

SUMMARY
Class I phosphoinositide-3-kinases (PI3Ks) phosphorylate PIP2 at its 3′ inositol position to generate PIP3, a second messenger that influences signaling cascades regulating cellular growth, survival, and proliferation. Previous studies have suggested that PI3Kα activation involves dislodging the p85α nSH2 domain from the p110α catalytic subunit by binding activated receptor tyrosine kinases. We carried out molecular dynamics simulations to determine, mechanistically and structurally, how PI3Kα conformations are influenced by physiological effectors and the nSH2 domain. We demonstrate that changes in protein dynamics mediated by allosteric regulation significantly increase the population of catalytically competent states without changing the enzyme ground-state structure. Furthermore, we demonstrate that modulation of active-site residue interactions with enzyme substrates can reciprocally influence nSH2 domain dynamics. Together, these results suggest that dynamic allostery plays a role in populating the catalytically competent conformation of PI3Kα, and provide a key platform for the design of novel chemotherapeutic PI3Kα inhibitors.
In Brief
PI3Kα is a phospholipid kinase that phosphorylates PIP2 to generate PIP3, a scaffold for many important downstream signaling pathways. In this paper, Chakrabarti et al. demonstrate that dynamic allostery can increase the population of PI3Kα states compatible with catalysis, which has implications for the development of new PI3K therapeutics.
Graphical Abstract
INTRODUCTION
Phosphoinositide 3-kinase (PI3K) is a lipid kinase that modifies phosphatidylinositol-4,5-bisphosphate (PIP2) phosphoinositide by transferring the γ-phosphate of ATP to the 3′ position of the PIP2 inositol ring, generating phosphatidylinositol-3,4,5-trisphosphate (PIP3), a docking site for many downstream proteins containing a pleckstrin homology domain (PH domain). These proteins include AKT and PDK1, which broadly regulate cell proliferation, survival, and cell death (Philp et al., 2001; Vogt et al., 2007). In mammalian cells, three classes of PI3K (I, II, and II) have been described on the basis of domain structure and substrate preference (Engelman et al., 2006; Walker et al., 1999). Within class I PI3Ks, there is a further subdivision into class IA and IB on the basis of regulatory subunit (Vadas et al., 2011). Three enzymes—PI3Kα, PI3Kβ, and PI3Kδ—belong to class IA. Our studies are focused on PI3Kα, the isoform comprising a p110α catalytic subunit and a p85α regulatory subunit. The p110α subunit consists of five domains: an N-terminal adaptor binding domain (ABD), which binds to the regulatory p85, a Ras-binding domain (RBD), a C2 domain, a helical domain, and a kinase domain (Huang et al., 2008). The C-terminal lobe of the kinase domain contains the activation loop (residues 933–958) and the catalytic loop (residues 909–920), highly flexible regions implicated in substrate binding and catalysis (Huang et al., 2007; Miller et al., 2014; Pirola et al., 2001). The p85α subunit consists of an Src-Homology 3 (SH3) domain, a domain with homology to a guanosine triphosphatase-activating domain (GAP), an nSH2 (N-terminal SH2 domain), iSH2 (inter-SH2 domain), and cSH2 (C-terminal SH2 domain). However, there are currently no available structures containing all five domains of the regulatory subunit. The full p110α and only the nSH2 and the iSH2 domains of p85α have been included in the most complete structure of PI3K (hereafter called p110α/niSH2) (Mandelker et al., 2009). Together, these two domains of p85α are necessary and sufficient for PI3K heterodimerization and regulation of catalytic activity (Dhand et al., 1994; Yu et al., 1998a, 1998b).
