

Summary
Lodging reduces grain yield in cereal crops. Understanding the genetic basis of lodging resistance (LR) benefits LR breeding. In the study, 524 accessions from a rice germplasm collection and 193 recombinant inbred lines were phenotyped for 17 LR‐related traits. Height and culm strength (the magnitude of applied force necessary to break the culm) were two major factors affecting LR. We conducted genome‐wide association study (GWAS) and identified 127 LR‐associated loci. Significant phenotypic correlations between culm‐strength traits and yield‐related traits were observed. To reveal the genetic relationship between them, we conducted GWAS of culm‐strength traits with adding yield‐related trait as a covariate and detected 63 loci linking culm strength and yield. As a proof, a near‐isogenic line for an association locus on chromosome 7 showed enhanced LR and yield. Strikingly, 58 additional loci were identified in the covariate‐added GWAS. Several LR‐associated loci had undergone divergent selection. Linkage analysis supported the GWAS results. We propose that introgression of alleles beneficial for both culm strength and panicle weight without negative effects on panicle number or pyramiding high‐yielding alleles and lodging‐resistant alleles without effects on yield can be employed for the post‐Green‐Revolution breeding.
Keywords: natural variation, genetic relationship, genome‐wide association study, linkage analysis, lodging resistance, culm strength, yield
Introduction
In cereal crops, especially for high‐yielding cultivars, lodging largely impairs grain yield and quality (Islam et al., 2007). In the 1960s, a breakthrough—‘Green Revolution’ in rice—enhanced lodging resistance (LR) by using gibberellin‐deficient semi‐dwarf varieties (Sasaki et al., 2002). Unfortunately, dwarfism limits canopy photosynthesis, biomass and thus grain yield (Islam et al., 2007). High‐yielding breeding programs promote large panicles of modern cultivars, which make culms vulnerable to lodging, especially when the rainfall occurs or the wind blows (Hirano et al., 2017). Regarding agronomical management, high planting density and heavy fertilizer input have been widely adopted to enhance grain yield, resulting in a high risk of lodging due to reduced culm strength (Shah et al., 2019).
One promising solution to address these problems is to enhance culm physical strength (Yano et al., 2015). Some breeding trials have tried to exploit strong‐culm alleles, but these efforts were not very successful due to the negative trade‐offs between culm strength and grain yield (Hirano et al., 2017). So, it is of significance to gain comprehensive insights into the genetic basis of culm strength and yield‐related traits, to identify some alleles that can enhance culm strength without negative effects or with positive effects on grain yield.
Genetic analyses and gene identification can accelerate precise and efficient breeding. Geneticists and biologists have made great efforts to identify QTLs and clone genes associated with LR in rice. A QTL prl5 was identified in a population of backcross inbred lines (BILs), which affects LR by changing the culm characteristics (Kashiwagi and Ishimaru, 2004). A total of 12 QTLs controlling LR‐related traits were identified in another BIL population (Yadav et al., 2017). Chromosome segment substitution lines (CSSLs) were constructed to identify QTLs associated with LR (Mulsanti et al., 2018; Ookawa et al., 2010). A population of recombinant inbred lines (RILs) was developed to identify favourable alleles of LR and yield in rice (Nomura et al., 2019). Besides the synthetic populations, a natural population for association mapping was used to mine loci and favourable alleles for five LR‐related traits using 262 SSR markers (Sowadan et al., 2018).
Regarding gene identification, several genes are associated with LR by affecting plant height, plant architecture, culm morphology, cell wall construction and culm silicon accumulation. The 'Green Revolution’ gene, SD1, encodes a GA synthesis enzyme, and the sd‐1 allele is a loss‐of‐function mutation, which reduces plant height and thus enhances LR (Sasaki et al., 2002). TAC1 is a major gene controlling tiller angle (Yu et al., 2007), which is related to LR (Li et al., 1999; Sasaki and Ashikari, 2018). Its expression polymorphism caused by variants in the 3'UTR underlies the tiller angle variation. The SCM2 gene controlling culm diameter, was identified using chromosome segment substitution lines (Ookawa et al., 2010). This gene is identical to APO1, which was previously reported to control panicle structure. The SCM2 allele from a donor parent can enhance both culm strength and spikelet number. SCM3, which controls culm morphology, was identified using 93 BILs (Yano et al., 2015). This gene is identical to OsTB1, a gene previously reported to control strigolactone (SL) signalling. The SCM3 allele from Chugoku117 can enhance both culm strength and spikelet number, with a negative effect on panicle number. Several genes encoding extensins (Fan et al., 2018) and sucrose synthases (Fan et al., 2017) have impacts on cell wall construction and thus culm physical strength. Silicon increases resistance to lodging in rice, so genes responsible for silicon accumulation can contribute to LR, such as silicon transporter gene Lsi1, Lsi2 and Lsi6 (Ma et al., 2006; Ma et al., 2007; Yamaji and Ma, 2009).
To our knowledge, few studies have exploited LR natural variations in a large rice germplasm collection to provide comprehensive insights into the genetic architecture of LR and the genetic relationship between culm strength and yield in rice (Hirano et al., 2017; Shah et al., 2019). In this study, we conducted GWAS based on millions of genotypic markers, linkage analysis using RILs and genome‐wide F ST analysis, to dissect the genetic architecture of LR. To reveal the genetic relationship between culm strength and yield, we extended the covariate‐added GWAS of culm‐strength traits using yield‐related trait as a covariate. The results from this study provide useful clues for post‐Green‐Revolution breeding.
Results
Variation of LR‐related traits in a rice germplasm collection
In our study, we phenotyped 17 lodging‐related traits across 524 diverse rice accessions at 25 days after the full heading stage in 2014 and 2015 (Figure 1a‐d; Table 1). Broad‐sense heritability of these traits ranged from 0.63 to 0.95, suggesting that these traits are mainly genetically controlled, and their genetic bases merited dissection (Table 1). To reduce the effect of environment on phenotypic data, the best linear unbiased prediction (BLUP) value of each trait was calculated for the following analyses. Great variations of these traits were observed and the coefficient of variation ranged from 0.14 to 0.51 (Figure 1e; Table 1). For example, lodging index (LI) was used to quantitatively evaluate LR (Hirano et al., 2017; Islam et al., 2007), and it varied in the population ranging from 0.38 to 3.88 in 2014 and from 0.36 to 3.79 in 2015, respectively. The traits were mainly classified into two groups: 6 traits associated with gravity‐centre height (termed as ‘height traits’) and 7 traits associated with culm strength (termed as ‘culm‐strength traits’) (Figure 1f). In the study, path analysis was conducted to explore direct influences of variables on LI (Table S1) and to draw the path diagram (Figure S1). Based on the correlation analysis and path analysis (Figure 1f; Table S1), LI was significantly correlated with gravity‐centre height (GH, Pearson correlation coefficient R = 0.55, P = 4.82 × 10‐43; path coefficient from GH to LI R′ = −0.10) and the breaking strength of the basal internode (BS, Pearson correlation coefficient R = −0.24, P = 2.12 × 10−8; path coefficient from BS to LI R′ = −0.96). GH and BS collectively explained 65.8% of variance of LI, suggesting that GH and BS play important roles in LR. GH was affected by many factors, such as plant height (PH, R = 0.97, P < 1.00 × 10−100), panicle weight (PW, R = 0.38, P = 7.50 × 10−20); BS was largely affected by culm diameter (CD1, R = 0.87, P < 1.00 × 10‐100) and thickness (CT1, R = 0.85, P < 1.00 × 10−100). We also found all the culm‐strength traits were significantly positively correlated with fresh panicle weight (PW) (R = 0.69, P = 9.66 × 10−75 between BS and PW) and were significantly negatively correlated with panicle number (PN) (R = −0.54, P = 3.71 × 10−40 between BS and PN; Figure 1f), indicating that trade‐offs existed between culm strength and panicle number, and there was a synergistic effect between culm strength and panicle weight.
Figure 1.
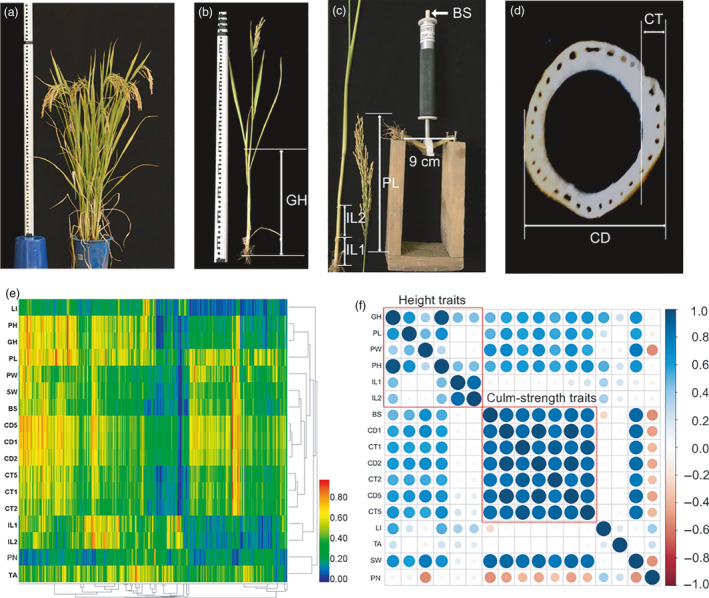
Phenotyping of lodging resistance (LR) related traits in the association mapping population. (a) Plant height measurement (the longest distance from the plant base to the tip of the highest leaf or panicle (whichever is longer)). (b) Culm gravity‐centre height measurement (GH, the distance from culm base to culm gravity‐centre). (c) Measurement of the 1st lowermost internode length from the plant base (IL1), the 2nd internode length (IL2), the panicle length (PL) and the basal culm breaking strength (BS). (d) Measurement of the culm outer diameter (CD) and culm thickness (CT). (e) Heatmap showing the values of 17 LR‐related traits across 524 accessions. The range of values for each trait was transformed to 0‐1 by linear normalization. (f) Correlation matrix diagram showing the correlation coefficients among the 17 traits. The circle with greater diameter and greater colour intensity represents a greater absolute correlation coefficient value. These traits are mainly classified into two groups–height traits and culm‐strength traits.
Table 1.
