

Abstract
Liver hepatocellular carcinoma (LIHC) is one of the major causes of cancer-related death worldwide with increasing incidences, however there are very few studies about the underlying mechanisms and pathways in the development of LIHC. We obtained LIHC samples from The Cancer Genome Atlas (TCGA) to screen differentially expressed mRNAs, lncRNAs, miRNAs and driver mutations. The Kyoto Encyclopedia of Genes and Genomes (KEGG) pathway, Gene ontology enrichment analyses and protein–protein interaction (PPI) network were performed. Moreover, we constructed a competing endogenous lncRNAs-miRNAs-mRNAs network. Finally, cox proportional hazards regression analysis was used to identify important prognostic differentially expressed genes. Total of 1284 mRNAs, 123 lncRNAs, 47 miRNAs were identified within different tissues of LIHC patients. GO analysis indicated that upregulated and downregulated differentially expressed mRNAs (DEmRNAs) were mainly associated with cell division, DNA replication, mitotic sister chromatid segregation and complement activation respectively. Meanwhile, KEGG terms revealed that upregulated and downregulated DEmRNAs were primarily involved in DNA replication, Metabolic pathways, cell cycle and Metabolic pathways, chemical carcinogenesis, retinol metabolism pathway respectively. Among the DERNAs, 542 lncRNAs-miRNAs-mRNAs pairs were predicted to construct a ceRNA regulatory network including 35 DElncRNAs, 26 DEmiRNAs and 112 DEmRNAs. In the Kaplan‐Meier analysis, total of 43 mRNAs, 14 lncRNAs and 3 miRNAs were screened out to be significantly correlated with overall survival of LIHC. The mutation signatures were analyzed and its correlation with immune infiltrates were evaluated using the TIMER in LIHC. Among the mutation genes, TTN mutation is often associated with poor immune infiltration and a worse prognosis in LIHC. This work conducted a novel lncRNAs-miRNAs-mRNAs network and mutation signatures for finding potential molecular mechanisms underlying the development of LIHC. The biomarkers also can be used for predicting prognosis of LIHC.
Introduction
Liver hepatocellular carcinoma (LIHC) has been one of the major causes of cancer mortality worldwide, which is the second highest cancer-related death disease in the world. There are 841,000 new cases of liver cancer and 782,000 deaths reports each year worldwide [1, 2]. Nowadays, the improvement of modern medicine treatment and combination of various therapeutic strategies can prolong the LIHC patient’s survival [3], however the outcome is still poor [4, 5]. Therefore, discovery of new means to diagnosis and prognosis is urgent and important.
Next generation sequencing is a breakthrough technology, which is redefining the landscape of human molecular genetic testing. Bioinformatics is a novelty cross discipline which develops and utilizes modern computational tools to analyze and interpret high dimensional biological data [6]. With the advances of next-generation sequencing technology, numerous disease-related genetic alterations have been revealed, bioinformatics has become an important component in clinical disease research. Progress in cancer genomics research over the past few decades has demonstrated that cancer is driven by various types of genomic alterations [7]. It has been reported that many types of tumourigenesis and development are closely associated with genomic alterations, such as papillary thyroid carcinomas [8], lung cancer [9], liver cancer [10], and etc.
Biomarker is designated as a parameter that can objectively be measured and evaluated as an indicator of normal biological processes, pathologic processes, or pharmacological response to therapeutic intervention [11]. Finding cancer biomarkers is available for early cancer detection, monitoring strategies, and tumor classification; so that the patient can receive the most appropriate therapy and that doctor can monitor the disease progression, regression, and recurrence [12]. Therefore, we urgently need to better understand the pathogenesis and progression of LIHC and to find specific biomarkers for diagnosis and prognosis.
Non-coding RNAs, such as micro RNAs (miRNAs) and long non-coding RNAs (lncRNAs), are defined as gene transcripts with little or no evident protein coding potential [13]. LncRNAs transcripts more than 200 nucleotides in length which are distinguished from other small ncRNAs such as tRNA, miRNA etc [14]. They are cell or tissue-specific in development processes or different disease. More and more evidence suggests that gene expression is regulated by lncRNAs at the transcriptional, post-transcriptional, and epigenetic levels [15]. Recently, it has increasing evidences indicated that many lncRNAs are involved in regulating tumorigenesis and progression via different biological processes, such as cell apoptosis [16], proliferation [17], and metastasis [18]. Those pathways in tumors are also regulated by lncRNAs and related to cancer diagnosis, prognosis, staging, and treatment [19–21].
MicroRNAs are small noncoding RNA molecule (about 22 nucleotides) that, by binding to complementary target mRNAs, can regulate cell proliferation, differentiation, and apoptosis [22]. The aberrantly expressed miRNA are found in many cancers [23], including lung cancer [24], neuroblastoma [25], and also liver cancer [26]. And it has diagnostic value for cancer diagnosis and therapy [27, 28]. There are many papers analyzed the differential expression and regulation of lncRNAs and miRNAs separately in LIHC [29, 30]. However, few researches have been studied on ceRNA of LIHC. Competitive endogenous RNAs (ceRNAs) are transcripts that can cross-regulate their ability by competing for shared miRNAs, thereby depredating or suppressing all target genes of the respective miRNA [31]. CeRNA networks link the function of mRNAs with microRNA, long non-coding RNA, and so on. A lot of ceRNAs have been reported successively, but few studies characterized such modulators of miRNA activity in LIHC [32].
