

Abstract
Polycystic ovary syndrome (PCOS) is a common form of anovulatory infertility with a strong hereditary component but no candidate genes have been found. The inheritance pattern may be due to in utero androgen programming on gene expression and mitochondria. Mitochondria are maternally inherited and alterations to mitochondria after fetal androgen exposure may explain one of the mechanisms of fetal programming in PCOS. Our aim was to investigate the role of excessive prenatal androgens in ovarian development by identifying how hyperandrogenemia affects gene expression and mitochondria in neonatal ovary. Pregnant dams were injected with dihydrotestosterone on days 16–18 of pregnancy. Day 0 ovaries were collected for gene expression and mitochondrial studies. RNAseq showed differential gene expressions which were related to mitochondrial dysfunction, fetal gonadal development, oocyte maturation, metabolism, angiogenesis, and PCOS. Top 20 up and downregulated genes were validated with qPCR and Western Blot. Transcriptional pathways involved in folliculogenesis and genes involved in ovarian and mitochondrial function were dysregulated. Further, DHT exposure altered mitochondrial ultrastructure and function by increasing mitochondrial oxygen consumption and decreasing mitochondrial efficiency with increased proton leak within the first day of life. Our data indicates that one path that leads to PCOS begins at birth and is programmed in utero by androgens.
Keywords: PCOS, ovary, androgens, mitochondria, genes
INTRODUCTION
Polycystic ovary syndrome (PCOS) is one of the most common causes of anovulation and infertility, affecting up to 10% of reproductive age women [1]. It is a multifactorial disease, with heterogonous clinical and biochemical phenotypes. There are a number of different diagnostic criteria, however, these criteria sometimes miss additional abnormalities that women often exhibit and do not distinguish between the degree of manifestations or severity of disease, such as metabolic syndrome, fatty liver, and infertility [2]. It is therefore no surprise that PCOS likely has a heterogeneous etiology arising from multiple genetic and environmental determinants. PCOS is known to have strong familial clustering, demonstrated in identical twin studies, exhibiting a heritable element, with hyperandrogenism the most heritable trait [2, 3]. Furthermore, daughters born to women with PCOS had a fivefold-increased risk of a PCOS diagnosis compared to daughters born to women without PCOS [4]. However, no gene candidates have been found, likely due to the wide phenotypic heterogeneity of the syndrome [2, 5].
Fetal androgen exposure leading to an adult PCOS phenotype has been shown in rats, mice, sheep, and rhesus monkeys [2, 4, 6–8]. Further, studies in humans have shown that in the second trimester amniotic fluid of PCOS patients have elevated testosterone compared to controls in female fetuses [9], and studies of cord blood also found elevated androgens in newborns of PCOS mothers compared to controls [10, 11]. This indicates that the increased androgen exposure in the developing fetus could be involved in the pathogenesis of PCOS [2, 7, 8, 12–19].
It is unclear if the PCOS phenotype arises from dysregulation of gene expression or abnormal mitochondrial function after androgen exposure or a combination of both. Mitochondrial dysfunction could explain much of PCOS pathophysiology. This includes the mechanism of inheritance, given that mitochondria are strictly maternally inherited, and androgen excess as it is the site of hormone synthesis. Further, mitochondrial role in insulin function could explain the increased risk of metabolic syndrome including insulin resistance in PCOS. Furthermore, aberrant follicular development and oocyte quality in PCOS can be related to mitochondrial function as appropriate mitochondrial content is vital for cellular energy and survival. Studies both during fetal development and postnatally of excess androgens in mice creating a PCOS phenotype found compromised mitochondria post treatment [19, 20]. To our knowledge this is the first study using a lean PCOS model to analyze real-time functional analysis of neonatal ovaries.
Our central hypothesis is that fetal ovaries exposed to a hyperandrogenemic environment are reprogrammed during fetal development which is evident at birth with altered gene regulation and mitochondrial dysfunction. We investigated this hypothesis through two objective. First we wanted to identify if fetal androgen exposure altered gene expression at birth using a lean PCOS mouse model. This was accomplished through RNA seq, bioinformatics, and targeted gene analysis. Our second aim was to characterize mitochondrial structure and function after in-utero androgen exposure. We evaluated gene mitochondrial structure and function with electron microscopy, mitochondrial DNA (mtDNA) copy number, ATP production, and real-time oxygen consumption studies in neonatal murine ovaries.
MATERIALS AND METHODS
Mouse model:
All experiments were approved through the Institutional Animal Care and Use Committee of Baylor College of Medicine (AN-7156). For the lean PCOS mouse model, 6 to 8-week-old C57/Bl6 female mice (n=75) were mated with male studs overnight. Mating was confirmed by the presence of copulatory plugs or sperm in vaginal smears the following morning, considered day 0.5 of pregnancy. Pregnant dams were injected with 250 μg of dihydrotestosterone (DHT, Sigma-Aldrich) prepared in sesame oil (Texas Lab Supply, Lubbock, TX, USA) or vehicle on 16.5, 17.5 and 18.5 days post coitus. At birth, pups were tentatively sexed by using anogenital distance and visualization of the ovary through the translucent body wall. Pups were euthanized on day 0, sex of the pups were confirmed by the visualization of the ovary under the dissection microscope, and the ovaries were harvested. The ovary was then placed in Formalde-Fresh™ (Buffered 4% formaldehyde) for histology, cacodylate buffered glutaraldehyde for TEM or frozen in −80C for further study and experiments. For, oxygen consumption studies, ovaries were quickly harvested and placed in DMEM for further analysis. One pup each from each dam was randomly chosen for every experiment and sisters were not used within the same experiment.
Histology:
After fixing in buffered formaldehyde, ovaries were dehydrated in ethanol, embedded in paraffin wax and sectioned. Five micron sections were stained with hematoxylin and eosin followed by imaging using a light microscope.
RNA Isolation:
Ovaries were individually disrupted and homogenized with RLT buffer (Qiagen, Germantown, MD) containing β-metcapernol using a disposable micro-pestle in a 1.5mL micro-centrifuge tube. RNA was isolated using the commercially available RNeasy Mini Kit (Qiagen, Germantown, MD) using the manufacturers’ instructions. RNA quantity was measured using the NanoDrop™ 2000 Spectrophotometer (Thermofisher, Waltham, MA).
