‐2 in mice impedes the degradation of not only 2‐methyl‐branched fatty acids and bile acid intermediates but also of very long chain fatty acids. J Biol Chem 275:16329–16336. [DOI] [PubMed] [Google Scholar]
- 7. Baes M, Van Veldhoven PP (2006) Generalised and conditional inactivation of Pex genes in mice. Biochim Biophys Acta 1763:1785–1793. [DOI] [PubMed] [Google Scholar]
- 8. Bailey K, Rahimi Balaei M, Mannan A, Del Bigio MR, Marzban H (2014) Purkinje cell compartmentation in the cerebellum of the lysosomal Acid phosphatase 2 mutant mouse (nax—naked‐ataxia mutant mouse). PLoS ONE 9:e94327.24722417 [Google Scholar]
- 9. Bams‐Mengerink AM, Majoie CB, Duran M, Wanders RJ, Van Hove J, Scheurer CD et al (2006) MRI of the brain and cervical spinal cord in rhizomelic chondrodysplasia punctata. Neurology 66:798–803. [DOI] [PubMed] [Google Scholar]
- 10. Bams‐Mengerink AM, Koelman JH, Waterham H, Barth PG, Poll‐The BT (2013) The neurology of rhizomelic chondrodysplasia punctata. Orphanet J Rare Dis 8:174. doi: 10.1186/1750-1172-8-174. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Barkovich AJ, Peck WW (1997) MR of Zellweger syndrome. AJNR Am J Neuroradiol 18:1163–1170. [PMC free article] [PubMed] [Google Scholar]
- 12. Barth PG, Wanders RJ, Schutgens RB, Staalman CR (1996) Variant rhizomelic chondrodysplasia punctata (RCDP) with normal plasma phytanic acid: clinico‐biochemical delineation of a subtype and complementation studies. Am J Med Genet 62:164–168. [DOI] [PubMed] [Google Scholar]
- 13. Bottelbergs A, Verheijden S, Van Veldhoven PP, Just W, Devos R, Baes M (2012) Peroxisome deficiency but not the defect in ether lipid synthesis causes activation of the innate immune system and axonal loss in the central nervous system. J Neuroinflammation 9:61. doi: 10.1186/1742-2094-9-61. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Braverman N, Zhang R, Chen L, Nimmo G, Scheper S, Tran T et al (2010) A Pex7 hypomorphic mouse model for plasmalogen deficiency affecting the lens and skeleton. Mol Genet Metab 99:408–416. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Braverman NE, Moser AB (2012) Functions of plasmalogen lipids in health and disease. Biochim Biophys Acta 1822:1442–1452. [DOI] [PubMed] [Google Scholar]
- 16. Braverman NE, D'Agostino MD, Maclean GE (2013) Peroxisome biogenesis disorders: biological, clinical and pathophysiological perspectives. Dev Disabil Res Rev 17:187–196. [DOI] [PubMed] [Google Scholar]
- 17. Brites P, Motley AM, Gressens P, Mooyer PA, Ploegaert I, Everts V et al (2003) Impaired neuronal migration and endochondral ossification in Pex7 knockout mice: a model for rhizomelic chondrodysplasia punctata. Hum Mol Genet 12:2255–2267. [DOI] [PubMed] [Google Scholar]
- 18. Buchert R, Tawamie H, Smith C, Uebe S, Innes AM, Al Hallak B et al (2014) A peroxisomal disorder of severe intellectual disability, epilepsy, and cataracts due to fatty Acyl‐CoA reductase 1 deficiency. Am J Hum Genet 95:602–610. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Busanello EN, Amaral AU, Tonin AM, Zanatta A, Viegas CM, Vargas CR, Wajner M (2013) Disruption of mitochondrial homeostasis by phytanic acid in cerebellum of young rats. Cerebellum 12:362–369. [DOI] [PubMed] [Google Scholar]
- 20. Busanello EN, Zanatta A, Tonin AM, Viegas CM, Vargas CR, Leipnitz G et al (2013) Marked inhibition of Na+, K(+)‐ ATPase activity and the respiratory chain by phytanic acid in cerebellum from young rats: possible underlying mechanisms of cerebellar ataxia in Refsum disease. J Bioenerg Biomembr 45:137–144. [DOI] [PubMed] [Google Scholar]
- 21. Carrozzo R, Bellini C, Lucioli S, Deodato F, Cassandrini D, Cassanello M et al (2008) Peroxisomal acyl‐CoA‐oxidase deficiency: two new cases. Am J Med Genet A 146A:1676–1681. [DOI] [PubMed] [Google Scholar]
- 22. Cervos‐Navarro J (1990) Heredopathia atactica polyneuritiformis (Refsum's disease). Histol Histopathol 5:439–450. [PubMed] [Google Scholar]
- 23. Chen H, McCaffery JM, Chan DC (2007) Mitochondrial fusion protects against neurodegeneration in the cerebellum. Cell 130:548–562. [DOI] [PubMed] [Google Scholar]
- 24. Clarke CE, Alger S, Preece MA, Burdon MA, Chavda S, Denis S et al (2004) Tremor and deep white matter changes in alpha‐methylacyl‐CoA racemase deficiency. Neurology 63:188–189. [DOI] [PubMed] [Google Scholar]
