

Abstract
COVID-19 is an infectious disease caused by a newly discovered corona virus SARS-COV-2. It is the most dangerous epidemic existing currently all over the world. To date, there is no licensed vaccine and not any particular efficient therapeutic agent available to prevent or cure the disease. So development of an effective vaccine is the urgent need of the time. The proposed study aims to identify potential vaccine candidates by screening the complete proteome of SARS-COV-2 using the computational approach. From 14 protein entries in UniProtKB, 4 proteins were screened for epitope prediction based on consensus antigenicity predictions and various physico-chemical criteria like transmembrane domain, allergenicity, GRAVY value, toxicity, stability index. Comprehensive analysis of these 4 antigens revealed that spike protein (P0DTC2) and nucleoprotein (P0DTC9) show the greatest potential for experimental immunogenicity analysis. These 2 proteins have several potential CD4+ and CD8+ T-cell epitopes, as well as high probability of B-cell epitope regions as compared to well-characterized antigen the matrix protein 1 [Influenza A virus (H5N1)]. In addition, the epitope SIIAYTMSL predicted from spike protein (P0DTC2) and epitope SPRWYFYYL predicted from nucleoprotein (P0DTC9) exhibited more than 60% population coverage in the target populations Europe, North America, South Asia, Northeast Asia taken in this study. These epitopes have also been found to exhibit highly significant TCR–pMHC interactions having a joint Z value of 4.51 and 4.37 respectively. Therefore, this analysis suggests that the predicted epitopes might be suitable vaccine candidates and should be subjected to further in-vivo and in-vitro studies.
Keywords: SARS-COV-2, Covid-19, Vaccine, Epitope, MHC
Introduction
COVID-19 is a deadly disease caused by SARS corona viruses world-wide. More than 59 million (59,481,31) confirmed cases and more than 1 million (1,404,542) deaths have been reported to WHO till 25 November 2020.A pneumonia of unknown cause detected in Wuhan, China was first reported to the WHO Country Office in China on 31 December 2019. The outbreak was declared a Public Health Emergency of International Concern on 30 January 2020. On 11 February 2020, WHO announced a name for the new coronavirus disease: COVID-19.
SARS-COV-2 has round or elliptic and often pleomorphic form, and a diameter of approximately 60–140 nm (Cascella et al. 2020). It is a positive sense ssRNA virus of about 30 kb genome size. This virus belongs to family coronaviridae and genus Betacoronavirus. SARS-COV-2 genome contains two flanking untranslated regions (UTRs) and a single long open reading frame encoding a polyprotein. The 2019-nCoV genome is arranged in the order of 5′-replicase (orf1/ab)-structural proteins [Spike (S)-Envelope (E)-Membrane (M)-Nucleocapsid (N)]-3′ (Chan et al. 2020). Two-thirds of viral RNA, mainly located in the first ORF (ORF1a/b) translates two polyproteins, pp1a and pp1ab, and encodes 16 non-structural proteins (NSP), while the remaining ORFs encode accessory and structural proteins. The rest part of virus genome encodes four essential structural proteins, including spike (S) glycoprotein, small envelope (E) protein, matrix (M) protein, and nucleocapsid (N) protein (Cui et al. 2019), and also several accessory proteins, that interfere with the host innate immune response.
Based on virus genome sequencing results and evolutionary analysis, bat has been suspected as natural host of virus origin, and SARS-COV-2 might be transmitted from bats via unknown intermediate hosts to infect humans. Direct contact with intermediate host animals or consumption of wild animals was suspected to be the main route of SARS-COV-2 transmission. However, the source(s) and transmission routine(s) of SARS-COV-2 remain elusive (Guo et al. 2020).
COVID-19 affects different people in different ways. Symptoms may appear 2–14 days after exposure. Serious symptoms include difficulty in breathing, chest pain and loss of speech or movement. The most common symptoms of COVID-19 are fever, dry cough, and tiredness. Other symptoms that are less common and may affect some patients include aches and pains, nasal congestion, headache, conjunctivitis, sore throat, diarrhoea, loss of taste or smell or a rash on skin or discoloration of fingers or toes.
Transmission of the disease occurs mainly through person to person. When the person infected with COVID-19 coughs, sneezes or speaks, small droplets expelled from them land on surfaces and objects around them. Other people then catch COVID-19 by touching these objects or surfaces, then touching their eyes, nose and mouth or by breathing these droplets. Major complications due to COVID-19 include acute respiratory failure, pneumonia, acute respiratory distress syndrome, acute kidney injury, acute liver injury, acute cardiac injury, septic shock, blood clots, rhabdomyolysis, disseminated intravascular coagulation, secondary infections (Zaim et al. 2020).
Researchers worldwide are working around the clock to find a vaccine against SARS-CoV-2, the virus causing the COVID-19 pandemic. There are no effective vaccines or specific antiviral drugs for COVID-19 (Dhama et al. 2020). Possible vaccines and some specific drug treatments are under investigation. Three vaccines, two adenoviral vector vaccines and a protein-based vaccine, have been given early or limited approval without waiting for the results of phase III trials. Sputnik V formerly known as Gam-COVID-Vac developed by the Gamaleya Research Institute in Moscow, Russia, was approved by the Ministry of Health of the Russian Federation on 11 August 2020. Another vaccine developed by the Chinese company CanSino Biologics, was approved by the Chinese military in June 2020 for a year as “a specially needed drug”. A second vaccine in Russia, EpiVacCorona, developed by the State Research Center of Virology and Biotechnology, has also been granted regulatory approval On 14 October 2020, also without entering Phase 3 clinical trials (Robinson 2020 online). According to WHO Draft landscape of COVID-19 vaccine candidates 12 November 2020, there are 48 vaccine candidates in clinical evaluation and 164 in preclinical evaluation.