The genome-scale screening analysis of LIHC was conducted to examine the relationship between the gene mutation signatures and its correlation with immune response. Several studies have shown that TP53 and CTNNB1 mutations tend to have a negative correlation with tumor immunity and immunotherapy response [33–35]. Besides TP53 and CTNNB1 mutations demonstrated a poor prognosis compared with wildtype. Titin (TTN) is a gene encoding a large abundant protein of striated muscle, which is divided into two regions, a N-terminal I-band and a C-terminal A-band. TTN mutation is frequently detected in LIHC, is associated with increased TMB and correlated with objective responsiveness to checkpoint blockades [36]. To detect the enrichment levels of immune signatures, we analyzed the LIHC cohorts from TCGA project and find the closed relationship between TTN mutation and immune infiltration.
In our study, LIHC samples from The Cancer Genome Atlas (TCGA) were obtained to screen differentially expressed genes (DEGs) between tumor and normal samples. We established a ceRNA-net signature. Univariable survival analyses were performed to identify prognostic genes of LIHC based on the intersection differentially expressed genes, miRNAs, and lncRNAs, mutation and clinical data.
Materials and methods
LIHC data sets collection
All the gene expression data (RNA-seqv2 & miRNA-seq) and corresponding clinical information were collected from The Cancer Genome Atlas (TCGA) database (https://portal.gdc.cancer.gov/). The screening criteria were as the following: (1) Studies with Liver Hepatocellular Carcinoma (LIHC) tissue samples; (2) the selected datasets containing lncRNA, mRNA and miRNA. (3) patients complicated without other tumors. Based on these criteria, a total of 333 tumor samples and 50 corresponding adjacent peritumoral samples were included. To investigate the lncRNA-miRNA-mRNA network associated with the pathogenesis and prognosis of LIHC and to identify novel molecular subtypes as potential biomarkers of LIHC. The LIHC data sets downloaded from the TCGA databases were integrated and analyzed by bioinformatics as the following workflow in Fig 1.
Fig 1. Flowchart of bioinformatics analysis for LIHC data from TCGA databases.

Data processing and identification of differential expression genes
To integrated dysregulated lncRNAs, mRNAs and miRNAs across samples, hierarchical clustering was performed. Firstly, we used R statistical software (http://www.rproject.org/) to correct, normalize and calculate the raw data expression of RNA-seqv2 & miRNA-seq data. Next, differential expression of lncRNAs, mRNAs and miRNAs between tumor tissues and adjacent peritumoral tissues were identified through limma R package, and P < 0.05 & false discovery rate (FDR) < 0.05 & |log2FC|> 2 were set as statistically significant criterion. Total of 1284 mRNAs, 123 lncRNAs, 47 miRNAs were selected out for next step analysis.
Functional enrichment analysis of differentially expressed mRNAs
To identify the meaningful functional annotation and pathway terms of differentially expressed mRNAs (DEmRNAs), we performed Gene Ontology (GO) classification and Kyoto Encyclopedia of Genes and Genomes (KEGG) pathway analysis among 1284 DEmRNAs using the online Bioinformatics Tool WebGestalt (http://www.webgestalt.org/) and KOBAS 3.0 (http://kobas.cbi.pku.edu.cn/index.php), respectively. Fisher test and Least Significant Difference-test were used to calculate the p value and False Discovery Rate. On the basis of test results, top 25 upregulated and downregulated GO and KEGG notable terms were generated. Then we identified the intersection genes of differentially expressed mRNAs included in both GO and KEGG pathways.
Construction of ceRNA network
LncRNAs as a critical class of regulatory RNAs modulated the expression of mRNAs through competitively sponging miRNAs. miRNAs targeting bind both lncRNAs and mRNAs through specific base sequence pairing. From the result set obtained in step 2, we identified 123 DElncRNAs and 47 DEmiRNAs. Integration of GO analysis and KEGG pathway in step 6 made up the 532 DEmRNAs. In order to construct the competing endogenous regulating network, the miRanda (http://www.microrna.org/) was applied to match DEmiRNAs-DElncRNAs pairs. Meanwhile, the miRanda (http://www.microrna.org/), Targetscan (http://www.targetscan.org/), and miRWalk (http://129.206.7.150/) were used to predict the potential interaction of DEmiRNAs and DEmRNAs. According to miRNA negatively regulating target theory and Pearson correlation coefficients, we screened inverse relationship of DEmiRNA-DEmRNA, DEmiRNA-DElncRNA pairs and constructed the ceRNA network (lncRNA-miRNA-mRNA-network). Finally, we used the Cytoscape software to make the ceRNA network visualization.
Protein-protein interaction network construction
To elucidate the underlying interaction of DEmRNAs involved in above ceRNA network, protein-protein interaction network was constructed using the STRING database (http://string-db.org/cgi/input.pl).
Survival analysis
To determine the prognostic value of differential expression genes involved in ceRNA network in LIHC patients, we selected the data containing complete clinical information to perform the Kaplan‐Meier analysis using R software survival package. Firstly, the patients were divided into high expression level and low expression level two groups according to median expression of the differentially expressed gene including lncRNA, miRNA and mRNA. Then all the DERNAs were subjected to Univariate Cox regression analysis and survival curves were further calculated. p < 0.05 was considered as statistically significant.
Mutation signatures and immune infiltrates analysis
We downloaded the Mutation annotation files (MAFs) from the GDC Data Portal (https://portal.gdc.cancer.gov), analyzed the mutation signatures containing top 30 mutated genes in LIHC. Among them, TTN ranking second only to TP53, demonstrated very high mutation rates. In addition, we explored the immune cell infiltration differences between TTN mutation and TTN wildtype, evaluated the correlation of OS and TTN mutation.