- 25. da Silva TF, Eira J, Lopes AT, Malheiro AR, Sousa V, Luoma A et al (2014) Peripheral nervous system plasmalogens regulate Schwann cell differentiation and myelination. J Clin Invest 124:2560–2570. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Darnell RB, Victor J, Rubin M, Clouston P, Plum F (1993) A novel antineuronal antibody in stiff‐man syndrome. Neurology 43:114–120. [DOI] [PubMed] [Google Scholar]
- 27. De Marcos Lousa C, van Roermund CW, Postis VL, Dietrich D, Kerr ID, Wanders RJ et al (2013) Intrinsic acyl‐CoA thioesterase activity of a peroxisomal ATP binding cassette transporter is required for transport and metabolism of fatty acids. Proc Natl Acad Sci USA 110:1279–1284. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Deon M, Garcia MP, Sitta A, Barschak AG, Coelho DM, Schimit GO et al (2008) Hexacosanoic and docosanoic acids plasma levels in patients with cerebral childhood and asymptomatic X‐linked adrenoleukodystrophy: Lorenzo's oil effect. Metab Brain Dis 23:43–49. [DOI] [PubMed] [Google Scholar]
- 29. Dick D, Horvath R, Chinnery PF (2011) AMACR mutations cause late‐onset autosomal recessive cerebellar ataxia. Neurology 76:1768–1770. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Dinkel K, Rickert M, Moller G, Adamski J, Meinck HM, Richter W (2002) Stiff‐man syndrome: identification of 17 beta‐hydroxysteroid dehydrogenase type 4 as a novel 80‐kDa antineuronal antigen. J Neuroimmunol 130:184–193. [DOI] [PubMed] [Google Scholar]
- 31. Dunne E, Hyman NM, Huson SM, Nemeth AH (1999) A novel point mutation in X‐linked adrenoleukodystrophy presenting as a spinocerebellar degeneration. Ann Neurol 45:652–655. [DOI] [PubMed] [Google Scholar]
- 32. Ebberink MS, Csanyi B, Chong WK, Denis S, Sharp P, Mooijer PA et al (2010) Identification of an unusual variant peroxisome biogenesis disorder caused by mutations in the PEX16 gene. J Med Genet 47:608–615. [DOI] [PubMed] [Google Scholar]
- 33. Eldjarn L, Try K, Stokke O, Munthe‐Kaas AW, Refsum S, Steinberg D et al (1966) Dietary effects on serum‐phytanic‐acid levels and on clinical manifestations in heredopathia atactica polyneuritiformis. Lancet 1:691–693. [DOI] [PubMed] [Google Scholar]
- 34. Evrard P, Caviness VS Jr, Prats‐Vinas J, Lyon G (1978) The mechanism of arrest of neuronal migration in the Zellweger malformation: an hypothesis bases upon cytoarchitectonic analysis. Acta Neuropathol 41:109–117. [DOI] [PubMed] [Google Scholar]
- 35. Farioli‐Vecchioli S, Moreno S, Ceru MP (2001) Immunocytochemical localization of acyl‐CoA oxidase in the rat central nervous system. J Neurocytol 30:21–33. [DOI] [PubMed] [Google Scholar]
- 36. Faust PL (2003) Abnormal cerebellar histogenesis in PEX2 Zellweger mice reflects multiple neuronal defects induced by peroxisome deficiency. J Comp Neurol 461:394–413. [DOI] [PubMed] [Google Scholar]
- 37. Faust PL, Banka D, Siriratsivawong R, Ng VG, Wikander TM (2005) Peroxisome biogenesis disorders: the role of peroxisomes and metabolic dysfunction in developing brain. J Inherit Metab Dis 28:369–383. [DOI] [PubMed] [Google Scholar]
- 38. Ferdinandusse S, Denis S, Mooijer PA, Zhang Z, Reddy JK, Spector AA, Wanders RJ (2001) Identification of the peroxisomal beta‐oxidation enzymes involved in the biosynthesis of docosahexaenoic acid. J Lipid Res 42:1987–1995. [PubMed] [Google Scholar]
- 39. Ferdinandusse S, Denis S, Mooyer PA, Dekker C, Duran M, Soorani‐Lunsing RJ et al (2006) Clinical and biochemical spectrum of D‐bifunctional protein deficiency. Ann Neurol 59:92–104. [DOI] [PubMed] [Google Scholar]
- 40. Ferdinandusse S, Kostopoulos P, Denis S, Rusch H, Overmars H, Dillmann U et al (2006) Mutations in the gene encoding peroxisomal sterol carrier protein X (SCPx) cause leukencephalopathy with dystonia and motor neuropathy. Am J Hum Genet 78:1046–1052. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Ferdinandusse S, Denis S, Hogenhout EM, Koster J, van Roermund CW, IJlst L et al (2007) Clinical, biochemical, and mutational spectrum of peroxisomal acyl‐coenzyme A oxidase deficiency. Hum Mutat 28:904–912. [DOI] [PubMed] [Google Scholar]
- 42. Ferdinandusse S, Zomer AW, Komen JC, van den Brink CE, Thanos M, Hamers FP et al (2008) Ataxia with loss of Purkinje cells in a mouse model for Refsum disease. Proc Natl Acad Sci U S A 105:17712–17717. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Ferdinandusse S, Barker S, Lachlan K, Duran M, Waterham HR, Wanders RJ, Hammans S (2010) Adult peroxisomal acyl‐coenzyme A oxidase deficiency with cerebellar and brainstem atrophy. J Neurol Neurosurg Psychiatry 81:310–312. [DOI] [PubMed] [Google Scholar]