The conventional approach to vaccine development is based on dissection of the pathogen using biochemical, immunological and microbiological methods. Although successful in several cases, this approach has several limitations. This method can employ many years to identify a protective and useful antigen, and has failed to provide a vaccine against those pathogens that did not have obvious immunodominant protective antigens. The availability of complete genome sequences in combination with novel advanced technologies, such as bioinformatics, microarrays and proteomics, have revolutionized the approach to vaccine development and provided a new impulse to microbial research (Capecchi et al. 2004). To use computers to rationally design vaccines starting with information present in the genome, without the need to grow the specific microbe, this new approach was denominated ‘reverse vaccinology’ (Rappuoli 2000). The first example of reverse vaccinology approach is the development of a vaccine against serogroup B Neisseria minigitidis (MenB), a pathogen that causes 50% of the meningococcal meningitis worldwide. It took less than 18 months to identify more and some novel vaccine candidates in MenB than had been discovered during the past 40 years by conventional methods (Pizza et al. 2000). Reverse vaccinology is now being applied to many bacterial, viral and eukaryotic pathogens and has been successful in all cases in providing novel antigens for the design of new vaccines (Bagnoli et al. 2011). Vaccine candidates identified from a pathogen's genome or proteome can then be expressed as recombinant proteins and tested in appropriate in vitro or in vivo models to assess immunogenicity and protection (Seib et al. 2000).
In the present study, SARS-COV-2 (NC_045512.2) reference strain, which is known to cause COVID-19 pandemic was undertaken to characterize its antigens as potential vaccine candidates.
Materials and Method
Retrieval of Proteome Data Set
The complete proteome sequence of SARS-COV-2 has been retrieved from Viralzone Expasy server (viralzone.expasy.org). The sequences have been stored as fasta file containing all 14 annotated UniProtKB protein entries. A well characterized viral antigen showing proper immune response in humans the matrix protein 1 [Influenza A virus (H5N1)] has been taken as control to compare and validate the outcomes. It has been tested as an adjuvanted virosomal H5N1 vaccine and found to induce a balanced Th1/Th2 CD4(+) T cell response in man (Pederson et al. 2014).
Antigenicity Prediction
Antigenicity prediction of all the protein sequences has been performed to determine their overall possible role in initiating an immune response. Consensus antigenicity predictions have been performed using Vaxijen and ANTIGENpro tools. VaxiJen is the first server for alignment-independent prediction of protective antigens. It was developed to allow antigen classification solely based on the physicochemical properties of proteins without recourse to sequence alignment. It is freely available through https://www.ddg-pharmfac.net/vaxijen/VaxiJen/VaxiJen.html (Doytchinova and Flower 2007). ANTIGENpro is a sequence-based, alignment-free and pathogen-independent predictor of protein antigenicity. The predictions are made by a two-stage architecture based on multiple representations of the primary sequence and five machine learning algorithms. ANTIGENpro is integrated in the SCRATCH suite of predictors available at: http://scratch.proteomics.ics.uci.edu (Magnan et al. 2010).
Characterization of Predicted Antigenic Proteins
Genome-wide characterization of vaccine candidates has been performed using various computational tools. Transmembrane regions have been predicted using TMHMM web server. It is based on hidden Markov model (Krogh et al. 2001). Assessment of allergenic potential has been carried out using AllerCatPro tool. It is entropy-adjusted hexamer hit approach as well as switching from a linear sequence window similarity to a B-cell epitope-like 3D surface similarity with predicted structures for 74% of all known allergens in a workflow guided by safety rationale (Maurer et al. 2019). Physical chemical parameters are calculated using ProtParam tool available at expasy. These parameters include the molecular weight, theoretical pI, instability index, aliphatic index and grand average of hydropathicity (GRAVY) (Gasteiger et al. 2005).
B Cell Epitope Prediction
The antigenic regions of protein recognized by the binding sites of immunoglobulin molecules are called B cell epitopes (Van Regenmortel 1993). B cell epitopes can be classified into two categories: conformational/ discontinuous epitope, where residues are distantly separated in the sequence and brought into physical proximity by protein folding and linear/continuous epitope comprised of a single continuous stretch of amino acids within a protein sequence that can react with anti-protein antibodies (Barlow et al. 1986). The designing of conformational epitopes is difficult and so experimental B cell epitopes largely include linear epitopes. A web server, BepiPred has been used to determine the probability of presence of linear B cell epitopes in the selected antigen sequences. It is based on a random forest algorithm trained on epitopes annotated from antibody-antigen protein structures. It is available at http://www.cbs.dtu.dk/services/BepiPred/ (Jespersen et al. 2017).
T Cell Epitope Prediction
T-cell epitope prediction aims to identify the shortest peptides within an antigen that are able to stimulate either CD4 or CD8 T-cells (Ahmed and Maeurer 2009). T-cell epitopes are presented on the surface of an antigen presenting cell (APC), where they are bound to major histocompatibility (MHC) molecules in order to induce immune response (Madden 1995). Cytotoxic T lymphocytes (CTL) epitope prediction has been performed using NetCTL, a web based tool designed for predicting human CTL epitopes in any given protein. It does so by integrating predictions of peptide MHC class I binding, proteasomal C terminal cleavage and TAP transport efficiency. MHC class I binding and proteasomal cleavage is performed using artificial neural networks. TAP transport efficiency is predicted using weight matrix. Peptides with a combined prediction score greater than or equal to default threshold value (0.75) are marked as potential HLA class I supertype CTL epitopes. NetCTL provides a comprehensive prediction about epitopes binding to 12 HLA class I supertypes including 5 HLA-A [A1, A2, A3, A24, A26] and 7 HLA-B [B7, B8, B27, B39, B44, B58, B62] (Larsen et al. 2007). It is available at http://www.cbs.dtu.dk/services/NetCTL. These predicted CTL epitopes have been again subjected to antigenicity prediction using Vaxijen server to assure the credibility. Furthermore, to predict binding of peptides to HLA-DR, MHC class II alleles, NetMHCII 2.2 server has been used. Predictions can be obtained for 25 HLA-Dr alleles, 20 HLA-DQ, 9 HLA-DP, and 7 mouse H2 class II alleles. It is based on artificial neural networks and publicly available at www.cbs.dtu.dk/services/NetMHCII (Nielsen and Lund 2009).