Structural characterizations of PI3Kα suggest that several interfacial contacts (salt bridges and hydrogen-bonding interactions) between the p85α and p110α subunits are responsible for inhibiting catalytic activity in the basal state (Mandelker et al., 2009; Miled et al., 2007; Miller et al., 2014). Nonetheless, PI3Kα has a low but detectable level of lipid kinase activity in the absence of activation (Hon et al., 2012), indicating that catalytically competent conformations in which catalytic residues are proximal to the substrates can still occur in the ground state. In response to cellular stimuli, the nSH2 domain (and possibly also the cSH2 domain) of P85α binds phosphorylated tyrosine (pY) residues found in the C-terminal region of activated receptor protein kinases (RTKs) or their adaptor proteins; this leads to alteration in the interfacial contacts between the PI3Kα subunits and enhances the activation of p110α by 2- to 4-fold (Carson et al., 2008; Hon et al., 2012; Nolte et al., 1996). Hydrogen/deuterium exchange experiments have been used to interrogate PI3K activation dynamics upon binding of a cognate pY peptide (a region of an activating protein containing the pY), as well as the dynamic effects of cancer-associated mutations in the catalytic subunit (Burke et al., 2012). Intriguingly, these experiments demonstrate that several oncogenic mutations (e.g., G106V, N345K, E545K) increase fluctuations in domains far from the mutation site, including the iSH2, ABD, C2, and helical domains, and influence interdomain contacts in the iSH2-C2 interface upon addition of a cognate pY peptide. This result suggests that dynamic allostery (previously observed experimentally in catabolite activator protein [CAP], PDZ domains, trp RNA-binding attenuation protein [TRAP], chromosomal zinc-regulated repressor [CzrA], and peptidyl-prolyl cis-trans isomerase, NIMA-interacting 1 [Pin1], among others) may be involved in the activation mechanism of PI3K (Campitelli et al., 2018; Capdevila et al., 2017; Cooper and Dryden, 1984; Fuentes et al., 2004; Malay et al., 2011; Popovych et al., 2006; Tsai et al., 2008; Weinkam et al., 2012). In previous work, we demonstrated using computational methods that oncogenic mutations spatially distant from the catalytic site of the enzyme can influence nSH2 domain fluctuations and disrupt key intermolecular inter-actions between the p85 and p110 subunits that inhibit catalytic activity (Echeverria et al., 2015).
Recently, we determined that mutations in residues H917 of the catalytic loop, H936 in the activation loop, and K776 in the phosphate-binding (P) loop of PI3K were found to have significant effects on both substrate-binding affinity and catalytic activity, leading to the hypothesis that these residues are involved in catalysis (Maheshwari et al., 2017). Based on the information obtained from these kinetics studies, we proposed a catalytic mechanism for PI3Kα (Maheshwari et al., 2017) whereby H936 acts as the catalytic base, H917 accepts the O3′ proton from the γ-phosphate of ATP, and K776 stabilizes the transition-state intermediate.
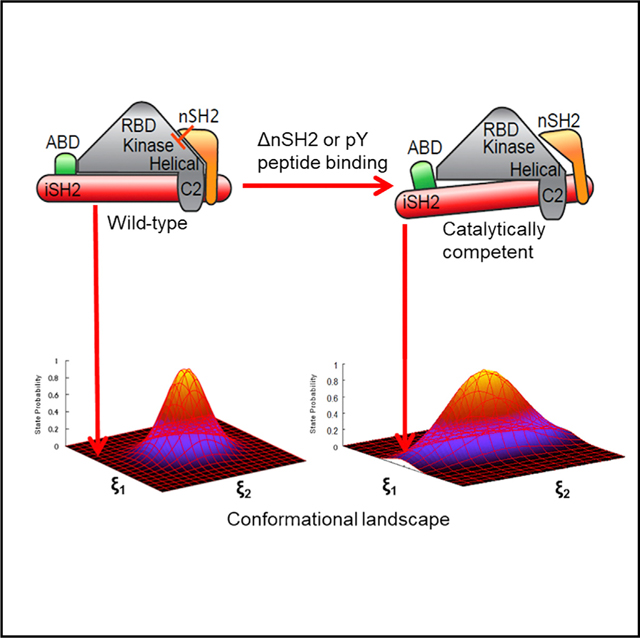
In the current work, we employed computational approaches to determine how binding of a cognate pY peptide or removal of the nSH2 domain increases the populations of catalytically competent states of the enzyme and, more specifically, how the conformational dynamics of PI3Kα influences the configuration of residues of the activation loop of the kinase domain. To this effect, we investigated three situations: (1) wild-type (WT) p110α/niSH2, (2) the same without the nSH2, and (3) WT p110α/niSH2 with bound cognate pY peptide (Nolte et al., 1996). We determined that the nSH2 domain, widely accepted as an autoinhibitory regulator of PI3Kα activation, may also be involved in mediating productive interactions of the ATP and PIP2 ligands that are important for catalysis, in part through its modulation of the activation-loop ensemble. Furthermore, we were able to observe significant changes in the activation-loop dynamics as well as a significant effect on large-scale motions of PI3Kα by binding cognate pY peptide. Together, these observations shed new light on the influence of allosteric perturbations on the activation mechanism of PI3Kα, and suggest that activation is a consequence of dynamically altering the population of competent conformations in the original ground-state ensemble. In this context, binding of an allosteric regulator (or other allosteric effects) results in a change in the conformational landscape of the protein that enhances the population of functionally competent configurations. Traditionally, in the modified landscape, the functionally competent conformation becomes the new ground state. In the case of dynamic allostery, the population of the functionally competent state increases by a small factor (e.g., 5–50), which increases the representation of this state in the ensemble distribution without significantly altering the population of the original ground state. The identification of these conformations opens new avenues for the design of PI3Kα-specific inhibitors.
