

Heterodimeric prodrug nanoparticles overcome the penetration limitations of the neighboring effect.
Abstract
Chemotherapeutic nanomedicines can exploit the neighboring effect to increase tumor penetration. However, the neighboring effect is limited, likely by the consumption of chemotherapeutic agents and resistance of internal hypoxic tumor cells. Here, we first propose and demonstrate that apoptotic bodies (ApoBDs) could carry the remaining drugs to neighboring tumor cells after apoptosis. To enhance the ApoBD-based neighboring effect, we fabricated disulfide-linked prodrug nanoparticles consisting of camptothecin (CPT) and hypoxia-activated prodrug PR104A. CPT kills external normoxic tumor cells to produce ApoBDs, while PR104A remains inactive. The remaining drugs could be effectively delivered into internal tumor cells via ApoBDs. Although CPT exhibits low toxicity to internal hypoxic tumor cells, PR104A could be activated to exert strong cytotoxicity, which further facilitates deep penetration of the remaining drugs. Such a synergic approach could overcome the limitations of the neighboring effect to penetrate deep into solid tumors for whole tumor destruction.
INTRODUCTION
Chemotherapeutic nanomedicines have been found to enhance drug penetration in solid tumors and amplify their therapeutic effects through the neighboring effect. The neighboring effect means that tumor cells may become in situ drug depots after taking up drug-loaded nanoparticles (NPs). When drug-loaded NPs induce cell apoptosis, the remaining drugs are liberated from dead or dying cells, which then infect surrounding cells (1–3). The detailed mechanism behind the transport of the drugs from the depot cells to other cells needs to be further elucidated. Yong and coworkers (4) reported that the neighboring effect of NPs might be modulated via lysosomal exocytosis. Zhou et al. (5) proposed that the cationization of NPs could effectively induce adsorption-mediated transcytosis, which was favorable for tumor penetration. However, we suppose that there may be other routes for the effective intercellular transport of large amounts of NPs after tumor cell death.
When tumor cells undergo apoptosis, the cell membrane shrinks, divides, and wraps the cytoplasm to produce apoptotic bodies (ApoBDs) (6). Therefore, the remaining drugs may be stored in ApoBDs. The elimination of apoptotic cells is mainly accomplished by “professional phagocytes” such as macrophages or “nonprofessional neighboring cells.” Professional phagocytes are often not abundant in the sites where apoptosis occurs. Therefore, nonprofessional neighbors usually clear apoptotic cells during development (7–9). In addition, given the nutrient-deprived conditions of tumor cells to proliferate, they generally adsorb nutrients through macropinocytosis with the scavenging of macromolecules from the microenvironment, such as ApoBDs (10, 11). Hence, we propose that the macropinocytosis of ApoBDs by neighboring tumor cells may contribute to the efficient intercellular drug delivery of the neighboring effect.
Theoretically, the neighboring effect could facilitate chemotherapeutic agents to continuously penetrate deep into the tumor until it kills all tumor cells like “peeling an onion” layer by layer. However, according to previous reports, the neighboring effect is an effect of limited tumor penetration distance (2). The limitations of penetration may be ascribed to the consumption of chemotherapeutic agents during the peeling an onion process and the resistance of internal hypoxic tumor cells (12, 13). Hypoxia-activated prodrugs (HAPs) are a class of drugs that maintain nontoxicity under normoxia but can be converted into toxic drugs by highly expressed reductases in the hypoxic tumor region (14–16). Therefore, combining a chemotherapeutic agent with an HAP may provide a synergic approach to overcome the limitations of the neighboring effect for a “relay race” tumor deep penetration and the entire tumor-killing effect.
Prodrug nanoassemblies are a new type of self-delivering nanoplatform by the drug conjugates, demonstrating a variety of advantages, such as facile fabrication, high drug-loading capacity, extended blood circulation, and reduced carrier-related toxicity (17–20). In addition, redox-responsive nanodrug delivery systems have attracted widespread attention due to the significantly different redox levels between tumor cells and normal cells (21, 22). Therefore, self-assembled prodrug NPs with redox-sensitive drug release behavior hold great potential for application in combination treatment.
Here, we prepared CSSP NPs through the self-assembly of the heterodimeric prodrug CPT-SS-PR104A consisting of camptothecin (CPT), HAP PR104A, and a disulfide linkage (Fig. 1A). As expected, the high-level cytosolic glutathione (GSH) could trigger the cleavage of the disulfide bond to release CPT and PR104A quickly. CPT killed external normoxic tumor cells to produce CPT- and PR104A-coloaded ApoBDs. ApoBDs can be engulfed by neighboring cells to deliver drugs into internal hypoxic tumor cells. The drug content of CPT was gradually consumed by the normoxic cells, and the hypoxic cells were also resistant to CPT, while PR104A with low consumption could be activated to exert cytotoxicity in the hypoxic cells for further drug penetration (Fig. 1B). Our research indicates that the ApoBD-mediated neighboring effect could facilitate CSSP NPs to achieve enhanced tumor penetration, thereby eliminating all tumor cell subgroups.
Fig. 1. Schematic diagram.
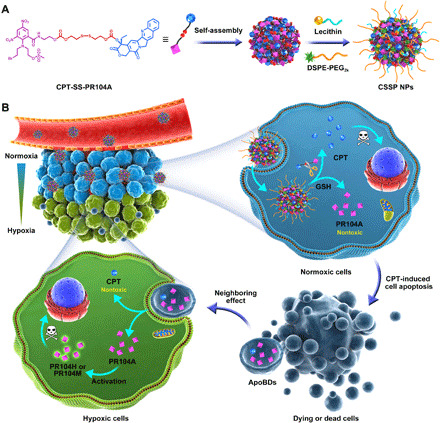
(A) Fabrication of self-assembled CSSP NPs. (B) CSSP NPs enhance drug penetration and whole tumor destruction through ApoBD-mediated neighboring effect.
RESULTS
Fabrication and characterization of CSSP NPs
First, we synthesized PR104A on the basis of previous reports (20, 23, 24). Then, we synthesized two previously unreported heterodimeric prodrugs by bonding CPT and PR104A with a disulfide or carbon chain as linkers (fig. S1). CPT was reacted with bis(2-hydroxyethyl) disulfide and 1,6-hexanediol using carbonate ester formation to produce CPT-SS-OH and CPT-CC-OH, respectively. The structure of the intermediate products was characterized by electrospray ionization mass spectrometry (ESI-MS) and 1H nuclear magnetic resonance (1H NMR) (figs. S2 and S3). CPT-SS-OH and CPT-CC-OH were further connected to PR104A through carbonate bonds to synthesize CPT-SS-PR104A and CPT-CC-PR104A, respectively. The structure of the end products was also characterized by ESI-MS and 1H NMR (figs. S4 and S5).
Next, prodrug NPs were fabricated through one-step nanoprecipitation. Both CPT-SS-PR104A and CPT-CC-PR104A were found to self-assemble into NPs (CSSP NPs and CP NPs) in deionized water. 1,2-distearoylsn-glycero-3-phosphoethanolamine-N-[methoxy(polyethyleneglycol)-2000] (DSPE-PEG2k) [20% (w/w)] and lecithin [10% (w/w)] were added to increase the stability of the nanoassemblies. In our previous studies, we found that the self-assembly process of prodrugs mainly benefits from π-π stacking, the properties of linkages, or structural flexibility (25). The CPT-SS-PR104A molecules were assembled in a spiral shape, which has a lower docking energy than the stacked form of CPT-CC-PR104A (−45.45 kcal mol−1 versus –37.91 kcal mol−1), so the self-assembled structure of CSSP NPs is relatively more compact and stable (fig. S6A). The resulting prodrug NPs had a uniform spherical morphology, with an average particle size of approximately 170 nm and a surface zeta potential value of approximately −30 mV (Fig. 2, A and B, and table S1). Because the prodrugs were both nanocarriers and payloads, the prodrug NPs showed higher drug-loading efficiency [25.4 to 26.3% for CPT and 36.4 to 37.7% for PR104A (w/w)] than the commonly encapsulated nanopreparations (usually <10%). This high drug-loading capacity could ensure high drug delivery efficiency while reducing carrier-related side effects. In addition, the prodrug NPs exhibited favorable colloidal stability and excellent long-term storage stability (fig. S6, B and C).