RNA sequencing and bioinformatics:
Quality control was performed to evaluate RNA integrity using Qubit (ThermoFisher Scientific, Waltham, MA). Extracted whole RNA was enriched for mRNA using NEBNext® Poly(A) mRNA magnetic isolation. The primed mRNA was then fragmented, cDNA library with 150bp paired end reads was prepared and amplified by PCR and then sequenced on Illumina HiSeq4000 platform by Novogene. Processing was carried out with Genialis RNAseq analysis pipeline using Trimmomatic, HiSAT2 was used for mapping, HTseq was used to normalize amplifications. DESeq2 was used for differential expression analysis allowing for a false discovery rate (FDR) of 0.05 and expression change of log2 fold-change. Pathway and Gene Ontology Analysis (Enrich R) and Gene Set Enrichment Analysis (GSEA) were carried out to evaluate gene expression and pathway signatures as well as principle component analysis of publicly available raw RNAseq data of murine ovarian tissue.
Quantitative Real Time PCR:
cDNA was prepared by reverse-transcription of RNA using the Omniscript kit (Qiagen, Germantown, MD) according to the manufacturer’s instructions. Target genes were amplified by qPCR using SsoAdvanced Universal SYBR Green Supermix (Bio-Rad, Hercules, CA). All reactions were performed in duplicate or triplicate, and tubulin was used as an internal control. For mtDNA copy number, DNA was extracted from mouse ovaries using a commercially available DNeasy Blood and Tissue kit (Qiagen, Germantown, MD) according to the manufacturers’ instructions. qPCR was performed to determine the ratio of mtDNA copy number of a single mtDNA gene relative to tubulin and run in duplicates.
ATP production:
Ovaries were homogenized with 50μl of lysis buffer (Promega, Madison, WI, USA) for 30 seconds on ice using a disposable micro-pestle in a 1.5mL micro-centrifuge tube, and supernatant was analyzed using an ATP kit utilizing a luciferase assay (ThermoFisher Scientific, Waltham, MA) following kit instructions.
Western Blot:
Given the small protein content of each neonatal ovary, protein estimation could not be done and protein was directly extracted using 20μl of Laemmli buffer (loading buffer) with DTT, homogenized on ice using a micro-pestle in a 1.5mL micro-centrifuge tube, followed by sonication for 10 seconds at 60% power (QSonica sonicators, Newton, CT). The supernatant was extracted, warmed to 95°C for 5 min. 10μL of protein obtained from two ovaries from a single pup was loaded into each lane, separated electrophoretically using Mini-PROTEAN TGX Gels (Bio-Rad, Hercules, CA) and transferred onto polyvinylidene difluoride (PVDF; Bio-Rad, Hercules, CA). The membrane was blocked with TBST containing 1% non-fat dried milk for 60 min at room temperature. Due to limited amount of sample, the membranes were cut at ~75 kDa or at ~25 KDa based on the molecular size markers and various proteins within the appropriate molecular sizes were probed with primary antibodies. In some cases where there was enough molecular size differences, two proteins were co-probed together. Primary antibodies (dilutions: 1:1000 for Cidec, 1:500 for Ang4 and Cox6a2 from Novus Biologicals; 1:500 for Actc1 antibody from Abcam; 1:250 for Retnlb and 1:500 Myom2 from Sigma; 1:500 for Crybb2 from ThermoFisher Scientific; 1:1000 for FosB (5G4) and Gapdh from Cell Signaling) in TBST containing 1% nonfat dried milk powder were incubated overnight at 4°C. After washing with TBST, the membranes were incubated with HRP-conjugated secondary antibodies (1:2000, Cell Signaling) in TBST at room temperature for 60 min and then washed. Immunoreactivity and signals were monitored by chemiluminescence (Immobilin ECL Ultra Western HRP Substrate, Millipore Sigma, Burlington, MA) and images were obtained using ChemiDOC (Bio-Rad). Protein bands were quantified using Bio-Rad Image Lab Software.
Transmission Electron Microscopy (TEM):
TEM was performed at the Image core facility of Baylor College of Medicine. Ovaries were fixed in 2.5% glutaraldehyde with cacodylate buffer to a pH 7.4 at 4 °C for 24 hours and then post-fixed with 1% osmium tetroxide in cacodylate buffer for 45 minutes at 4°C, and then dehydrated with increasing ethanol concentrations. They were embedded in resin Epon 812 and cured at 62°C for two days. Blocks were then cut with an ultra-microtome at 50 nanometers. The sections were placed on a copper grid and stained with heavy metal. The images were viewed under the electron beam of the transmission electron microscope (Hitachi H-7500) and captured at 7,000x and 40,000x.
Ovarian oxygen consumption:
The Seahorse XFe96 analyzer™ (Agilent, Santa Clara, CA) was utilized to measure oxygen consumption of fresh day 0 murine ovaries. The XFe96 measures oxygen in a transient micro-chamber, formed at the bottom of the XFe96 spheroid plate, using solid state fluorescent sensors imbedded in a sensor cartridge. Oxygen concentration is measured over a 3 minute period, and the rate of oxygen consumption (OCR) is determined by Seahorse WAVE software. Briefly, the XFe96 spheroid microplates were used along with hydrated sensor cartridges and reagents prepared according to manufacturer’s instructions. Next, 10μl DMEM medium was placed in each well of the XFe96 spheroid microplate and then ovaries were placed onto the center well. An additional 170μl of media was added and the plate was spun. Reagent ports for each well were filled with 20μl of 5μM oligomycin, 22μl of 1μM FCCP and 25μl of 2.5μM Antimycin/Rotenone. The analyzer was programmed to take 5 baseline measurements, 10 measurements post oligomycin injection, and 5 measurements each after FCCP and antimycin/rotenone injections. Oxygen consumption (OCR) measurements were normalized with protein content of each well. The concentrations of the compounds along with the timing and the number of reads were optimized by prior pilot experiments.