RESULTS AND DISCUSSION
The nSH2 Domain Inhibits PI3Kα Activation and Alters Activation-Loop Dynamics
Recent computational studies on PI3Kα have suggested that the loss of nSH2 autoinhibition—the first step necessary for the enzyme to achieve its catalytically competent conformation—is a mechanistically enabling event that can be observed on the timescale of current molecular simulations (Leontiadou et al., 2018). In this work, we conducted three simulations of WT PI3Kα in systems with and without the nSH2 domain (PI3Kα ΔnSH2) (4.5 μs total sampling per system). Absence of nSH2 is used to model the effect of nSH2 dislodgment from its interaction with the p110α catalytic subunit of PI3Kα, an approach also taken in a recent study (Zhang et al., 2019). To determine the influence of the absence of the nSH2 domain on interactions that may lead to the catalytically competent state, we examined the distribution of distances between the di-C4 PIP2 3′ inositol oxygen (O3) and the ATP γ-phosphate (PG) substrate in six combinations (three replicates with the nSH2 versus three replicates without). The trajectories with the nSH2 domain (WT-PI3Kα) contained substrate-bound conformations in which the diC4-PIP2 O3′-ATP PG distance was shifted toward closer proximity. The combined results for all simulation replicates are shown in Figure 1. However, these proximal interactions were not coupled to interactions of ATP or PIP2 with residues in the active site that have been shown to be important for catalysis. Low-frequency interactions of ATP and PIP2 with the active-site H917, H936, and H940 residues were only observed in systems not containing the nSH2 domain (Table 1). In one such snapshot taken from a simulation without the nSH2 domain, the distance between the ATP PG and diC4-PIP2 O3′ was 4.9 Å, with the nitrogen of the putative catalytic His 936 only 4.1 Å away from the diC4-PIP2 O3′ (Figure 2). In a second snapshot obtained from the same simulation, the distance between the ATP PG and diC4-PIP2 O3′ was only 3.6 Å. In this case, His940 in the activation loop is positioned favorably to be the putative catalytic base, with its nitrogen located only 3.4 Å away from the diC4-PIP2 O3′ (Figure 3). These observations suggest that the absence of the nSH2 domain may be a prerequisite to the existence of catalytically competent states, despite reducing the frequency of ATP PG-diC4 PIP2 interactions that might appear catalytically significant. That is, binding of substrates takes place in the nSH2-inhibited configuration (Figure 1 and Table 1), but release of the nSH2 is needed for gaining significant access to the catalytically competent form (Figures 2 and 3). This implies that loop conformations compatible with catalysis arise through dynamic sampling resulting from nSH2 domain removal. Our results illustrate that that the population enhancement of catalytically competent conformations arising from nSH2 removal is sufficient to permit their observation on the limited timescales of our molecular dynamics (MD) simulations.
Figure 1. Distance Distribution of ATP PG to di-C4 PIP2 O3′.