Population Coverage Analysis
T cells recognize a complex between a specific major histocompatibility complex (MHC) molecule and a particular pathogen-derived epitope. A given epitope will elicit a response only in individuals that express an MHC molecule capable of binding that particular epitope. MHC molecules are extremely polymorphic and over a thousand different human MHC (HLA) alleles are known (Bui et al. 2006). Specific HLA alleles are expressed at dramatically different frequencies in different ethnicities (Gjertson and Lee 1998; Imanishi et al. 1992). A web based tool, IEDB population coverage, has been used for population coverage analysis. This method calculates the fraction of individuals predicted to respond to a given epitope or epitope set on the basis of HLA genotypic frequencies and on the basis of MHC binding and/or T cell restriction data (Bui et al. 2006). It can be accessed through http://tools.iedb.org/population/. COVID-19 has affected all over the world, in this study Europe, North America, South Asia, Northeast Asia have been taken as target populations. The analysis focused on MHC I because of the fact that viral peptides are presented only on MHC I via the endogenous pathway (Srivastava et al. 2016).
pMHC-TCR Interaction Analysis
Proper interaction of peptide-MHC complex with TCR is very important for adaptive immune responses. PAComplex server has been utilized for this purpose. The PAComplex is a web server for predicting TCR-pMHC interactions and inferring antigen families across organisms, of a query protein or a set of peptides. This server first identifies significantly similar TCR–pMHC templates (joint Z-value ≥ 4.0) of the query by using antibody–antigen and protein–protein interacting scoring matrices for peptide-TCR and pMHC interfaces, respectively (Liu et al. 2011). The joint Z-value (Jz) is defined as:
√Jz = Z MHC × Z TCR (Marrack et al. 2008)
Here, J z ≥ 4.0 is considered a significant similarity according to the statistical analysis.
PAComplex then identifies the homologous peptide antigens of these hit templates from complete pathogen genome databases and experimental peptide databases. Finally, the server outputs peptide antigens and homologous peptide antigens of the query and displays detailed interacting models of hit TCR-pMHC templates (Liu et al. 2011). The PAComplex server is available at http://PAcomplex.life.nctu.edu.tw. Here, the CTL epitope set predicted by NetCTL and optimized by IEDB for the different target population, has been used as the target peptide set and TCR-pMHC interactions have been analyzed.
Results and Discussion
Selection of Antigens
The complete protein repertoire of SARS-COV-2 has been screened for proteins having sufficient antigenicity property. Consensus predictions have been made using Vaxijen and ANTIGENpro tools at pre-defined threshold value 0.4 for both. Out of 14 proteins, 7 have shown antigenic probability ≥ 0.4. Therefore, based on consensus prediction these 7 antigenic proteins have been taken for further analysis (Table 1). Control antigen has been found to be antigenic by both the tools.
Table 1.
List of proteins predicted to be antigenic with corresponding antigenic probabilities
| Protein no | UniProtKB id | Protein name | Antigenic Probability | No. of TM regions predicted using TMHMM | |
|---|---|---|---|---|---|
| VaxiJen | ANTIGENpro | ||||
| 1 | P0DTC1 | Replicase polyprotein 1a (pp1a) | 0.47 | 0.64 | 14 |
| 2 | P0DTD1 | Replicase polyprotein 1ab (pp1ab) | 0.46 | 0.68 | 14 |
| 3 | P0DTC2 | Spike glycoprotein (S) | 0.46 | 0.71 | 1 |
| 4 | P0DTC3 | ORF3a protein (NS3a) | 0.49 | 0.40 | 3 |
| 5 | P0DTC7 | ORF7a protein | 0.64 | 0.40 | 1 |
| 6 | P0DTC9 | Nucleoprotein (N) | 0.50 | 0.93 | 0 |
| 7 | P0DTD2 | ORF9b protein | 0.90 | 0.74 | 0 |
| Control | Q9Q0L8 | Matrix protein 1 | 0.47 | 0.86 | 0 |
Characterization of Selected Antigens
Proteins with more than one transmembrane (TM) region have been found to be difficult to clone, express and purify; thus 7 antigenic proteins predicted in the previous step have been subjected to predict presence of transmembrane domains using TMHMM server. Out of 7 antigenic proteins, 2 antigens (P0DTC1, P0DTD1) have been predicted to contain 14 TM regions, 1 antigen (P0DTC3) with 3 TM regions, 2 antigens (P0DTC2, P0DTC7) with 1 TM region and 2 antigens (P0DTC9, P0DTD2) with no TM regions (Table 1). So, these 4 antigens (P0DTC2, P0DTC7, P0DTC9, P0DTD2) are taken for further analysis. The control antigen has also not shown any TM regions. In allergenicity prediction using AllerCatPro tool, all the 4 antigenic proteins have been predicted as non- allergen. The control antigen has also been found to be non-allergen. The physical chemical parameters calculated using ProtParam tool has been shown in Table 2. Antigen P0DTC7 has been shown instability index > 40 i.e. 48.66, GRAVY value positive i.e. 0.318 so it has been removed from further analysis. Thus, based on screening so far, finally 3 candidate antigens (P0DTC2, P0DTC9, P0DTD2) have been selected for epitope prediction.