Data Analysis:
Student t-test and analysis of variance (ANOVA) were used to compare continuous variables as appropriate, chi square or Fisher’s exact was used to compare categorical variables. A p-value of < 0.05 was considered significant. Data was analyzed using GraphPad Prism version 6.0 (San Francisco, CA, USA).
RESULTS
DHT exposure showed differential gene expression:
RNAseq data from the ovaries exposed to DHT showed differential clustering of mRNA (Figure 1A) and principle component analysis (Figure 1B) showed a clear delineation between the two groups while the replicates were similar. Further, on heat map analysis of differentially expressed genes the controls were very distinct from the DHT exposed ovaries (Figure 1C) with 241 upregulated and 112 downregulated genes in the DHT exposed ovaries compared to controls (Supplementary Table 1 & 2). Top 20 upregulated and downregulated genes were further assessed for confirmation using qPCR (n=6–8 per group) (Table 1). Most downregulated genes on RNAseq were also significantly down regulated on qPCR, only two were concordantly upregulated (Table 1). We filtered the genes that were both differentially expressed on qPCR and RNAseq by evaluating their biological or clinical relevance to ovarian development, androgen interaction, or mitochondrial changes and found 8 relevant genes through a literature search. These genes were further investigated by evaluating changes in protein expression in neonatal ovaries through Western blot. Retnlb, Cox6a2 and Actc1 protein levels were significantly downregulated in the DHT exposed ovaries compared to controls, Ang4 and Crybb2 also showed a similar trend but did not reach statistical significance (Figure 2). FosB did not show any difference. Myom2 and Cidec expression were very low and could not be reliably quantified.
Figure 1:

Differential grouping of mRNA of 3 control and 3 DHT exposed mouse ovaries after RNAseq on cluster analysis (A) and principal component analysis (B). Heat map of individual genes in DHT exposed ovaries compared to controls with an FDR of 0.05, log2 scale (C).
Table 1:
Top 20 up and downregulated genes on RNAseq followed by qPCR analysis
| Upregulated | Downregulated | ||||
|---|---|---|---|---|---|
| Gene | Fold | qPCR | Gene | Fold | qPCR |
| Mb | 8.1 | Down | Cryaa | −11.0 | Up |
| Myl2 | 5.3 | NS | Crybb2 | −10.6 | Down |
| Lmod2 | 5.1 | Down | Cryba1 | −10.1 | NS |
| Ckmt2 | 4.6 | NS | Crygf | −8.3 | Down |
| Zscan5b | 4.4 | NS | Cryba2 | −8.2 | NS |
| Actc1 | 4.4 | Down | Apoa4 | −8.1 | Down |
| Myoz2 | 4.3 | Down | Crygd | −7.7 | Down |
| Myl3 | 4.3 | NS | Fxyd4 | −7.5 | Down |
| Krt31 | 4.2 | NS | Fos | −6.9 | NS |
| Krt5 | 4.1 | Down | Cfd | −6.9 | NS |
| Trdn | 3.9 | NS | Lim2 | −6.6 | Down |
| Cox6a2 | 3.9 | Down | Clca1 | −6.5 | Down |
| Myh6 | 3.7 | NS | Cidec | −6.3 | Down |
| Adprhl1 | 3.7 | NS | Anxa13 | −6.2 | Down |
| 2310069G16Rik | 3.0 | Up | H2-T3 | −6.2 | Down |
| Myom2 | 2.9 | Up | Ang4 | −6.0 | Down |
| Eif4e1b | 2.8 | Down | Retnlb | −5.9 | Down |
| 4930453L07Rik | 2.8 | NS | Fosb | −5.6 | Down |
| Khdc3 | 2.8 | NS | Clec2h | −5.1 | Down |
| Eef1a2 | 2.8 | Down | Clrn3 | −4.4 | Down |
Legend: Fold indicates fold change in differential expression; qPCR results are represented under the qPCR column indicating significantly upregulated (Up), downregulated (Down), or non-significant (NS) findings.
Figure 2:

RNA expression on qPCR of top up and downregulated genes found on RNAseq analysis of DHT exposed ovaries (red) and controls (blue) indicated by fold change (A). Western blot detection of selected genes comparing DHT exposed ovaries (DHT) to controls (CON) (B). Quantification of protein level expression on Western blot (C) (*p<0.05, **p<0.01, ***p<0.001, ****p<0.0001); n=6–8 in each group.
DHT exposure altered ovarian and mitochondria genes:
We also evaluated the expression of 60 validated target genes that were well known to be involved in various ovarian and mitochondria functions using qPCR. Prenatal DHT exposure downregulated 25 genes and upregulated 2 genes, Ddx4 and Esr2 (Figure 3). Key genes such as integrins (Itgav, Itga1 and Itga4), gap junction protein (Gja1) and hormone receptors (Amhr2, Pr, Prmc1, Lhcgr, Oxtr and Ar) as well as mitochondrial encoded genes (mtNd5, mtCo1) and mitochondrial function genes (Mfn1 and Opa1) were downregulated (Figure 3). No difference in expression was seen in both controls and the DHT exposed ovaries for somatic genes Atrx, Cep70, Diaph2, Ddm1, Ect2, Igfr, Itga6, Fgfr1, Fox12, Gata4, Kt8, Lkbkb, Mfn2, Mbn, Nobox, Nr2f2, Oct4, Pcm1, Pp2ca, Spast, Xrcc1, and mitochondrial genes mtAtp6, mtAtp8, Co2, mtCytb, mtNd1, mtNd2, mtNd3, mtNd4, mtNd4l, mtNd6, mtRnr1, mtRnr2 (data not shown). Functional enrichment analysis based on data from the Kyoto Encyclopedia of Genes and Genomes (KEGG) revealed that differentially expressed genes in neonatal ovaries that were exposed to androgens in-utero were mainly involved in the ovarian steroidogenesis pathway, cell adhesion molecule pathway and PI3K-Akt signaling pathway.
Figure 3:

RNA expression of target genes on qPCR analysis of DHT exposed ovaries (red) and controls (blue) indicated by fold change (*p<0.05, **p<0.01, ***p<0.001, ****p<0.0001); n= 5–6 in each group.