Detailed information of 17 lodging resistance (LR) related traits
| Trait | Full names of traits | Year | Minimum | Maximum | Mean | SD | CV | N | Corr | H 2 |
|---|---|---|---|---|---|---|---|---|---|---|
| LI | Lodging index | 2014 | 0.38 | 3.88 | 1.20 | 0.53 | 0.44 | 522 | 0.66 | 0.79 |
| 2015 | 0.36 | 3.79 | 1.15 | 0.45 | 0.39 | 522 | ||||
| PH | Plant height(cm) | 2014 | 63.13 | 215.11 | 120.96 | 31.90 | 0.26 | 522 | 0.92 | 0.95 |
| 2015 | 69.93 | 210.03 | 128.32 | 29.48 | 0.23 | 522 | ||||
| GH | Gravity‐centre height(cm) | 2014 | 30.21 | 94.48 | 52.69 | 12.06 | 0.23 | 522 | 0.87 | 0.93 |
| 2015 | 30.63 | 84.75 | 55.36 | 11.23 | 0.20 | 522 | ||||
| IL1 | Length of the 1st internode from plant base (cm) | 2014 | 2.54 | 14.18 | 6.13 | 1.87 | 0.30 | 522 | 0.46 | 0.63 |
| 2015 | 2.47 | 13.55 | 5.84 | 2.00 | 0.34 | 522 | ||||
| IL2 | Length of the 2nd internode from plant base (cm) | 2014 | 6.05 | 32.00 | 14.46 | 5.20 | 0.36 | 522 | 0.79 | 0.88 |
| 2015 | 4.48 | 26.75 | 13.00 | 4.58 | 0.35 | 522 | ||||
| PL | Panicle length(cm) | 2014 | 14.31 | 35.71 | 24.32 | 3.53 | 0.15 | 522 | 0.79 | 0.88 |
| 2015 | 14.53 | 37.40 | 26.60 | 3.72 | 0.14 | 521 | ||||
| PW | Panicle weight(g) | 2014 | 1.79 | 11.01 | 4.91 | 1.62 | 0.33 | 522 | 0.69 | 0.82 |
| 2015 | 1.20 | 12.87 | 5.34 | 1.74 | 0.33 | 522 | ||||
| BS | Breaking strength of 10‐cm basal culm (g) | 2014 | 70.83 | 2758.33 | 875.35 | 447.42 | 0.51 | 522 | 0.74 | 0.85 |
| 2015 | 87.50 | 3400.00 | 1171.49 | 522.59 | 0.45 | 522 | ||||
| CD5 | Culm outer diameter of cross‐section of 5‐cm distance from plant base (mm) | 2014 | 3.45 | 9.07 | 6.13 | 1.10 | 0.18 | 522 | 0.85 | 0.92 |
| 2015 | 3.17 | 9.87 | 6.54 | 1.03 | 0.16 | 522 | ||||
| CT5 | Culm thickness of the cross‐section of 5‐cm distance from the plant base(mm) | 2014 | 0.50 | 1.27 | 0.84 | 0.14 | 0.17 | 522 | 0.75 | 0.86 |
| 2015 | 0.53 | 1.48 | 0.91 | 0.16 | 0.17 | 522 | ||||
| CD1 | Culm outer diameter of cross‐section of the 1st internode from plant base (mm) | 2014 | 3.58 | 9.45 | 6.43 | 1.15 | 0.18 | 522 | 0.83 | 0.90 |
| 2015 | 3.31 | 9.97 | 6.75 | 1.06 | 0.16 | 522 | ||||
| CT1 | Culm thickness of cross‐section of the 1st internode from plant base (mm) | 2014 | 0.54 | 1.36 | 0.91 | 0.15 | 0.17 | 522 | 0.71 | 0.83 |
| 2015 | 0.52 | 1.58 | 0.98 | 0.17 | 0.17 | 522 | ||||
| CD2 | Culm outer diameter of cross‐section of the 2nd internode from plant base (mm) | 2014 | 3.03 | 9.21 | 5.72 | 1.06 | 0.19 | 522 | 0.84 | 0.91 |
| 2015 | 2.92 | 9.42 | 6.24 | 1.01 | 0.16 | 522 | ||||
| CT2 | Culm thickness of cross‐section of the 2nd internode from plant base (mm) | 2014 | 0.41 | 1.12 | 0.68 | 0.12 | 0.17 | 522 | 0.71 | 0.83 |
| 2015 | 0.42 | 1.25 | 0.76 | 0.13 | 0.17 | 522 | ||||
| PN | Panicle number | 2014 | 7.50 | 45.25 | 17.13 | 4.79 | 0.28 | 522 | 0.74 | 0.85 |
| 2015 | 7.00 | 49.00 | 17.64 | 4.78 | 0.27 | 522 | ||||
| SW | Shoot dry weight of three culms harvested from one plant (g) | 2014 | 6.30 | 56.94 | 23.77 | 8.46 | 0.36 | 477 | 0.82 | 0.90 |
| 2015 | 5.98 | 75.33 | 27.92 | 9.09 | 0.33 | 522 | ||||
| TA | Tiller angle(°) | 2014 | 13.00 | 45.00 | 28.07 | 4.72 | 0.17 | 506 | 0.56 | 0.72 |
| 2015 | 15.84 | 47.50 | 29.65 | 5.41 | 0.18 | 424 |
Minimum, Maximum, Mean, SD and CV indicate the minimum value, maximum value, mean value, standard deviation and coefficient of variation of a trait, respectively.
N, Corr and H 2 indicate the number of accessions, correlation coefficient of two‐year phenotypic data and broad‐sense heritability, respectively.
We conducted in‐depth analysis for the three LR‐related traits: LI, GH (from the height traits) and BS (from the culm‐strength traits). The bimodal distribution of GH suggested that there may be a major locus controlling GH; the nearly normal distribution of LI and BS suggested they may be controlled by multiple minor loci (Figure 2a). We observed accessions of four types at the maturity stage, including accessions which had large height and lodged (type I in Figure 2b), accessions which had large height but were still upright (type II), accessions which had small height and were upright (type III), and accessions which had small height but lodged (type IV). Based on the phenotypic data, we found that large BS could protect the accessions with large GH from lodging (such as the accession named ‘Jinzhinuo’ of type II in Figure 2b) and that accessions with small GH still lodged because of small BS (such as the accession named ‘IR 2071‐625‐1‐252’ of type IV in Figure 2b), suggesting the important role of BS in LR. The scatter plot of GH and BS suggested a polynomial regression relationship between them (Figure 2b and Figure S2).
Figure 2.
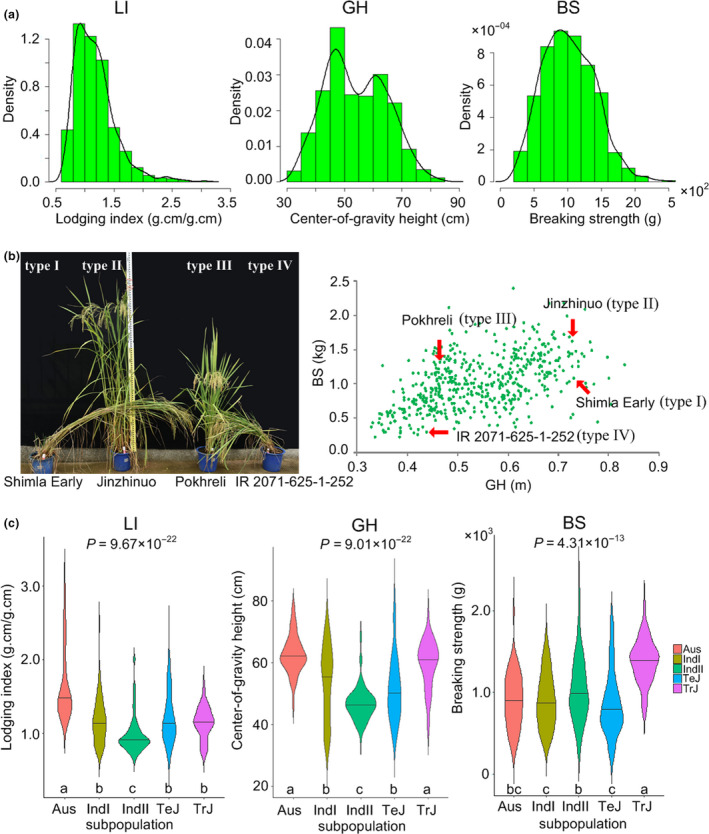
Three representative LR‐related traits‐lodging index (LI), culm gravity‐centre height (GH) and breaking strength of the basal culm (BS). (a) Histograms showing phenotypic distribution of LI, GH and BS across 524 rice accessions. (b) Images of four representative accessions at the maturity stages (left panel) and a scatter plot showing GH and BS values for all the accessions (right panel), of which the red arrows point to the four accessions shown in the left panel. Accessions of type I, II, III and IV represent accessions which had large height and lodged, accessions which had large height but were still upright, accessions which had small height and were upright, and accessions which had small height but lodged, respectively. The names of representative accessions are shown at the bottom of the corresponding images. (c) Violin plots showing phenotypic distributions of different subpopulations for LI, GH and BS, respectively. P values are calculated using Kruskal–Wallis one‐way ANOVA. The different letters indicate significant difference at the P < 0.05 level. [Correction added on 06 January 2021, after first online publication: an incorrect scale value was used in x‐axis of BS of Figure 2(a), and this has been corrected.]
Further, we found phenotypic differentiation of the three traits among different subpopulations (P = 9.67 × 10−22 for LI, P = 9.01 × 10−22 for GH, P = 4.31 × 10−13 for BS, Kruskal–Wallis one‐way ANOVA). Similar to Aus accessions in BS, indica‐II (IndII) accessions showed significantly lower LI levels (low risk of lodging) due to lower GH levels; similar to Aus accessions in GH, tropical japonica (TrJ) accessions showed significantly lower LI levels (low risk of lodging) due to greater BS levels; and there also exists differentiation in GH and BS between indica‐I (IndI) and IndII for indica accessions, and between temperate japonica (TeJ) and TrJ for japonica accessions (Figure 2c). These results indicate that LR‐related genetic loci may have undergone selection.