Fig. 2. Characterization of reduction-sensitive CSSP NPs.
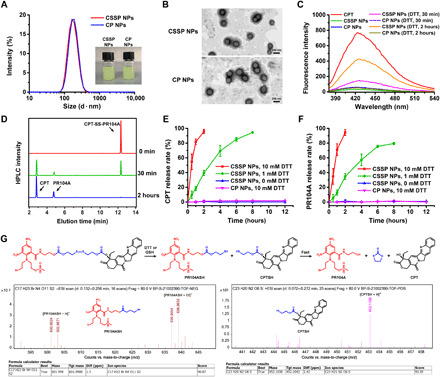
(A) Hydrodynamic sizes and photos of prodrug NPs. (B) Transmission electron microscopy (TEM) images of prodrug NPs. (C) Fluorescence spectral changes of prodrug NPs (1 μg ml−1, CPT equivalent) after incubation with 10 mM dithiothreitol (DTT). (D) High-performance liquid chromatography (HPLC) determination of CSSP NPs incubated with 10 mM DTT. In vitro (E) CPT and (F) PR104A release of prodrug NPs incubated with different concentrations of DTT (n = 3). (G) ESI-MS of CSSP NPs when incubated with 1 mM DTT for approximately 2 hours and the drug release mechanism. ppm, parts per million. Photo credit: Dongyang Zhao, Department of Pharmaceutics, Wuya College of Innovation, Shenyang Pharmaceutical University, Shenyang 110016, China.
Selective drug release at the tumor site is important for the safety and effectiveness of the treatment. Here, dithiothreitol (DTT), a common GSH simulant, was used to study the reduction-sensitive drug release ability of CSSP NPs. Compared with free CPT, the fluorescence of prodrug NPs was quenched because of the aggregation-caused quenching effect (26). However, the fluorescence of CSSP NPs increased after incubation with 10 mM DTT, indicating reduction-responsive drug release (Fig. 2C). In addition, the high-performance liquid chromatography (HPLC) profiles indicated that the released compounds were free CPT and PR104A (Fig. 2D). As shown in Fig. 2 (E and F), less than 5% of both CPT and PR104A were released from CSSP NPs within 12 hours in the absence of DTT, but 95.7% of CPT and 94.4% of PR104A were released within 2 hours when incubated with 10 mM DTT. The release ratio of CPT and PR104A was always close to 1:1, indicating simultaneous drug release. By comparison, almost no CPT or PR104A was released from the CP NPs after 12 hours of incubation with 10 mM DTT. To further investigate the drug release mechanism, the precise molecular weight changes were determined by high-resolution mass spectrometry (HRMS) after CSSP NPs were incubated with 1 mM DTT for approximately 2 hours. The molecular weight of PR104ASH and CPTSH indicated the fracture of the disulfide bond and the formation of free thiols. Then, the generated free thiol compounds could catalyze the fast release of CPT and PR104A via intramolecular nucleophilic substitution (Fig. 2G) (27, 28).
Intercellular drug delivery via ApoBD-mediated neighboring effect after apoptosis
Cellular uptake was investigated by observing the intracellular fluorescence intensity of CPT. As shown in Fig. 3 (A to C), both CP NPs and CSSP NPs showed stronger intracellular fluorescence than free CPT at 12 and 24 hours, and the fluorescence increased with time. Such a result indicated that prodrug NPs have notably higher cellular uptake than the free drug. It is worth noting that the intracellular fluorescence of CSSP NPs was higher than that of CP NPs, despite their similar nanostructures. Compared with CP NPs, due to the reductive responsiveness of CSSP NPs, CSSP NPs disassembled and released free CPT effectively after internalization into tumor cells to enhance fluorescence.
Fig. 3. Cellular uptake, cytotoxicity, and ApoBD-mediated neighboring effect of CSSP NPs.
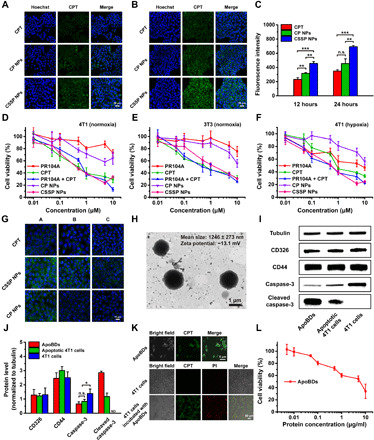
Fluorescence images of 4T1 cells after incubation with free CPT or prodrug NPs (10 μM, CPT equivalent) for (A) 12 and (B) 24 hours. (C) Flow cytometry of 4T1 cells treated with various formulations (n = 3). n.s., not significant. Cytotoxicity against (D) 4T1 or (E) 3T3 cells under normoxia after 48-hour treatment (n = 6). (F) Cytotoxicity against 4T1 cells under hypoxia after 48-hour treatment (n = 6). (G) Migration of drugs from the infected 4T1 cells to the untreated cells. A, B, and C represented coverslips. (H) Representative TEM image of ApoBDs. (I) Western blot characterization and (J) the semiquantitative analysis by Image-Pro Plus software (n = 3). ND means “not detected.” (K) Confocal laser scanning microscopy (CLSM) images of ApoBDs, 4T1 cells, and 4T1 cells incubated with ApoBDs. Dead cells were labeled with propidium iodide (PI) (red). (L) Cytotoxicity of ApoBDs after 48-hour incubation with 4T1 cells under normoxia (n = 6). Statistical significance: *P < 0.05, **P < 0.01, and ***P < 0.001.
Breast cancer 4T1 cells and fibroblast 3T3 cells were used to study the differential drug release of prodrug NPs between tumor cells and normal cells. As shown in fig. S7, much more CPT and PR104A were released when CSSP NPs were incubated with 4T1 cells than 3T3 cells at 6 and 12 hours. This result was ascribed to a higher GSH level in 4T1 cells (2.7-fold) than in 3T3 cells (fig. S7A) (29, 30). In contrast with CSSP NPs, CP NPs exhibited less drug release in 4T1 cells. We also found that CP NPs released more drugs in 4T1 cells than in 3T3 cells, indicating that high carboxylesterase expression in tumor cells may also promote the drug release of prodrug NPs (31, 32). Such selective drug release in tumor cells enables the effective tumor-killing ability of CSSP NPs.
Next, cytotoxicity against 4T1 and 3T3 cells under normoxic or hypoxic conditions was tested. CSSP NPs were found to be more cytotoxic to 4T1 cells than 3T3 cells under normoxia [median inhibitory concentration (IC50): 0.66 μM versus 1.64 μM], which was attributed to the faster drug release in 4T1 cells (Fig. 3, D and E, and table S2). Compared to normoxia, PR104A had enhanced toxicity under hypoxia, while CPT showed weakened toxicity, which was ascribed to the activation of PR104A and the drug resistance of CPT (Fig. 3F). In addition, both the CSSP NPs and PR104A + CPT groups exhibited a strong inhibitory effect on growth for normoxic and hypoxic tumor cells. This indicates that the combination of the two drugs is expected to eliminate all cell populations in the tumor. CP NPs had a slow intracellular drug release rate and therefore showed low cytotoxicity.