Mitochondrial structure was compromised but ovaries had normal histology:
Stark differences were observed on TEM of ovarian mitochondria. DHT exposed mice had disorganized cristae, vacuoles, and were less electron dense whereas the controls had normal mitochondrial ultrastructure in both granulosa cells and oocytes (Figure 4) Histological assessment revealed no difference in the ovary between the two groups. Both groups showed unassembled clusters of oocytes beneath the surface epithelium, and some primordial follicles in the inner ovarian cortices (Supplementary Figure 1).
Figure 4:
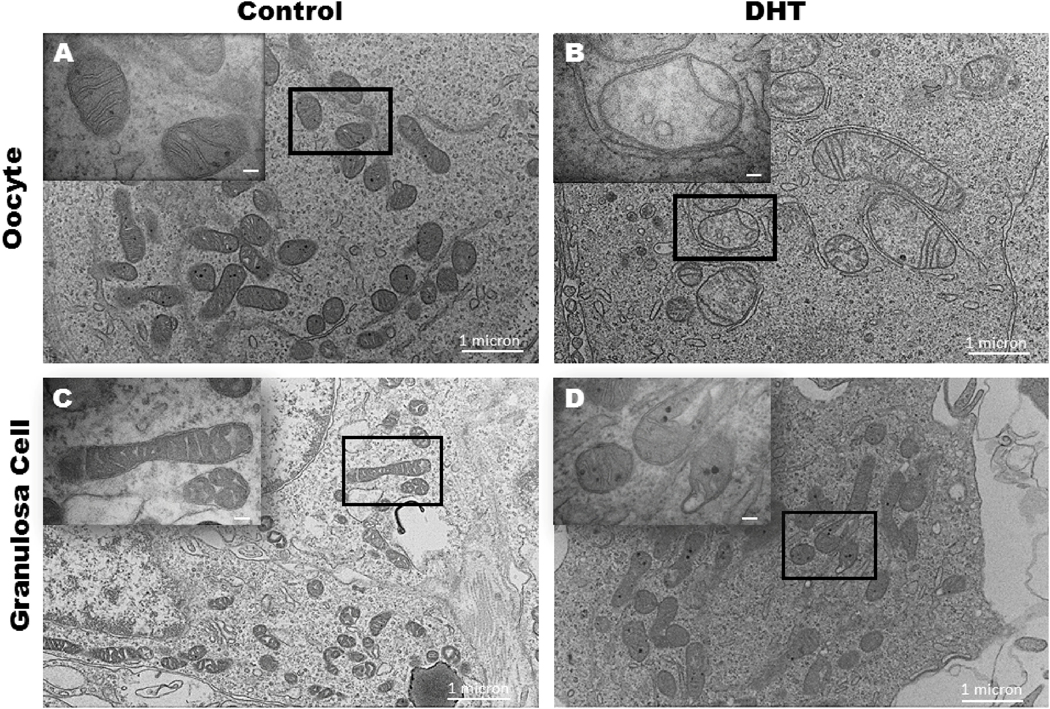
Electron microscopy imaging of control (A, C) and DHT exposed (B, D) mouse granulosa cell mitochondria (A, B) and oocyte mitochondria (C,D) at 7000X magnification with inserts at 40,000X magnification; n=3 controls, 4 DHT.
DHT treatment did not alter mtDNA copy number:
Mitochondrial DNA copy number was assessed by qPCR of mitochondrial genes (mtCo1, mtCo2, mtCo3) compared with a somatic reference gene (tubulin) in DHT exposed ovaries versus controls. There was no difference noted in mtDNA copy number of DHT exposed ovaries when compared to controls for mtCo1 (DHT 107 ± 23.14 vs. Control 111 ± 30), for mtCo2 (DHT 69 ± 15 vs. Control 70 ± 16), and for mtCo3 (DHT treated 76 ± 17 vs. Control 79 ± 21) (Supplementary Figure 2).
DHT exposed ovaries consumed more oxygen
Ovaries exposed to DHT in utero had significantly higher oxygen consumption rate when compared with controls at all measured time points (Figure 5A). DHT exposure increased basal respiration (DHT 571± 85 vs. Control 183 ± 57 pmol/min/μg protein, p< 0.01; Figure 5B), maximum respiration (DHT 429 ± 62 vs. Control 125 ± 43 pmol/min/μg protein, p<0.01; Figure 5C), ATP linked respiration (DHT 414 ± 63 vs. Control 135 ± 43 pmol/min/μg protein, p<0.05; Figure 5D), and proton leak (DHT 157 ± 24 vs. Control 48 ± 14 pmol/min/μg protein, p< 0.01; Figure 5E) when compared to control ovaries. Nonmitochondrial respiration tended to be higher in DHT exposed mouse ovaries and controls but did not reach statistical significance (DHT 106 ± 26 vs. Control 45 ±18; Figure 5F). Neither the DHT exposed ovaries nor the controls had any spare capacity.
Figure 5:

Normalized oxygen consumption rate (OCR) measurements by Seahorse XFe96 analyzer™ of mouse ovaries exposed to DHT (red) vs controls (blue) including basal measurements before inhibitor compound injection followed by arrows pointing to time at which the different compounds oligomycin, FCCP, and antimycin/rotenone were injected (A). Normalized OCR rates for basal respiration (B), maximum respiration (C), ATP-linked respiration (D), proton leak (E), and non-mitochondrial respiration (F). Data represent mean +/− standard deviation (*p<0.05, **p<0.01); n= 10 controls, 25 DHT.
Total ATP levels similar between the groups:
Total ATP levels were assed utilizing a luciferase assay in whole ovaries. There were no overall differences in ATP levels between DHT exposed ovaries and controls (16.04 ± 11 in DHT vs. 23.72 ± 7 nM/μg protein in controls).