Genome‐wide association study of LR traits
Linear mixed model (LMM) was adopted to perform GWAS of the 17 lodging‐related traits. After Bonferroni correction based on an effective SNP number, genome‐wide thresholds were set to 1.21 × 10−6, 1.66 × 10−6 and 3.81 × 10−6 for the whole population, indica subpopulation and japonica subpopulation, respectively (Guo et al., 2018). Considering the linkage disequilibrium (LD) decay distance in rice (see Methods), adjacent SNPs spanning less than 300 kb were defined as a single locus to reduce the redundancy of association signals of different traits (Chen et al., 2014; Guo et al., 2018; Wang et al., 2015). In total, 263 associations between 212 lead SNPs (corresponding to 127 loci) and 17 traits were identified in the whole population or at least one of the two subpopulations (Table S2). Some known LR‐related genes were identified. The rice Green Revolution gene, SD1, was identified by GH GWAS in the indica subpopulation (P LMM = 9.05 × 10−9 for the lead SNP sf0138431058) (Figure 3a). Another LR‐related gene controlling tiller angle, TAC1, was identified by tiller angle (TA) GWAS in the whole population (P LMM = 2.96 × 10−11 for the lead SNP sf0920735688) (Figure 3b). Besides, we identified many unreported loci with strong and clear association signals for LR traits (Figure 3c‐f; Table S2). For example, a lead SNP sf0722312558 was significantly associated with BS (P LMM = 4.17 × 10−7), culm outer diameter of the 1st internode cross‐section (CD1, P LMM = 7.03 × 10−9), the 2nd internode (CD2, P LMM = 3.26 × 10‐8), and the 5‐cm distance from the plant base (CD5, P LMM = 1.48 × 10−8), and shoot dry weight of culms (SW, P LMM = 4.60 × 10−7) in the indica subpopulation. Moreover, we explored the SD1 and TAC1 haplotypes. Four haplotypes of SD1 were discovered (Figure 3g). Compared with H1 (haplotype of large GH), H4 (haplotype of small height) corresponds to the null sd1 allele with a 383‐bp deletion, resulting in a frame shift and a premature stop codon (Monna et al., 2002; Sasaki et al., 2002); H3 (haplotype of moderate height) corresponds to the weak sd1 allele with two known functional non‐synonymous SNPs, resulting in amino acid changes (glycine → glutamic acid at residue 100, arginine → glutamine at residue 340) (Asano et al., 2011); no known functional variants were discovered in H2, which showed similar height with H1. Extremely low GA levels decrease not only plant height (thus enhancing LR), but also grain yield (Hirano et al., 2017), so it is necessary to balance the trade‐off by utilizing the SD1 haplotype with moderate effect size, such as H3 in our study. For TAC1, a reported functional SNP has been discovered in 3′‐splicing site of the fourth intron, of which A → G results in decreased mRNA level (Yu et al., 2007). H1 and H2 correspond to allele A and G, respectively. H1 with greater TA comprised most of the indica accessions and all the Aus associations; H2 with less TA comprised almost all the japonica accessions (Figure 3h). So TAC1 may contribute to the divergence between the japonica subpopulation and the indica and Aus subpopulations. Based on the GWAS results, we constructed a comprehensive association network. Of the 127 genetic loci, each locus was associated with 1‐7 traits (Figure 3i). For example, the hub locus 84 was associated with BS, CD1, CD2, CD5, CT1, CT5 and SW (Figure 3j). In total, 73 loci and 34 loci were associated with height and culm traits, respectively, and nine loci were detected for both the height and culm traits. For the culm traits, of the ten loci associated with BS, six loci were also identified by culm diameter and thickness, suggesting that morphological culm traits are highly related to culm physical strength (Table S2). Further, we re‐run GWAS in each environment separately (Table S2; Figure S3). By comparing the GWAS results of two environments, we revealed 180 stable associations between 94 loci and 17 traits (P values < 1.00 × 10−4 in both years), suggesting stable effects of these loci in different environments. For example, the SNP sf0717021526 was associated with TA in both years with the P value 4.27 × 10−8 in 2014 and 5.10 × 10−9 in 2015. Besides, we also revealed 66 loci showing interactions with environment. For example, the SNP sf0431592816 was associated with CT2 with the P value 9.49 × 10−3 in 2014 and 2.70 × 10−8 in 2015.
Figure 3.
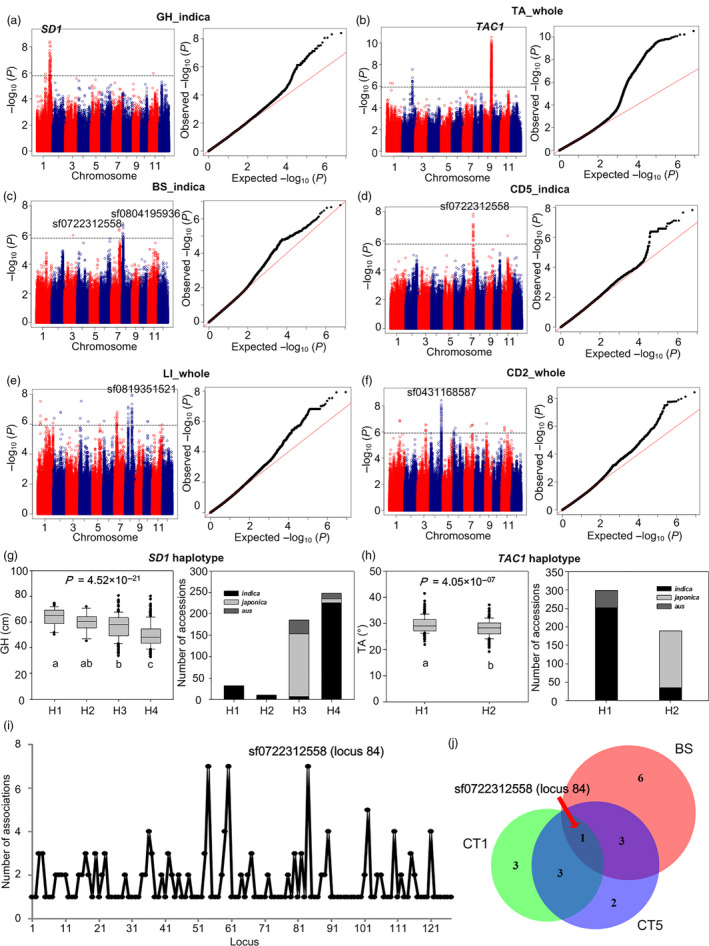
Genome‐wide association study of LR‐related traits. (a) Manhattan plot (left) and quantile–quantile plot (right) for GH in the indica subpopulation. (b) Manhattan plot (left) and quantile–quantile plot (right) for tiller angle (TA) in the whole population. (c) Manhattan plot (left) and quantile–quantile plot (right) for BS in the indica subpopulation. (d) Manhattan plot (left) and quantile–quantile plot (right) for culm outer diameter at the cross‐section at 5‐cm distance from the plant base (CD5) in the indica subpopulation. (e) Manhattan plot (left) and quantile–quantile plot (right) for the lodging index (LI) in the whole population. (f) Manhattan plot (left) and quantile–quantile plot (right) for the culm outer diameter of the 2nd internode (CD2) in the whole population. (g) For each SD1 haplotype, box plot (left) showing the GH phenotypic distribution and histogram (right) showing the number of accessions from different subpopulations. (h) For each TAC1 haplotype, box plot (left) showing the TA phenotypic distribution and histogram (right) showing the number of accessions from different subpopulations. (i) Number of associated traits for each locus in the association network. (j) Venn plot showing the number of co‐localized loci among three traits—BS, culm thickness of the cross‐section of the 1st internode from the plant base (CT1), and culm thickness of the cross‐section at 5‐cm distance from the plant base (CT5). In the Manhattan plot, ‐log10 P values from the genome‐wide association study using linear mixed model are plotted against the position of SNPs, and the horizontal grey dashed line indicates the genome‐wide threshold; the name of a priori gene or lead SNP is shown above the corresponding association signal. In quantile–quantile plot, ‐log10‐transformed observed P values are plotted against ‐log10‐transformed expected P values. In the box plot, P values are calculated based on Kruskal–Wallis one‐way ANOVA for SD1 (g) and t‐test for TAC1 (h); different letters indicate a significant difference of the phenotypic values between haplotypes (P < 0.05).
Genetic relationship between culm and yield‐related traits
We observed that the culm‐strength traits and yield‐related traits were significantly correlated (Figure 4a). To further investigate the genetic relationship between them, we conducted a GWAS of culm traits using panicle weight (PW) or panicle number (PN) as a covariate. We identified a total of 92 loci which were significantly associated with culm‐strength traits, including 34 loci which were detected in GWAS without a covariate and 58 newly identified loci which were detected in GWAS with PW or PN as a covariate (Tables S3 and S4). Of the 92 loci, when adding PW/PN as a covariate, 63 loci showed changed significance with |∆(−log10 P LMM)| ≧ 2 (Figure 4b). Of the 63 loci, when adding PW as a covariate, 32 loci and 28 loci showed decreased and increased significance, respectively; when adding PN as a covariate, 15 loci and 7 loci showed decreased and increased significance, respectively (Figure 4b‐c).
Figure 4.
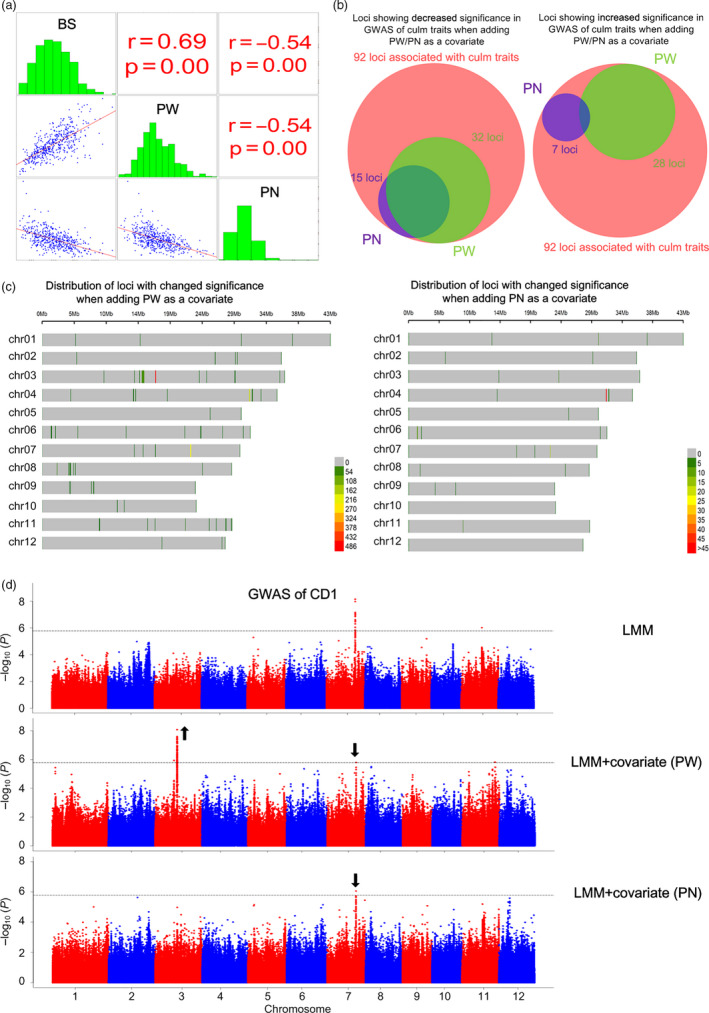
Genetic relationships between culm strength and yield‐related traits. (a) Phenotypic distribution and correlation of BS, PW and PN. The Pearson correlation coefficients and P values are shown. (b) Venn diagrams showing the number of genetic loci linking culm strength and yield‐related traits. The pink circle represents all the loci significantly associated with culm‐strength traits. The purple and green circles represent loci of changed significance (|∆(−log10 P)| ≧ 2, linear mixed model) when adding PN and PW as a covariate, respectively. The left and right panel corresponds to loci with decreased significance (∆(−log10 P) ≦ −2) and increased significance (∆(−log10 P) ≧ 2) in GWAS of culm‐strength traits when adding a covariate, respectively. (c) Chromosome distribution of SNPs of |∆(−log10 P)| ≧ 2 with adding PW (left panel) and PN (right panel) as a covariate. The colour of the vertical line indicates SNP density in a 1 Mb window. (d) GWAS of CD1 using linear mixed model in the indica subpopulation without a covariate (top panel), with adding PW as a covariate (middle panel), and with adding PN as a covariate (bottom panel)..
On one hand, the decreased significance by adding PW/PN as a covariate in the GWAS was attributed to the pleiotropic relationship between culm strength and PW/PN. The effects of these loci were synergistic between culm traits and PW, but were the opposite between culm traits and PN (Table S4). For example, when adding PW/PN as a covariate in GWAS of CD1, the association signal on chromosome 7 was attenuated due to the locus’s pleiotropic effect on CD1 and PW/PN (Figure 4d). And the high‐CD1 allele of the locus could enhance culm strength and PW but reduce PN. Further, we identified 19 loci which had synergistic effects on culm strength and PW without negative effect on PN. These loci may be very valuable for LR and high‐yield breeding.