Histograms of distributions are shown for the WT system containing the nSH2 domain (left) and the system in which the nSH2 is absent (right). The combined data of all replicates for each system is presented.
Table 1.
Interactions of ATP PG-diC4 PIP2 Complex with Putative Catalytic Residues
| Interaction (Distance ≤ 5.0 Å) | Frequency in Simulation with nSH2 (%) | Interaction (Distance ≤ 5.0 Å) | Frequency in Simulation without nSH2 (%) |
|---|---|---|---|
| PG of ATP to | 9.59 | PG of ATP to | 1.00 |
| O3 of di-C4 PIP2 | O3 of di-C4 PIP2 | ||
| PG to O3 and | 6.43 | PG to O3 and | 0.10 |
| HZ1 of K776 to | NE2 of H940 to | ||
| O3 of di-C4 PIP2 | O3 of di-C4 PIP2 | ||
| PG to O3 and | 6.40 | PG to O3 and | 0.017 |
| HZ2 of K776 to | NE2 of H936 to | ||
| O3 of di-C4 PIP2 | O3 of di-C4 PIP2 | ||
| PG to O3 and | 6.30 | PG to O3 and | 0.006 |
| HZ3 of K776 to | NE2 of H917 to | ||
| O3 of di-C4 PIP2 | O1G of ATP | ||
The frequencies of interactions are tabulated for the systems in which the nSH2 is present (left) versus absent (right).
Figure 2. Putative Active Conformation of PI3Kα Obtained from Molecular Simulation.

A conformation from the simulation in which the nSH2 domain was absent is shown. Distances between the ATP PG, di-C4 PIP2, and other important active-site residues are labeled. The Nϵ H936 is only 4.1 Å away from the diC4 PIP2 O3. Mg2+ ions are represented by gray spheres.
Figure 3. Putative Active Conformation of PI3Kα Obtained from Molecular Simulation.

A conformation from the simulation in which the nSH2 domain was absent is shown. Distances between the ATP PG, di-C4 PIP2, and other important active-site residues are labeled. The Nϵ of H940 is only 3.4 Å away from the diC4 PIP2 O3. Mg2+ ions are represented by gray spheres.
Using the same six simulations (three for WT PI3Kα and three for PI3Kα ΔnSH2), the influence of the presence of the nSH2 domain on activation-loop dynamics was investigated. Analysis of the data shows that the absence of the nSH2 domain causes the conformational variability of the activation loop to diminish, as evidenced by reduced backbone root-mean-square fluctuations (RMSFs) (Figure 4). This result has an important implication: it suggests that the conformational ensemble of the activation loop is converging toward a population of states compatible with catalysis, through a process of conformational selection resulting from the absence of the nSH2 domain. To fully explore the differences in the conformational ensemble of the activation-loop residues in the absence and presence of the nSH2 domain, we performed clustering using the Gromos algorithm. The center of each cluster is used as a representative conformation. The presence of the nSH2 domain directly influences the conformational states sampled by the activation-loop residues in the active site (Figure 5), suggesting that disruption of the interactions of the nSH2 domain with the catalytic subunit allosterically activate PI3Kα not by shifting the enzyme to a new ground state, but instead by changing the dynamics of the activation loop.
Figure 4. Comparison of Activation-Loop Root-Mean-Square Fluctuation Profiles.

The backbone root-mean-square fluctuation (RMSF) was computed for the WT PI3Kα system, and the system without nSH2 (PI3Kα ΔnSH2). Data from all replicate simulations are shown.
Figure 5. Clustering Analysis of Activation-Loop Conformations in PI3Kα.