- 44. Ferdinandusse S, Jimenez‐Sanchez G, Koster J, Denis S, Van Roermund CW, Silva‐Zolezzi I et al (2015) A novel bile acid biosynthesis defect due to a deficiency of peroxisomal ABCD3. Hum Mol Genet 24:361–370. [DOI] [PubMed] [Google Scholar]
- 45. Ferrer I, Kapfhammer JP, Hindelang C, Kemp S, Troffer‐Charlier N, Broccoli V et al (2005) Inactivation of the peroxisomal ABCD2 transporter in the mouse leads to late‐onset ataxia involving mitochondria, Golgi and endoplasmic reticulum damage. Hum Mol Genet 14:3565–3577. [DOI] [PubMed] [Google Scholar]
- 46. Fertl E, Foldy D, Vass K, Auff E, Vass C, Molzer B, Bernheimer H (2001) Refsum's disease in an Arabian family. J Neurol Neurosurg Psychiatry 70:564–565. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Fiumara A, Sorge G, Toscano A, Parano E, Pavone L, Opitz JM (2004) Perrault syndrome: evidence for progressive nervous system involvement. Am J Med Genet A 128A:246–249. [DOI] [PubMed] [Google Scholar]
- 48. Forss‐Petter S, Werner H, Berger J, Lassmann H, Molzer B, Schwab MH et al (1997) Targeted inactivation of the X‐linked adrenoleukodystrophy gene in mice. J Neurosci Res 50:829–843. [DOI] [PubMed] [Google Scholar]
- 49. Fouquet F, Zhou JM, Ralston E, Murray K, Troalen F, Magal E et al (1997) Expression of the adrenoleukodystrophy protein in the human and mouse central nervous system. Neurobiol Dis 3:271–285. [DOI] [PubMed] [Google Scholar]
- 50. Francisco T, Rodrigues TA, Pinto MP, Carvalho AF, Azevedo JE, Grou CP (2014) Ubiquitin in the peroxisomal protein import pathway. Biochimie 98:29–35. [DOI] [PubMed] [Google Scholar]
- 51. Funato M, Shimozawa N, Nagase T, Takemoto Y, Suzuki Y, Imamura Y et al (2006) Aberrant peroxisome morphology in peroxisomal beta‐oxidation enzyme deficiencies. Brain Dev 28:287–292. [DOI] [PubMed] [Google Scholar]
- 52. Galea E, Launay N, Portero‐Otin M, Ruiz M, Pamplona R, Aubourg P et al (2012) Oxidative stress underlying axonal degeneration in adrenoleukodystrophy: a paradigm for multifactorial neurodegenerative diseases? Biochim Biophys Acta 1822:1475–1488. [DOI] [PubMed] [Google Scholar]
- 53. Galino J, Ruiz M, Fourcade S, Schluter A, Lopez‐Erauskin J, Guilera C et al (2011) Oxidative damage compromises energy metabolism in the axonal degeneration mouse model of X‐adrenoleukodystrophy. Antioxid Redox Signal 15: 2095–2107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Gootjes J, Skovby F, Christensen E, Wanders RJ, Ferdinandusse S (2004) Reinvestigation of trihydroxycholestanoic acidemia reveals a peroxisome biogenesis disorder. Neurology 62:2077–2081. [DOI] [PubMed] [Google Scholar]
- 55. Gordon N, Hudson RE (1959) Refsum's syndrome; heredopathia atactica polyneuritiformis; a report of three cases, including a study of the cardiac pathology. Brain 82:41–55. [DOI] [PubMed] [Google Scholar]
- 56. Gorgas K, Teigler A, Komljenovic D, Just WW (2006) The ether lipid‐deficient mouse: tracking down plasmalogen functions. Biochim Biophys Acta 1763:1511–1526. [DOI] [PubMed] [Google Scholar]
- 57. Gottschalk ME, Coker SB, Fox LA (1996) Neurologic anomalies of Perrault syndrome. Am J Med Genet 65:274–276. [DOI] [PubMed] [Google Scholar]
- 58. Gronemeyer T, Wiese S, Ofman R, Bunse C, Pawlas M, Hayen H et al (2013) The proteome of human liver peroxisomes: identification of five new peroxisomal constituents by a label‐free quantitative proteomics survey. PLoS ONE 8:e57395. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. Haugarvoll K, Johansson S, Tzoulis C, Haukanes BI, Bredrup C, Neckelmann G et al (2013) MRI characterisation of adult onset alpha‐methylacyl‐coA racemase deficiency diagnosed by exome sequencing. Orphanet J Rare Dis 8:1. doi: 10.1186/1750-1172-8-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60. Hein S, Schonfeld P, Kahlert S, Reiser G (2008) Toxic effects of X‐linked adrenoleukodystrophy‐associated, very long chain fatty acids on glial cells and neurons from rat hippocampus in culture. Hum Mol Genet 17:1750–1761. [DOI] [PubMed] [Google Scholar]