The neighboring effect–mediated intercellular trafficking of free CPT and prodrug NPs was investigated using coverslips. 4T1 cells on coverslip A were preincubated with free CPT or prodrug NPs for 12 hours, and then, coverslip A was removed and cocultured with blank cells on coverslip B for 24 hours. Then, coverslip B was removed and cocultured with blank cells on coverslip C for 24 hours. As shown in Fig. 3G, compared with free CPT, CSSP NPs could more effectively infect neighboring cells. The reason might be that NPs have a higher cell uptake efficiency, and compared with free drug solutions, the delayed release of NPs makes them have a relatively longer duration of action so that they could continue to infect the neighboring cells. To determine the possible mechanism of the neighboring effect, after incubating 4T1 cells with CSSP NPs, we collected the supernatants after culture again and separated the ApoBDs by centrifugation (fig. S8A). The mean size of ApoBDs was 1.2 μm, and the membrane zeta potential was −13.1 mV, consistent with the literature (Fig. 3H and fig. S8B) (6). ApoBDs had the same membrane markers CD326 and CD44 as 4T1 cells, and the high level of cleaved caspase-3 indicated the successful isolation of ApoBDs (Fig. 3, I and J). In addition, ApoBDs were observed to show green CPT fluorescence, indicating that after CSSP NPs killed tumor cells, the remaining drugs were stored in ApoBDs produced by apoptotic cells. When incubated with 4T1 cells, ApoBDs could be effectively taken up by 4T1 cells and then exerted cytotoxicity, with an IC50 value of protein concentration (3.61 μg ml−1) (Fig. 3, K and L). Therefore, it was confirmed that ApoBDs could play a crucial role in intercellular drug delivery of the neighboring effect.
CSSP NPs overcome the penetration limitations of the neighboring effect
As hypothesized in Fig. 4A, PR104A had a limited killing distance due to its weak toxicity to normoxic tumor cells. CPT could kill external normoxic tumor cells and achieve further penetration through the neighboring effect mediated by ApoBDs. However, due to strong binding with DNA-topoisomerase in normoxic tumor cells, the content of free CPT decreased with the production of cytotoxicity (Fig. 4B). Internal hypoxic tumor cells have a certain tolerance to CPT, so the penetration and killing ability of CPT is limited (12, 13, 33). When in combination, PR104A is delivered inward along with CPT-induced ApoBDs. The activation of PR104A in hypoxic tumor cells could compensate for the insufficient toxicity of CPT. After killing the hypoxic tumor cells, the drugs could continue to be delivered inward through the neighboring effect, thereby achieving a comprehensive tumor-killing effect. Compared with a single drug, the combination of the two drugs, whether in a mixed solution or CSSP NPs, could more effectively achieve better penetration in three-dimensional (3D) tumor spheroids by overcoming the limitations of the neighboring effect (Fig. 4, C and D). In addition, the combined effect also showed the best overall tumor-killing ability (Fig. 4, E and F). Therefore, the combined administration exhibited the best growth inhibition effect on 3D tumor spheroids (Fig. 4, G and H).
Fig. 4. In vitro enhanced penetration of CSSP NPs in 4T1 tumor spheroids.
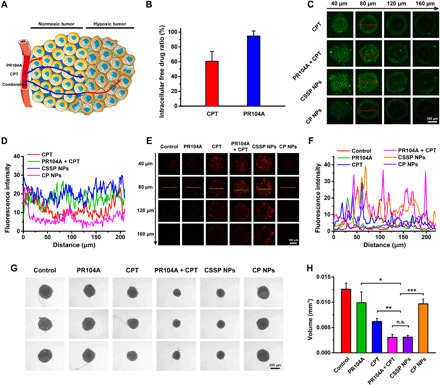
(A) Schematic diagram of a combination of CPT and PR104A to overcome the limitations of the neighboring effect for increased drug penetration and tumor-killing ability. Arrows indicate the distance of drug penetration and killing. (B) The proportion of free drugs in total drugs in 4T1 cells under normoxia (n = 3). (C) Z-stack images and (D) quantitative results for drug penetration in three-dimensional (3D) tumor spheroids. (E) CLSM images of dead viability assay and (F) quantitative results after various treatments. Dead cells were labeled with PI (red). (G) Optical images and (H) volume size of 3D tumor spheroids after treated with different formulations (10 μM) for 6 days (n = 3). Statistical significance: *P < 0.05, **P < 0.01, and ***P < 0.001.
In vivo enhanced penetration after tumor accumulation of CSSP NPs
We investigated the pharmacokinetic profile of prodrug NPs in blood circulation after intravenous administration. The plasma concentration–time profiles of the total CPT are presented in Fig. 5A; in addition, the pharmacokinetic parameters are listed in table S3. Free CPT was rapidly cleared from the blood, with a short half-life (t1/2) and small area under the curve (AUC0–24h). In contrast, prodrug NPs had a obviously longer circulation time than free CPT. Compared with free CPT, the AUC0–24h of CSSP NPs and CP NPs were increased by 6.8- and 11.7-fold, respectively. Furthermore, CP NPs delayed the elimination of CPT due to their inert chemical structure; therefore, their plasma clearance (CL) was slower than that of CSSP NPs.
Fig. 5. In vivo enhanced penetration of CSSP NPs in 4T1 tumor–bearing mice.
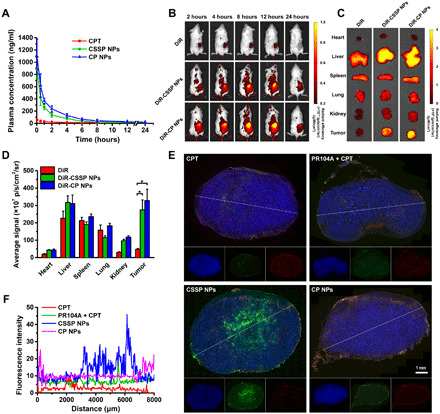
(A) Plasma concentration–time profiles of total CPT in rats after intravenous injection of free CPT or prodrug NPs (2 mg kg−1, CPT equivalent) (n = 3). (B) In vivo near-infrared (NIR) fluorescence images of the heterotopic 4T1 tumor–bearing mice at 2, 4, 8, 12, and 24 hours after intravenous injection of free DiR or DiR-labeled prodrug NPs (1 mg kg−1, DiR equivalent). (C) Ex vivo NIR fluorescence images and (D) quantitative results of tumor and major organs collected at 24 hours. (n = 3). (E) Fluorescence images of 4T1 tumor sections and (F) quantitative results of CPT penetration after intravenous injection of various formulations. 4′,6-diamidino-2-phenylindole (DAPI), blue; CPT, green; CD31, red. Statistical significance: *P < 0.05.
Then, the tumor accumulation and biodistribution of prodrug NPs were studied in heterotopic 4T1 tumor–bearing mice. Free 1,1-dioctadecyl-3,3,3,3-tetramethylindotricarbocyanine iodide (DiR) was rapidly eliminated in the body and showed high fluorescence intensity in the liver, spleen, and lung, but a negligible amount of dye was detected in the tumor. In contrast, after treatment with DiR-labeled CSSP NPs and DiR-labeled CP NPs, bright fluorescence signals were present in tumors, indicating that prodrug NPs could accumulate in tumors through the enhanced permeability and retention (EPR) effect (Fig. 5, B to D). In addition, the tumor accumulation of prodrug NPs was consistent with the pharmacokinetic characteristics. CP NPs, with longer blood circulation, exhibited greater tumor accumulation than CSSP NPs. After CSSP NPs accumulated in tumors, their enhanced penetration in solid tumors was also evaluated. As shown in Fig. 5 (E and F), CSSP NPs achieved the best solid tumor penetration effect as in the in vitro tumor spheroid penetration study due to their ability to overcome the limitations of the neighboring effect. In contrast, although CP NPs could also accumulate via the EPR effect, they were mainly distributed around the blood vessels at the edge of the tumor due to their poor penetration ability. It is worth noting that due to the poor blood circulation and the inability to guarantee the simultaneous delivery of two drugs to the tumor site, the tumor penetration of the PR104A + CPT mixture was as poor as that of the CPT solution.