DISCUSSION
PCOS affects ~10% of women, and the disease has a complex and diverse phenotype with a poorly understood pathogenesis. Findings from genome wide association studies accounts for only less than 10% of the syndrome’s heritability [4], and therefore we must search for other factors that could elucidate an increase in the likelihood a woman maybe susceptible to the syndrome, such as the hyperandrogenemic uterine environment found in PCOS mothers. Huang et al treated 7–8 week mice with androgens and found alterations in oocyte mitochondria which correlated with decreased fertilization and blastocyst formation rates [20]. Chappell at al. utilized the well-established lean PCOS mouse model and found that inner mitochondrial membrane function, ROS, and mitochondrial ultrastructure were altered. However, we wanted to explore further the timing of these mitochondrial alterations [19]. In a study by Tata et al., AMH was given prenatally to mice, which induced a lean PCOS phenotype with higher testosterone, hyperactivity of GnRH, disruption of estrous cyclicity with oligoanovulation, and impaired fertility [17]. Notably, dams treated with AMH had female pups that expressed higher testosterone and LH two hours postpartum, identifying alterations in sex steroids in the neonatal period [17]. In this study neonatal elevations of testosterone and increased LH surges after dams were treated with AMH were corrected with prenatal GnRH antagonist administration [17]. This indicates that hormonal reprogramming starts in utero and not in the neonatal period.
Using a lean PCOS model with DHT treatment in utero, we found that prenatal androgen exposure has many downstream effects on the ovaries of offspring as early as postnatal day 0. These findings support the hypothesis that some PCOS phenotypes can be programmed in utero through an altered androgen environment [2, 14–16, 21–28] causing changes in gene expression and mitochondria which could possibly then be inherited in future generations and can be further exacerbated by excess androgens.
We utilized two different approaches to identify genes in the ovary that are differentially regulated by DHT treatment, RNAseq to identify and characterize genes as well as targeted qPCR for genes regulated in PCOS and those involved in ovarian development. We found that the ovaries exposed to DHT had many changes in gene expression on RNAseq and that some of these genes were related to folliculogenesis and female infertility. Specifically, Cox6A2, Retnlb, and Actc1 were differentially expressed and down regulated on protein analysis. Both Cox6A2, a subunit of the cytochrome c oxidase complex (Complex IV), and Actc1, previously found to be two fold elevated in quadriceps of COPD patients [29], were differentially expressed and are related to mitochondrial function. While Retnlb, or Resistin Like beta, is involved in metabolism and cell proliferation with resistin found to be involved in modulating glucose tolerance and insulin action [30]. Furthermore, it is interesting to note that resistin was found to be a promising candidate gene for PCOS based on linkage and association studies related to the PCOS INSR gene on chromosome 19p13.3 [30]. These genes were previously found to be related to mitochondrial function and glucose metabolism, were found to be differently regulated in neonatal ovaries after in utero androgen exposure. The precise roles of these genes in the pathogenesis of PCOS, and how they affect oxidative phosphorylation and metabolism is not clearly understood currently and should be explored further.
On targeted analysis, we found that genes were down-regulated related to cell-cell communication such as the integrins and gap junction protein with cell adhesion molecule pathways altered on KEGG analysis. This would likely affect the communication between oocytes and granulosa cells, both of which we found had altered mitochondria on TEM. Androgen exposure in utero affected not only the individual cells of the ovary but also how these cells interact (granulosa and oocyte), vital to folliculogenesis. Insulin signaling pathway was also implicated, a known contributor to the PCOS phenotype contributing both to the insulin resistance and androgen excess experienced by PCOS patients. While genes in ovarian steroidogenesis pathways were also differentially expressed, interestingly, the androgen receptor was found to be downregulated while Esr2 was upregulated. Such a scenario has been observed in cardiometabolic studies where estrogen deficiency increases ROS, mitochondrial proteins, and ATP while androgen exposure induces ROS and cardiac dysfunction [31–34]. It is well known that sex steroid signaling are dysregulated in metabolic syndrome [35, 36]. We speculate that increased androgen exposure in utero downregulates AR [37] and upregulates ER to compensate for the increased androgens in circulation to be protective from possible detrimental effects. It would be important to evaluate these genes over time as well as their downstream targets to better understand the metabolic profile of PCOS and even target therapeutics.
We also found important changes in the mitochondria. Mitochondrial cristae are folds of the mitochondrial inner membrane that provide an increase in the surface area which allows the mitochondria to have more locations for ATP production [38]. In our study of neonatal ovaries, we saw that hyperandrogenemia in utero altered the mitochondria of both granulosa cells and oocytes with abnormal architecture containing decreased number of cristae which are essential for ATP production. While data on metabolism in PCOS mouse models is well established, evaluation of oocytes and their mitochondria is specifically at birth is lacking [28, 39, 40]. We looked to the obesity literature to correlate our findings with other fetal programming studies, where offspring oocytes from diabetic or obese mice fed high fat diets have altered mitochondria ultrastructure, mtDNA copy number, abnormal function, and spindle abnormalities [41, 42]. Previous work looking at an obese mouse model has demonstrated that maternal obesity drastically affect mitochondrial structure and function and is transmitted to the offspring [43, 44]. Mitochondria were noted to have abnormal ultrastructure, depressed respiration, and decreased concentration of electron transport system proteins [43, 44]. They also showed that damaged mitochondria were passed onto the progeny via maternal inheritance. We sought out to investigate this in terms of androgen exposure in a lean PCOS model by eliminating the obesity confounder in PCOS while evaluating the timing of mitochondrial change in ovarian tissue. We found that as early as day 0 of life, mitochondrial ultrastructure and function were impaired in granulosa cells and oocytes.
Based on their metabolic activity, different tissues have varying numbers of mitochondria and mitochondrial DNA (mtDNA) copy number [38]. In an adult human oocyte there are about 100,000 mitochondria which are transcriptionally mostly silent until fertilization [38]. The quiescent state is vital to keeping mutations to a minimum and help protect future offspring. However, these thousands of mitochondria are formed from only a few hundred in primordial germ cells. These mitochondria support early embryonic development as there is no replication of mtDNA until the hatched blastocyst stage. Therefore, mitochondria derived from the oocyte are the major source of ATP during preimplantation embryonic development, with threshold mtDNA copy number of 40,000–50,000 in the oocyte required for embryonic development following implantation [38, 45]. Interestingly in this study and our previous work on post-pubertal lean PCOS mice [19], the mtDNA copy number was not different in neonatal ovaries or post-pubertal oocytes after in utero DHT treatment. This indicates that the total amount of mitochondrial DNA copies are the same. But it is well known that mtDNA copy number is not the entire story as it does not relay the total biomass. Not only did we demonstrate alterations in morphology as above, but we also found decreased expression of some mitochondrial DNA encoded genes and genomic DNA encoded genes involved in mitochondrial function (mtNd5, mtCo1, Mfn1 and Opa1).