Molecular simulation data from the WT PI3Kα system (A, three replicates) and the system in which the nSH2 domain is absent (B, three replicates) were clustered using the Gromos algorithm. The conformations of the activation loop (residues 935–958) were clustered with a cutoff of 2.5 Å.
nSH2 Domain Allosterically Perturbs p110α Subunit Dynamics
The presence or absence of the nSH2 domain significantly influences the network of dynamic residue interactions that mediate correlated motions in the catalytic domain of the protein. For example, the presence of the nSH2 domain mediates dynamic protein-substrate interactions in configurations that are not catalytically competent, while in the absence of nSH2, although the protein-substrate interactions are less frequent, they do adopt catalytically competent configurations. To determine which residues appear to be important in mediating these interactions in the absence and in the presence of the nSH2 domain, we used a network analysis approach. This approach treats the protein as a graph, with each residue of the protein represented by a node and contacts between nodes represented by lines. Lines are only drawn between nodes whose heavy atoms satisfy a certain distance cutoff during the trajectory. In this work, we used Cα atoms as nodes in the network representation, and considered two nodes as interacting when the heavy atoms of their corresponding residues were within a cutoff distance of 4.5 Å for at least 75% of the trajectory. Interactions between nodes that are adjacent in sequence were ignored. The distance between two nodes j and k is given by , the negative log of the element of the covariance matrix that describes the cross-correlation between the fluctuations of the Cα atoms representing j and k, as obtained from MD simulations. The shortest path that can be drawn between two nodes is a path where the sum of the distances between the nodes is a minimum, and is determined by the Floyd-Warshall algorithm (Floyd, 1962). We use a particular network metric, known as “node betweenness” (Tse and Verkhivker, 2015) (for a detailed definition, see STAR Methods), which quantifies the involvement of specific residues in mediating correlated motions in the protein, with a higher value signifying that a residue is present more frequently on the shortest paths connecting residue pairs. When comparing the WT PI3Kα with PI3Kα ΔnSH2, a shift in the involvement of residues of the C2 domain (between 325 and 493) and of the ABD (between 24 and 109) was observed, together with a change in the involvement of residues in the helical and kinase domains (525–804) of the ΔnSH2 simulation. The shift involves C2 domain residues that are in close proximity to the helical domain (424–493 region), the helical domain itself (525–684 region), and the N-terminal residues of the kinase domain (744–804), as well as ABD residues that interface with both the kinase and iSH2 domains (64–104) in the PI3Kα ΔnSH2 (Figure 6). These changes in the involvement of residues in connecting multiple domains of the protein support the concept that allosteric activation of PI3Kα (Echeverria et al., 2015) is affected by changes in protein dynamics that result in allosteric communication among distant regions of the protein in a way that can increase the population of kinetically competent states.
Figure 6. Normalized Node Betweenness Comparison of PI3Kα Systems.

(A) Structural representation of domains (in red) exhibiting altered node betweenness. The location of the cell membrane is indicated by the blue dotted line.
(B) The node betweenness profile is shown for a replicate of WT PI3Kα (top) and of the PI3Kα ΔnSH2. The vertical red line indicates the end of the p110α domain.
Cognate pY Peptide Binding Affects PI3Kα Dynamics, and in Particular that of the Activation Loop
Simulations containing the cognate pY peptide bound to the nSH2, compared with simulations without a peptide bound, show decreases in the backbone fluctuations of residues in the activation loop (Figure 7). The change in dynamics is also reflected in the average solvent-accessible surface area (SASA) of activation-loop residues when the cognate pY peptide is bound versus when it is absent. In multiple replicate simulations, binding of the cognate pY peptide occludes activation-loop residues 940–954 compared with their positions in the unbound WT PI3Kα (Figure S1).
Figure 7. RMSF of the PI3Kα Activation Loop.

The backbone RMSF is shown for activation-loop residues in systems where the cognate pY peptide was included (black) versus where it was absent (red). Data from all replicate simulations are shown.