Statistical analysis
Statistical analysis was performed using R (version 3.5.1, Auckland, NZ). The statistical analysis of categorical data was conducted with Fisher test and Least Significant Difference-test. The differences between two groups were assessed using “Deseq2” package with |log2 (fold change)| > 1 and FDR < 0.05. The Univariate Cox regression analysis was used for Kaplan‐Meier analysis of patient. p < 0.05 was considered to be statistically significant.
Results
Differential expression profile of RNAs in LIHC
To study the Differential expression profile of RNAs in LIHC, we corrected and normalized the RNA-seqv2 & miRNA-seq data using R statistical software (http://www.rproject.org/) before differential expression analysis. Then we performed unsupervised hierarchical clustering analysis to compare the difference expression RNAs between LIHC tumor tissues and adjacent normal tissues. A total of 123 differentially expressed lncRNAs (24 up-regulated and 99 down-regulated), 47 differentially expressed miRNAs (19 up-regulated and 28 down-regulated) and 1284 differentially expressed mRNAs (412 up-regulated and 872 down-regulated) were identified in RNA-seqv2 & miRNA-seq (p < 0.05 & |log2FC|> 2). DERNAs were visualized in heatmap (Fig 2).
Fig 2. Differential expression profile of RNAs in LIHC.

(A) Heat map of differentially expressed mRNAs. (B) Heat map of differentially expressed lncRNAs. (C) Heat map of differentially expressed miRNAs. The red represented the higher expression levels and the green represented lower expression levels. The rows and the columns represented the DERNAs and samples respectively.
Functional enrichment analysis of the differentially expressed mRNAs
To illustrates the meaningful functional pattern of differentially expressed mRNAs (DEmRNAs), a total of 1284 DEmRNAs were subjected to GO annotation and KEGG pathway analysis including 412 upregulated genes and 872 downregulated genes. We showed the top 25 GO analyses results (Fig 3A and 3B). GO analysis indicated that upregulated DEmRNAs were mainly associated with cell division, DNA replication, mitotic sister chromatid segregation. In contrast, downregulated DEmRNAs were mainly involved in complement activation, classical pathway, regulation of complement activation. The top 25 KEGG pathway terms were shown in Fig 3C and 3D. KEGG pathway terms revealed that upregulated DEmRNAs were primarily enriched in DNA replication, Metabolic pathways, cell cycle. While downregulated DEmRNAs were primarily involved in Metabolic pathways, chemical carcinogenesis, retinol metabolism.
Fig 3. Functional enrichment analysis of the differentially expressed mRNAs.

(A) Top 25 GO analyses of upregulated DEmRNAs. (B) Top 25 GO analyses of downregulated DEmRNAs. (C) Top 25 GO KEGG pathways of upregulated DEmRNAs. (D) Top 25 GO KEGG pathways of downregulated DEmRNAs.
CeRNA network
LncRNAs modulated the expression of mRNAs through competitively sponging miRNAs. miRNAs bind to the 3′untranslated region (3′- UTR) of lncRNAs and mRNAs through specific base sequence pairing leading to RNAs degradation. According to miRNA negatively regulating target theory and co-expression relationship, first we identified 123 DElncRNAs, 47 DEmiRNAs from step 2 and 532 DEmRNAs from step 6. Next, we predicted the target binding of mRNAs, lncRNAs with miRNAs using miRanda, Targetscan and miRWalk websites. Then we screened inverse relationship of DEmiRNA-DEmRNA, DEmiRNA-DElncRNA pairs and constructed the ceRNA network (lncRNA-miRNA-mRNA-network) using Cytoscape software. As a result, 542 lncRNAs-miRNAs-mRNAs pairs were predicted to construct a ceRNA regulatory network including 35 DElncRNAs, 26 DEmiRNAs and 112 DEmRNAs. Among them, has-miR-93-5p, has-miR-34a-5p, has-miR-20a-5p, has-miR-103a-3p, has-miR-183a-5p, has-miR-182a-5p displayed stronger targeting function than other miRNAs, which indicated that they had important functions in regulating other genes (Fig 4).
Fig 4. LncRNA-miRNA-mRNA network of DERNAs.

Red stands for upregulated genes and green stands for downregulated genes. circles represent mRNAs, quadrates represent miRNAs, diamonds represent lncRNAs. The point size corresponds to regulatory ability of genes. Larger point means stronger regulatory ability.
Protein-Protein Interaction (PPI) network construction
Based on ceRNA network, we get 112 differentially expressed mRNAs including 21 upregulated mRNAs and 91 downregulated mRNAs. To elucidate the underlying interaction of DEmRNAs involved in above ceRNA network, mRNAs with interconnected relationships were selected to construct the protein-protein interaction network using the STRING website. The PPI network was composed of links and nodes, whose size represented the regulatory capacity of mRNAs. Among them, the upregulated gene CDKN2A and downregulated genes ESR1, AR had the closest connection to other proteins (Fig 5).
Fig 5. Protein–protein interaction network.

All the points are differentially expressed mRNAs, node size represents regulatory capacity of mRNAs. The regulatory capacity was stronger with larger size. Green represents downregulated mRNAs. Red represents upregulated mRNAs.