- 61. Ho JK, Moser H, Kishimoto Y, Hamilton JA (1995) Interactions of a very long chain fatty acid with model membranes and serum albumin. Implications for the pathogenesis of adrenoleukodystrophy. J Clin Invest 96:1455–1463. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62. Horiuchi I, Yamada T, Imaiso Y, Hara H, Taniwaki T, Kira J (1998) A case of stiff‐man syndrome with an antineuronal autoantibody against an 80 kDa protein]. Rinsho Shinkeigaku 38:936–940. [PubMed] [Google Scholar]
- 63. Horn MA, van den Brink DM, Wanders RJ, Duran M, Poll‐The BT, Tallaksen CM et al (2007) Phenotype of adult Refsum disease due to a defect in peroxin 7. Neurology 68:698–700. [DOI] [PubMed] [Google Scholar]
- 64. Hourez R, Servais L, Orduz D, Gall D, Millard I, de Kerchove d'Exaerde A et al (2011) Aminopyridines correct early dysfunction and delay neurodegeneration in a mouse model of spinocerebellar ataxia type 1. J Neurosci 31:11795–11807. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65. Huyghe S, Schmalbruch H, Hulshagen L, Veldhoven PV, Baes M, Hartmann D (2006) Peroxisomal multifunctional protein‐2 deficiency causes motor deficits and glial lesions in the adult central nervous system. Am J Pathol 168:1321–1334. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66. Jansen GA, Ofman R, Ferdinandusse S, Ijlst L, Muijsers AO, Skjeldal OH et al (1997) Refsum disease is caused by mutations in the phytanoyl‐CoA hydroxylase gene. Nat Genet 17:190–193. [DOI] [PubMed] [Google Scholar]
- 67. Jansen GA, Waterham HR, Wanders RJ (2004) Molecular basis of Refsum disease: sequence variations in phytanoyl‐CoA hydroxylase (PHYH) and the PTS2 receptor (PEX7). Hum Mutat 23:209–218. [DOI] [PubMed] [Google Scholar]
- 68. Kahlert S, Schonfeld P, Reiser G (2005) The Refsum disease marker phytanic acid, a branched chain fatty acid, affects Ca2+ homeostasis and mitochondria, and reduces cell viability in rat hippocampal astrocytes. Neurobiol Dis 18:110–118. [DOI] [PubMed] [Google Scholar]
- 69. Kaufmann WE, Theda C, Naidu S, Watkins PA, Moser AB, Moser HW (1996) Neuronal migration abnormality in peroxisomal bifunctional enzyme defect. Ann Neurol 39:268–271. [DOI] [PubMed] [Google Scholar]
- 70. Kemp S, Berger J, Aubourg P (2012) X‐linked adrenoleukodystrophy: clinical, metabolic, genetic and pathophysiological aspects. Biochim Biophys Acta 1822:1465–1474. [DOI] [PubMed] [Google Scholar]
- 71. Khan A, Wei XC, Snyder FF, Mah JK, Waterham H, Wanders RJ (2010) Neurodegeneration in D‐bifunctional protein deficiency: diagnostic clues and natural history using serial magnetic resonance imaging. Neuroradiology 52:1163–1166. [DOI] [PubMed] [Google Scholar]
- 72. Kobayashi T, Shinnoh N, Kondo A, Yamada T (1997) Adrenoleukodystrophy protein‐deficient mice represent abnormality of very long chain fatty acid metabolism. Biochem Biophys Res Commun 232:631–636. [DOI] [PubMed] [Google Scholar]
- 73. Komatsu M, Kominami E, Tanaka K (2006) Autophagy and neurodegeneration. Autophagy 2:315–317. [DOI] [PubMed] [Google Scholar]
- 74. Komen JC, Distelmaier F, Koopman WJ, Wanders RJ, Smeitink J, Willems PH (2007) Phytanic acid impairs mitochondrial respiration through protonophoric action. Cell Mol Life Sci 64:3271–3281. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75. Kono S, Miyajima H, Sugimoto M, Suzuki Y, Takahashi Y, Hishida A (2001) Stiff‐person syndrome associated with cerebellar ataxia and high glutamic acid decarboxylase antibody titer. Intern Med 40:968–971. [DOI] [PubMed] [Google Scholar]
- 76. Krysko O, Hulshagen L, Janssen A, Schutz G, Klein R, De Bruycker M et al (2007) Neocortical and cerebellar developmental abnormalities in conditions of selective elimination of peroxisomes from brain or from liver. J Neurosci Res 85:58–72. [DOI] [PubMed] [Google Scholar]
- 77. Krysko O, Bottelbergs A, Van Veldhoven P, Baes M (2010) Combined deficiency of peroxisomal beta‐oxidation and ether lipid synthesis in mice causes only minor cortical neuronal migration defects but severe hypotonia. Mol Genet Metab 100:71–76. [DOI] [PubMed] [Google Scholar]
- 78. Kulkarni KS, Baranano KW, Lin DD, Raymond GV (2011) Contrast enhancement of brainstem tracts in Zellweger spectrum disorder: evidence of inflammatory demyelination? Neuropediatrics 42:32–34. [DOI] [PubMed] [Google Scholar]
- 79. Kumar AJ, Kohler W, Kruse B, Naidu S, Bergin A, Edwin D, Moser HW (1995) MR findings in adult‐onset adrenoleukodystrophy. AJNR Am J Neuroradiol 16:1227–1237. [PMC free article] [PubMed] [Google Scholar]