Clustering analysis was performed using the Gromos algorithm (2.5 Å cutoff) to precisely characterize the conformations of the activation loop when the cognate pY peptide is bound. In two independent replicate simulations, the conformation of the activation loop was found to differ from the WT simulation replicates, but strikingly adopted a conformation similar to that of the simulations in which the nSH2 domain was removed (Figure 8). It may be argued that the similarity of behavior of the dynamics of PI3Kα without the nSH2 and that with the bound cognate pY peptide is an artifact caused by both simulations exploring similar trajectories. We investigated whether the two trajectories were similar by combining the frames of both trajectories into one set (using p110 backbone atom coordinates), generating the covariance matrix of the displacements of the x, y, and z coordinates of the backbone atoms with respect to a reference structure, determining the principal components of the matrix, and projecting the displacements of each frame onto the first two principal components (PC1 and PC2). This procedure has been documented in previous work to address questions about the sampling of conformational space from an ensemble of observed structures (Bakan and Bahar, 2011). The result of our analysis is a graph in which each frame is represented by a point in the PC1/PC2 plane (Figure S2). The figure shows that the frames from the PI3Kα ΔnSH2 trajectory (153,000 blue dots) and those from the PI3Kα with bound cognate pY peptide (153,000 red dots) occupy different regions of the PC1/PC2 space, indicating that the simulations did not sample similar trajectories.
Figure 8. Clustering Analysis of Activation-Loop Conformations in PI3Kα.

Molecular simulation data from the PI3Kα system without the nSH2 domain (three replicates, top) and system in which phosphotyrosine peptide is bound (two replicates, bottom) were clustered using the Gromos algorithm.
The correlated motions between the nSH2 domain and the residues of the activation loop indicate that binding of the cognate pY peptide to the nSH2 domain is directly coupled to changes in activation-loop motions: it increases the degree of correlation between these regions (Figure 9). When activation-loop distance restraints and binding of the cognate pY peptide are used simultaneously in the simulation, the nSH2 domain dynamics are seen to be directly perturbed: the correlated motions of multiple regions in this domain change relative to their motions during cognate pY peptide binding in the absence of active-site restraints (Figure S3). These results indicate that binding of physiological effectors to PI3Kα causes activation of the enzyme through an allosteric effect on the dynamics of the residues that surround the catalytic site: they sample a larger fraction of catalytically competent configurations.
Figure 9. Dynamic Cross-Correlation Matrices of PI3Kα Domains.

Correlated motions were examined between the nSH2 domain (residues 322–430 of p85, x axis) and the catalytic residues of the kinase domain (residues 909–958 in p110, y axis) for a system containing the cognate pY peptide (top) and a system without the peptide (bottom). See also Figure S3.
Binding of a cognate pY peptide to PI3Kα also produces noticeable effects on more global aspects of the protein dynamics. We performed principal component analysis on a system containing a cognate pY peptide, visualizing the first two modes using “porcupine plots” in which arrows drawn from the Cα atom of each residue represent the magnitude and direction of the eigenvector component corresponding to that Cα. For example, when comparing the first mode for the first replicates of the WT PI3Kα system without the cognate pY peptide and the system containing the peptide, we observe that the bound cognate pY peptide system favors a “clamping” motion between the kinase and nSH2 that is not evident in the absence of the cognate pY peptide (Figure S4A). The second principal component also suggests changes in the motion of the helical domain in the presence of the cognate pY peptide that are not evident in the system without the peptide (Figure S4B). When active-site distance restraints are added to the system with the cognate pY peptide, significant differences in the helical domain are observed in the first modes, especially in the nSH2 and kinase domains (Figure S5A), again suggesting that the dynamics of these domains are directly linked. The second mode is largely similar between the two systems (Figure S5B), with small changes seen in the motion of the kinase domain in the system containing the cognate pY peptide. A summary of the percentage of the variance explained by each of the first five principal components for the WT, WT + pY peptide, and WT + pY peptide + restraint simulations is shown in Table S2. Our results illustrate that perturbations due to cognate pY peptide binding are manifested in the global dynamics and coupled motions of domains in spatially distinct regions of the protein, in addition to altering the activation-loop ensemble.