This is the first study, to our knowledge, to evaluate neonatal ovarian oxygen consumption after in utero DHT treatment. Functionally, while the controls at baseline behaved much like other cell lines investigated using the Seahorse analyzer [46, 47] the DHT exposed ovaries consumed more oxygen across the entirety of the experiment, showing the striking effect of excess androgen on mitochondrial function. The baseline oxygen consumption was almost four times higher in DHT exposed ovaries. Other measures including maximum respiration, ATP linked respiration, and proton leak induced oxygen consumption were also significantly increased, all while the ATP total production stayed the same. Therefore, day 0 ovaries after prenatal DHT exposure are working much harder and are less efficient to produce the same amount of ATP. The amount of ATP generated per unit of oxygen consumed can vary significantly and not all mitochondrial oxygen consumption translates to ATP production, especially with changes in proton leak [48]. We found that DHT exposed ovaries had increased proton leak induced oxygen consumption. Therefore, oxygen is being consumed at a higher rate, with increased proton leak and ATP linked respiration, indicating increased mitochondrial function in DHT exposed ovaries. The total ATP levels were shown to be similar between control and DHT exposed ovaries, suggesting there is another source for ATP in control ovaries so they may meet the ATP demand. This source is most likely increased glycolytic activity in controls, which is an area of needed future research. What was also evident was that neonatal ovaries, whether treated in utero with DHT or not, lack spare respiratory capacity as the maximum respiration did not change from the basal respiration. Spare respiratory capacity is utilized by the cells to adapt to increased energy demands if there is a change in workload or cellular activity. Previous studies have shown that cells that have pluripotent potential and stem cells have significantly lower or no spare respiratory capacity [49, 50]. Therefore, it is possible that the mouse neonatal ovary, which is not yet finished in developing the primordial oocyte pool at birth, could behave more like pluripotent cells. These neonatal ovaries, control or DHT exposed, are working at maximum efficiency, and consuming oxygen at the highest threshold.
CONCLUSIONS:
Our study shows that excess androgen exposure in fetal life can alter gene pathways and mitochondria at birth leading to a lean PCOS phenotype later in life. The mechanisms behind these changes are still unknown. It is possible that in utero androgen exposure causes heritable epi-genetic changes or there is a propagation of impaired mitochondria from one generation to another but how this occurs is unclear. While many questions are still left unanswered, these experiments have shown that androgen exposure in utero affect gene pathways, and mitochondrial structure and function at birth.
Supplementary Material
Supplementary Figure 1: Representative images of hematoxylin and eosin stained histological section of control and DHT exposed ovary under 200x magnification.
Supplementary Figure 2: mtDNA copy number for mtCo1 (A), mtCo2 (B), and mtCo3 (C) compared to tubulin for relative fluorescence for day 0 ovaries exposed to DHT in utero (red) vs controls (blue).
Highlights:
Prenatal androgen exposure causes an adult lean PCOS phenotype and alters gene expression, and mitochondria structure and function at birth in mice.
Acknowledgements:
The authors would like to acknowledge the Department of Obstetrics and Gynecology at the Baylor College of Medicine for their support in this project as well as the Family Fertility Center embryologists Dr. Richard Cochran, Khalied Kaskar, Amanda David, Daneeka Hamilton and Dr. Yana Kisarova. The authors would also like to acknowledge the Baylor College of Medicine Integrative Microscopy Core and the Animal Facility for their assistance.
Funding: This work was supported by training grants by the Department of Obstetrics and Gynecology, Baylor College of Medicine (MB, JM, PH, LY and JBG.) and R-01 research grant (Grant # DK114689) for CSB from National Institutes of Health.
Footnotes
Disclosure: The authors have nothing to disclose.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References:
- [1].Palomba S, Santagni S, Falbo A, La Sala GB, Complications and challenges associated with polycystic ovary syndrome: current perspectives, International journal of women’s health, 7 (2015) 745–763. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [2].Abbott DH, Barnett DK, Bruns CM, Dumesic DA, Androgen excess fetal programming of female reproduction: a developmental aetiology for polycystic ovary syndrome?, Human reproduction update, 11 (2005) 357–374. [DOI] [PubMed] [Google Scholar]
- [3].Legro RS, Driscoll D, Strauss JF 3rd, Fox J, Dunaif A, Evidence for a genetic basis for hyperandrogenemia in polycystic ovary syndrome, Proceedings of the National Academy of Sciences of the United States of America, 95 (1998) 14956–14960. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [4].Risal S, Pei Y, Lu H, Manti M, Fornes R, Pui H-P, Zhao Z, Massart J, Ohlsson C, Lindgren E, Crisosto N, Maliqueo M, Echiburú B, Ladrón de Guevara A, Sir-Petermann T, Larsson H, Rosenqvist MA, Cesta CE, Benrick A, Deng Q, Stener-Victorin E, Prenatal androgen exposure and transgenerational susceptibility to polycystic ovary syndrome, Nat Med, 25 (2019) 1894–1904. [DOI] [PubMed] [Google Scholar]
- [5].Franks S, Berga SL, Does PCOS have developmental origins?, Fertility and sterility, 97 (2012) 2–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [6].Barraclough CA, Gorski RA, Evidence that the hypothalamus is responsible for androgen-induced sterility in the female rat, Endocrinology, 68 (1961) 68–79. [DOI] [PubMed] [Google Scholar]