Table 2.
Physical chemical parameters calculated using ProtParam tool
| UniprotKB id | Protein name | Molecular weight (KDa) | Theoretical pI | Instability Index | Aliphatic Index | GRAVY |
|---|---|---|---|---|---|---|
| P0DTC2 | Spike glycoprotein (S) | 141.17 | 6.24 | 33.01 | 84.67 | − 0.079 |
| P0DTC7 | ORF7a protein | 13.74 | 8.23 | 48.66 | 100.74 | 0.318 |
| P0DTC9 | Nucleoprotein (N) | 45.62 | 10.07 | 55.09 | 52.53 | − 0.971 |
| P0DTD2 | ORF9b protein | 10.79 | 6.56 | 33.11 | 105.46 | − 0.085 |
|
(Control) |
Matrix protein 1 | 27.85 | 9.42 | 38.72 | 82.90 | − 0.246 |
B Cell Epitope Prediction
According to BepiPred linear B cell epitope predictions at threshold 0.45, high probability of B cell epitope has been found in all the three antigens. Antigens P0DTC2, P0DTC9 and P0DTD2 have been predicted to have 25, 9 and 2 regions respectively as probable B cell epitopes regions. Similar criteria set has been used for control antigen and 6 regions have been predicted as probable B cell epitope regions.
T Cell Epitope Prediction
For HLA class I supertypes, based on highest value of combined score obtained using NetCTL, a total of 419 putative CTL epitopes have been predicted for antigen P0DTC2, 104 putative CTL epitopes have been predicted for antigen P0DTC9 and 33 putative CTL epitopes have been predicted for antigen P0DTD2. The control antigen has been predicted to show 88 putative CTL epitopes. Antigenicity analysis of these predicted CTL epitopes using Vaxijen server at threshold 0.4 has shown that many of them have been found to be non-antigen. So those non-antigenic peptides have been removed and peptides predicted to bind more than one HLA class I supertype have been selected. Thus, 53 putative CTL epitopes have been selected from antigen P0DTC2, 10 putative CTL epitopes have been selected from antigen P0DTC9 and 8 putative CTL epitopes have been selected from antigen P0DTD2 for further analysis as listed in Table 3.
Table 3.
Selected CTL epitopes and their binding to different MHC class I supertypes
| Protein | Epitope | MHC I supertypes |
|---|---|---|
|
Spike glycoprotein (S) |
AALQIPFAM | B7, B58 |
| AIVMVTIML | A2, B7 | |
| DEDDSEPVL | B39, B44 | |
| EPVLKGVKL | B7, B8 | |
| ESNKKFLPF | A26, B62 | |
| FAMQMAYRF | B58, B62 | |
| FEYVSQPFL | B39, B44 | |
| FLHVTYVPA | A2, B8 | |
| FRKSNLKPF | B8, B27 | |
| FTISVTTEI | A2, A26, B58 | |
| FVFLVLLPL | A2, A26, B8, B62 | |
| GAAAYYVGY | A1, B58, B62 | |
| GAEHVNNSY | A1, B62 | |
| GQTGKIADY | B27, B62 | |
| IAIPTNFTI | A24, B58 | |
| IGAGICASY | B58, B62 | |
| IGIVNNTVY | B58, B62 | |
| ITDAVDCAL | A1, B39, B58 | |
| KGIYQTSNF | B58, B62 | |
| KIADYNYKL | A2, B39 | |
| KIYSKHTPI | A2, B8 | |
| KTSVDCTMY | A1, A3, B58, B62 | |
| KVTLADAGF | B58, B62 | |
| LLALHRSYL | A2, B8 | |
| LPFFSNVTW | B7, B58 | |
| LSETKCTLK | A1, A3 | |
| MTSCCSCLK | A1, A3 | |
| NGVEGFNCY | A26, B62 | |
| NLLLQYGSF | B8, B62 | |
| NTSNQVAVL | A26, B39 | |
| QIITTDNTF | A24, A26, B58, B62 | |
| QLTPTWRVY | A1, B62 | |
| RVVVLSFEL | A2, B7, B58, B62 | |
| SIIAYTMSL | A2, A26, B62 | |
| SLSSTASAL | A2, B7, B62 | |
| SPRRARSVA | B7, B8 | |
| STECSNLLL | A1, B39 | |
| STQDLFLPF | A1, A24, A26, B62 | |
| TFEYVSQPF | A24, B62 | |
| TLDSKTQSL | A2, B39 | |
| TLLALHRSY | A3, B62 | |
| TSNQVAVLY | A1, A3, A26, B58, B62 | |
| VLKGVKLHY | A1, A3, B62 | |
| VLPFNDGVY | A1, B62 | |
| VRFPNITNL | B27, B39 | |
| VVNQNAQAL | B7, B62 | |
| VYDPLQPEL | A24, B39 | |
| WTAGAAAYY | A1, A26, B58, B62 | |
| WTFGAGAAL | A26, B62 | |
| YLQPRTFLL | A2, B39, B58, B62 | |
| YQDVNCTEV | A1, A2, B39 | |
| YQPYRVVVL | A2, A24, B8, B39, B62 | |
| YVPAQEKNF | A26, B62 | |
|
Nucleoprotein (N) |
DLSPRWYFY | A1, A3, A26 |
| FPRGQGVPI | B7, B8 | |
| KAYNVTQAF | A24, B7, B8, B58, B62 | |
| KMKDLSPRW | B58, B62 | |
| LSPRWYFYY | A1, A3, A26, B58, B62 | |
| QFAPSASAF | A24, B62 | |
| QKKQQTVTL | B8, B39 | |
| QRQKKQQTV | B8, B27 | |
| SPRWYFYYL | B7, B8 | |
| SSPDDQIGY | A1, A26, B62 | |
|
ORF9b protein |
GPKVYPIIL | B7, B8 |
| KISEMHPAL | A2, B7, B8, B39, B58, B62 | |
| KVYPIILRL | A2, A3, B58 | |
| LRLGSPLSL | B27, B39 | |
| MARKTLNSL | B7, B8 | |
| RLVDPQIQL | A2, B62 | |
| SEMHPALRL | B39, B44 | |
| SLEDKAFQL | A2, B39 |
For HLA class II supertypes using NetMHC II algorithm, 341, 79 and 33 putative HTL epitopes have been predicted for P0DTC2, P0DTC9 and P0DTD2 respectively. The control antigen has been predicted 67 HTL epitopes binding to 15 HLA-DR supertypes.