Kaplan-Meier curve analysis of DERNAs
Liver hepatocellular carcinoma (LIHC) is the second leading cause of cancer death in the world. Prolonging overall survival is considered as the ultimate goal of LIHC patients. It is very important to comprehensively investigate the utility of lncRNAs, mRNAs and miRNAs in LIHC as potential biomarkers for prognosis. Therefore we selected the data containing complete clinical information to perform the Kaplan‐Meier analysis using R software survival package. Then, DERNAs involved in ceRNA network were subjected to Univariate Cox regression analysis and survival curves were further calculated. Total of 43 mRNAs, 14 lncRNAs and 3 miRNAs were screened out to be significantly correlated with overall survival (OS) of LIHC. Among the DEmRNAs, the upregulated gene of LPCAT1 and CDCKN2A were the most significantly negatively correlated with OS (p = 0.0001 and p = 0.0003 respectively), the downregulated gene of ALDH6A1 was the most significantly positively correlated with OS (p = 0.0035). For DEmiRNAs, the upregulated has-miR-589-5p and has-miR-500a-3p most significantly suggested a shorter survival (p = 0.0498 and p = 0.0261 respectively). The downregulated has-miR-125b-5p most significantly suggested a longer survival (p = 0.021). A low expression of LINC00152 linked most significantly to the longer OS (p = 0.0001). Meanwhile, the high expression of RP11-290F5.1 and TMEM220-AS1 linked significantly to the shorter OS (p = 0.032 and p = 0.0001 respectively). Survival curves results included lncRNAs, mRNAs and miRNAs (Fig 6). Cox regression analysis was used to identify prognostic ceRNAs (Table 1). We verified the expression of prognostic ceRNAs in our own samples, which was consistent with the results of TCGA (S1 Fig).
Fig 6. Kaplan-Meier curves of the 12 DERNAs associated with overall survival.

(A) DEmRNAs associated with OS, (B) DEmiRNAs associated with OS, and (C) DElncRNAs associated with OS.
Table 1. Identify the biomarkers to predict prognosis based on multiple variate COX model.
| Characteristics | Univariate cox | Multivariate cox | ||||
|---|---|---|---|---|---|---|
| Hazard.Ratio | CI95 | P. Value | Hazard.Ratio | CI95 | P. Value | |
| ALDH6A1 | 0.58 | 0.4–0.84 | 0.00394 | 0.68 | 0.44–1.04 | 0.077 |
| AR | 0.64 | 0.44–0.93 | 0.01834 | 0.92 | 0.6–1.43 | 0.723 |
| FTCD | 0.63 | 0.44–0.92 | 0.01549 | 0.64 | 0.43–0.94 | 0.024 |
| hsa.miR.500a.3p | 1.52 | 1.05–2.19 | 0.02717 | 1.44 | 0.98–2.1 | 0.061 |
| RP11.119D9.1 | 0.64 | 0.44–0.93 | 0.01785 | 0.92 | 0.59–1.43 | 0.709 |
| SOCS2 | 0.47 | 0.32–0.69 | 0.00012 | 0.51 | 0.34–0.76 | 0.001 |
Comparisons of the mutation signatures and its correlation with immune infiltrates
We analyzed the characteristic pattern of mutated genes of 345 LIHC from TCGA, among them, TP53, TTN, CTNNB1, MUC16 and PCLO had the highest mutation frequency (Fig 7A). We further compared the immune signatures (B cell, CD8+ T cell, CD4+ T cell, Macrophage, Neutrophil and Dendritic cell) between mutated and wildtype genes in LIHC. we found that the immune signatures (CD8+ T cells, CD4+ T cell, Macrophage, Neutrophil and Dendritic cell) showed significantly higher enrichment levels in TTN-wildtype than in TTN- mutated cancers in LIHC (P < 0.01) (Fig 7B). Previous studies have indicated that TP53 mutations play a negative role in antitumor immunity. However, immune infiltrates showed no significantly difference between TP53-mutated and TP53-wildtype in LIHC (P < 0.01). Then, we examined the correlation between TTN mutations and five years OS, we found that TTN mutation weas significantly associated with a worse OS in LIHC (Fig 7C).
Fig 7. Identification of mutation signatures and its correlation with the immune infiltration and overall survival.