- 80. Kurian MA, Ryan S, Besley GT, Wanders RJ, King MD (2004) Straight‐chain acyl‐CoA oxidase deficiency presenting with dysmorphia, neurodevelopmental autistic‐type regression and a selective pattern of leukodystrophy. J Inherit Metab Dis 27:105–108. [DOI] [PubMed] [Google Scholar]
- 81. Lee TC (1998) Biosynthesis and possible biological functions of plasmalogens. Biochim Biophys Acta 1394:129–145. [DOI] [PubMed] [Google Scholar]
- 82. Lieber DS, Hershman SG, Slate NG, Calvo SE, Sims KB, Schmahmann JD, Mootha VK (2014) Next generation sequencing with copy number variant detection expands the phenotypic spectrum of HSD17B4‐deficiency. BMC Med Genet 15:30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83. Liegel R, Chang B, Dubielzig R, Sidjanin DJ (2011) Blind sterile 2 (bs2), a hypomorphic mutation in Agps, results in cataracts and male sterility in mice. Mol Genet Metab 103:51–59. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84. Lines MA, Jobling R, Brady L, Marshall CR, Scherer SW, Rodriguez AR et al (2014) Peroxisomal D‐bifunctional protein deficiency: three adults diagnosed by whole‐exome sequencing. Neurology 82:963–968. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85. Loes DJ, Fatemi A, Melhem ER, Gupte N, Bezman L, Moser HW, Raymond GV (2003) Analysis of MRI patterns aids prediction of progression in X‐linked adrenoleukodystrophy. Neurology 61:369–374. [DOI] [PubMed] [Google Scholar]
- 86. Lopez‐Erauskin J, Galino J, Ruiz M, Cuezva JM, Fabregat I, Cacabelos D et al (2013) Impaired mitochondrial oxidative phosphorylation in the peroxisomal disease X‐linked adrenoleukodystrophy. Hum Mol Genet 22:3296–3305. [DOI] [PubMed] [Google Scholar]
- 87. Lu JF, Barron‐Casella E, Deering R, Heinzer AK, Moser AB, deMesy Bentley KL et al (2007) The role of peroxisomal ABC transporters in the mouse adrenal gland: the loss of Abcd2 (ALDR), Not Abcd1 (ALD), causes oxidative damage. Lab Invest 87:261–272. [DOI] [PubMed] [Google Scholar]
- 88. MacBrinn MC, O'Brien JS (1968) Lipid composition of the nervous system in Refsum's disease. J Lipid Res 9:552–561. [PubMed] [Google Scholar]
- 89. McMillan HJ, Worthylake T, Schwartzentruber J, Gottlieb CC, Lawrence SE, Mackenzie A et al (2012) Specific combination of compound heterozygous mutations in 17beta‐hydroxysteroid dehydrogenase type 4 (HSD17B4) defines a new subtype of D‐bifunctional protein deficiency. Orphanet J Rare Dis 7:90. doi: 10.1186/1750-1172-7-90. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90. Mignarri A, Vinciguerra C, Giorgio A, Ferdinandusse S, Waterham H, Wanders R et al (2012) Zellweger spectrum disorder with mild phenotype caused by PEX2 gene mutations. JIMD Rep 6:43–46. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91. Moller G, Leenders F, van Grunsven EG, Dolez V, Qualmann B, Kessels MM et al (1999) Characterization of the HSD17B4 gene: D‐specific multifunctional protein 2/17beta‐hydroxysteroid dehydrogenase IV. J Steroid Biochem Mol Biol 69:441–446. [DOI] [PubMed] [Google Scholar]
- 92. Morita M, Imanaka T (2012) Peroxisomal ABC transporters: structure, function and role in disease. Biochim Biophys Acta 1822:1387–1396. [DOI] [PubMed] [Google Scholar]
- 93. Muller CC, Nguyen TH, Ahlemeyer B, Meshram M, Santrampurwala N, Cao S et al (2011) PEX13 deficiency in mouse brain as a model of Zellweger syndrome: abnormal cerebellum formation, reactive gliosis and oxidative stress. Dis Model Mech 4:104–119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94. Nagan N, Zoeller RA (2001) Plasmalogens: biosynthesis and functions. Prog Lipid Res 40:199–229. [DOI] [PubMed] [Google Scholar]
- 95. Nakano K, Zhang Z, Shimozawa N, Kondo N, Ishii N, Funatsuka M et al (2001) D‐bifunctional protein deficiency with fetal ascites, polyhydramnios, and contractures of hands and toes. J Pediatr 139:865–867. [DOI] [PubMed] [Google Scholar]
- 96. Nishi Y, Hamamoto K, Kajiyama M, Kawamura I (1988) The Perrault syndrome: clinical report and review. Am J Med Genet 31:623–629. [DOI] [PubMed] [Google Scholar]
- 97. Normand T, Husen B, Leenders F, Pelczar H, Baert JL, Begue A et al (1995) Molecular characterization of mouse 17 beta‐hydroxysteroid dehydrogenase IV. J Steroid Biochem Mol Biol 55:541–548. [DOI] [PubMed] [Google Scholar]
- 98. Paprocka J, Jamroz E, Adamek D, Stradomska TJ, Gluszkiewicz E, Grzybowska‐Chlebowczyk U, Marszal E (2007) Clinical and neuropathological picture of familial encephalopathy with bifunctional protein deficiency. Folia Neuropathol 45:213–219. [PubMed] [Google Scholar]