Population Coverage Analysis
Epitope vaccines trigger an immune response by confronting the immune system with immunogenic peptides. Binding of these peptides to proteins from the major histocompatibility complex (MHC) is crucial for immune system activation. However, since the MHC is highly polymorphic, crucial step in design of a peptide vaccine is the selection of the set of epitopes which yields the best immune response in a given population or individual (Jain et al. 2019). It has been demonstrated that a correlation exists between immunogenicity and MHC class I binding affinity (Sette et al. 1994). It is, therefore, reasonable to use MHC class I binding affinity prediction methods for the prediction of immunogenicity.
CTL epitope sets obtained in the previous step have been taken as input for population coverage analysis. IEDB population coverage server outputs percentage population coverage of individual epitope in the epitope set for all the target populations taken. Table 4 shows the top scoring epitopes and their respective population coverage percentage.
Table 4.
Population coverage analysis of optimized top scoring CTL epitopes for different target populations
| Protein | Target population | Epitope | Percentage coverage | Total HLA hits |
|---|---|---|---|---|
|
Spike glycoprotein (S) |
Europe | RVVVLSFEL | 80.55% | 31 |
| SLSSTASAL | 77.12% | 31 | ||
| YQPYRVVVL | 75.44% | 24 | ||
| AIVMVTIML | 72.91% | 18 | ||
| FVFLVLLPL | 68.03% | 20 | ||
| TSNQVAVLY | 63.35% | 26 | ||
| YLQPRTFLL | 63.31% | 26 | ||
| SIIAYTMSL | 60.53% | 33 | ||
| North America | RVVVLSFEL | 80.83% | 31 | |
| SLSSTASAL | 80.83% | 31 | ||
| SIIAYTMSL | 78.10% | 33 | ||
| YQPYRVVVL | 74.61% | 24 | ||
| AIVMVTIML | 69.55% | 18 | ||
| TSNQVAVLY | 67.13% | 26 | ||
| VVNQNAQAL | 64.63% | 23 | ||
| YLQPRTFLL | 64.56% | 26 | ||
| South Asia | TSNQVAVLY | 80.64% | 26 | |
| KTSVDCTMY | 76.42% | 23 | ||
| VLKGVKLHY | 71.64% | 17 | ||
| LSETKCTLK | 66.63% | 10 | ||
| MTSCCSCLK | 66.63% | 10 | ||
| TLLALHRSY | 66.62% | 13 | ||
| SIIAYTMSL | 65.03% | 33 | ||
| RVVVLSFEL | 61.94% | 31 | ||
| North East Asia | TSNQVAVLY | 82.88% | 26 | |
| KTSVDCTMY | 81.61% | 23 | ||
| RVVVLSFEL | 79.33% | 31 | ||
| VLKGVKLHY | 78.68% | 17 | ||
| TLLALHRSY | 76.97% | 13 | ||
| SLSSTASAL | 73.78% | 31 | ||
| VVNQNAQAL | 68.07% | 23 | ||
| SIIAYTMSL | 66.22% | 33 | ||
|
Nucleoprotein (N) |
Europe | KAYNVTQAF | 77.43% | 27 |
| LSPRWYFYY | 63.35% | 26 | ||
| SPRWYFYYL | 60.10% | 14 | ||
| FPRGQGVPI | 59.75% | 12 | ||
| North America | KAYNVTQAF | 76.89% | 27 | |
| LSPRWYFYY | 67.13% | 26 | ||
| DLSPRWYFY | 54% | 13 | ||
| SPRWYFYYL | 51.20% | 14 | ||
| South Asia | LSPRWYFYY | 80.64% | 26 | |
| DLSPRWYFY | 72.60% | 13 | ||
| KAYNVTQAF | 63.14% | 27 | ||
| SPRWYFYYL | 32.99% | 14 | ||
| North East Asia | LSPRWYFYY | 82.88% | 26 | |
| KAYNVTQAF | 76.27% | 27 | ||
| DLSPRWYFY | 64% | 13 | ||
| SPRWYFYYL | 25.03% | 14 | ||
|
ORF9b protein |
Europe | KISEMHPAL | 88.15% | 38 |
| KVYPIILRL | 80.84% | 20 | ||
| GPKVYPIIL | 60.10% | 15 | ||
| RLVDPQIQL | 55.65% | 14 | ||
| North America | KISEMHPAL | 85.77% | 38 | |
| KVYPIILRL | 77.80% | 20 | ||
| RLVDPQIQL | 55.50% | 15 | ||
| GPKVYPIIL | 51.20% | 14 | ||
| South Asia | KVYPIILRL | 76.77% | 20 | |
| KISEMHPAL | 65.26% | 38 | ||
| GPKVYPIIL | 32.99% | 14 | ||
| RLVDPQIQL | 31.48% | 15 | ||
| North East Asia | KVYPIILRL | 81.77% | 20 | |
| KISEMHPAL | 81.09% | 38 | ||
| RLVDPQIQL | 64.31% | 15 | ||
| SLEDKAFQL | 37.92% | 13 |
pMHC-TCR Interaction Analysis
T cells do not recognize soluble native antigen but rather recognize antigen that has been processed into antigenic peptides, which are presented in combination with MHC molecules. T-cell epitopes must be viewed in terms of their ability to interact with both a T-cell receptor and an MHC molecule. The interaction between the T-cell receptor and an antigen bound to an MHC molecule is central to both humoral and cell-mediated responses (Goldsby et al. 2007). The results obtained in TCR-pMHC interaction analysis using PAComplex are described below.