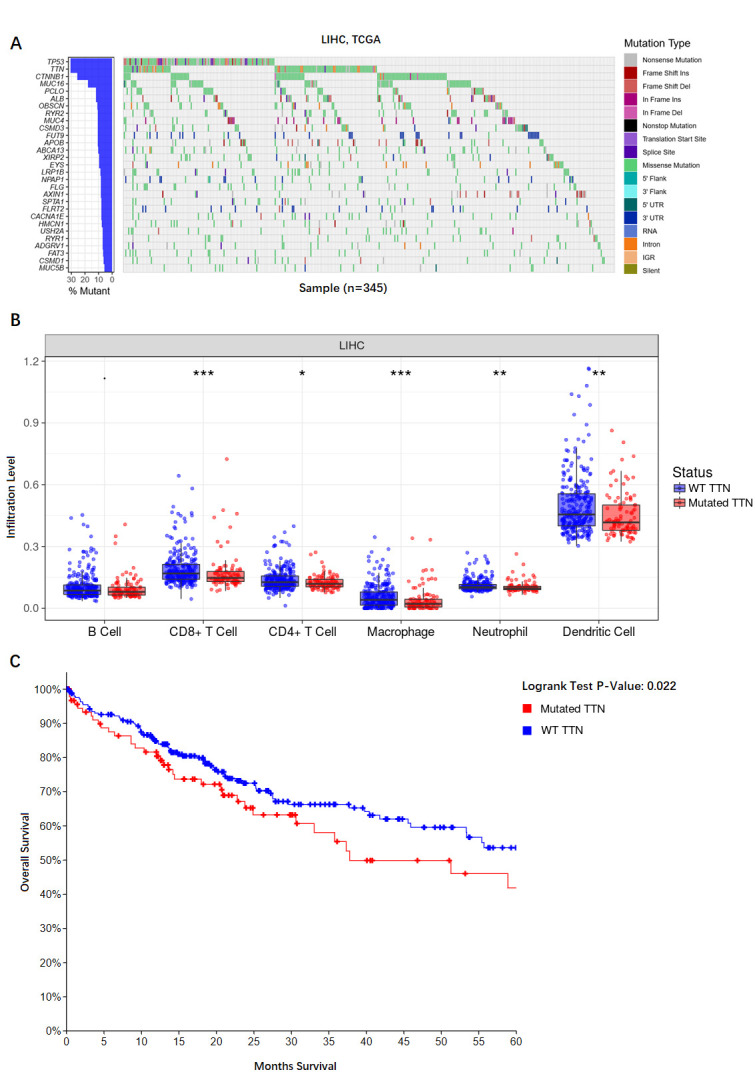
(A) Heatmap representation of the distribution of driver genes mutations in patients, (B) The immune signatures (CD8+ T cells, Macrophage, Neutrophil, and Dendritic Cells) show significantly higher enrichment levels in TTN- wildtype than in TTN- mutated cancers in LIHC, (C) Kaplan-Meyer survival curves showed that TTN mutation have a negative correlation with OS in LIHC.
Discussion
LIHC is the second highest cancer-related death in the world, with a steady rise of the incidence every year [37]. Nowadays, surgical resection, ablation by radiofrequency or ethanol injection, and liver transplantation are used on early stage LIHC patients, however the overall five-year survival rate still ranges between 50% and 70% [38]. Therefore, there is an urgent need to find genes that associated with the pathogenesis and progression in LIHC and to search biomarkers for diagnosis and prognosis. Many studies have reported that LIHC patients have certain dysregulated expression of mRNAs [2], miRNAs [39], and lncRNAs [40]. However, there are few studies about the crosstalk between mRNAs, miRNAs, and lncRNAs. Single-cell transcriptome-based multilayer network biomarker for predicting prognosis and therapeutic response of gliomas [41]. Recently, ceRNAs were be identified that play important roles in the physiology and development of cancer [32]. And ceRNA interactions are different in different kind of cancers [42]. Systematic studies that evaluate ceRNAs in LIHC are need.
In this study, mRNA, miRNA, and lncRNA data from TCGA were used to identify the differential expression profiles. Gene oncology analysis revealed that the upregulated genes were primarily enriched in cell division, DNA replication, and mitotic sister chromatid segregation. The downregulated genes were primarily involved in complement activation, classical pathway, complement activation, regulation of complement activation. According to the results from KEGG pathway analysis, upregulated genes play an important role in DNA replication, metabolic pathways, and cell cycle. The top three down-regulated gene enrichment pathways are metabolic pathways, chemical carcinogenesis, and retinol metabolism. We constructed a ceRNA interplay networks to search out RNAs that regulates each other. Based on the mRNA in this regulatory network, PPI network analysis was constructed to annotate the correlations between the differentially expressed genes, and we found that ESR1, AR, HGF, CDKN2A, and CXCL12 are essential in regulatory networks. We are recommitted to pursuing the biomarkers that more accurately predict the prognosis of LIHC to improve personalized cancer therapy. For this purpose, we analysed the clinical data and possibilities of using those RNAs as diagnostic biomarkers or therapeutic targets.
Many ceRNAs have been found to play vital roles in tumour progression. The abundance of other RNA transcripts can be altered by ceRNAs. In order to understand the biological implications of the miRNAs which were involved in the ceRNA network, we conducted lncRNA-miRNA-mRNA network analyses and identified 25 ceRNAs, including up-regulated miR-103a-3p, miR-1269b, miR-1269a, miR-34a-5p, miR-532-5p, miR-93-5p, miR-500a-3p, miR-20a-5p, miR-183-5p, miR-589-5p, miR-182-5p, miR-221-3p, and down-regulated miR-10a-5p, miR-378a-5p, miR-199a-5p, miR-122-5p, miR-145-5p, miR-195-5p, miR-125b-5p, miR199a-3p, miR199b-3p, miR-424-5p, miR-134-5p, miR-142-3p. To further determine the correlations between the differentially expressed genes in ceRNA network and survival, we used Kaplan-Meier survival analysis to evaluate the prediction prognostic biomarkers of LIHC. We found 3 miRNAs that are strongly related to prognosis, including miR-589-5p, miR-500a-3p, and miR-125b-5p. There have been some studies on these miRNAs in tumour-associated research. The overexpressed miR-589-5p maintains the stemness of hepatocellular carcinoma cells and promotes chemoresistance [43]. However, in another study, miR-589-5p was found to inhibit the stemness of LIHC cancer stem cells (CSCs) through MAP3K8 and miR-589-5p down-regulation in LIHC is associated with a poor clinical prognosis [44]. We found that miR-589-5p is overexpressed in LIHC and associated with a poor clinical prognosis. MiR-500a-3p was shown to promote the stemness maintenance of CSCs via JAK/STAT3 signaling pathway in LIHC [45]. MiR-125b-5p functions as an oncogene and a prognostic biomarker in LIHC [46]. All the studies on miRNA in LIHC demonstrated the importance of those miRNAs, but the regulatory relationships between these miRNAs and mRNAs or lncRNAs are still unclear. This is the first report that includes annotated ceRNAs in LIHC, which may provide new therapeutic possibilities for patients.