In vivo antitumor and antimetastatic effects of CSSP NPs
The in vivo therapeutic effect of prodrug NPs was studied by using heterotopic 4T1 tumor–bearing mice. As illustrated in Fig. 6 (A to C), the saline-treated group had the fastest tumor growth, while a certain tumor growth inhibition was observed in the free PR104A and free CPT groups. There was no obvious difference in tumor suppression between the PR104A + CPT and single CPT treatment groups. The poor combination therapy of drug mixtures should be ascribed to the inability to ensure the simultaneous drug delivery of the two drugs in vivo. CSSP NPs had high tumor accumulation and could quickly release the two drugs in tumor cells simultaneously, therefore exploiting the ApoBD-mediated neighboring effect to penetrate and destroy the tumor to the maximum extent for unique superiority in tumor suppression. Notably, CP NPs exhibited weak antitumor activity due to the slow release of CPT and PR104A. Except for the mice in the CPT and PR104A + CPT groups with notable body weight loss during the administration period, the other groups had no obvious changes (Fig. 6D). There was no notable difference in hepatorenal function and no apparent hematoxylin and eosin (H&E) staining histological damage in major organs, indicating the safety of prodrug NPs (Fig. 6, E and F, and fig. S9). However, H&E, terminal deoxynucleotidyl transferase–mediated deoxyuridine triphosphate nick end labeling (TUNEL), and Ki67 staining of tumor tissue slices indicated that CSSP NPs could induce widespread apoptosis, inhibit cell proliferation, and elicit potent antitumor efficacy (Fig. 6G).
Fig. 6. In vivo therapeutic efficacy of CSSP NPs against heterotopic 4T1 tumors.
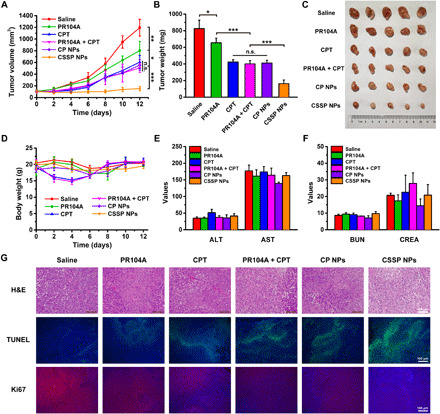
(A) The tumor growth profiles, (B) tumor weights, and (C) tumor photos after various treatments (5.7 mg kg−1, PR104A equivalent; 4 mg kg−1, CPT equivalent) (n = 5). (D) Body weight changes of mice after administration of different formulations (n = 5). (E) Hepatic and (F) renal function parameters of mice after treatments (n = 3). ALT (unit per liter), alanine aminotransferase; AST (unit per liter), aspartate aminotransferase; BUN (millimolar), blood urea nitrogen; CREA (micromolar), creatinine. (G) Hematoxylin and eosin (H&E), terminal deoxynucleotidyl transferase–mediated deoxyuridine triphosphate nick end labeling (TUNEL), and Ki67 staining of tumors after mice receiving different treatments. Statistical significance: *P < 0.05, **P < 0.01, and ***P < 0.001. Photo credit: Dongyang Zhao, Department of Pharmaceutics, Wuya College of Innovation, Shenyang Pharmaceutical University, Shenyang 110016, China.
Tumor hypoxia might drive tumor metastasis (34). CSSP NPs could effectively achieve tumor penetration and kill internal hypoxic tumor cells through the neighboring effect. Therefore, we investigated the antimetastatic ability of CSSP NPs on orthotopic 4T1 tumor–bearing mice. As shown in Fig. 7 (A to C), CSSP NPs exhibited the best tumor growth inhibition. In addition, compared with other groups, CSSP NPs effectively inhibited tumor metastasis to lung and liver tissues (Fig. 7, D and E). Similarly, the weight loss of mice in the CPT and PR104A + CPT groups was relatively notable, while the other groups had no obvious changes (fig. S10A). There was also no notable difference in hepatorenal function (fig. S10B). H&E staining showed that CSSP NPs could cause large-scale apoptosis of tumor cells, and except for the CPT and PR104A + CPT groups with clear histological damage to the liver, the other groups had no obvious toxicity (Fig. 7D and fig. S10C).
Fig. 7. In vivo antimetastatic capacity of CSSP NPs against orthotopic 4T1 tumors.
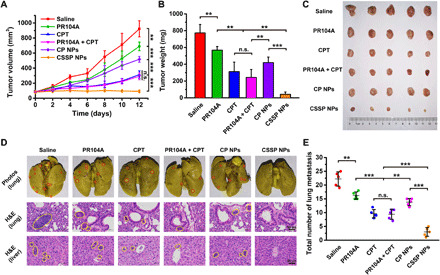
(A) The tumor growth profiles, (B) tumor weights, and (C) tumor photos after various treatments (5.7 mg kg−1, PR104A equivalent; 4 mg kg−1, CPT equivalent) (n = 5). (D) Representative photographs of whole lungs of mice and H&E staining of the lung and liver. Red and yellow circles denote metastases. (E) The average number of surface lung metastases (n = 5). Statistical significance: **P < 0.01 and ***P < 0.001. Photo credit: Dongyang Zhao, Department of Pharmaceutics, Wuya College of Innovation, Shenyang Pharmaceutical University, Shenyang 110016, China.
DISCUSSION
Chemotherapeutic nanomedicines offer broad prospects for cancer therapy, such as clinically approved Abraxane and Doxil. Despite EPR effect–based passive tumor accumulation, NPs were predominantly located near the blood vessels of the tumor periphery due to abnormal tumor vasculature, elevated interstitial fluid pressure, and dense tumor extracellular matrix (35). Several strategies to enhance tumor permeability have been reported, including the structural design of NPs and regulation of the tumor microenvironment (36). In addition, it was reported that the neighboring effect could also enhance the drug penetration and cytotoxicity of NPs in tumors (1–3). After drug-induced apoptosis, the remaining drugs could be released from dead or dying cells to infect surrounding cells. During repeated cell-to-cell delivery, the drugs gradually moved close to the center of the tumor. Therefore, with the help of the neighboring effect, we could design nanomedicines with high tumor permeability from a new perspective. However, before that, we need to clarify two essential issues: (i) How does intercellular drug delivery of the neighboring effect work? (ii) Why is the neighboring effect–based enhanced tumor penetration of NPs not obvious and how can it be amplified? Here, we first proposed and attempted to verify that the neighboring effect could be mediated by ApoBDs. In addition, we put forward that the consumption of chemotherapeutic agents and the resistance of internal hypoxic tumor cells might limit the persistence of neighboring effect–mediated tumor penetration. To extend the effect, we integrated CPT and the HAP PR104A to design CSSP NPs. In normoxic tumor cells, CPT had strong cytotoxicity, and PR104A had low toxicity, while in hypoxic tumor cells, PR104A cytotoxicity became stronger and CPT toxicity decreased. Because of the normoxic tumor cell killing of CPT, the resulting ApoBDs carried the remaining drugs and were engulfed by internal hypoxic tumor cells via the neighboring effect. PR104A killed hypoxic tumor cells to generate ApoBDs, which further delivered the drugs to deeper tumor cells. CSSP NPs overcame the obstacles of the neighboring effect, amplified the tumor penetration effect in vitro and in vivo, and achieved superior tumor growth suppression and antimetastatic ability.