For peptide set from antigen P0DTC2, the same hit peptide has been obtained for all the four target populations. The epitope SIIAYTMSL has been found to have a joint Z value of 4.51, illustrating that this peptide demonstrates highly significant pMHC-TCR interactions (Fig. 1). This hit peptide is homologous to template peptide GILGFVFTL (PDB: 1oga), which is a linear peptidic epitope of matrix protein 1 from influenza A virus as recorded in IEDB and shows 40 peptides in peptide antigen family of template 1oga across 25 organisms.
Fig. 1.

PAComplex server showing pMHC-TCR interactions and homologous peptide for antigen P0DTC2
The peptide set from antigen P0DTC9 has also shown the same hit peptide for all the four target populations. The epitope SPRWYFYYL has been found to have a joint Z value of 4.37, indicating that this peptide exhibits immensely valuable pMHC-TCR interactions (Fig. 2). This hit peptide is homologous to template peptide GILGFVFTL (PDB: 2vlr), which is a linear peptidic epitope from matrix protein 1 of influenza A virus as recorded in IEDB and shows 61 peptides in peptide antigen family of template 2vlr across 34 organisms.
Fig. 2.

PAComplex server showing pMHC-TCR interactions and homologous peptide for antigen P0DTC9
Peptide set from antigen P0DTD2 has not shown any hit peptide for any of the four target populations.
The hit peptide antigen SIIAYTMSL from P0DTC2 matches the profile of the homologous antigen family on positions 2, 4, 5, 8 and 9 (Fig. 3). The homologous peptide antigens prefers the nonpolar residues on second and fourth position (Met, Ile, Leu and Gly, Ala respectively) and the second position of the hit peptide is nonpolar residue Ile forming five VDW interactions with residues Tyr99, Val67, Met45, Tyr7, Phe9 and two hydrogen bonds with residues Lys 66, Glu63 on MHC molecule; fourth position of hit peptide is nonpolar residue Ala forming hydrogen bond with residue Gln52 in TCR. Position 5 of homologous peptide antigens prefers the aromatic residues (Phe, Tyr and Trp) and fifth position of hit peptide is aromatic residue Tyr forming strong VDW interaction with residue Leu156 on MHC molecule. Additionally position 8 of homologous peptide antigens prefers the polar residues (Ser, Thr and Asp) and Ser at position 8 in hit peptide forms VDW interaction with residue Thr73 and two hydrogen bonds with residues Trp147, Lys146 on MHC molecule and one hydrogen bond with residue Asp32 in TCR. Position 9 of homologous peptide antigens prefers the nonpolar residues (Leu, Ile) and Leu at position 9 in hit peptide forms three VDW interactions with residue Leu81, Ile124, Trp147 and three hydrogen bonds with residue Asp77, Tyr84, Thr143 on MHC molecule.
Fig. 3.

Frequency logo for the peptide antigen family of homologous template peptide 1oga (GILGFVFTL) of top hit peptide (SIIAYTMSL)
Furthermore, the hit peptide antigen SPRWYFYYL from P0DTC9 relates the profile of the homologous antigen family on positions 2, 5, 7 and 9 (Fig. 4). Position 2 of homologous peptide antigens prefers the nonpolar residues (Ile, Leu, Met) and second position in the hit peptide is nonpolar residue Pro forming five VDW interactions with residue Tyr99, Val67, Met45, Tyr7, Phe9 and two hydrogen bonds with residue Lys66, Glu63 on MHC molecule. Position 5 and 7 of homologous peptide antigens prefers the aromatic residues (Phe, Tyr); fifth and seventh position of hit peptide are also aromatic residue Tyr forming strong VDW interaction with residue Leu156 and residue Leu156, Val152, Tyr166, Trp147 on MHC molecule respectively. Additionally position 9 of homologous peptide antigens prefers the nonpolar residues (Leu, Ile, Val, Met) and position 9 in the hit peptide is nonpolar residue Leu forming three VDW interactions with residue Leu81, Ile124, Trp147 and three hydrogen bonds with residue Asp77, Tyr84, Thr143 on MHC molecule.
Fig. 4.
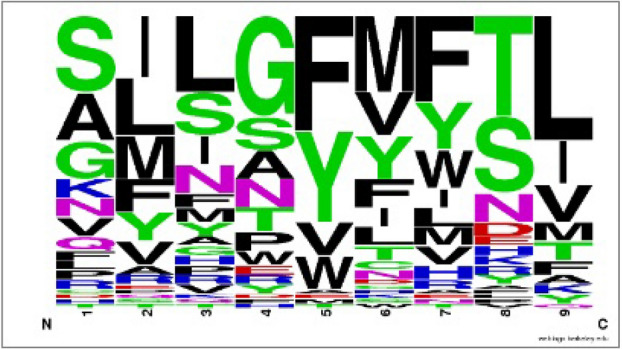
Frequency logo for the peptide antigen family of homologous template peptide 2vlr (GILGFVFTL) of top hit peptide (SPRWYFYYL)
Therefore, these two peptides can be considered as potential vaccine candidates and can be capable of evoking significant immune response. Further in-vivo/in-vitro assessment should facilitate the effectiveness, development of polytopic vaccines and immune modulatory effects of the predicted peptides.