- 99. Pierce SB, Walsh T, Chisholm KM, Lee MK, Thornton AM, Fiumara A et al (2010) Mutations in the DBP‐deficiency protein HSD17B4 cause ovarian dysgenesis, hearing loss, and ataxia of Perrault Syndrome. Am J Hum Genet 87:282–288. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100. Poll‐The BT, Roels F, Ogier H, Scotto J, Vamecq J, Schutgens RB et al (1988) A new peroxisomal disorder with enlarged peroxisomes and a specific deficiency of acyl‐CoA oxidase (pseudo‐neonatal adrenoleukodystrophy). Am J Hum Genet 42:422–434. [PMC free article] [PubMed] [Google Scholar]
- 101. Poulos A, Sheffield L, Sharp P, Sherwood G, Johnson D, Beckman K et al (1988) Rhizomelic chondrodysplasia punctata: clinical, pathologic, and biochemical findings in two patients. J Pediatr 113:685–690. [DOI] [PubMed] [Google Scholar]
- 102. Powers JM, Moser HW (1998) Peroxisomal disorders: genotype, phenotype, major neuropathologic lesions, and pathogenesis. Brain Pathol 8:101–120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103. Powers JM, Kenjarski TP, Moser AB, Moser HW (1999) Cerebellar atrophy in chronic rhizomelic chondrodysplasia punctata: a potential role for phytanic acid and calcium in the death of its Purkinje cells. Acta Neuropathol 98:129–134. [DOI] [PubMed] [Google Scholar]
- 104. Pujol A, Hindelang C, Callizot N, Bartsch U, Schachner M, Mandel JL (2002) Late onset neurological phenotype of the X‐ALD gene inactivation in mice: a mouse model for adrenomyeloneuropathy. Hum Mol Genet 11:499–505. [DOI] [PubMed] [Google Scholar]
- 105. Raas‐Rothschild A, Wanders RJ, Mooijer PA, Gootjes J, Waterham HR, Gutman A et al (2002) A PEX6‐defective peroxisomal biogenesis disorder with severe phenotype in an infant, versus mild phenotype resembling Usher syndrome in the affected parents. Am J Hum Genet 70:1062–1068. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106. Reese H, Bareta J (1950) Heredopathia atactica polyneuritiformis. J Neuropathol Exp Neurol 9:385–395. [DOI] [PubMed] [Google Scholar]
- 107. Regal L, Ebberink MS, Goemans N, Wanders RJ, De Meirleir L, Jaeken J et al (2010) Mutations in PEX10 are a cause of autosomal recessive ataxia. Ann Neurol 68:259–263. [DOI] [PubMed] [Google Scholar]
- 108. Reiser G, Schonfeld P, Kahlert S (2006) Mechanism of toxicity of the branched‐chain fatty acid phytanic acid, a marker of Refsum disease, in astrocytes involves mitochondrial impairment. Int J Dev Neurosci 24:113–122. [DOI] [PubMed] [Google Scholar]
- 109. Rodemer C, Thai TP, Brugger B, Kaercher T, Werner H, Nave KA et al (2003) Inactivation of ether lipid biosynthesis causes male infertility, defects in eye development and optic nerve hypoplasia in mice. Hum Mol Genet 12:1881–1895. [DOI] [PubMed] [Google Scholar]
- 110. Ronicke S, Kruska N, Kahlert S, Reiser G (2009) The influence of the branched‐chain fatty acids pristanic acid and Refsum disease‐associated phytanic acid on mitochondrial functions and calcium regulation of hippocampal neurons, astrocytes, and oligodendrocytes. Neurobiol Dis 36:401–410. [DOI] [PubMed] [Google Scholar]
- 111. Rosewich H, Waterham HR, Wanders RJ, Ferdinandusse S, Henneke M, Hunneman D, Gartner J (2006) Pitfall in metabolic screening in a patient with fatal peroxisomal beta‐oxidation defect. Neuropediatrics 37:95–98. [DOI] [PubMed] [Google Scholar]
- 112. Salisachs P (1982) Is the “cerebellar” incoordination of Refsum's disease due to structural lesions in the cerebellum? J Neurol Neurosurg Psychiatry 45:473–474. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113. Savolainen K, Kotti TJ, Schmitz W, Savolainen TI, Sormunen RT, Ilves M et al (2004) A mouse model for alpha‐methylacyl‐CoA racemase deficiency: adjustment of bile acid synthesis and intolerance to dietary methyl‐branched lipids. Hum Mol Genet 13:955–965. [DOI] [PubMed] [Google Scholar]
- 114. Schmahmann JD, Caplan D (2006) Cognition, emotion and the cerebellum. Brain 129:290–292. [DOI] [PubMed] [Google Scholar]
- 115. Schonfeld P, Kahlert S, Reiser G (2004) In brain mitochondria the branched‐chain fatty acid phytanic acid impairs energy transduction and sensitizes for permeability transition. Biochem J 383:121–128. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116. Schroder JM, Hackel V, Wanders RJ, Gohlich‐Ratmann G, Voit T (2004) Optico‐cochleo‐dentate degeneration associated with severe peripheral neuropathy and caused by peroxisomal D‐bifunctional protein deficiency. Acta Neuropathol 108:154–167. [DOI] [PubMed] [Google Scholar]