Extracellular vesicles (EVs) are cell-derived membrane vesicles that can be divided into exosomes, microvesicles, and ApoBDs according to different sources (37). Initially, EVs were considered as garbage bags. In recent decades, it was revealed that EVs play an important role in intercellular communication. Among them, the most studied are exosomes, which originate from the intraluminal budding of multivesicular endosomes. In contrast, microvesicles are generated through direct budding from the plasma membrane. It has been shown that exosomes and microvesicles play a major role in the process of antigen presentation, immune suppression, antitumor immunity, and autoimmunity (6). ApoBDs, as products released by the decomposition of apoptotic cells, have failed to arouse wide attention from researchers. In our study, we linked ApoBDs with intercellular drug delivery of the already found neighboring effect and proved that, after chemotherapeutic drugs induce apoptosis, apoptotic cell–divided ApoBDs wrap the remaining unacted drugs and are swallowed by neighboring tumor cells. Nevertheless, whether exosomes and microvesicles also contribute to the intercellular delivery of chemotherapeutic drugs still needs further exploration and verification in the future. However, there is no doubt that even these two also have certain contributions, which may be insignificant compared to ApoBDs, because ApoBDs are the largest proportion of EVs produced after cell death caused by chemotherapeutic drugs (6). Regarding the mechanism of the neighboring effect, it has also been reported that it might be mediated by lysosomal exocytosis or possible transcytosis involving active cellular efflux of NPs (4, 5). Therefore, we tentatively inferred that intercellular drug delivery should be a multipath process. Regarding which method is dominant, we speculate that it may be related to the state of the cells after the drug is applied. The surviving cells may secrete drugs by means of active efflux, while cells in the apoptotic state are mainly dominated by the ApoBD pathway.
In summary, we established a new strategy for deep tumor penetration by exploiting the intercellular drug delivery of the neighboring effect. This strategy needs attention to the following points in its implementation: First, chemotherapeutic drugs should preferably be in the form of delayed release. Compared with the solution form, the delayed release will extend the action time of the drug, allowing it to repeatedly infect more tumor cells to achieve the best penetration enhancing effect. In addition, when chemotherapeutic drugs contact internal hypoxic tumor cells, the neighboring effect may be terminated. Therefore, appropriate supplementary strategies should be adopted to reverse the drug resistance of hypoxic cells, such as hypoxia-activated therapy or drug resistance inhibitors. Alternatively, this strategy can be designed to selectively release large amounts of chemotherapeutic drugs in the hypoxic zone. Although the toxicity of chemotherapeutic drugs is reduced, a high drug concentration in the hypoxic zone is sufficient to kill hypoxic tumors and continue to achieve penetration via ApoBDs. Last, the killing of normoxic tumor cells is not limited to conventional chemotherapeutics. Radiotherapy, photodynamic therapy, and other therapies that can cause cell apoptosis can be replaced. In short, we provided new insights for achieving deep tumor penetration through the neighboring effect mediated by ApoBDs. In future studies, this approach may improve the therapeutic efficiency of clinically approved nanomedicines and boost the clinical transformation of nanomedicines under nonclinical research.
MATERIALS AND METHODS
Materials
CPT, bis(2-hydroxyethyl) disulfide, 1,6-hexanediol, triphosgene, and 4-dimethylaminopyridine (DMAP) were purchased from Aladdin Co. Ltd. (Shanghai, China). Egg yolk lecithin and DSPE-PEG2k were obtained from Shanghai Advanced Vehicle Technology Co. Ltd. (Shanghai, China). Cell culture dishes/plates, round coverslips, 20-mm glass-bottom dishes, and centrifuge tubes were obtained from NEST Biotechnology Co. Ltd. (Wuxi, China). The total GSH assay kit was purchased from the Nanjing Jiancheng Bioengineering Institute (Nanjing, China). DTT, Hoechst 33342, 4′,6-diamidino-2-phenylindole (DAPI), and TUNEL apoptosis assay kits were purchased from Beijing Solarbio Science & Technology Co. Ltd. (Beijing, China). The anti-Ki67 antibody was obtained from Biosynthesis Biotechnology Inc. (Beijing, China). The anti-CD44 antibody, anti-CD326 antibody, anti–caspase3 antibody, anti–β-tubulin antibody, and anti-CD31 antibody were purchased from ABclonal Biotechnology Co. Ltd. (Wuhan, China). CCK-8, DiR, and Bouin’s fluid were purchased from Dalian Meilun Biotechnology Co. Ltd. (Dalian, China). All other reagents used were of analytical grade.
Synthesis of CPT-SS-OH and CPT-CC-OH
DMAP (234.4 mg, 1.92 mmol) in anhydrous dichloromethane (DCM) (5 ml) was added dropwise to the mixture of CPT (281.9 mg, 0.81 mmol) and triphosgene (83 mg, 0.28 mmol) in anhydrous DCM (100 ml) under ice bath with stirring. After 1 hour of reaction, bis(2-hydroxyethyl) disulfide (249.5 mg, 1.62 mmol) or 1,6-hexanediol (191.2 mg, 1.62 mmol) in anhydrous DCM (5 ml) was added, with stirring at room temperature overnight. Then, the crude product was purified by preparative liquid chromatography to obtain CPT-SS-OH or CPT-CC-OH as a yellow solid with a yield of 85.6 or 81.5%, respectively. The compounds were confirmed by SolariX 7.0 T ESI-MS (Bruker, Germany) and Bruker AV-400 NMR Spectroscopy (Bruker, Germany). HRMS (ESI) mass/charge ratio (m/z): [M + H]+ calcd for C25H25N2O7S2, 529.10977; found, 529.11009. 1H NMR (400 MHz, CDCl3, δ): 8.42 (s, 1H), 8.23 (d, J = 8.5 Hz, 1H), 7.96 (d, J = 8.2 Hz, 1H), 7.86 (t, J = 7.0 Hz, 1H), 7.69 (t, J = 6.9 Hz, 1H), 7.44 (s, 1H), 5.71 (d, J = 17.3 Hz, 1H), 5.39 (d, J = 17.2 Hz, 1H), 5.31 (s, 2H), 4.44 to 4.28 (m, 2H), 3.96 to 3.84 (m, 2H), 3.05 to 2.80 (m, 4H), 2.33 to 2.13 (m, 2H), and 1.02 (t, J = 7.5 Hz, 3H). HRMS (ESI) m/z: [M + H]+ calcd for C27H29N2O7, 493.19693; found, 493.19744. 1H NMR (400 MHz, CDCl3, δ): 8.46 (s, 1H), 8.32 (d, J = 8.5 Hz, 1H), 7.97 (d, J = 8.1 Hz, 1H), 7.88 (t, J = 7.7 Hz, 1H), 7.71 (t, J = 7.4 Hz, 1H), 7.51 (s, 1H), 5.70 (d, J = 17.3 Hz, 1H), 5.39 (d, J = 17.3 Hz, 1H), 5.32 (s, 2H), 4.21 to 4.08 (m, 2H), 3.60 (t, J = 6.4 Hz, 2H), 2.41 to 2.05 (m, 2H), 1.74 to 1.64 (m, 2H), 1.59 to 1.49 (m, 2H), 1.43 to 1.34 (m, 4H), and 1.00 (t, J = 7.3 Hz, 3H).