- [7].Sullivan SD, Moenter SM, Prenatal androgens alter GABAergic drive to gonadotropin-releasing hormone neurons: implications for a common fertility disorder, Proceedings of the National Academy of Sciences of the United States of America, 101 (2004) 7129–7134. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [8].West C, Foster DL, Evans NP, Robinson J, Padmanabhan V, Intra-follicular activin availability is altered in prenatally-androgenized lambs, Molecular and cellular endocrinology, 185 (2001) 51–59. [DOI] [PubMed] [Google Scholar]
- [9].Palomba S, Marotta R, Di Cello A, Russo T, Falbo A, Orio F, Tolino A, Zullo F, Esposito R, La Sala GB, Pervasive developmental disorders in children of hyperandrogenic women with polycystic ovary syndrome: a longitudinal case-control study, Clinical endocrinology, 77 (2012) 898–904. [DOI] [PubMed] [Google Scholar]
- [10].Daan NM, Koster MP, Steegers-Theunissen RP, Eijkemans MJ, Fauser BC, Endocrine and cardiometabolic cord blood characteristics of offspring born to mothers with and without polycystic ovary syndrome, Fertility and sterility, 107 (2017) 261–268.e263. [DOI] [PubMed] [Google Scholar]
- [11].Mehrabian F, Kelishadi R, Comparison of the metabolic parameters and androgen level of umbilical cord blood in newborns of mothers with polycystic ovary syndrome and controls, Journal of research in medical sciences : the official journal of Isfahan University of Medical Sciences, 17 (2012) 207–211. [PMC free article] [PubMed] [Google Scholar]
- [12].Abbott DH, Bruns CR, Barnett DK, Dunaif A, Goodfriend TL, Dumesic DA, Tarantal AF, Experimentally induced gestational androgen excess disrupts glucoregulation in rhesus monkey dams and their female offspring, American journal of physiology. Endocrinology and metabolism, 299 (2010) E741–751. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [13].Hogg K, McNeilly AS, Duncan WC, Prenatal androgen exposure leads to alterations in gene and protein expression in the ovine fetal ovary, Endocrinology, 152 (2011) 2048–2059. [DOI] [PubMed] [Google Scholar]
- [14].Manikkam M, Crespi EJ, Doop DD, Herkimer C, Lee JS, Yu S, Brown MB, Foster DL, Padmanabhan V, Fetal programming: prenatal testosterone excess leads to fetal growth retardation and postnatal catch-up growth in sheep, Endocrinology, 145 (2004) 790–798. [DOI] [PubMed] [Google Scholar]
- [15].Ortega HH, Rey F, Velazquez MM, Padmanabhan V, Developmental programming: effect of prenatal steroid excess on intraovarian components of insulin signaling pathway and related proteins in sheep, Biology of reproduction, 82 (2010) 1065–1075. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [16].Ortega HH, Salvetti NR, Padmanabhan V, Developmental programming: prenatal androgen excess disrupts ovarian steroid receptor balance, Reproduction (Cambridge, England), 137 (2009) 865–877. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [17].Tata B, Mimouni NEH, Barbotin AL, Malone SA, Loyens A, Pigny P, Dewailly D, Catteau-Jonard S, Sundstrom-Poromaa I, Piltonen TT, Dal Bello F, Medana C, Prevot V, Clasadonte J, Giacobini P, Elevated prenatal anti-Mullerian hormone reprograms the fetus and induces polycystic ovary syndrome in adulthood, Nat Med, (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- [18].Xita N, Tsatsoulis A, Review: fetal programming of polycystic ovary syndrome by androgen excess: evidence from experimental, clinical, and genetic association studies, The Journal of clinical endocrinology and metabolism, 91 (2006) 1660–1666. [DOI] [PubMed] [Google Scholar]
- [19].Chappell NR, Zhou B, Schutt AK, Gibbons WE, Blesson CS, Prenatal androgen induced lean PCOS impairs mitochondria and mRNA profiles in oocytes, Endocrine connections, (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- [20].Huang Y, Yu Y, Gao J, Li R, Zhang C, Zhao H, Zhao Y, Qiao J, Impaired oocyte quality induced by dehydroepiandrosterone is partially rescued by metformin treatment, PloS one, 10 (2015) e0122370. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [21].Almond D, Currie J, Killing Me Softly: The Fetal Origins Hypothesis, The journal of economic perspectives : a journal of the American Economic Association, 25 (2011) 153–172. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [22].Barker DJ, The fetal and infant origins of adult disease, BMJ (Clinical research ed.), 301 (1990) 1111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [23].Barker DJ, The origins of the developmental origins theory, Journal of internal medicine, 261 (2007) 412–417. [DOI] [PubMed] [Google Scholar]
- [24].Dumesic DA, Goodarzi MO, Chazenbalk GD, Abbott DH, Intrauterine environment and polycystic ovary syndrome, Seminars in reproductive medicine, 32 (2014) 159–165. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [25].Jaeger K, Saben JL, Moley KH, Transmission of Metabolic Dysfunction Across Generations, Physiology (Bethesda, Md.), 32 (2017) 51–59. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [26].Painter RC, Osmond C, Gluckman P, Hanson M, Phillips DI, Roseboom TJ, Transgenerational effects of prenatal exposure to the Dutch famine on neonatal adiposity and health in later life, BJOG : an international journal of obstetrics and gynaecology, 115 (2008) 1243–1249. [DOI] [PubMed] [Google Scholar]
- [27].Stocker CJ, Arch JR, Cawthorne MA, Fetal origins of insulin resistance and obesity, The Proceedings of the Nutrition Society, 64 (2005) 143–151. [DOI] [PubMed] [Google Scholar]
- [28].Stener-Victorin E, Padmanabhan V, Walters KA, Campbell RE, Benrick A, Giacobini P, Dumesic DA, Abbott DH, Animal Models to Understand the Etiology and Pathophysiology of Polycystic Ovary Syndrome, Endocr Rev, 41 (2020) 538–576. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [29].Willis-Owen SAG, Thompson A, Kemp PR, Polkey MI, Cookson W, Moffatt MF, Natanek SA, COPD is accompanied by co-ordinated transcriptional perturbation in the quadriceps affecting the mitochondria and extracellular matrix, Scientific reports, 8 (2018) 12165. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [30].Urbanek M, Du Y, Silander K, Collins FS, Steppan CM, Strauss JF 3rd, Dunaif A, Spielman RS, Legro RS, Variation in resistin gene promoter not associated with polycystic ovary syndrome, Diabetes, 52 (2003) 214–217. [DOI] [PubMed] [Google Scholar]