- 117. Seedorf U, Raabe M, Ellinghaus P, Kannenberg F, Fobker M, Engel T et al (1998) Defective peroxisomal catabolism of branched fatty acyl coenzyme A in mice lacking the sterol carrier protein‐2/sterol carrier protein‐x gene function. Genes Dev 12:1189–1201. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118. Sevin C, Ferdinandusse S, Waterham HR, Wanders RJ, Aubourg P (2011) Autosomal recessive cerebellar ataxia caused by mutations in the PEX2 gene. Orphanet J Rare Dis 6:8. doi: 10.1186/1750-1172-6-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 119. Singh I, Pujol A (2010) Pathomechanisms underlying X‐adrenoleukodystrophy: a three‐hit hypothesis. Brain Pathol 20:838–844. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 120. Skjeldal OH, Stokke O, Refsum S, Norseth J, Petit H (1987) Clinical and biochemical heterogeneity in conditions with phytanic acid accumulation. J Neurol Sci 77:87–96. [DOI] [PubMed] [Google Scholar]
- 121. Smith EH, Gavrilov DK, Oglesbee D, Freeman WD, Vavra MW, Matern D, Tortorelli S (2010) An adult onset case of alpha‐methyl‐acyl‐CoA racemase deficiency. J Inherit Metab Dis 33 (Suppl. 3):S349–S353. [DOI] [PubMed] [Google Scholar]
- 122. Soorani‐Lunsing RJ, van Spronsen FJ, Stolte‐Dijkstra I, Wanders RJ, Ferdinandusse S, Waterham HR et al (2005) Normal very‐long‐chain fatty acids in peroxisomal D‐bifunctional protein deficiency: a diagnostic pitfall. J Inherit Metab Dis 28:1172–1174. [DOI] [PubMed] [Google Scholar]
- 123. Spranger JW, Opitz JM, Bidder U (1971) Heterogeneity of Chondrodysplasia punctata. Humangenetik 11:190–212. [DOI] [PubMed] [Google Scholar]
- 124. Steinberg SJ, Dodt G, Raymond GV, Braverman NE, Moser AB, Moser HW (2006) Peroxisome biogenesis disorders. Biochim Biophys Acta 1763:1733–1748. [DOI] [PubMed] [Google Scholar]
- 125. Suzuki Y, Iai M, Kamei A, Tanabe Y, Chida S, Yamaguchi S et al (2002) Peroxisomal acyl CoA oxidase deficiency. J Pediatr 140:128–130. [DOI] [PubMed] [Google Scholar]
- 126. Sztriha L, Al‐Gazali LI, Wanders RJ, Ofman R, Nork M, Lestringant GG (2000) Abnormal myelin formation in rhizomelic chondrodysplasia punctata type 2 (DHAPAT‐deficiency). Dev Med Child Neurol 42:492–495. [DOI] [PubMed] [Google Scholar]
- 127. Sztriha LS, Nork MP, Abdulrazzaq YM, Al‐Gazali LI, Bakalinova DB (1997) Abnormal myelination in peroxisomal isolated dihydroxyacetonephosphate acyltransferase deficiency. Pediatr Neurol 16:232–236. [DOI] [PubMed] [Google Scholar]
- 128. ten Brink HJ, van den Heuvel CM, Poll‐The BT, Wanders RJ, Jakobs C (1993) Peroxisomal disorders: concentrations of metabolites in cerebrospinal fluid compared with plasma. J Inherit Metab Dis 16:587–590. [DOI] [PubMed] [Google Scholar]
- 129. Takemoto Y, Suzuki Y, Horibe R, Shimozawa N, Wanders RJ, Kondo N (2003) Gas chromatography/mass spectrometry analysis of very long chain fatty acids, docosahexaenoic acid, phytanic acid and plasmalogen for the screening of peroxisomal disorders. Brain Dev 25:481–487. [DOI] [PubMed] [Google Scholar]
- 130. Teigler A, Komljenovic D, Draguhn A, Gorgas K, Just WW (2009) Defects in myelination, paranode organization and Purkinje cell innervation in the ether lipid‐deficient mouse cerebellum. Hum Mol Genet 18:1897–1908. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 131. Thompson SA, Calvin J, Hogg S, Ferdinandusse S, Wanders RJ, Barker RA (2008) Relapsing encephalopathy in a patient with alpha‐methylacyl‐CoA racemase deficiency. J Neurol Neurosurg Psychiatry 79:448–450. [DOI] [PubMed] [Google Scholar]
- 132. Tobin AJ, Signer ER (2000) Huntington's disease: the challenge for cell biologists. Trends Cell Biol 10:531–536. [DOI] [PubMed] [Google Scholar]
- 133. Try K (1969) Herdopathia atactica polyneuritiformis (Refsum's disease). The diagnostic value of phytanic acid determination in serum lipids. Eur Neurol 2:296–314. [DOI] [PubMed] [Google Scholar]
- 134. van den Brink DM, Brites P, Haasjes J, Wierzbicki AS, Mitchell J, Lambert‐Hamill M et al (2003) Identification of PEX7 as the second gene involved in Refsum disease. Am J Hum Genet 72:471–477. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 135. van der Knaap MS, Wassmer E, Wolf NI, Ferreira P, Topcu M, Wanders RJ et al (2012) MRI as diagnostic tool in early‐onset peroxisomal disorders. Neurology 78:1304–1308. [DOI] [PubMed] [Google Scholar]