Conclusions
The world is in the midst of a COVID-19 pandemic. Vaccines can prevent infectious diseases and save millions of lives each year. Vaccines work by training and preparing the body’s natural defences, the immune system, to recognize and fight off the viruses and bacteria they target. If the body is exposed to those disease-causing germs later, the body is immediately ready to destroy them, preventing illness. In recent years, peptide based vaccines have emerge as very convenient and crucial protection against infectious diseases. Immunoinformatics is a branch of bioinformatics that involves application of computational algorithms to analyse immunological data and problems. Advances in the field of immunoinformatics have led the development and widely distribution of hundreds of new vaccine design algorithms for exploration of proteomics. Prediction and analysis of antigenic peptides recognized by T helper and cytotoxic T lymphocytes from protein repertoire of pathogen followed by refined focus on the resulting set of peptides is central to modern vaccine development. The development of an effective and affordable vaccine against COVID-19 is the necessity of the hour for global public health. The present study involves application of various available bioinformatics tools for prediction of promising vaccine candidates by comprehensive mining of the proteome of SARS-COV-2. The pMHC-TCR interaction analysis in-silico demonstrated that the predicted peptides show homology to well-known potential antigens. Therefore, the present work is a very prominent strategy for rational antigen identification with further in-vivo/in-vitro experimentation required to emphasize the importance of the epitopes.
Acknowledgements
The authors sincerely thank the anonymous Editor and the Reviewers for their valuable comments and suggestions in improving the quality of manuscript.
Funding
The study was conducted using resources available online and no additional funding was involved.
Data Availability
Data are available on request to the corresponding author.
Declarations
Conflict of interest
Authors declare that they have no conflict of interest.
Footnotes
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
References
- Ahmed RK, Maeurer MJ. T-cell epitope mapping. Methods Mol Biol. 2009;524:427–438. doi: 10.1007/978-1-59745-450-6_31. [DOI] [PubMed] [Google Scholar]
- Bagnoli F, Baudner B, Mishra RP, Bartolini E, Fiaschi L, Mariotti P, Nardi-Dei V, Boucher P, Rappuoli R. Designing the next generation of vaccines for global public health. OMICS. 2011;15(9):545–566. doi: 10.1089/omi.2010.0127. [DOI] [PubMed] [Google Scholar]
- Barlow DJ, Edwards MS, Thornton JM. Continuous and discontinuous protein antigenic determinants. Nature. 1986;322(6081):747–748. doi: 10.1038/322747a0. [DOI] [PubMed] [Google Scholar]
- Bui HH, Sidney J, Dinh K, Southwood S, Newman MJ, Sette A. Predicting population coverage of T-cell epitope-based diagnostics and vaccines. BMC Bioinform. 2006;7:153. doi: 10.1186/1471-2105-7-153. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Capecchi B, Serruto D, Adu-Bobie J, Rappuoli R, Pizza M. The genome revolution in vaccine research. Curr Issues Mol Biol. 2004;6(1):17–27. [PubMed] [Google Scholar]
- Cascella M, Rajnik M, Cuomo A, et al (2020) Features, evaluation, and treatment of coronavirus. [Updated 2020 Oct 4]. In: StatPearls [Internet]. Treasure Island (FL): StatPearls Publishing. https://www.ncbi.nlm.nih.gov/books/NBK554776/ [PubMed]
- Chan JF, Kok KH, Zhu Z, Chu H, To KK, Yuan S, Yuen KY (2020) Genomic characterization of the 2019 novel human-pathogenic coronavirus isolated from a patient with atypical pneumonia after visiting Wuhan. Emerg Microbes Infect 9(1):221–236. doi: 10.1080/22221751.2020.1719902. Erratum in: Emerg Microbes Infect. 2020; 9(1):540 [DOI] [PMC free article] [PubMed]
- Cui J, Li F, Shi Z. Origin and evolution of pathogenic coronaviruses. Nat Rev Microbiol. 2019;17:181–192. doi: 10.1038/s41579-018-0118-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dhama K, Sharun K, Tiwari R, Dadar M, Malik YS, Singh KP, Chaicumpa W. COVID-19, an emerging coronavirus infection: advances and prospects in designing and developing vaccines, immunotherapeutics, and therapeutics. Hum Vaccin Immunother. 2020;16(6):1232–1238. doi: 10.1080/21645515.2020.1735227. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Doytchinova IA, Flower DR. VaxiJen : a server for prediction of protective antigens, tumour antigens and subunit vaccines. BMC Bioinform. 2007;8:4. doi: 10.1186/1471-2105-8-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gasteiger E, Hoogland C, Gattiker A, Duvaud S, Wilkins MR, Appel RD, Bairoch A. Protein identification and analysis tools on the ExPASy server. In: Walker JM, editor. The proteomics protocols handbook. Totowa: Humana Press; 2005. pp. 571–607. [Google Scholar]
- Gjertson DW, Lee S-H. HLA-A/B and -DRB1/DQB1 allele-level haplotype frequencies. In: Terasaki PI, editor. HLA 1998. Lenexa: American Society for Histocompatibility and Immunogenetics; 1998. pp. 365–450. [Google Scholar]
- Goldsby R, Kindt T, Kuby J, Osborne B. T-cell receptor. In: Goldsby R, Kindt T, Kuby J, Osborne B, editors. Kuby immunology. 5. New York: W. H. Freeman; 2007. p. 217. [Google Scholar]
