
Figure 3.
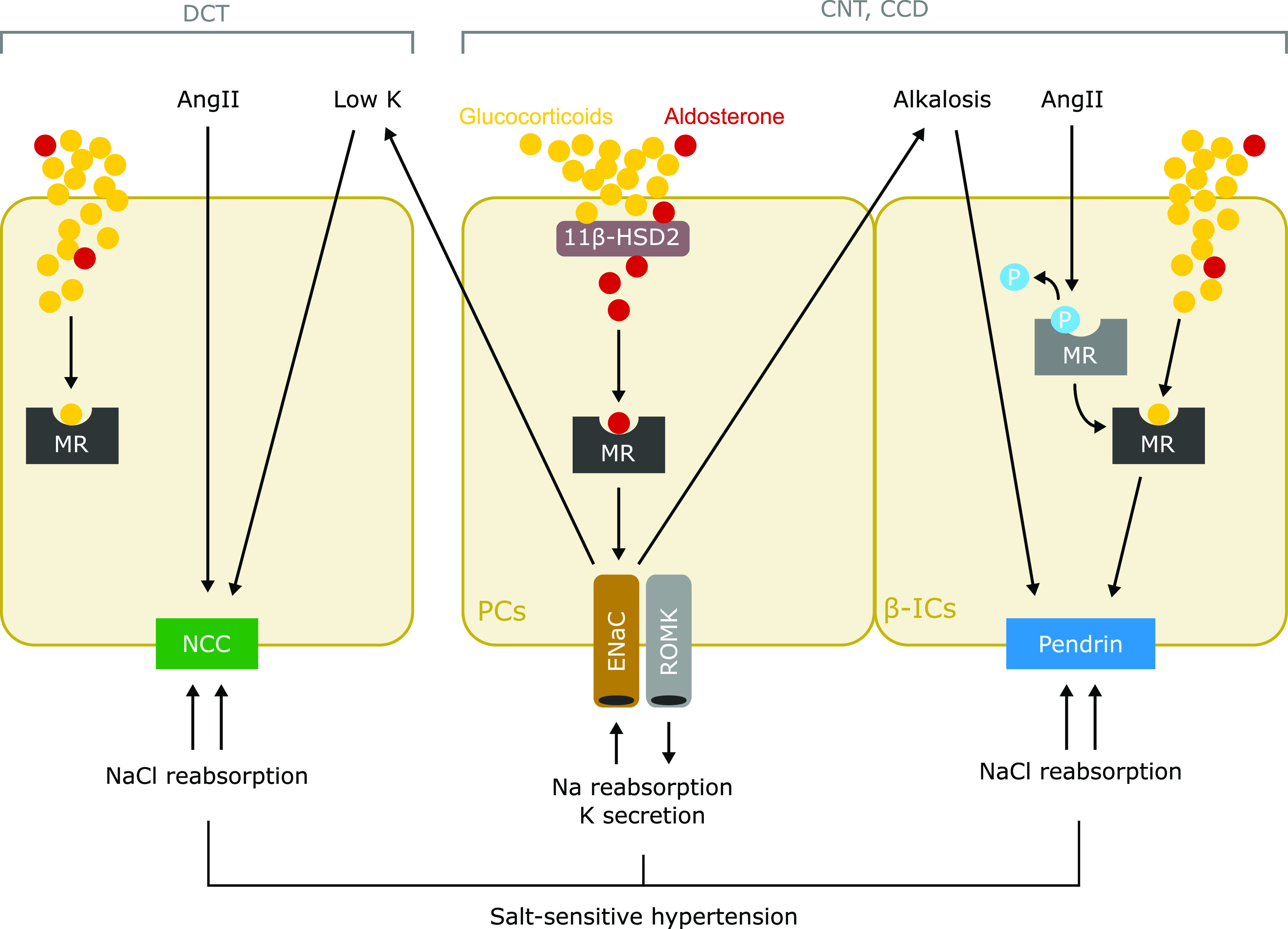
Putative mechanism of salt-sensitive hypertension in primary aldosteronism. Cell type–specific mechanisms of sodium reabsorption are mediated by MR in the ASDN. Classically, aldosterone activates the MR-ENaC pathway in principal cells (PCs) of the connecting tubules (CNTs) and cortical collecting ducts (CCDs) to reabsorb sodium and secrete potassium. The interaction between aldosterone and MR in PCs is guaranteed by 11β-HSD2 that converts glucocorticoids to inactive metabolites. NCC expressed in DCT is directly activated by AngII and hypokalemia, which is induced by the aldosterone-induced activation of MR-ENaC in PCs. In β-ICs in the CNTs and CCDs, AngII induces dephosphorylation of MR, which increases affinity of ligands, and resultantly causes MR activation, leading to upregulation of pendrin. In addition, metabolic alkalosis, which is induced by activation of MR-ENaC in PCs, directly induces pendrin upregulation in β-ICs. The contrasting activation of NCC and pendrin induced by aldosterone excess, in a coordinated manner, mediates sodium chloride reabsorption, leading to the development of salt-sensitive hypertension. P, phosphorylation of MR; ROMK, renal outer medullary potassium.