Synthesis of CPT-SS-PR104A and CPT-CC-PR104A
DMAP (234.4 mg, 1.92 mmol) in anhydrous DCM (5 ml) was added dropwise to the mixture of CPT-SS-OH (430 mg, 0.81 mmol) or CPT-CC-OH (400.7 mg, 0.81 mmol) and triphosgene (83 mg, 0.28 mmol) in anhydrous DCM (100 ml) under ice bath with stirring. After 1 hour of reaction, PR104A (401 mg, 0.81 mmol) in anhydrous DCM (5 ml) was added, with stirring at room temperature overnight. Then, the crude product was purified by preparative liquid chromatography to obtain CPT-SS-PR104A or CPT-CC-PR104A as a yellow solid with a yield of 82.3 or 78.6%, respectively. The compounds were confirmed by SolariX 7.0 T ESI-MS (Bruker, Germany) and Bruker AV-400 NMR Spectroscopy (Bruker, Germany). HRMS (ESI) m/z: [M + H]+ calcd for C40H42BrN6O17S3, 1053.09465; found, 1053.09476. 1H NMR (400 MHz, CDCl3, δ): 8.60 (d, J = 2.8 Hz, 1H), 8.55 (d, J = 2.8 Hz, 1H), 8.43 (s, 1H), 8.23 (d, J = 8.5 Hz, 1H), 7.96 (d, J = 7.4 Hz, 1H), 7.85 (t, J = 7.1 Hz, 1H), 7.68 (t, J = 7.0 Hz, 1H), 7.57 (t, J = 5.8 Hz, 1H), 7.36 (s, 1H), 5.66 (d, J = 17.2 Hz, 1H), 5.35 (d, J = 17.2 Hz, 1H), 5.29 (s, 2H), 4.46 to 4.21 (m, 8H), 3.76 (t, J = 5.2 Hz, 2H), 3.66 to 3.49 (m, 6H), 3.01 (s, 3H), 2.98 to 2.86 (m, 4H), 2.30 to 2.07 (m, 2H), and 0.99 (t, J = 7.5 Hz, 3H). HRMS (ESI) m/z: [M + H]+ calcd for C42H46BrN6O17S, 1017.18180; found, 1017.18416. 1H NMR (400 MHz, CDCl3, δ): 8.62 (d, J = 2.7 Hz, 1H), 8.56 (d, J = 2.8 Hz, 1H), 8.42 (s, 1H), 8.23 (d, J = 8.5 Hz, 1H), 7.96 (d, J = 8.1 Hz, 1H), 7.85 (t, J = 7.5 Hz, 1H), 7.68 (t, J = 7.5 Hz, 1H), 7.41 (t, J = 5.7 Hz, 1H), 7.35 (s, 1H), 5.68 (d, J = 17.2 Hz, 1H), 5.38 (d, J = 17.2 Hz, 1H), 5.30 (s, 2H), 4.49 to 4.28 (m, 4H), 4.22 to 4.02 (m, 4H), 3.82 to 3.71 (m, 2H), 3.66 to 3.50 (m, 6H), 3.01 (s, 3H), 2.34 to 2.08 (m, 2H), 1.70 to 1.66 (m, 2H), 1.64 to 1.57 (m, 2H), 1.42 to 1.32 (m, 4H), and 0.99 (t, J = 7.4 Hz, 3H).
Preparation of prodrug NPs
Heterodimeric prodrugs (CPT-SS-PR104A or CPT-CC-PR104A) (8 mg), DSPE-PEG2k (1.6 mg), and lecithin (0.8 mg) in acetone (2 ml) were added dropwise with stirring (1000 rpm) into deionized water (8 ml). Then, the acetone was removed by a rotary evaporator under vacuum, and the prepared prodrug NPs (CSSP NPs and CP NPs) were stored in a refrigerator at 4°C. Characterization of prodrug NPs was carried out using a Nano ZS Zetasizer instrument (Malvern, UK) and a JEM-2100 transmission electron microscope (JEOL, Japan).
Colloidal stability
CSSP or CP NPs (0.25 mg ml−1) were incubated in phosphate-buffered saline (PBS; pH 7.4) supplemented with 10% fetal bovine serum at 37°C. At predetermined time points (0, 1, 2, 4, 6, 8, 12, and 24 hours), the mean particle size was recorded. In addition, the long-term stability was also studied by storing CSSP NPs or CP NPs (1 mg ml−1) at 4°C for 2 weeks.
In vitro drug release
CSSP or CP NPs (300 nmol) were incubated with 10, 1, or 0 mM DTT in 30 ml of PBS (pH 7.4) [30% ethanol (v/v)] at 37°C (n = 3). The released CPT and PR104A were simultaneously determined by HPLC. In addition, the molecular weight change of prodrugs was measured by Agilent 1290-6545 Q-TOF (Agilent, USA) to investigate the release mechanism after CSSP NPs were incubated with 1 mM DTT for approximately 2 hours.
Cellular uptake
4T1 cells (1 × 105 cells per well) were seeded in 12-well plates and cultured for 24 hours. Then, cells were incubated with free CPT, CSSP NPs, or CP NPs (10 μM, CPT equivalent) for 12 or 24 hours (n = 3). After incubation, the cells were washed, fixed with 4% paraformaldehyde, and counterstained with Hoechst 33342. The round coverslips were observed by TCS SP2/AOBS confocal laser scanning microscopy (CLSM) (Leica, Germany). For flow cytometry analysis, after incubation with different formulations, cells were washed, digested, and suspended in PBS. The intracellular fluorescence intensity was determined by a FACSCalibur flow cytometer (BD, USA).
Detection of intracellular GSH
4T1 cells or 3T3 cells were cultured in culture dishes for 24 hours. Then, the cells were washed, digested, and suspended in PBS (5 × 105 cells per ml). Cells were broken by freezing and thawing, and intracellular GSH was measured by using a total GSH assay kit (n = 3).
Intracellular drug release
4T1 cells (1 × 105 cells per well) or 3T3 cells (2 × 105 cells per well) were seeded in 12-well plates and cultured for 24 hours. Then, cells were incubated with CSSP NPs or CP NPs (10 μM, CPT equivalent) for 6 or 12 hours (n = 3). After incubation, the cells and the drug-containing medium were collected together, and the cells were also broken by ultrasonication. The released CPT and PR104A were simultaneously determined by HPLC after protein precipitation.
Cytotoxicity assay
4T1 cells (1000 cells per well) or 3T3 cells (2000 cells per well) were seeded in 96-well plates and cultured under normoxic conditions for 12 hours. Then, the cells were cultured in normoxic or hypoxic atmospheres for another 12 hours. After culture, the cells were incubated with serial dilutions of free CPT, free PR104A, PR104A + CPT mixture, CSSP NPs, or CP NPs in normoxic or hypoxic atmospheres for 48 hours (n = 6). Then, 10 μl of CCK-8 was added into each well and incubated for 2 hours, and the absorbance at 450 nm was measured by using a microplate reader (BioTek, USA).
Intercellular delivery of CSSP NPs
4T1 cells seeded on coverslip A were pretreated with free CPT, CSSP NPs, or CP NPs (10 μM, CPT equivalent) for 12 hours. Coverslip A was washed and then coincubated with fresh cells on coverslip B for 24 hours. After coincubation, coverslip B was withdrawn and coincubated with fresh cells on another coverslip C for 24 hours. The cells (A, B, and C) were washed with PBS, stained with Hoechst 33342, and observed by CLSM.
To explore the mechanism of intercellular delivery of CSSP NPs, after incubating 4T1 cells with CSSP NPs (10 μM, CPT equivalent) for 12 hours, the cells were washed and cultured with fresh medium for another 24 hours. Then, the supernatant medium was sucked and centrifuged twice at 50g (5 min) to remove apoptotic cells and debris. The ApoBDs were collected by centrifugation at 1000g (10 min). The size and morphology of ApoBDs were observed using a particle size analyzer and transmission electron microscopy (TEM), respectively. Western blot was also performed to characterize the protein constitution of ApoBDs. In addition, after incubating 4T1 cells with ApoBDs (approximately 10 μM, CPT equivalent) for 24 hours, the cells were washed, stained with propidium iodide (PI), and observed by CLSM. To further verify the cytotoxicity of ApoBDs, the collected ApoBDs of different protein concentrations were incubated with 4T1 cells under normoxia for 48 hours, and then the cell survival rate was measured (n = 6).
Determination of intracellular free drug ratio
4T1 cells (4 × 105 cells per well) were seeded in six-well plates and cultured for 24 hours. Then, cells were incubated with free PR104A or free CPT (10 μM) for 12 hours (n = 3). After incubation, the cells were washed, collected, and broken by freezing and thawing. The free drug ratio was determined by ultrafiltration.
Drug penetration and tumor inhibition in 3D tumor spheroids
4T1 cells (1 × 104 cells) were mixed with 15 μl of culture medium (0.24% methylcellulose, w/v). The droplet-shaped cell suspension was then placed on the lid of a 96-well plate, and the lid was inverted on the plate. After incubation for 24 hours, the resulting cell aggregates on the lid were transferred to a round bottom 96-well plate by centrifugation (2800 rpm, 3 min). Then, the fresh medium was changed every 2 days until the sixth day. After that, 3D tumor spheroids were incubated with different formulations (10 μM) for 24 hours. CPT fluorescence was detected using Z-stack imaging at 40-μm intervals from the top of the spheroids by CLSM. In addition, apoptotic cells were also stained with PI and observed by CLSM. The fluorescence intensity of CPT and PI at 80-μm cross section was quantified by ImageJ software. For the 3D tumor spheroid growth inhibition study, tumor spheroids were added with various preparations (10 μM) every 2 days, and the volume size was recorded by an optical microscope on the sixth day.