Genomic alterations are hallmarks of many types of cancers, the same to LIHC. We detected 43 mRNAs that are obviously related to prognosis, including CDKN2A, ALDH6A1, LPCAT1, and so on. Although these genes perform well in LIHC prognosis prediction, due to the limitation of the samples in the datasets, larger cohorts and experimental validation are needed. We use the TIMER database to explore the relationship between TTN mutation and immune infiltration in LIHC. We find the negative relationship of TTN mutation and immune cells infiltration including CD8+ T cell, CD4+ T cell, Macrophage, Neutrophil and Dendritic cell. In addition, our survival analysis revealed TTN mutation predicts a poor prognosis. Thus we infer that TTN plays a pivotal role in the regulation and recruitment of immune cell infiltration in LIHC and can serve as a therapeutic target.
Overall, lncRNA-miRNA-mRNA network analyses were performed for prediction of important ceRNAs in LIHC. Cox proportional hazards regression analysis was used to identify significant prognostic differentially expressed genes. Immune infiltration analysis showed the importance of mutation signature in tumor immunity. between mutation These findings could advance the accuracy of diagnosis and prognosis and might aid in the development of targeted therapy for LIHC.
Supporting information
(DOCX)
Acknowledgments
The authors express their gratitude to the authors who uploaded the databases online in this study.
Data Availability
All the gene expression data (RNA-seqv2 & miRNA-seq) and corresponding clinical information were collected from The Cancer Genome Atlas (TCGA) database (https://portal.gdc.cancer.gov/).
Funding Statement
The authors received no specific funding for this work.
References
- 1.Bray F, Ferlay J, Soerjomataram I, Siegel RL, Torre LA, Jemal A. Global cancer statistics 2018: GLOBOCAN estimates of incidence and mortality worldwide for 36 cancers in 185 countries. CA: a cancer journal for clinicians. 2018;68(6):394–424. Epub 2018/09/13. 10.3322/caac.21492 . [DOI] [PubMed] [Google Scholar]
- 2.Zheng Y, Liu Y, Zhao S, Zheng Z, Shen C, An L, et al. Large-scale analysis reveals a novel risk score to predict overall survival in hepatocellular carcinoma. Cancer management and research. 2018;10:6079–96. Epub 2018/12/13. 10.2147/CMAR.S181396 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Raza A, Sood GK. Hepatocellular carcinoma review: current treatment, and evidence-based medicine. World journal of gastroenterology. 2014;20(15):4115–27. Epub 2014/04/26. 10.3748/wjg.v20.i15.4115 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Byam J, Renz J, Millis JM. Liver transplantation for hepatocellular carcinoma. Hepatobiliary surgery and nutrition. 2013;2(1):22–30. Epub 2014/02/27. 10.3978/j.issn.2304-3881.2012.11.03 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Frenette C, Gish R. Targeted systemic therapies for hepatocellular carcinoma: clinical perspectives, challenges and implications. World journal of gastroenterology. 2012;18(6):498–506. 10.3748/wjg.v18.i6.498 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Oliver GR, Hart SN, Klee EW. Bioinformatics for clinical next generation sequencing. Clinical chemistry. 2015;61(1):124–35. Epub 2014/12/03. 10.1373/clinchem.2014.224360 . [DOI] [PubMed] [Google Scholar]
- 7.MacConaill LE. Existing and emerging technologies for tumor genomic profiling. Journal of clinical oncology: official journal of the American Society of Clinical Oncology. 2013;31(15):1815–24. Epub 2013/04/17. 10.1200/JCO.2012.46.5948 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Cohen Y, Xing M, Mambo E, Guo Z, Wu G, Trink B, et al. BRAF mutation in papillary thyroid carcinoma. Journal of the National Cancer Institute. 2003;95(8):625–7. Epub 2003/04/17. 10.1093/jnci/95.8.625 . [DOI] [PubMed] [Google Scholar]
- 9.Ding L, Getz G, Wheeler DA, Mardis ER, McLellan MD, Cibulskis K, et al. Somatic mutations affect key pathways in lung adenocarcinoma. Nature. 2008;455(7216):1069–75. Epub 2008/10/25. 10.1038/nature07423 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Sanyal AJ, Yoon SK, Lencioni R. The etiology of hepatocellular carcinoma and consequences for treatment. The oncologist. 2010;15 Suppl 4:14–22. Epub 2010/12/09. 10.1634/theoncologist.2010-S4-14 . [DOI] [PubMed] [Google Scholar]
- 11.Golubnitschaja O, Flammer J. What are the biomarkers for glaucoma? Survey of ophthalmology. 2007;52 Suppl 2:S155-61. Epub 2007/12/06. 10.1016/j.survophthal.2007.08.011 . [DOI] [PubMed] [Google Scholar]
- 12.Wu L, Qu X. Cancer biomarker detection: recent achievements and challenges. Chemical Society reviews. 2015;44(10):2963–97. Epub 2015/03/06. 10.1039/c4cs00370e . [DOI] [PubMed] [Google Scholar]