On the other hand, when adding PW/PN as a covariate in the GWAS, the increased significance can indicate the improvement of statistical power due to reduced residual variance by adjusting for heritable covariate (Mefford and Witte, 2012). While only 34 loci were detected in the GWAS of culm‐strength traits without a covariate, 58 additional loci were identified when using PW or PN as a covariate (Table S4). For CD1 in the indica subpopulation (Figure 4d), for example, there is no significant association signal on chromosome 3, but when adding PW as a covariate, a new strong association signal appeared (P value of the lead SNP sf0317021899 was 8.83 × 10−9 when using PW as a covariate) (Figure 4d). These results indicate that adding heritable covariate in GWAS can provide a more complete landscape of genetic basis. To validate the GWAS results of culm‐strength traits with adding PW/PN as a covariate, we conducted GWAS of PW and PN by adding culm‐strength trait as a covariate. The P value changes of loci, which were caused by adding a covariate in the GWAS, were highly correlated between the bi‐directional covariate‐added GWAS (Pearson correlation coefficient R = 0.99, P < 0.001; Table S5). To reduce false‐negative associations brought by the LMM due to overcompensation for relatedness, we also adopted a linear regression model (LRM), which took population structure into consideration, to conduct GWAS of culm‐strength traits with and without PW/PN as a covariate (Table S6). As a result, we identified a total of 400 loci linking culm‐strength traits with PW/PN, which could cover true loci that were masked by LMM.
It is of significance for LR breeding to explore beneficial alleles for dual‐purposes: enhanced culm strength and high yield. We found SNP sf0729627489 was significantly associated with BS when using PW as a covariate (P LRM = 1.70 × 10−11), but was not associated without a covariate (P LRM = 8.78 × 10−4). The lead SNP was located in a gene and caused amino acid change (TAC → CAC, Lyr → His) (Figure 5a; Table S7). The major and minor allele of the SNP was T and C, respectively, and the minor allele frequency (MAF) was 0.06. Further, we found that this gene was allelic to OsPRR37 (LOC_Os07g49460), which encodes pseudo‐response regulator 7‐like protein and enhances yield via increasing PW (Yan et al., 2013; Zhang et al., 2019). To verify the effect of allelic variation at this locus on culm strength, a near‐isogenic line NILTQ harbouring high‐yielding allele was obtained. We observed the culm cross‐section of NILTQ and its recurrent parent Zhenshan 97 (Oryza sativa L. ssp. indica) at the whole plant level at a high spatial resolution using computed tomography (Figure 5b). Significant differences in BS (P = 3.20 × 10−3, t‐test), CT1 (P = 7.10 × 10−3), CT2 (P = 2.28 × 10−2), CT5 (P = 2.01 × 10−3), CD2 (P = 2.54 × 10−2) and CD5 (P = 1.63 × 10−3) between the NILTQ and Zhenshan 97 were found (Figure 5c), demonstrating that the allele from the donor parent (Teqing) enhances both culm strength and yield, which is valuable for post‐Green‐Revolution breeding. To explore possible causal variants, we compared the OsPRR37 sequences from the two parents. Compared to high‐culm‐strength allele (reference allele) harboured by the donor parent (Teqing), an 8‐bp deletion (GAACGTTG) in the seventh exon of OsPRR37 was found for the low‐culm‐strength allele harboured by the recurrent parent (Zhenshan 97), which causes frame shift and thus premature stop codon, producing a truncated protein without CCT domain. Moreover, the genotypes of the 8‐bp INDEL were obtained by variant calling from whole‐genome re‐sequencing data (Table S8). The INDEL was significantly associated with BS (P LRM = 5.63 × 10−7), and when adding heading date as a covariate, the association signal was largely attenuated (P LRM = 6.26 × 10−2; Table S9). Considering that the 8‐bp INDEL is the causal variant of OsPRR37 in controlling heading date (Yan et al., 2013), we proposed that the 8‐bp INDEL of OsPRR37 may have a pleiotropic effect on culm strength and heading date.
Figure 5.
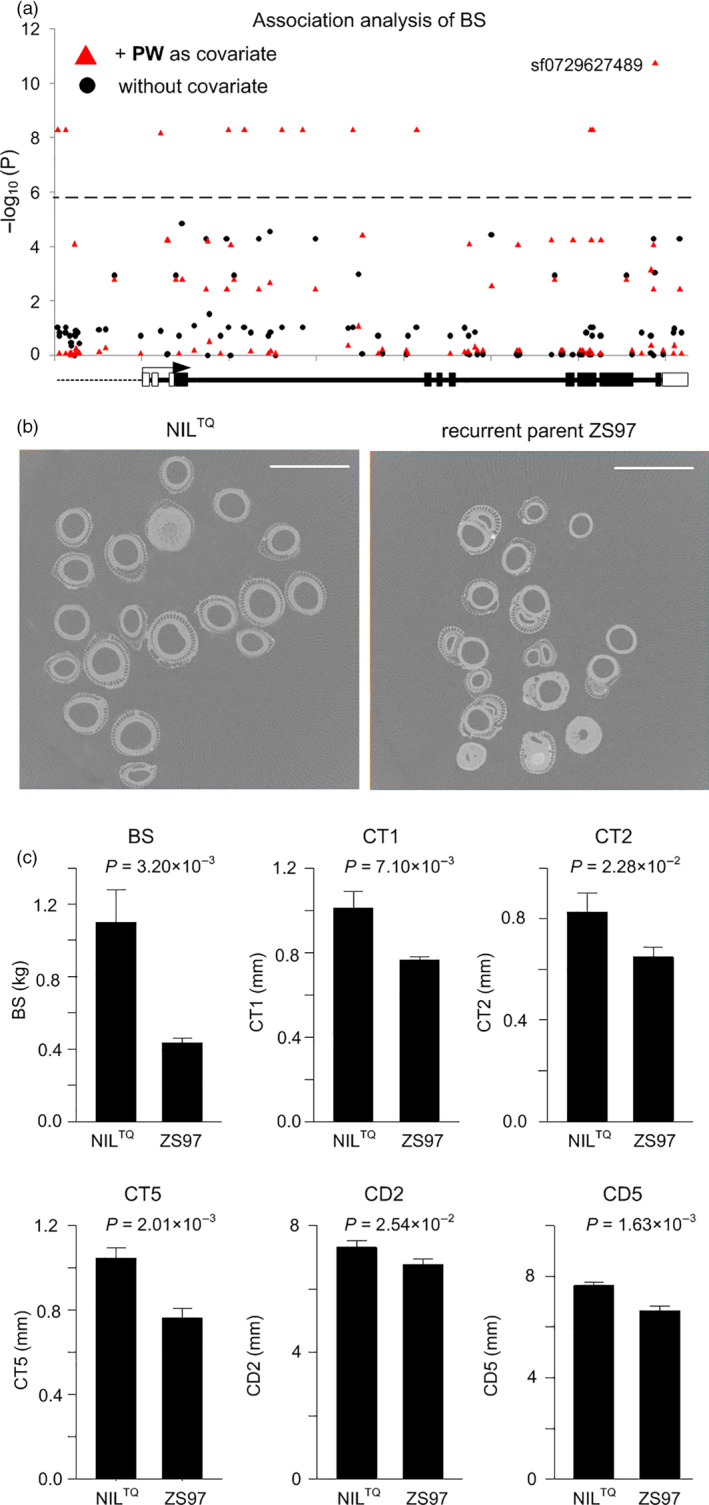
Associations between culm strength and the candidate gene OsPRR37. (a) Local Manhattan plot showing the associations between BS and SNPs across the gene body and the 2‐kb upstream region for the candidate gene OsPRR37 (LOC_Os07g49460) without a covariate (black dots), and with adding PW as a covariate (red triangles) using a linear regression model taking the population structure into account. For gene structure, dotted line, solid lines, white rectangles, black rectangles and black arrow indicate a 2‐kb promoter region, introns, UTRs, exons and transcription direction, respectively. (b) Culm cross‐sections of NILTQ (near‐isogenic line constructed by consecutive backcrossing with Zhenshan 97 as a recurrent parent and TQ as a donor) and its recurrent parent Zhenshan 97, observed by high‐resolution computed tomography. Scale bars, 10 mm. (c) Comparison of phenotypic values of BS, CT1, CT2, CT5, CD2 and CD5 between the NILTQ and Zhenshan 97. P values from the t‐test are shown.
Genetic basis underlying LR subpopulation differentiation
Subpopulation differentiation in LR was obviously observed (Figure 2c). To reveal its genetic basis, we firstly performed the genome‐wide population differentiation analysis (F ST) between the IndI and IndII subpopulations (IndI‐IndII), between TeJ and TrJ (TeJ‐TrJ), between Aus and IndII (Aus‐IndII), and between Aus and TrJ (Aus‐TrJ). The greatly divergent regions (top 5% F ST) (F ST ≥ 0.2254, 0.5019, 0.4724, 0.6718, for IndI‐IndII, TeJ‐TrJ, Aus‐IndII and Aus‐TrJ, respectively) were identified (Table S10). In this study, 11 GWAS loci were co‐localized with the greatly divergent regions, and the allele frequency difference at the lead SNP site between the corresponding subpopulations was more than 0.40 (Table S11). For example, significant differences of LI and CD5 were observed for IndI‐IndII (P = 1.97 × 10−6, t‐test) and TeJ‐TrJ (P = 1.13 × 10−16), respectively. The lead SNP sf0142424010 was significantly associated with LI and localized in an IndI‐IndII greatly divergent region (the SNP major alleles in IndI and IndII were T and C respectively), and the SNP allele frequency difference between the two subpopulations was 0.54 (Figure 6a). The lead SNP sf0129912226 was significantly associated with CD5, was located in a TeJ‐TrJ greatly divergent region (the major allele in TeJ and TrJ was A and C, respectively), and the allele frequency difference between the two subpopulations was 0.43 (Figure 6b). IndI and IndII are two putative heterotic groups and have undergone independent breeding efforts (Xie et al., 2015), and TeJ and TrJ accessions diverge due to geo‐environmental adaptation (Civan et al., 2015). We speculate that the LR‐related loci may have undergone divergent selection during local breeding or geographic adaptation.
Figure 6.