In vivo pharmacokinetics
Animal experiments were conducted according to the Guide for the Care and Use of Laboratory Animals issued by the Institutional Animal Ethical Care Committee of Shenyang Pharmaceutical University. Male Sprague-Dawley rats (220 to 250 g) were intravenously administered free CPT, CSSP NPs, or CP NPs (2 mg kg−1, CPT equivalent) (n = 3). Blood samples were then collected at 0.083, 0.5, 1, 2, 4, 6, 8, 12, and 24 hours after injection, and the blood in heparin tubes was centrifuged to obtain the plasma. The plasma samples were incubated with organic alkali to liberate CPT from prodrug form. After protein precipitation, the plasma concentrations of total CPT were measured by using a microplate reader (BioTek, USA) (excitation, 365 nm; emission, 430 nm).
In vivo biodistribution
4T1 cells (5 × 106) were injected subcutaneously into the right back of female BALB/c mice. When tumors reached approximately 300 mm3, mice were intravenously injected with free DiR, DiR-labeled CSSP NPs, or DiR-labeled CP NPs (1 mg kg−1, DiR equivalent) (n = 3). At 2, 4, 8, 12, and 24 hours after injection, mice were anesthetized and then imaged using an IVIS Lumina III Small Animal Imaging System (PerkinElmer, USA). Mice were sacrificed at 24 hours after injection, and organs, including the heart, liver, spleen, lung, kidney, and tumor, were collected and subjected to fluorescence imaging. In addition, 4T1 tumor–bearing mice were injected with free CPT, PR104A + CPT mixture, CSSP NPs, or CP NPs (5.7 mg kg−1, PR104A equivalent; 4 mg kg−1, CPT equivalent). After 24 hours, the mice were sacrificed, and tumor sections were subjected to DAPI and CD31 staining and analyzed by CLSM to evaluate in vivo tumor penetration. In addition, the CPT fluorescence distribution of tumor slices was quantified by ImageJ software.
In vivo combination therapy
4T1 cells (5 × 106) were injected subcutaneously into the right back of female BALB/c mice to establish a heterotopic tumor–bearing mouse model. When tumors reached approximately 100 mm3, mice were intravenously administered saline, free PR104A, free CPT, PR104A + CPT mixture, CSSP NPs, or CP NPs (5.7 mg kg−1, PR104A equivalent; 4 mg kg−1, CPT equivalent) on days 0, 2, 4, 6, and 8 (n = 5). Body weight and tumor size were recorded on days 0, 2, 4, 6, 8, 10, and 12. Tumor growth was measured by using a digital caliper, and the tumor volume was calculated as tumor volume = 0.5 × length × width2. On day 12, the mice were sacrificed, and serum was obtained for hepatorenal function analysis. In addition, tumor weight was recorded, and histopathological analysis of major organs was performed by H&E staining. The tumor samples were also stained with TUNEL and Ki67. Furthermore, 4T1 cells (5 × 105) were injected into the mammary fat pad of female BALB/c mice to establish an orthotopic tumor–bearing mouse model. The dosage regimen was consistent with the heterotopic tumor model. After the experiment, the lungs were fixed in Bouin’s solution, and the surface tumors in pulmonary lobes were counted and recorded. In addition, the inhibition of metastasis was also investigated by H&E staining of lung and liver tissues.
Statistical analysis
The results were presented as means ± SD. Comparison between groups was performed using one-way analysis of variance (ANOVA) and Student’s t test, and P < 0.05 was considered significant.
Acknowledgments
Funding: This research was supported by the National Natural Science Foundation of China (no. 82073777), Liaoning Revitalization Talents Program (no. XLYC1808017), and Shenyang Youth Science and Technology Innovation Talents Program (no. RC190454). Author contributions: D.Z., W.T., Z.H., and J.S. designed research; D.Z., W.T., S.L., and Y.C. performed research; D.Z., W.T., Y.S., Z.H., B.S., and J.S. analyzed data; and D.Z., W.T., and J.S. wrote the paper. Competing interests: J.S., Z.H., D.Z., and W.T. are inventors on a pending patent related to this work filed by Shenyang Pharmaceutical University (no. CN202011130485.8, filed 21 October 2020, published 22 January 2021). The authors declare that they have no other competing interests. Data and materials availability: All data needed to evaluate the conclusions in the paper are present in the paper and/or the Supplementary Materials. Additional data related to this paper may be requested from the authors.
SUPPLEMENTARY MATERIALS
Supplementary material for this article is available at http://advances.sciencemag.org/cgi/content/full/7/16/eabg0880/DC1
REFERENCES AND NOTES
- 1.Guo S., Wang Y., Miao L., Xu Z., Lin C. M., Zhang Y., Huang L., Lipid-coated cisplatin nanoparticles induce neighboring effect and exhibit enhanced anticancer efficacy. ACS Nano 7, 9896–9904 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Ju C., Mo R., Xue J., Zhang L., Zhao Z., Xue L., Ping Q., Zhang C., Sequential intra-intercellular nanoparticle delivery system for deep tumor penetration. Angew. Chem. Int. Ed. 53, 6253–6258 (2014). [DOI] [PubMed] [Google Scholar]
- 3.Wang C., Chen S., Wang Y., Liu X., Hu F., Sun J., Yuan H., Lipase-triggered water-responsive “pandora’s box” for cancer therapy: Toward induced neighboring effect and enhanced drug penetration. Adv. Mater. 30, e1706407 (2018). [DOI] [PubMed] [Google Scholar]
- 4.Yong T., Hu J., Zhang X., Li F., Yang H., Gan L., Yang X., Domino-like intercellular delivery of undecylenic acid-conjugated porous silicon nanoparticles for deep tumor penetration. ACS Appl. Mater. Interfaces 8, 27611–27621 (2016). [DOI] [PubMed] [Google Scholar]
- 5.Zhou Q., Shao S., Wang J., Xu C., Xiang J., Piao Y., Zhou Z., Yu Q., Tang J., Liu X., Gan Z., Mo R., Gu Z., Shen Y., Enzyme-activatable polymer–drug conjugate augments tumour penetration and treatment efficacy. Nat. Nanotechnol. 14, 799–809 (2019). [DOI] [PubMed] [Google Scholar]
- 6.Caruso S., Poon I. K. H., Apoptotic cell-derived extracellular vesicles: More than just debris. Front. Immunol. 9, 1486 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Gregory C. D., Pound J. D., Cell death in the neighbourhood: Direct microenvironmental effects of apoptosis in normal and neoplastic tissues. J. Pathol. 223, 177–194 (2011). [DOI] [PubMed] [Google Scholar]
- 8.Shklover J., Levy-Adam F., Kurant E., Apoptotic cell clearance in development. Curr. Top. Dev. Biol. 114, 297–334 (2015). [DOI] [PubMed] [Google Scholar]
- 9.C. D. Gregory, C. A. Ford, J. J. L. P. Voss, Microenvironmental effects of cell death in malignant disease, in Apoptosis in Cancer Pathogenesis and Anti-cancer Therapy: New Perspectives and Opportunities, C. D. Gregory, Ed. (Springer, 2016), pp. 51–88. [Google Scholar]
- 10.Finicle B. T., Jayashankar V., Edinger A. L., Nutrient scavenging in cancer. Nat. Rev. Cancer 18, 619–633 (2018). [DOI] [PubMed] [Google Scholar]