- 13.Matsui M, Corey DR. Non-coding RNAs as drug targets. Nature reviews Drug discovery. 2017;16(3):167–79. Epub 2016/11/04. 10.1038/nrd.2016.117 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Ulitsky I, Bartel DP. lincRNAs: genomics, evolution, and mechanisms. Cell. 2013;154(1):26–46. Epub 2013/07/06. 10.1016/j.cell.2013.06.020 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Xiong XD, Ren X, Cai MY, Yang JW, Liu X, Yang JM. Long non-coding RNAs: An emerging powerhouse in the battle between life and death of tumor cells. Drug resistance updates: reviews and commentaries in antimicrobial and anticancer chemotherapy. 2016;26:28–42. Epub 2016/05/18. 10.1016/j.drup.2016.04.001 . [DOI] [PubMed] [Google Scholar]
- 16.Chen X, Yan CC, Zhang X, You ZH. Long non-coding RNAs and complex diseases: from experimental results to computational models. Briefings in bioinformatics. 2017;18(4):558–76. Epub 2016/06/28. 10.1093/bib/bbw060 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Chen X, Sun YZ, Guan NN, Qu J, Huang ZA, Zhu ZX, et al. Computational models for lncRNA function prediction and functional similarity calculation. Briefings in functional genomics. 2019;18(1):58–82. Epub 2018/09/25. 10.1093/bfgp/ely031 . [DOI] [PubMed] [Google Scholar]
- 18.Li H, Ma SQ, Huang J, Chen XP, Zhou HH. Roles of long noncoding RNAs in colorectal cancer metastasis. Oncotarget. 2017;8(24):39859–76. Epub 2017/04/19. 10.18632/oncotarget.16339 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Su M, Xiao Y, Ma J, Cao D, Zhou Y, Wang H, et al. Long non-coding RNAs in esophageal cancer: molecular mechanisms, functions, and potential applications. Journal of hematology & oncology. 2018;11(1):118. Epub 2018/09/19. 10.1186/s13045-018-0663-8 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Zhou CX, Wang X, Yang N, Xue SK, Li WC, Xie PP. LncRNA LET function as a tumor suppressor in breast cancer development. European review for medical and pharmacological sciences. 2018;22(18):6002–7. Epub 2018/10/04. 10.26355/eurrev_201809_15935 . [DOI] [PubMed] [Google Scholar]
- 21.Quan J, Pan X, Zhao L, Li Z, Dai K, Yan F, et al. LncRNA as a diagnostic and prognostic biomarker in bladder cancer: a systematic review and meta-analysis. OncoTargets and therapy. 2018;11:6415–24. Epub 2018/10/17. 10.2147/OTT.S167853 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Rupaimoole R, Slack FJ. MicroRNA therapeutics: towards a new era for the management of cancer and other diseases. Nature reviews Drug discovery. 2017;16(3):203–22. Epub 2017/02/18. 10.1038/nrd.2016.246 . [DOI] [PubMed] [Google Scholar]
- 23.Bracken CP, Scott HS, Goodall GJ. A network-biology perspective of microRNA function and dysfunction in cancer. Nature reviews Genetics. 2016;17(12):719–32. Epub 2016/11/01. 10.1038/nrg.2016.134 . [DOI] [PubMed] [Google Scholar]
- 24.Wiggins JF, Ruffino L, Kelnar K, Omotola M, Patrawala L, Brown D, et al. Development of a lung cancer therapeutic based on the tumor suppressor microRNA-34. Cancer research. 2010;70(14):5923–30. Epub 2010/06/24. 10.1158/0008-5472.CAN-10-0655 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Mestdagh P, Bostrom AK, Impens F, Fredlund E, Van Peer G, De Antonellis P, et al. The miR-17-92 microRNA cluster regulates multiple components of the TGF-beta pathway in neuroblastoma. Molecular cell. 2010;40(5):762–73. Epub 2010/12/15. 10.1016/j.molcel.2010.11.038 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Pineau P, Volinia S, McJunkin K, Marchio A, Battiston C, Terris B, et al. miR-221 overexpression contributes to liver tumorigenesis. Proceedings of the National Academy of Sciences of the United States of America. 2010;107(1):264–9. Epub 2009/12/19. 10.1073/pnas.0907904107 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Chen X, Xie D, Zhao Q, You ZH. MicroRNAs and complex diseases: from experimental results to computational models. Briefings in bioinformatics. 2019;20(2):515–39. Epub 2017/10/19. 10.1093/bib/bbx130 . [DOI] [PubMed] [Google Scholar]
- 28.Chen X, Guan NN, Sun YZ, Li JQ, Qu J. MicroRNA-small molecule association identification: from experimental results to computational models. Briefings in bioinformatics. 2018. Epub 2018/10/17. 10.1093/bib/bby098 . [DOI] [PubMed] [Google Scholar]
- 29.Xiong H, Ni Z, He J, Jiang S, Li X, He J, et al. LncRNA HULC triggers autophagy via stabilizing Sirt1 and attenuates the chemosensitivity of HCC cells. Oncogene. 2017;36(25):3528–40. Epub 2017/02/07. 10.1038/onc.2016.521 . [DOI] [PubMed] [Google Scholar]
- 30.Huang X, Gao Y, Qin J, Lu S. lncRNA MIAT promotes proliferation and invasion of HCC cells via sponging miR-214. American journal of physiology Gastrointestinal and liver physiology. 2018;314(5):G559–g65. Epub 2017/11/04. 10.1152/ajpgi.00242.2017 . [DOI] [PubMed] [Google Scholar]