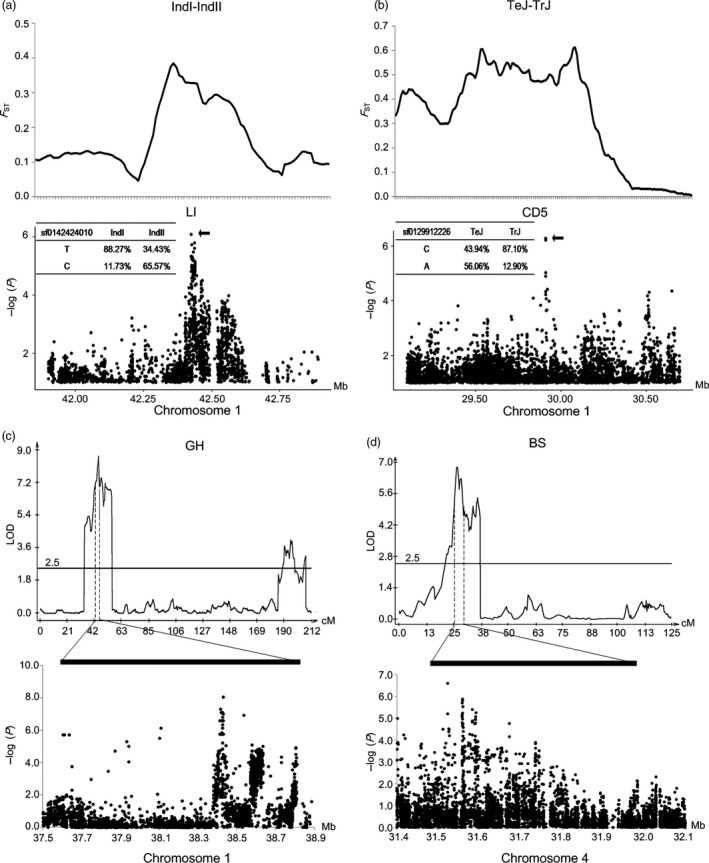
GWAS loci co‐localized with greatly divergent regions identified by F ST and QTLs identified by linkage analyses. (a) A greatly divergent region between the IndI and IndII subpopulations identified by F ST (first row) covers a GWAS locus associated with LI (second row). (b) A greatly divergent region between the TeJ and TrJ subpopulations identified by F ST (first row) covers a GWAS locus associated with CD5 (second row). (c) A QTL on chromosome 1 controlling GH is identified by both linkage analysis (first row) and association analysis (second row). (d) A QTL on chromosome 4 controlling BS is identified by both linkage analysis (first row) and association analysis (second row).
Linkage analysis in bi‐parental mapping population
To further verify the GWAS results, we phenotyped LR in a bi‐parental mapping population grown in the field. The population was derived from a cross between the japonica variety IRAT109 and the indica variety Zhenshan 97, and was comprised of 193 recombinant inbred lines. We found that nine QTLs identified by linkage analysis were overlapped with GWAS loci, which were distributed on chromosomes 1, 4, 7, 8 and 11 (Table S12). For example, a strong QTL on chromosome 1 (peak at 45.21 cM, logarithm of odds [LOD] = 8.68) controlling GH, was co‐localized with the GWAS locus 20 associated with GH (lead SNP sf0138431058, P LMM = 9.05 × 10‐9) (Figure 6c). The QTL also controlled plant height (PH) in both linkage analysis and the GWAS. Another strong QTL on chromosome 4 (peak at 26.41 cM, LOD = 6.80) controlling BS, was co‐localized with the GWAS locus 54 associated with BS (lead SNP sf04315237458, P LMM = 2.65 × 10−7) (Figure 6d). The QTL also controlled CD1, CD2 and CD5, which was confirmed by both linkage analyses and GWAS. For co‐localized QTLs, the lengths of candidate regions of GWAS were smaller than those of linkage analysis (Figure 6c‐d; Table S12), suggesting that the mapping resolution of GWAS is greater than that of linkage analysis in the study.
Discussion
In this study, we phenotyped a large rice natural population for 17 LR‐related traits across two years, which were comprised of height‐, culm‐strength‐ and yield‐related traits. Great heritability and great phenotypic variations in LR ensure the dissection of genetic basis and exploration of natural genetic variations. Regarding the relevance of LR‐related traits under pot and field growing conditions, we performed a field trial of LR‐related traits for 96 accessions from the natural population, of which agronomic practices were the same as the RIL population in the field mentioned above. High correlation coefficients of LR‐related traits, such as for BS (R = 0.75, P < 0.001) under pot and field growing conditions were observed (Figure S4), suggesting the genetic loci under pot‐growing condition may be useful for LR breeding in the field. A number of genetic loci associated with LR were identified by GWAS. For each locus, multiple alleles of varying effect sizes can be identified from germplasm collections. For example, the ‘Green Revolution’ gene SD1 was significantly associated with height traits, and four major haplotypes with different effect sizes were identified. Lodging is a well‐known problem of tall varieties harbouring a dominant SD1 allele, and dwarf varieties harbouring an sd1 allele which largely disrupts GA synthesis would have higher LR level but lower grain yield due to smaller biomass. Therefore, exploration of other sd1 alleles which only weaken GA synthesis may be an effective strategy to balance the negative trade‐off. To validate our GWAS results, linkage analysis was carried out using a RIL population phenotyped in the field. Although the RIL population was phenotyped in one season, a large proportion of the identified QTLs overlapped with GWAS loci, which further supported the reliability of the genetic loci of LR identified by GWAS. Significant differences in LR across subpopulations were observed. Through GWAS and genome‐wide F ST analysis, we revealed genetic loci which could underlie the subpopulation differentiation in LR (Huang et al., 2012; Xie et al., 2015).
Most genetic studies on LR only focused on genetic loci associated with LR itself and neglected the relationship between LR and yield in rice. In crop breeding, the relationship between traits should be considered to enhance desirable correlated traits and simultaneously to reduce undesirable trade‐offs (Chen and Lubberstedt, 2010). A published study on panicle traits in rice found that some association signals of panicle traits were attenuated when adding flowering time as a covariate in GWAS, which was attributed to the relationship between flowering time and panicle traits (Crowell et al., 2016). In our study, considering positive correlation between culm strength and PW and negative trade‐off between culm strength and PN based on the phenotypic data, we aimed to identify genetic loci with pleiotropic effects on culm strength and yield by adding PW/PN as a covariate in GWAS of culm‐strength traits, which is of significance to breed for lodging‐resistant and high‐yielding rice cultivars. Association signals of 32 loci associated with culm traits were attenuated when adding PW as a covariate and these loci had synergistic effects on culm strength and PW, suggesting that favourable alleles for these loci could be used to enhance both culm strength and PW. Of the 32 loci, however, 13 loci had negative effects on PN. So the remaining 19 loci had synergistic effects on culm strength and PW but without negative effects on PN, which are very useful for the dual‐purpose breeding of enhanced LR and high yield. As a proof, it was demonstrated that an allele from the rice variety Teqing could enhance both culm strength and yield using a NIL, though the candidate gene OsPRR37 remains to be further confirmed for its function in LR. Regarding the loci associated with culm strength but without effects on yield, the favourable alleles of the loci can be introgressed to high‐yielding rice cultivars or pyramiding of the favourable alleles and high‐yielding alleles can be employed.
On the other hand, new association signals could be detected when adding PW/PN as a covariate, which resulted in 58 additional loci controlling culm‐strength traits. Adjusting for heritable covariates in GWAS can reduce residual variance of a trait of interest and thus improve statistical power (Mefford and Witte, 2012), which provides a more complete genetic landscape. Though it was not the theme of our study, we performed GWAS of 17 LR‐related traits by adding heading date as a covariate to understand the relationship between LR‐related traits and phenology. As a result, we found 42 loci with changed significance of |∆(‐log10 P LMM)| ≧2. Of the 42 loci, while 33 loci linked height traits with heading date, only nine loci linked culm‐strength traits with heading date (Table S13). Considering the most recently published GWAS studies in plants were performed without adjusting for heritable covariate, our study provides insights into the important role of heritable covariate in GWAS.
Fast and accurate quantification of LR is critical for genetic and breeding studies. To visualize the culm cross‐section structure of the NIL and its recurrent parent in‐situ at the whole plant level, we used high‐resolution computed tomography instead of a traditional destructive approach (tissue sampling, staining and observation by digital microscope). In the study, it takes 0.6 s to acquire one CT image, and 380 X‐ray projected images of different orientations for a plant can be obtained within 4 min. In the future, with an automatic conveyer for transport, the images of 300 plants will be acquired in one day (20 running hours per day), which is estimated based on the current efficiency of image acquisition and the required number of images for a plant. After reconstructing and segmenting all the images by a series of algorithm, we shall extract a number of culm traits of interest for a rice mapping population. New phenotyping technologies based on optoelectronics and imaging, with non‐destructive and high‐throughput characteristics, will facilitate crop genetic and genomic research and breeding.
Experimental procedures
Plant material and genotype data
The rice natural population used in the study was comprised of 524 accessions (Table S14), which has been described in previous reports (Chen et al., 2014; Xie et al., 2015) and is available at National Key Laboratory of Crop Genetic Improvement, Huazhong Agricultural University, Wuhan, China. A total of 188 637 evenly distributed SNPs were selected and the population structure was inferred using ADMIXTURE based on these SNPs. For the SNPs’ selection, the genome was divided into 100 000 regions (~3.8 kb in length for each region), and then, SNPs with minor allele frequencies (MAF) ≥0.01 were randomly selected in each region. The number of ancient cluster K was set from two to seven to infer population structure. The possible suitable K value was estimated using ADMIXTURE (Alexander and Lange, 2011) (Figure S5) and STRUCTURE coupled with StructureHarvester (http://taylor0.biology.ucla.edu/struct_harvest/ or https://github.com/dentearl/structure‐Harvester) (Evanno et al., 2005) (Figure S6), respectively. When K = 2, accessions were divided into indica and japonica groups; when K = 3, the aus cluster appeared within the indica group; when K = 4, the indica were further divided into two sub groups (IndI and IndII), which were two putative heterotic groups (Xie et al., 2015); when K = 5, japonica were divided into two sub groups, corresponding to tropical japonica and temperate japonica; when K = 6, an independent group (VI) emerged, which is an intermediate group between indica and japonica; only 14 accessions belonged to VI and nine of them harboured mutated fragrance gene fgr, suggesting that VI corresponds to Aromatic group (Garris et al., 2005; Glaszmann, 1987); when K = 7, a new group appeared within the japonica group but the group was not a known rice subpopulation and there was no a priori knowledge about it. K = 6 was the minimum value to cover all previously known groups (tropical japonica, temperate japonica, IndI, IndII, Aus and Aromatic). Considering the clear biological interpretation of K value (Wang et al., 2018), the number of ancient cluster K was set to six in the study. Each accession was classified based on the maximum subpopulation component value. Accessions with the difference between maximum and secondary subpopulation component value less than 0.4, were classified as intermediate. As a result, the population was mainly classified into five major subpopulations: Aus (46 accessions), IndI (indica‐I, 98 accessions), IndII (indica‐II, 104 accessions), TeJ (temperate japonica, 89 accessions) and TrJ (tropical japonica, 44 accessions). IndI and IndII are two putative heterotic groups caused by independent breeding efforts, and have South China and International Rice Research Institute (IRRI) origins, respectively (Xie et al., 2015). TeJ and TrJ are two divergent japonica versions due to adaptation to distinct climate conditions; Aus was domesticated independently of indica and japonica, and has its distinct geographic origins (Civan et al., 2015).