- 136. van Geel BM, Poll‐The BT, Verrips A, Boelens JJ, Kemp S, Engelen M (2015) Hematopoietic cell transplantation does not prevent myelopathy in X‐linked adrenoleukodystrophy: a retrospective study. J Inherit Metab Dis 38:359–361. [DOI] [PubMed] [Google Scholar]
- 137. Valianpour F, Selhorst JJ, van Lint LE, van Gennip AH, Wanders RJ, Kemp S (2003) Analysis of very long‐chain fatty acids using electrospray ionization mass spectrometry. Mol Genet Metab 79:189–196. [DOI] [PubMed] [Google Scholar]
- 138. Van Maldergem L, Espeel M, Wanders RJ, Roels F, Gerard P, Scalais E et al (1992) Neonatal seizures and severe hypotonia in a male infant suffering from a defect in peroxisomal beta‐oxidation. Neuromuscul Disord 2:217–224. [DOI] [PubMed] [Google Scholar]
- 139. Van Veldhoven PP (2010) Biochemistry and genetics of inherited disorders of peroxisomal fatty acid metabolism. J Lipid Res 51:2863–2895. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 140. Verheijden S, Bottelbergs A, Krysko O, Krysko DV, Beckers L, De Munter S et al (2013) Peroxisomal multifunctional protein‐2 deficiency causes neuroinflammation and degeneration of Purkinje cells independent of very long chain fatty acid accumulation. Neurobiol Dis 58:258–269. [DOI] [PubMed] [Google Scholar]
- 141. Volpe JJ, Adams RD (1972) Cerebro‐hepato‐renal syndrome of Zellweger: an inherited disorder of neuronal migration. Acta Neuropathol 20:175–198. [DOI] [PubMed] [Google Scholar]
- 142. Walkley SU, Sikora J, Micsenyi M, Davidson C, Dobrenis K (2010) Lysosomal compromise and brain dysfunction: examining the role of neuroaxonal dystrophy. Biochem Soc Trans 38:1436–1441. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 143. Wanders RJ (2013) Peroxisomes in human health and disease: metabolic pathways, metabolite transport, interplay with other organelles and signal transduction. Subcell Biochem 69:23–44. [DOI] [PubMed] [Google Scholar]
- 144. Wanders RJ, Waterham HR (2006) Biochemistry of mammalian peroxisomes revisited. Annu Rev Biochem 75:295–332. [DOI] [PubMed] [Google Scholar]
- 145. Wanders RJ, Waterham HR (2006) Peroxisomal disorders: the single peroxisomal enzyme deficiencies. Biochim Biophys Acta 1763:1707–1720. [DOI] [PubMed] [Google Scholar]
- 146. Wanders RJ, Schelen A, Feller N, Schutgens RB, Stellaard F, Jakobs C et al (1990) First prenatal diagnosis of acyl‐CoA oxidase deficiency. J Inherit Metab Dis 13:371–374. [DOI] [PubMed] [Google Scholar]
- 147. Wanders RJ, Jansen GA, Skjeldal OH (2001) Refsum disease, peroxisomes and phytanic acid oxidation: a review. J Neuropathol Exp Neurol 60:1021–1031. [DOI] [PubMed] [Google Scholar]
- 148. Wanders RJ, Komen J, Ferdinandusse S (2011) Phytanic acid metabolism in health and disease. Biochim Biophys Acta 1811:498–507. [DOI] [PubMed] [Google Scholar]
- 149. Wang RY, Monuki ES, Powers J, Schwartz PH, Watkins PA, Shi Y et al (2014) Effects of hematopoietic stem cell transplantation on acyl‐CoA oxidase deficiency: a sibling comparison study. J Inherit Metab Dis 37:791–799. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 150. Waterham HR, Ebberink MS (2012) Genetics and molecular basis of human peroxisome biogenesis disorders. Biochim Biophys Acta 1822:1430–1441. [DOI] [PubMed] [Google Scholar]
- 151. Weller S, Rosewich H, Gartner J (2008) Cerebral MRI as a valuable diagnostic tool in Zellweger spectrum patients. J Inherit Metab Dis 31:270–280. [DOI] [PubMed] [Google Scholar]
- 152. Wierzbicki AS, Lloyd MD, Schofield CJ, Feher MD, Gibberd FB (2002) Refsum's disease: a peroxisomal disorder affecting phytanic acid alpha‐oxidation. J Neurochem 80:727–735. [DOI] [PubMed] [Google Scholar]
- 153. Wiesinger C, Kunze M, Regelsberger G, Forss‐Petter S, Berger J (2013) Impaired very long‐chain acyl‐CoA beta‐oxidation in human X‐linked adrenoleukodystrophy fibroblasts is a direct consequence of ABCD1 transporter dysfunction. J Biol Chem 288:19269–19279. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 154. Zarrouk A, Vejux A, Nury T, El Hajj HI, Haddad M, Cherkaoui‐Malki M et al (2012) Induction of mitochondrial changes associated with oxidative stress on very long chain fatty acids (C22:0, C24:0, or C26:0)‐treated human neuronal cells (SK‐NB‐E). Oxid Med Cell Longev 2012:623257. doi: 10.1155/2012/623257. Epub 2012 Aug 5. [DOI] [PMC free article] [PubMed] [Google Scholar]