- 11.Jayashankar V., Edinger A. L., Macropinocytosis confers resistance to therapies targeting cancer anabolism. Nat. Commun. 11, 1121 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Teicher B. A., Hypoxia and drug resistance. Cancer Metastasis Rev. 13, 139–168 (1994). [DOI] [PubMed] [Google Scholar]
- 13.Rohwer N., Cramer T., Hypoxia-mediated drug resistance: Novel insights on the functional interaction of HIFs and cell death pathways. Drug Resist. Update 14, 191–201 (2011). [DOI] [PubMed] [Google Scholar]
- 14.Phillips R. M., Targeting the hypoxic fraction of tumours using hypoxia-activated prodrugs. Cancer Chemother. Pharmacol. 77, 441–457 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Baran N., Konopleva M., Molecular pathways: Hypoxia-activated prodrugs in cancer therapy. Clin. Cancer Res. 23, 2382–2390 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.McKeage M. J., Jameson M. B., Ramanathan R. K., Rajendran J., Gu Y., Wilson W. R., Melink T. J., Tchekmedyian N. S., PR-104 a bioreductive pre-prodrug combined with gemcitabine or docetaxel in a phase Ib study of patients with advanced solid tumours. BMC Cancer 12, 496 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Luo C., Sun J., Sun B., He Z., Prodrug-based nanoparticulate drug delivery strategies for cancer therapy. Trends Pharmacol. Sci. 35, 556–566 (2014). [DOI] [PubMed] [Google Scholar]
- 18.Feng B., Zhou F., Hou B., Wang D., Wang T., Fu Y., Ma Y., Yu H., Li Y., Binary cooperative prodrug nanoparticles improve immunotherapy by synergistically modulating immune tumor microenvironment. Adv. Mater. 30, e1803001 (2018). [DOI] [PubMed] [Google Scholar]
- 19.Luo C., Sun B., Wang C., Zhang X., Chen Y., Chen Q., Yu H., Zhao H., Sun M., Li Z., Zhang H., Kan Q., Wang Y., He Z., Sun J., Self-facilitated ROS-responsive nanoassembly of heterotypic dimer for synergistic chemo-photodynamic therapy. J. Control. Release 302, 79–89 (2019). [DOI] [PubMed] [Google Scholar]
- 20.Zhao D., Tao W., Li S., Li L., Sun Y., Li G., Wang G., Wang Y., Lin B., Luo C., Wang Y., Cheng M., He Z., Sun J., Light-triggered dual-modality drug release of self-assembled prodrug-nanoparticles for synergistic photodynamic and hypoxia-activated therapy. Nanoscale Horiz. 5, 886–894 (2020). [DOI] [PubMed] [Google Scholar]
- 21.Zhang X., Han L., Liu M., Wang K., Tao L., Wan Q., Wei Y., Recent progress and advances in redox-responsive polymers as controlled delivery nanoplatforms. Mater. Chem. Front. 1, 807–822 (2017). [Google Scholar]
- 22.Tian Y., Guo R., Jiao Y., Sun Y., Shen S., Wang Y., Lu D., Jiang X., Yang W., Redox stimuli-responsive hollow mesoporous silica nanocarriers for targeted drug delivery in cancer therapy. Nanoscale Horiz. 1, 480–487 (2016). [DOI] [PubMed] [Google Scholar]
- 23.Atwell G. J., Denny W. A., Synthesis of 3H- and 2H4-labelled versions of the hypoxia-activated pre-prodrug 2-[(2-bromoethyl)-2,4-dinitro-6- [[[2-(phosphonooxy)ethyl]amino]carbonyl]anilino]ethyl methanesulfonate (PR-104). J. Labelled Comp. Radiopharm. 50, 7–12 (2007). [Google Scholar]
- 24.Yang S., Atwell G. J., Denny W. A., Synthesis of asymmetric halomesylate mustards with aziridineethanol/alkali metal halides: Application to an improved synthesis of the hypoxia prodrug PR-104. Tetrahedron 63, 5470–5476 (2007). [Google Scholar]
- 25.Sun B., Luo C., Zhang X., Guo M., Sun M., Yu H., Chen Q., Yang W., Wang M., Zuo S., Chen P., Kan Q., Zhang H., Wang Y., He Z., Sun J., Probing the impact of sulfur/selenium/carbon linkages on prodrug nanoassemblies for cancer therapy. Nat. Commun. 10, 3211 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Zhou D., Li D., Jing P., Zhai Y., Shen D., Qu S., Rogach A. L., Conquering aggregation-induced solid-state luminescence quenching of carbon dots through a carbon dots-triggered silica gelation process. Chem. Mater. 29, 1779–1787 (2017). [Google Scholar]
- 27.Zhang H., Wang K., Na K., Li D., Li Z., Zhao D., Zhong L., Wang M., Kou L., Luo C., Zhang H., Kan Q., Ding H., He Z., Sun J., Striking a balance between carbonate/carbamate linkage bond- and reduction-sensitive disulfide bond-bearing linker for tailored controlled release: In situ covalent-albumin-binding gemcitabine prodrugs promote bioavailability and tumor accumulation. J. Med. Chem. 61, 4904–4917 (2018). [DOI] [PubMed] [Google Scholar]
- 28.Xu Z., Hou M., Shi X., Gao Y.-E., Xue P., Liu S., Kang Y., Rapidly cell-penetrating and reductive milieu-responsive nanoaggregates assembled from an amphiphilic folate-camptothecin prodrug for enhanced drug delivery and controlled release. Biomater. Sci. 5, 444–454 (2017). [DOI] [PubMed] [Google Scholar]
- 29.Lin H., Chen Y., Shi J., Nanoparticle-triggered in situ catalytic chemical reactions for tumour-specific therapy. Chem. Soc. Rev. 47, 1938–1958 (2018). [DOI] [PubMed] [Google Scholar]
- 30.Lee M. H., Yang Z., Lim C. W., Lee Y. H., Dongbang S., Kang C., Kim J. S., Disulfide-cleavage-triggered chemosensors and their biological applications. Chem. Rev. 113, 5071–5109 (2013). [DOI] [PubMed] [Google Scholar]
- 31.Huang T. L., Székács A., Uematsu T., Kuwano E., Parkinson A., Hammock B. D., Hydrolysis of carbonates, thiocarbonates, carbamates, and carboxylic esters of α-naphthol, β-naphthol, and p-nitrophenol by human, rat, and mouse liver carboxylesterases. Pharm. Res. 10, 639–648 (1993). [DOI] [PubMed] [Google Scholar]
- 32.Xu G., Zhang W., Ma M. K., McLeod H. L., Human carboxylesterase 2 is commonly expressed in tumor tissue and is correlated with activation of irinotecan. Clin. Cancer Res. 8, 2605–2611 (2002). [PubMed] [Google Scholar]
- 33.Gokduman K., Strategies targeting DNA topoisomerase I in cancer chemotherapy: Camptothecins, nanocarriers for camptothecins, organic non-camptothecin compounds and metal complexes. Curr. Drug Targets 17, 1928–1939 (2016). [DOI] [PubMed] [Google Scholar]
- 34.Nobre A. R., Entenberg D., Wang Y., Condeelis J., Aguirre-Ghiso J. A., The different routes to metastasis via hypoxia-regulated programs. Trends Cell Biol. 28, 941–956 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Jain R. K., Stylianopoulos T., Delivering nanomedicine to solid tumors. Nat. Rev. Clin. Oncol. 7, 653–664 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Ding J., Chen J., Gao L., Jiang Z., Zhang Y., Li M., Xiao Q., Lee S. S., Chen X., Engineered nanomedicines with enhanced tumor penetration. Nano Today 29, 100800 (2019). [Google Scholar]
- 37.Vader P., Mol E. A., Pasterkamp G., Schiffelers R. M., Extracellular vesicles for drug delivery. Adv. Drug Deliv. Rev. 106, 148–156 (2016). [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary material for this article is available at http://advances.sciencemag.org/cgi/content/full/7/16/eabg0880/DC1