The whole genomes of all the accessions have been re‐sequenced using an Illumina Hiseq 2000 platform. Approximately 1 Gb high‐quality sequences were obtained for each accession, with depth >2.5 × per genome. Paired‐end 90‐bp reads were aligned to the rice reference genome Nipponbare (Oryza sativa L. ssp. japonica) MSU version 6.1 using software BWA (Li and Durbin, 2009). SNP calling was conducted using SAMtools and BCFtools, with parameters of ‐C50 ‐Q10 ‐q40 for the mpileup subcommand of SAMtools (Li et al., 2009). Genotype imputation was performed based on k nearest neighbour algorithm (Roberts et al., 2007). SNPs were annotated using SnpEff (Cingolani et al., 2012). All the sequencing data could be downloaded from NCBI Sequence Read Archive under accession number PRJNA171289. The detailed genotype data (BCF format) can be downloaded from the RiceVarMap database (http://ricevarmap.ncpgr.cn/v1) (Zhao et al., 2015).
Phenotyping
For LR phenotyping of the 524 rice accessions, clean seeds were soaked in water for 1 d and then incubated for 1 d. The pre‐germinated seeds were sown in the nurseries, and four healthy and uniform 20‐day‐old seedlings for each accession were transplanted to pots at 2 cm depth. Each pot was filled with 5 kg soil, which was air‐dried, pulverized and well‐mixed with organic fertilizer in advance. Irrigation was applied to each pot to keep soil saturated with water for one week before seedlings transplanting and 150 mL nutrient resolution (5.4 g urea: 3.2 g potassium dihydrogen phosphate: 2.2 g potassium chlorate/L) was applied to each pot one day before transplanting. After transplanting, irrigation was applied to keep standing water of 3–5 cm depth in pots, and the irrigation frequency depended on weather, environmental temperature and transpiration of plants. Topdressing 150 mL nutrient resolution (2.7 g urea: 1.6 g potassium dihydrogen phosphate: 1.1 g potassium chlorate/L) to each pot when most accessions entered active tillering stage and panicle initiation stage, respectively. Weeding was performed manually, and pests were controlled by spraying pesticides, and diseases were intensively controlled by spraying chemicals. The pot experiments were conducted outdoors and plants were grown in the natural rice‐growing season at the experimental station in Huazhong Agricultural University, Wuhan, Hubei Province, China (30°28′N 114°20′E). The experiment followed a randomized complete block design with four replications and each replication contained one plant for each accession. For each accession, the heading process was traced and recorded from the start of heading to the date on which panicle emergence was completed. The date on which 80% of the total panicles were emerged from the flag leaf sheath, was defined as the full heading date (Table S14). The phenotyping was started at 25 days after the full heading date for each accession.
Plant height (the longest length from the plant base to the top of the highest leaf or panicle, whichever is longer), panicle number and tiller angle were measured. A protractor was used to measure the angle between the most distant tillers bearing panicles on the two sides of the culm base, and half of the angle was treated as the tiller angle of an individual plant (Dong et al., 2016). For each accession, a total of 12 culms with leaf, leaf sheath and panicle (three largest culms were harvested from each plant and four plants were harvested) were collected in 2014; a total of six culms with leaf, leaf sheath and panicle (three largest culms were harvested from each plant and two plants were harvested) were collected in 2015. For each culm, the gravity‐centre height (the distance from culm base to gravity‐centre) was measured using a ruler, and the balance fulcrum of a horizontally put culm with panicle, leaf and leaf sheath was marked as the gravity‐centre. The internodes were counted from culm base upward (Pan et al., 2019; Zhong et al., 2020). The first internode length (IL1), the second internode length (IL2) and the panicle length (PL) were measured using a ruler (Figure 1a‐c). Then each culm was cut to measure the panicle fresh weight (PW), breaking strength (BS) of a 10‐cm basal culm with leaf sheath using DIK‐7401 (Japan) (Figure 1c). Culm outer diameter (CD) and culm thickness (CT) of a cross‐section of the middle point of the 1st internode, 2nd internode, and 10‐cm basal culm were measured using a caliper (Digital Caliper 0–150 mm, Guanglu111‐101B, China) after removing leaf sheath (Figure 1d). Because the culm cross‐section is not a standard circle, the outer diameter was calculated as the mean of the major axis length and minor axis length. Referring to a previous study (Ookawa & Ishihara, 1992), the lodging index (LI) was calculated as LI = bending moment/ (BS × the distance between fulcra (9 cm in this study) × 1/4), of which the bending moment was length × fresh weight from the basal culm breaking point (where BS was measured) to the panicle top. Then the culm was dried in an oven at 105 °C for 30 min and at 80 °C for 72 h to measure the shoot dry weight for three culms harvested from one plant (SW). The trait mean values of culms for each accession (12 culms in 2014 and six culms in 2015 for each accession) were calculated. To reduce the effect of environment on phenotypic data, best linear unbiased prediction (BLUP) value for each trait was calculated based on the phenotypic data across two years using R package ‘lme4’ (Fang et al., 2017; Liu et al., 2020; Wang et al., 2019).
To visualize the culm cross‐section structure in‐situ at the whole plant level for NILTQ and its recurrent parent Zhenshan 97, high‐resolution computed tomography was used to horizontally scan the culm structure (Wu et al., 2019). The micro‐CT imaging facility used in the study includes an X‐ray source (Nova600, Oxford Instruments, UK), an X‐ray source chiller (Nova600, Oxford Instruments, UK), an X‐ray flat panel detector (PaxScan 2520DX, Varian Medical Systems, Inc). The image acquisition and processing pipeline was developed using LabVIEW 8.6 (National Instruments, Inc.). A total of 380 X‐ray projected images of different orientations for a plant were obtained within 4 min (0.6 s per image). Based on the images, a filtered back‐projection (FBP) algorithm was adopted to acquire reconstructed transverse section images of rice culms. After image segmentation, the culm cross‐section structure of a plant was shown.
Phenotypic data analysis
We calculated the trait mean values of all the harvested culms for each accession in each year and then calculated the broad‐sense heritability (H 2) as: H 2 = V G/(V G + V e/N), where V G and V e represented genetic and residual variance, respectively; N was the number of years (N = 2 in this study) (Liu et al., 2020). The VG and Ve were estimated by treating genotype and environment as random effects in the mixed linear model.
A heatmap was drawn using HemI software (Deng et al., 2014) after linear normalization of the phenotypic data was conducted. Linear normalization: y = (x‐min)/(max–min) in which x, y, max and min indicated raw phenotypic data, normalized data, maximum and minimum, respectively. Pearson correlation analysis, independent‐sample t‐test and Kruskal–Wallis one‐way ANOVA were conducted using IBM SPSS version 19 (IBM, Armonk). Path analysis was conducted to predict the direct and indirect effects of traits on lodging index (LI) using R packages ‘agricolae’. Polynomial regression was conducted using the poly function in R. The correlation coefficient matrix diagram and violin plot of phenotypic data across subpopulations were drawn using R packages of ‘corrplot’ and ‘ggplot2’.
Genome‐wide association study
Either SNPs with the minor allele frequency (MAF) less than 0.05 or SNPs with the minor allele count (defined as number of accessions harbouring the minor allele) less than 6 were removed from association analysis. A total of 4 358 600, 2 863 169 and 1 959 460 SNPs remained for GWAS in the whole, indica, and japonica populations, respectively. With the factored spectrally transformed linear mixed models software (FsST‐LMM) (Lippert et al., 2011), GWAS was performed using a linear mixed model considering the kinship estimated by genetic similarities as random effect. Based on an effective SNP number (Ne) calculated by a GEC tool (Li et al., 2012), the genome‐wide thresholds were set to 1.21 × 10‐6, 1.66 × 10‐6, 3.81 × 10‐6 in the whole, indica, and japonica populations, respectively.
LD decay varies among different populations, different chromosomes, and different genomic regions in the same chromosome, so for a certain locus, the resolution is determined by local LD decay level. In the study, independent lead SNPs were determined by removing redundant SNPs using ‘—clump’ function of Plink, and candidate regions of association signals were determined based on local LD level using ‘‐‐clump‐range’ function of Plink (Purcell et al., 2007). However, for convenience of presentation and summary of GWAS results, a locus was usually defined as a chromosomal region at which adjacent pairs of associated SNPs are less than a certain physical distance without considering varying LD levels (Yang et al., 2012). The median physical distance when r 2 decay to 0.2 was approximately 150 kb (167 kb, 93 kb, 171 kb in the whole population, indica subpopulation and japonica subpopulation, respectively), which is in agreement with previous reports about LD in rice (Huang et al., 2010; Xie et al., 2015). Thus association signals could appear within 150 kb upstream or 150 kb downstream of the causative variant/gene, so we chose 300‐kb distance as the range for identifying overlapping marker‐trait association signals, which is the same with previous reports of the same mapping population (Chen et al., 2014; Guo et al., 2018; Wang et al., 2015). An association network based on significant associations between loci and LR‐related traits was constructed (Guo et al., 2018).
Haplotypes were determined based on the SNPs with a MAF ≥ 0.05 in the genomic region covering the gene body and 1‐kb upstream region. The haplotype group comprised of less than ten accessions was removed. The comparison of phenotypic values across multiple haplotype groups was conducted using Kruskal–Wallis one‐way ANOVA (for ≥ three haplotypes) or t‐test (for two haplotypes) using SPSS software version 19 (IBM).
To study the genetic relationships between culm traits and yield components, GWAS of culm‐strength traits using PW/PN as a covariate was performed by adding the function ‘‐covar’ in the FaST‐LMM program. To reduce the false negatives brought by the linear mixed model due to overcompensation for relatedness (kinship), the linear regression model considering the population structure (Q matrix) as a fixed effect was also adopted for culm‐strength traits using the functions ‘‐linreg’ and ‘‐covar’ in the FaST‐LMM program. The Q matrix was calculated based on 188,165 randomly distributed SNPs on the rice genome using the ADMIXTURE program (Alexander et al., 2009; Zhao et al., 2015).
To confirm the association between the association locus and culm strength, the NILTQ (BC6F2) was obtained and investigated for culm‐strength traits, which was constructed by consecutive backcrossing with Zhenshan 97 (Oryza sativa L. ssp. indica, containing the non‐functional allele for the gene) as the recurrent parent and Teqing (Oryza sativa L. ssp. indica, containing the high‐yielding allele for the gene) as the donor (Yan et al., 2013; Zhang et al., 2019).
Genome‐wide population differentiation analysis
To identify subpopulation divergent regions, genome‐wide population differentiation (F ST) analysis was performed using VCFtools (Danecek et al., 2011) with a 100‐kb window and 10‐kb step size. The genomic regions with the top 5% F ST were defined as greatly divergent regions.
Linkage analysis
A linkage mapping population comprised of 193 F9 RILs was developed from a cross between the indica variety Zhenshan 97 and the japonica variety IRAT109 (Zou et al., 2005). The linkage map length is 1567 centimorgans (cM) and the number of bins is 2499, with 0.63 cM per bin on average.