- Guo Y, Cao Q, Hong Z, Tan Y, Chen S, Jin H, Tan K, Wang D, Yan Y. The origin, transmission and clinical therapies on coronavirus disease 2019 (COVID-19) outbreak—an update on the status. Mil Med Res. 2020;7:11. doi: 10.1186/s40779-020-00240-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Imanishi T, Akaza T, Kimura A, Tokunaga K, Gojoubori T (1992) Allele and haplotype frequencies for HLA and complement loci in various ethnic groups In: Tsuji K MA, Sasazuki T (eds) HLA 1991: Proceedings of the Eleventh International Histocompatibility Workshop and Conference. Oxford University Press, Oxford, pp 1065–1220
- Jain R, Singh S, Verma SK, Jain A. Genome-wide prediction of potential vaccine candidates for campylobacter jejuni using reverse vaccinology. Interdiscip Sci. 2019;11(3):337–347. doi: 10.1007/s12539-017-0260-5. [DOI] [PubMed] [Google Scholar]
- Jespersen MC, Peters B, Nielsen M, Marcatili P. BepiPred-20: improving sequence-based B-cell epitope prediction using conformational epitopes. Nucleic Acids Res. 2017 doi: 10.1093/nar/gkx352. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Krogh A, Larsson È, Heijne GV, Sonnhammer ELL. Predicting transmembrane protein topology with a hidden markov model : application to complete genomes. J Mol Biol. 2001;305:567–580. doi: 10.1006/jmbi.2000.4315. [DOI] [PubMed] [Google Scholar]
- Larsen MV, Lundegaard C, Lamberth K, Buus S, Lund O, Nielsen M. Large-scale validation of methods for cytotoxic T-lymphocyte epitope prediction. BMC Bioinform. 2007;8:424. doi: 10.1186/1471-2105-8-424. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu I, Lo Y, Yang J. PAComplex : a web server to infer peptide antigen families and binding models from TCR – pMHC complexes. Nucleic Acids Res. 2011 doi: 10.1093/nar/gkr434. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Madden DR. The three-dimensional structure of peptide-MHC complexes. Annu Rev Immunol. 1995;13:587–622. doi: 10.1146/annurev.iy.13.040195.003103. [DOI] [PubMed] [Google Scholar]
- Magnan CN, Zeller M, Kayala MA, Vigil A, Randall A, Felgner PL, Baldi P. High-throughput prediction of protein antigenicity using protein microarray data. Bioinformatics. 2010;26(23):2936–2943. doi: 10.1093/bioinformatics/btq551. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marrack P, Scott-Browne JP, Dai S, Gapin L, Kappler JW. Evolutionarily conserved amino acids that control TCR-MHC interaction. Annu Rev Immunol. 2008;26:171–203. doi: 10.1146/annurev.immunol.26.021607.090421. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maurer-Stroh S, Krutz NL, Kern PS, Gunalan V, Nguyen MN, Limviphuvadh V, Eisenhaber F, Gerberick GF. AllerCatPro-prediction of protein allergenicity potential from the protein sequence. Bioinformatics. 2019;35(17):3020–3027. doi: 10.1093/bioinformatics/btz029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nielsen M, Lund O. NN-align. An artificial neural network-based alignment algorithm for MHC class II peptide binding prediction. BMC Bioinform. 2009;10:296. doi: 10.1186/1471-2105-10-296. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pederson GK, Sjursen H, Nostbakken JK, Jul-Larsen A, Hoschler K, Cox RJ. Matrix M(TM) adjuvanted virosomal H5N1 vaccine induces balanced Th1/Th2 CD4(?) T cell responses in man. Hum Vaccin Immunother. 2014;10(8):2408–2416. doi: 10.4161/hv.29583. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pizza M, Scarlato V, Masignani V, et al. Identification of vaccine candidates against serogroup B meningococcus by whole genome sequencing. Science. 2000;287(5459):1816–1820. doi: 10.1126/science.287.5459.1816. [DOI] [PubMed] [Google Scholar]
- Rappuoli R. Reverse vaccinology. Curr Opin Microbiol. 2000;3(5):445–450. doi: 10.1016/s1369-5274(00)00119-3. [DOI] [PubMed] [Google Scholar]
- Robinson J. Ten things pharmacists should know about COVID-19 vaccines. Pharm J. 2020 doi: 10.1211/PJ.2020.20208429. [DOI] [Google Scholar]
- Seib KL, Zhao X, Rappuoli R. Developing vaccines in the era of genomics: a decade of reverse vaccinology. Clin Microbiol Infect. 2000;18(5):109–116. doi: 10.1111/j.1469-0691.2012.03939.x. [DOI] [PubMed] [Google Scholar]
- Sette A, Vitiello A, Reherman B, Fowler P, Nayersina R, Kast WM, Melief CJ, Oserof C, Yuan L, Ruppert J. The relationship between class I binding affinity and immunogenicity of potential cytotoxic T cell epitopes. J Immunol Res. 1994;153:5586–5592. doi: 10.1186/1471-2105-15-241. [DOI] [PubMed] [Google Scholar]
- Srivastava PN, Jain R, Dubey SD, Bhatnagar S, Ahmad N. Prediction of epitope-based peptides for vaccine development from coat proteins GP2 and VP24 of ebola virus using immunoinformatics. Int J Pept Res Ther. 2016;22:119–133. doi: 10.1007/s10989-015-9492-6. [DOI] [Google Scholar]
- Van Regenmortel MH. Synthetic peptides versus natural antigens in immunoassays. Ann Biol Clin (Paris) 1993;51(1):39–41. [PubMed] [Google Scholar]
- Zaim S, Chong JH, Sankaranarayanan V, Harky A. COVID-19 and multiorgan response. Curr Prob Cardiol. 2020;45(8):100618. doi: 10.1016/j.cpcardiol.2020.100618. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Data Availability Statement
Data are available on request to the corresponding author.