- 31.Salmena L, Poliseno L, Tay Y, Kats L, Pandolfi PP. A ceRNA hypothesis: the Rosetta Stone of a hidden RNA language? Cell. 2011;146(3):353–8. Epub 2011/08/02. 10.1016/j.cell.2011.07.014 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Qi X, Zhang DH, Wu N, Xiao JH, Wang X, Ma W. ceRNA in cancer: possible functions and clinical implications. Journal of medical genetics. 2015;52(10):710–8. Epub 2015/09/12. 10.1136/jmedgenet-2015-103334 . [DOI] [PubMed] [Google Scholar]
- 33.Li L, Li M, Wang X. Cancer type-dependent correlations between TP53 mutations and antitumor immunity. DNA repair. 2020;88:102785. Epub 2020/02/03. 10.1016/j.dnarep.2020.102785 . [DOI] [PubMed] [Google Scholar]
- 34.Ruiz de Galarreta M, Bresnahan E, Molina-Sanchez P, Lindblad KE, Maier B, Sia D, et al. beta-Catenin Activation Promotes Immune Escape and Resistance to Anti-PD-1 Therapy in Hepatocellular Carcinoma. Cancer discovery. 2019;9(8):1124–41. Epub 2019/06/13. 10.1158/2159-8290.CD-19-0074 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Luke JJ, Bao R, Sweis RF, Spranger S, Gajewski TF. WNT/beta-catenin Pathway Activation Correlates with Immune Exclusion across Human Cancers. Clinical cancer research: an official journal of the American Association for Cancer Research. 2019;25(10):3074–83. Epub 2019/01/13. 10.1158/1078-0432.CCR-18-1942 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Jia Q, Wang J, He N, He J, Zhu B. Titin mutation associated with responsiveness to checkpoint blockades in solid tumors. JCI insight. 2019;4(10). Epub 2019/05/17. 10.1172/jci.insight.127901 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Li Z, Zhang H, Han J, Chen Y. Surface Nanopore Engineering of 2D MXenes for Targeted and Synergistic Multitherapies of Hepatocellular Carcinoma. 2018;30(25):e1706981. 10.1002/adma.201706981 . [DOI] [PubMed] [Google Scholar]
- 38.Bruix J, Llovet JM. Major achievements in hepatocellular carcinoma. Lancet (London, England). 2009;373(9664):614–6. Epub 2009/02/24. 10.1016/S0140-6736(09)60381-0 . [DOI] [PubMed] [Google Scholar]
- 39.Han B, Zheng Y, Wang L, Wang H, Du J, Ye F, et al. A novel microRNA signature predicts vascular invasion in hepatocellular carcinoma. 2019. 10.1002/jcp.28690 . [DOI] [PubMed] [Google Scholar]
- 40.Liu J, Lu C, Xiao M, Jiang F, Qu L, Ni R. Long non-coding RNA SNHG20 predicts a poor prognosis for HCC and promotes cell invasion by regulating the epithelial-to-mesenchymal transition. Biomedicine & pharmacotherapy = Biomedecine & pharmacotherapie. 2017;89:857–63. Epub 2017/03/12. 10.1016/j.biopha.2017.01.011 . [DOI] [PubMed] [Google Scholar]
- 41.Zhang J, Guan M, Wang Q, Zhang J, Zhou T, Sun X. Single-cell transcriptome-based multilayer network biomarker for predicting prognosis and therapeutic response of gliomas. Brief Bioinform. 2020;21(3):1080–97. Epub 2019/07/23. 10.1093/bib/bbz040 . [DOI] [PubMed] [Google Scholar]
- 42.Karreth FA, Pandolfi PP. ceRNA cross-talk in cancer: when ce-bling rivalries go awry. Cancer discovery. 2013;3(10):1113–21. Epub 2013/09/28. 10.1158/2159-8290.CD-13-0202 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Long J, Jiang C, Liu B, Dai Q, Hua R, Chen C, et al. Maintenance of stemness by miR-589-5p in hepatocellular carcinoma cells promotes chemoresistance via STAT3 signaling. Cancer letters. 2018;423:113–26. Epub 2017/12/03. 10.1016/j.canlet.2017.11.031 . [DOI] [PubMed] [Google Scholar]
- 44.Zhang X, Jiang P, Shuai L, Chen K, Li Z, Zhang Y, et al. miR-589-5p inhibits MAP3K8 and suppresses CD90(+) cancer stem cells in hepatocellular carcinoma. Journal of experimental & clinical cancer research: CR. 2016;35(1):176. Epub 2016/11/12. 10.1186/s13046-016-0452-6 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Jiang C, Long J, Liu B, Xu M, Wang W, Xie X, et al. miR-500a-3p promotes cancer stem cells properties via STAT3 pathway in human hepatocellular carcinoma. Journal of experimental & clinical cancer research: CR. 2017;36(1):99. Epub 2017/07/29. 10.1186/s13046-017-0568-3 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Giray BG, Emekdas G, Tezcan S, Ulger M, Serin MS, Sezgin O, et al. Profiles of serum microRNAs; miR-125b-5p and miR223-3p serve as novel biomarkers for HBV-positive hepatocellular carcinoma. Molecular biology reports. 2014;41(7):4513–9. Epub 2014/03/07. 10.1007/s11033-014-3322-3 . [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
(DOCX)
Data Availability Statement
All the gene expression data (RNA-seqv2 & miRNA-seq) and corresponding clinical information were collected from The Cancer Genome Atlas (TCGA) database (https://portal.gdc.cancer.gov/).