The experiment followed a randomized complete block design with two biological replications in the field. For each line of one replication, 20 healthy and uniform 20‐day‐old seedlings were transplanted in a four‐row plot at 2‐cm depth with 20‐cm distance between adjacent plants and 27‐cm distance between adjacent rows. Regarding fertilization, 450 kg/ha commercial compound fertilizer (N/P2O5/K2O = 15:15:15) was applied one day before transplanting as basal fertilizer, and 150 kg/ha and 75 kg/ha urea, as top‐dressed fertilizer, were applied when most accessions entered middle tillering stage and panicle initiation stage, respectively. For the water management, 5–10 cm water depth was kept in the field from transplanting to two weeks before harvest. Weeding was performed manually, and pests were controlled by spraying pesticides, and diseases were intensively controlled by spraying chemicals. At 25 days after the full heading date, the largest culm with leaf, leaf sheath and panicles was harvested from one plant. Four plants in a plot were investigated, and thus, four culms were collected for each line of one replication. The phenotyping procedure of the RIL population was the same as that of the natural population mentioned above. For each plot, the trait mean values of culms from four plants were calculated. The best linear unbiased prediction (BLUP) values based on the mean values of LR‐related traits across two biological replications were calculated for QTL mapping.
QTL mapping was performed with WinQTLcart v2.5 using the composite interval mapping method (Model 6: Standard Model) (Silva et al., 2012). The backward regression method and 0.5 cM walking speed were chosen. To control the background, the control marker number and window size were set to 5 and 10 cM, respectively. The genome‐wide LOD threshold was set to 2.5, and a two‐LOD drop support interval was determined as a QTL.
Conflict of interest
The authors declare no conflicts of interest.
Author contributions
Z.G. designed the experiments, conducted all the data analysis and wrote the manuscript; X.L. and X.Y. performed the phenotyping; B.Z. and Y.X. participated in the construction of the near‐isogenic line. H.L. and L.L. provided seeds and genotypic data of the RIL population. G.C. and L.X. designed the experiment, conceived the project and supervised the study; L.X. wrote the manuscript. Z.G. and X.L. contributed equally. All the authors carefully read and discussed the manuscript. The authors declare no competing financial interests.
Supporting information
Figure S1. Path diagram showing the effects of PH and BS on LI.
Figure S2. Polynomial regression of GH and BS.
Figure S3. Manhattan plots of LR‐related traits in 2014 and 2015.
Figure S4. Scatter plots showing the correlations of LR‐related traits for 96 accessions under pot and field growing conditions.
Figure S5. Cross‐validation errors of successive K values using ADMIXTURE.
Figure S6. Statistic ΔK values of successive K values using STRUCTURE and Structure Harvester.
Table S1. Coefficients and residual effect of path analysis from 16 traits to lodging index (LI).
Table S2. GWAS results of 17 LR‐related traits without a covariate using a linear mixed model.
Table S3. GWAS results of culm‐strength traits with PW or PN as a covariate using a linear mixed model for the 34 loci identified by GWAS without a covariate.
Table S4. GWAS results of culm‐strength traits with and without adding PW or PN as a covariate using a linear mixed model.
Table S5. Bi‐directional validation of covariate‐added GWAS using a linear mixed model.
Table S6. GWAS results of culm‐strength traits with and without adding PW or PN as a covariate using a linear regression model considering the population structure.
Table S7. Association results between the candidate gene OsPRR37 and BS with and without adding PW as a covariate using a linear regression model considering the population structure.
Table S8. Genotypes of the 8‐bp INDEL in OsPRR37 (vf0729626365) obtaind by variant calling from whole‐genome re‐sequencing data.
Table S9. Association results between the candidate gene OsPRR37 and BS with and without adding heading date as a covariate using a linear regression model considering the population structure.
Table S10. Genome‐wide divergent regions of the top 5% F ST..
Table S11. GWAS loci overlapping with greatly divergent regions identified by F ST.
Table S12. LR‐related QTL overlapping with GWAS loci.
Table S13. GWAS results of LR‐related traits with adding heading date as a covariate using a linear mixed model.
Table S14. Detailed information of 524 accessions in the study.
Acknowledgements
This work was supported by grants from the National Natural Science Foundation of China (31771359, 31821005), and the National Key Research and Development Program of China (2016YFD0100600).
Guo, Z. , Liu, X. , Zhang, B. , Yuan, X. , Xing, Y. , Liu, H. , Luo, L. , Chen, G. and Xiong, L. (2021) Genetic analyses of lodging resistance and yield provide insights into post‐Green‐Revolution breeding in rice. Plant Biotechnol J, 10.1111/pbi.13509
Contributor Information
Guoxing Chen, Email: chenguoxing@mail.hzau.edu.cn.
Lizhong Xiong, Email: lizhongx@mail.hzau.edu.cn.
References
- Alexander, D.H. and Lange, K. (2011) Enhancements to the ADMIXTURE algorithm for individual ancestry estimation. BMC Bioinform. 12, 246. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alexander, D.H. , Novembre, J. and Lange, K. (2009) Fast model‐based estimation of ancestry in unrelated individuals. Genome Res. 19, 1655–1664. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Asano, K. , Yamasaki, M. , Takuno, S. , Miura, K. , Katagiri, S. , Ito, T. , Doi, K. et al. (2011) Artificial selection for a green revolution gene during japonica rice domestication. Proc. Natl Acad. Sci. USA, 108, 11034–11039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen, W. , Gao, Y. , Xie, W. , Gong, L. , Lu, K. , Wang, W. , Li, Y. et al. (2014) Genome‐wide association analyses provide genetic and biochemical insights into natural variation in rice metabolism. Nat. Genet. 46, 714–721. [DOI] [PubMed] [Google Scholar]
- Chen, Y. and Lubberstedt, T. (2010) Molecular basis of trait correlations. Trends Plant Sci. 15, 454–461. [DOI] [PubMed] [Google Scholar]
- Cingolani, P. , Platts, A. , le Wang, L. , Coon, M. , Nguyen, T. , Wang, L. , Land, S.J. et al. (2012) A program for annotating and predicting the effects of single nucleotide polymorphisms, SnpEff: SNPs in the genome of Drosophila melanogaster strain w1118; iso‐2; iso‐3. Fly, 6, 80–92. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Civan, P. , Craig, H. , Cox, C.J. and Brown, T.A. (2015) Three geographically separate domestications of Asian rice. Nature Plants, 1, 15164. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Crowell, S. , Korniliev, P. , Falcao, A. , Ismail, A. , Gregorio, G. , Mezey, J. and McCouch, S. (2016) Genome‐wide association and high‐resolution phenotyping link Oryza sativa panicle traits to numerous trait‐specific QTL clusters. Nat. Commun. 7, 10527. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Danecek, P. , Auton, A. , Abecasis, G. , Albers, C.a , Banks, E. , DePristo, M.a , Handsaker, R.e et al. (2011) The variant call format and VCFtools. Bioinformatics, 27, 2156–2158. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Deng, W.K. , Wang, Y.B. , Liu, Z.X. , Cheng, H. and Xue, Y. (2014) HemI: A toolkit for illustrating heatmaps. PLoS One, 9, e111988. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dong, H. , Zhao, H. , Xie, W. , Han, Z. , Li, G. , Yao, W. , Bai, X. et al. (2016) A novel tiller angle gene, TAC3, together with TAC1 and D2 largely determine the natural variation of tiller angle in rice cultivars. PLoS Genet. 12, e1006412. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Evanno, G. , Regnaut, S. And Goudet, J. (2005) Detecting the number of clusters of individuals using the software STRUCTURE: a simulation study. Mol. Ecol. 14, 2611–2620. [DOI] [PubMed] [Google Scholar]
- Fan, C.F. , Feng, S.Q. , Huang, J.F. , Wang, Y.T. , Wu, L.M. , Li, X.K. , Wang, L.Q. et al. (2017) atcesa8‐driven ossus3 expression leads to largely enhanced biomass saccharification and lodging resistance by distinctively altering lignocellulose features in rice. Biotechnol. Biofuels, 10, 1‐12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fan, C.F. , Li, Y. , Hu, Z. , Hu, H.Z. , Wang, G.Y. , Li, A. , Wang, Y.M. et al. (2018) Ectopic expression of a novel OsExtensin‐like gene consistently enhances plant lodging resistance by regulating cell elongation and cell wall thickening in rice. Plant Biotechnol. J. 16, 254–263. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fang, C. , Ma, Y. , Wu, S. , Liu, Z. , Wang, Z. , Yang, R. , Hu, G. et al. (2017) Genome‐wide association studies dissect the genetic networks underlying agronomical traits in soybean. Genome Biol. 18, 161. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Garris, A.J. , Tai, T.H. , Coburn, J. , Kresovich, S. and McCouch, S. (2005) Genetic structure and diversity in Oryza sativa L. Genetics, 169, 1631–1638. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Glaszmann, J.C. (1987) Isozymes and classification of Asian rice varieties. Theoret. Appl. Genet. 74, 21–30. [DOI] [PubMed] [Google Scholar]
- Guo, Z. , Yang, W. , Chang, Y. , Ma, X. , Tu, H. , Xiong, F. , Jiang, N. et al. (2018) Genome‐wide association studies of image traits reveal genetic architecture of drought resistance in rice. Mol. Plant, 11, 789–805. [DOI] [PubMed] [Google Scholar]
- Hirano, K. , Ordonio, R.L. and Matsuoka, M. (2017) Engineering the lodging resistance mechanism of post‐Green Revolution rice to meet future demands. Proc. Japan Acad. Series B, Phys. Biol. Sci. 93, 220–233. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang, X. , Kurata, N. , Wei, X. , Wang, Z.X. , Wang, A. , Zhao, Q. , Zhao, Y. et al. (2012) A map of rice genome variation reveals the origin of cultivated rice. Nature, 490, 497–501. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang, X. , Wei, X. , Sang, T. , Zhao, Q. , Feng, Q. , Zhao, Y. , Li, C. et al. (2010) Genome‐wide association studies of 14 agronomic traits in rice landraces. Nat. Genet. 42, 961–967. [DOI] [PubMed] [Google Scholar]
- Islam, M.S. , Peng, S. , Visperas, R.M. , Ereful, N. , Bhuiya, M.S.U. and Julfiquar, A.W. (2007) Lodging‐related morphological traits of hybrid rice in a tropical irrigated ecosystem. Field. Crop. Res. 101, 240–248. [Google Scholar]