- [31].Hou M, Gu HC, Wang HH, Liu XM, Zhou CL, Yang Q, Jiang ZR, Lin J, Wu YM, Wu YT, Sheng JZ, Huang HF, Prenatal exposure to testosterone induces cardiac hypertrophy in adult female rats through enhanced Pkcdelta expression in cardiac myocytes, Journal of molecular and cellular cardiology, 128 (2019) 1–10. [DOI] [PubMed] [Google Scholar]
- [32].Jonker SS, Louey S, Roselli CE, Cardiac myocyte proliferation and maturation near term is inhibited by early gestation maternal testosterone exposure, American journal of physiology. Heart and circulatory physiology, 315 (2018) H1393–h1401. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [33].Lopes RA, Neves KB, Pestana CR, Queiroz AL, Zanotto CZ, Chignalia AZ, Valim YM, Silveira LR, Curti C, Tostes RC, Testosterone induces apoptosis in vascular smooth muscle cells via extrinsic apoptotic pathway with mitochondria-generated reactive oxygen species involvement, American journal of physiology. Heart and circulatory physiology, 306 (2014) H1485–1494. [DOI] [PubMed] [Google Scholar]
- [34].Ventura-Clapier R, Piquereau J, Veksler V, Garnier A, Estrogens, Estrogen Receptors Effects on Cardiac and Skeletal Muscle Mitochondria, Front Endocrinol (Lausanne), 10 (2019) 557–557. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [35].Mauvais-Jarvis F, Estrogen and androgen receptors: regulators of fuel homeostasis and emerging targets for diabetes and obesity, Trends Endocrinol Metab, 22 (2011) 24–33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [36].Walters KA, Polycystic ovary syndrome: Is it androgen or estrogen receptor?, Current Opinion in Endocrine and Metabolic Research, 12 (2020) 1–7. [Google Scholar]
- [37].Tetsuka M, Hillier SG, Androgen receptor gene expression in rat granulosa cells: the role of follicle-stimulating hormone and steroid hormones, Endocrinology, 137 (1996) 4392–4397. [DOI] [PubMed] [Google Scholar]
- [38].Babayev E, Seli E, Oocyte mitochondrial function and reproduction, Current opinion in obstetrics & gynecology, 27 (2015) 175–181. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [39].Moore AM, Prescott M, Campbell RE, Estradiol negative and positive feedback in a prenatal androgen-induced mouse model of polycystic ovarian syndrome, Endocrinology, 154 (2013) 796–806. [DOI] [PubMed] [Google Scholar]
- [40].Roland AV, Moenter SM, Reproductive neuroendocrine dysfunction in polycystic ovary syndrome: insight from animal models, Frontiers in neuroendocrinology, 35 (2014) 494–511. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [41].Luzzo KM, Wang Q, Purcell SH, Chi M, Jimenez PT, Grindler N, Schedl T, Moley KH, High fat diet induced developmental defects in the mouse: oocyte meiotic aneuploidy and fetal growth retardation/brain defects, PloS one, 7 (2012) e49217. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [42].Wang Q, Ratchford AM, Chi MM-Y, Schoeller E, Frolova A, Schedl T, Moley KH, Maternal diabetes causes mitochondrial dysfunction and meiotic defects in murine oocytes, Molecular endocrinology, 23 (2009) 1603–1612. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [43].Saben JL, Boudoures AL, Asghar Z, Thompson A, Drury A, Zhang W, Chi M, Cusumano A, Scheaffer S, Moley KH, Maternal Metabolic Syndrome Programs Mitochondrial Dysfunction via Germline Changes across Three Generations, Cell reports, 16 (2016) 1–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [44].Ferey JLA, Boudoures AL, Reid M, Drury A, Scheaffer S, Modi Z, Kovacs A, Pietka T, DeBosch BJ, Thompson MD, Diwan A, Moley KH, A maternal high-fat, high-sucrose diet induces transgenerational cardiac mitochondrial dysfunction independently of maternal mitochondrial inheritance, American journal of physiology. Heart and circulatory physiology, 316 (2019) H1202–h1210. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [45].Chappel S, The role of mitochondria from mature oocyte to viable blastocyst, Obstetrics and gynecology international, 2013 (2013) 183024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [46].Cox CS, McKay SE, Holmbeck MA, Christian BE, Scortea AC, Tsay AJ, Newman LE, Shadel GS, Mitohormesis in Mice via Sustained Basal Activation of Mitochondrial and Antioxidant Signaling, Cell metabolism, 28 (2018) 776–786.e775. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [47].Boland ML, Oldham S, Boland BB, Will S, Lapointe JM, Guionaud S, Rhodes CJ, Trevaskis JL, Nonalcoholic steatohepatitis severity is defined by a failure in compensatory antioxidant capacity in the setting of mitochondrial dysfunction, World journal of gastroenterology, 24 (2018) 1748–1765. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [48].Salin K, Auer SK, Rey B, Selman C, Metcalfe NB, Variation in the link between oxygen consumption and ATP production, and its relevance for animal performance, Proc Biol Sci, 282 (2015) 20151028–20151028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [49].Varum S, Momcilovic O, Castro C, Ben-Yehudah A, Ramalho-Santos J, Navara CS, Enhancement of human embryonic stem cell pluripotency through inhibition of the mitochondrial respiratory chain, Stem cell research, 3 (2009) 142–156. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [50].Zhou Y, Al-Saaidi RA, Fernandez-Guerra P, Freude KK, Olsen RK, Jensen UB, Gregersen N, Hyttel P, Bolund L, Aagaard L, Bross P, Luo Y, Mitochondrial Spare Respiratory Capacity Is Negatively Correlated with Nuclear Reprogramming Efficiency, Stem cells and development, 26 (2017) 166–176. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary Figure 1: Representative images of hematoxylin and eosin stained histological section of control and DHT exposed ovary under 200x magnification.
Supplementary Figure 2: mtDNA copy number for mtCo1 (A), mtCo2 (B), and mtCo3 (C) compared to tubulin for relative fluorescence for day 0 ovaries exposed to DHT in utero (red) vs controls (blue).