- Kashiwagi, T. and Ishimaru, K. (2004) Identification and functional analysis of a locus for improvement of lodging resistance in rice. Plant Physiol. 134, 676–683. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li, H. and Durbin, R. (2009) Fast and accurate short read alignment with Burrows‐Wheeler transform. Bioinformatics, 25, 1754–1760. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li, H. , Handsaker, B. , Wysoker, A. , Fennell, T. , Ruan, J. , Homer, N. , Marth, G. et al. (2009) The sequence alignment/Map format and SAMtools. Bioinformatics, 25, 2078–2079. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li, Z. , Paterson, A.H. , Pinson, S.R.M. and Stansel, J.W. (1999) RFLP facilitated analysis of tiller and leaf angles in rice (Oryza sativa L.). Euphytica, 109, 79–84. [Google Scholar]
- Li, M.X. , Yeung, J.M. , Cherny, S.S. and Sham, P.C. (2012) Evaluating the effective numbers of independent tests and significant p‐value thresholds in commercial genotyping arrays and public imputation reference datasets. Hum. Genet. 131, 747–756. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lippert, C. , Listgarten, J. , Liu, Y. , Kadie, C.M. , Davidson, R.I. and Heckerman, D. (2011) FaST linear mixed models for genome‐wide association studies. Nat. Methods, 8, 833–835. [DOI] [PubMed] [Google Scholar]
- Liu, H.J. , Wang, X.Q. , Xiao, Y.J. , Luo, J.Y. , Qiao, F. , Yang, W.Y. , Zhang, R.Y. et al. (2020) CUBIC: an atlas of genetic architecture promises directed maize improvement. Genome Biol. 21, 20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ma, J.F. , Tamai, K. , Yamaji, N. , Mitani, N. , Konishi, S. , Katsuhara, M. , Ishiguro, M. et al. (2006) A silicon transporter in rice. Nature, 440, 688–691. [DOI] [PubMed] [Google Scholar]
- Ma, J.F. , Yamaji, N. , Mitani, N. , Tamai, K. , Konishi, S. , Fujiwara, T. , Katsuhara, M. et al. (2007) An efflux transporter of silicon in rice. Nature, 448, 209–U212. [DOI] [PubMed] [Google Scholar]
- Mefford, J. and Witte, J.S. (2012) The covariate’s dilemma. PLoS Genet. 8, e1003096. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Monna, L. , Kitazawa, N. , Yoshino, R. , Suzuki, J. , Masuda, H. , Maehara, Y. , Tanji, M. et al. (2002) Positional cloning of rice semidwarfing gene, sd‐1: Rice "Green revolution gene" encodes a mutant enzyme involved in gibberellin synthesis. DNA Res. 9, 11–17. [DOI] [PubMed] [Google Scholar]
- Mulsanti, I.W. , Yamamoto, T. , Ueda, T. , Samadi, A.F. , Kamahora, E. , Rumanti, I.A. , Thanh, V.C. et al. (2018) Finding the superior allele of japonica‐type for increasing stem lodging resistance in indica rice varieties using chromosome segment substitution lines. Rice, 11, 1‐14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nomura, T. , Arakawa, N. , Yamamoto, T. , Ueda, T. , Adachi, S. , Yonemaru, J. , Abe, A. et al. (2019) Next generation long‐culm rice with superior lodging resistance and high grain yield, Monster Rice 1. PLoS One, 14, e0221424. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ookawa, T. , Hobo, T. , Yano, M. , Murata, K. , Ando, T. , Miura, H. , Asano, K. et al. (2010) New approach for rice improvement using a pleiotropic QTL gene for lodging resistance and yield. Nat. Commun. 1, 132. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pan, J.F. , Zhao, J.L. , Liu, Y.Z. , Huang, N.R. , Tian, K. , Shah, F. , Liang, K.M. et al. (2019) Optimized nitrogen management enhances lodging resistance of rice and its morpho‐anatomical, mechanical, and molecular mechanisms. Sci. Rep. 9, 1‐13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Purcell, S. , Neale, B. , Todd‐Brown, K. , Thomas, L. , Ferreira, M.A. , Bender, D. , Maller, J. et al. (2007) PLINK: a tool set for whole‐genome association and population‐based linkage analyses. Am. J. Hum. Genet. 81, 559–575. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roberts, A. , McMillan, L. , Wang, W. , Parker, J. , Rusyn, I. and Threadgill, D. (2007) Inferring missing genotypes in large SNP panels using fast nearest‐neighbor searches over sliding windows. Bioinformatics, 23, i401–407. [DOI] [PubMed] [Google Scholar]
- Sasaki, T. and Ashikari, M. , (eds). (2018) Rice Genomics, Genetics and Breeding. Singapore: Ltd: Springer. 10.1007/978-981-10-7461-5. [DOI] [Google Scholar]
- Sasaki, A. , Ashikari, M. , Ueguchi‐Tanaka, M. , Itoh, H. , Nishimura, A. , Swapan, D. , Ishiyama, K. et al. (2002) Green revolution: a mutant gibberellin‐synthesis gene in rice. Nature, 416, 701–702. [DOI] [PubMed] [Google Scholar]
- Shah, L. , Yahya, M. , Shah, S.M.A. , Nadeem, M. , Ali, A. , Ali, A. , Wang, J. et al. (2019) Improving lodging resistance: using wheat and rice as classical examples. Int. J. Mol. Sci. 20, 4211. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Silva, L.E. , Wang, S. and Zeng, Z.B. (2012) Composite interval mapping and multiple interval mapping: procedures and guidelines for using Windows QTL Cartographer. Methods Mol. Biol. 871, 75–119. [DOI] [PubMed] [Google Scholar]
- Sowadan, O. , Li, D.L. , Zhang, Y.Q. , Zhu, S.S. , Hu, X.X. , Bhanbhro, L.B. , Edzesi, W.M. et al. (2018) Mining of favorable alleles for lodging resistance traits in rice (Oryza sativa) through association mapping. Planta, 248, 155–169. [DOI] [PubMed] [Google Scholar]
- Wang, Y. , Chen, J. , Guan, Z. , Zhang, X. , Zhang, Y. , Ma, L. , Yao, Y. et al. (2019) Combination of multi‐locus genome‐wide association study and QTL mapping reveals genetic basis of tassel architecture in maize. Mol. Genet. Genom. 294, 1421–1440. [DOI] [PubMed] [Google Scholar]
- Wang, W. , Mauleon, R. , Hu, Z. , Chebotarov, D. , Tai, S. , Wu, Z. , Li, M. et al. (2018) Genomic variation in 3,010 diverse accessions of Asian cultivated rice. Nature, 557, 43–49. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang, Q.X. , Xie, W.B. , Xing, H.K. , Yan, J. , Meng, X.Z. , Li, X.L. , Fu, X.K. et al. (2015) Genetic architecture of natural variation in rice chlorophyll content revealed by a genome‐wide association study. Mol. Plant, 8, 946–957. [DOI] [PubMed] [Google Scholar]
- Wu, D. , Guo, Z. , Ye, J. , Feng, H. , Liu, J. , Chen, G. , Zheng, J. et al. (2019) Combining high‐throughput micro‐CT‐RGB phenotyping and genome‐wide association study to dissect the genetic architecture of tiller growth in rice. J. Exp. Bot. 70, 545–561. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xie, W. , Wang, G. , Yuan, M. , Yao, W. , Lyu, K. , Zhao, H. , Yang, M. et al. (2015) Breeding signatures of rice improvement revealed by a genomic variation map from a large germplasm collection. Proc. Natl Acad. Sci. USA, 112, E5411–5419. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yadav, S. , Singh, U.M. , Naik, S.M. , Venkateshwarlu, C. , Ramayya, P.J. , Raman, K.A. , Sandhu, N. et al. (2017) Molecular mapping of QTLs associated with lodging resistance in dry direct‐seeded rice (Oryza sativa L.). Front. Plant Sci. 8, 1431. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yamaji, N. and Ma, J.F. (2009) A transporter at the node responsible for intervascular transfer of silicon in rice. Plant Cell, 21, 2878–2883. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yan, W. , Liu, H. , Zhou, X. , Li, Q. , Zhang, J. , Lu, L. , Liu, T. et al. (2013) Natural variation in Ghd7.1 plays an important role in grain yield and adaptation in rice. Cell Res. 23, 969–971. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang, J. , Ferreira, T. , Morris, A.P. , Medland, S.E. , Madden, P.A.F. , Heath, A.C. , Martin, N.G. et al. (2012) Conditional and joint multiple‐SNP analysis of GWAS summary statistics identifies additional variants influencing complex traits. Nat. Genet. 44, 369–375. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yano, K. , Ookawa, T. , Aya, K. , Ochiai, Y. , Hirasawa, T. , Ebitani, T. , Takarada, T. et al. (2015) Isolation of a novel lodging resistance QTL gene involved in strigolactone signaling and its pyramiding with a QTL gene involved in another mechanism. Mol. Plant, 8, 303–314. [DOI] [PubMed] [Google Scholar]
- Yu, B. , Lin, Z. , Li, H. , Li, X. , Li, J. , Wang, Y. , Zhang, X. et al. (2007) TAC1, a major quantitative trait locus controlling tiller angle in rice. Plant J. Cell Mol. Biol. 52, 891–898. [DOI] [PubMed] [Google Scholar]
- Zhang, B. , Liu, H. , Qi, F. , Zhang, Z. , Li, Q. , Han, Z. and Xing, Y. (2019) Genetic interactions among Ghd7, Ghd8, OsPRR37 and Hd1 contribute to large variation in heading date in rice. Rice, 12, 48. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhao, H. , Yao, W. , Ouyang, Y. , Yang, W. , Wang, G. , Lian, X. , Xing, Y. et al. (2015) RiceVarMap: a comprehensive database of rice genomic variations. Nucleic Acids Res. 43, D1018–1022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhong, X.H. , Liang, K.M. , Peng, B.L. , Tian, K. , Li, X.J. , Huang, N.R. , Liu, Y.Z. et al. (2020) Basal internode elongation of rice as affected by light intensity and leaf area. Crop J. 8, 62–70. [Google Scholar]
- Zou, G.H. , Mei, H.W. , Liu, H.Y. , Liu, G.L. , Hu, S.P. , Yu, X.Q. , Li, M.S. et al. (2005) Grain yield responses to moisture regimes in a rice population: association among traits and genetic markers. Theoret. Appl. Genet. 112, 106–113. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Figure S1. Path diagram showing the effects of PH and BS on LI.
Figure S2. Polynomial regression of GH and BS.
Figure S3. Manhattan plots of LR‐related traits in 2014 and 2015.
Figure S4. Scatter plots showing the correlations of LR‐related traits for 96 accessions under pot and field growing conditions.
Figure S5. Cross‐validation errors of successive K values using ADMIXTURE.
Figure S6. Statistic ΔK values of successive K values using STRUCTURE and Structure Harvester.
Table S1. Coefficients and residual effect of path analysis from 16 traits to lodging index (LI).
Table S2. GWAS results of 17 LR‐related traits without a covariate using a linear mixed model.
Table S3. GWAS results of culm‐strength traits with PW or PN as a covariate using a linear mixed model for the 34 loci identified by GWAS without a covariate.
Table S4. GWAS results of culm‐strength traits with and without adding PW or PN as a covariate using a linear mixed model.
Table S5. Bi‐directional validation of covariate‐added GWAS using a linear mixed model.
Table S6. GWAS results of culm‐strength traits with and without adding PW or PN as a covariate using a linear regression model considering the population structure.
Table S7. Association results between the candidate gene OsPRR37 and BS with and without adding PW as a covariate using a linear regression model considering the population structure.
Table S8. Genotypes of the 8‐bp INDEL in OsPRR37 (vf0729626365) obtaind by variant calling from whole‐genome re‐sequencing data.
Table S9. Association results between the candidate gene OsPRR37 and BS with and without adding heading date as a covariate using a linear regression model considering the population structure.
Table S10. Genome‐wide divergent regions of the top 5% F ST..
Table S11. GWAS loci overlapping with greatly divergent regions identified by F ST.
Table S12. LR‐related QTL overlapping with GWAS loci.
Table S13. GWAS results of LR‐related traits with adding heading date as a covariate using a linear mixed model.
Table S14. Detailed information of 524 accessions in the study.