Summary and Conclusions
It has been shown that even though the iSH2 domain is sufficient for stabilizing the p110α subunit (Yu et al., 1998a), the nSH2 domain is necessary for regulation of the kinase activity: in the absence of the nSH2, the enzyme shows constitutive activity similar to that observed in the presence of the cognate pY peptide. We have examined the effects of nSH2 domain removal and cognate pY peptide binding on the dynamics of PI3Kα, with a particular interest in determining how these changes influence the active site of the protein, especially the conformational ensemble of the activation loop. Prior experimental work showed that binding of a cognate pY peptide results in a 2- to 4-fold activation of PI3K, while removal of the nSH2 domain can result in more than 10-fold activation (Carpenter et al., 1993; Carson et al., 2008; Hon et al., 2012). An alignment of p110α/p85α niSH2 structures to p110α/p85α iSH2 structures provides some mechanistic insight: in structures without the nSH2 domain, a clockwise rotation of the p110α subunit about the p85 iSH2 coiled-coil axis is observed, with significant displacements of the C-terminal lobe of the kinase domain and the helical domain (Hon et al., 2012). This suggests that the loss of nSH2 allosterically modulates the conformation of the p110α catalytic subunit in a way that may facilitate catalysis. Indeed, we find that removal of the nSH2 domain influences the conformational ensemble of the activation loop and increases the frequency of interactions of the ATP and lipid substrates. In the case of cognate pY peptide binding, hydrogen-deuterium exchange (HDX) experiments were revealing: peptide binding, in addition to disrupting interactions of the nSH2 domain with the C2 and helical domains, also influences the linker residues between the ABD and RBD domains and residues at the C2-iSH2 interface (Burke et al., 2012). These large changes may reflect a change in the conformational landscape in a way that allows new conformations to be accessed. At the same time, the nSH2 domain itself undergoes very little structural alteration when bound to a cognate pY peptide: there is less than a 0.3-Å backbone RMS difference between the ligand-bound and apo conformations (Nolte et al., 1996). Together, these observations strongly suggest that cognate pY peptide binding to the p85 domain influences the activation of the enzyme primarily through changes in the dynamics of domains distant from the peptide-binding site. The simulations suggest that cognate pY peptide binding causes significant changes to the dynamic motions of the nSH2, kinase and helical domains while simultaneously altering activation-loop fluctuations and solvent accessibility, indicating that the active site must shift toward a more solvent-occluded (hydrophobic) environment to facilitate lipid phosphoryl transfer.
In earlier work, we proposed that allosteric changes resulting from p85α nSH2 displacement from the p110α subunit shifts the conformational ensemble of the kinase domain in a way that increases the population of the active conformation (Echeverria et al., 2015). Here, we report simulations that show evidence of such a change in model systems that mimic nSH2 displacement, in agreement with the mechanism proposed by our group based on kinetics measurements (Maheshwari et al., 2017). The conformations of the activation loop that are accessed by the increased dynamics involve residues that are important in catalysis, including active-site residue H940. In two catalytically competent structures (Figures 2 and 3), there is disruption of a previously observed iSH2-kinase domain interface, involving a hydrophobic stacking interaction between L598 (p85) and F945 (p110) and a hydrogen-bonding interaction between Q591 (p85) and K948 (p110) (Miller et al., 2014). We had proposed that the loss of this interface results in the release of an inhibitory interaction, which would further weaken the inhibition of kinase activity by the p85 domain (Miller et al., 2014). According to earlier literature, deletions or truncations in this region of p85 can result in enzyme activation by disrupting the iSH2-C2 interface (Wu et al., 2009). Furthermore, we found that the activation loops in these two structures (accounted for in the conformational ensemble of the first replicate simulation without the nSH2 domain [Figure 5B]) share striking similarity to activation-loop cluster conformations obtained from the simulations containing bound cognate pY peptide, with residues 935–946 adopting a similar orientation. We observe that the removal of the nSH2 domain or binding of cognate pY peptide (without restraints applied at the active site) result in similar conformational ensembles of the kinase domain activation loop (Figure 8), implying that both these perturbations, which are known to result in enzyme activation, may be allosterically influencing the dynamics of the active site toward a population of states that are competent for catalysis. Our proposed mechanism differs from that of Zhang et al. (2019), who recently proposed a mechanism of PI3Kα activation that consists of a significant conformational change in its ground-state structure involving the C2, kinase, and helical domains. Upon dissociation of the nSH2 domain, they observed movement of the kinase domain relative to the C2 domain and suggest that the surface of the kinase domain becomes accessible to membrane interaction due to loss of interactions with the iSH2. It is worth pointing out that no such structure was observed experimentally, even when the nSH2 domain was absent. Furthermore, they posit that nSH2 domain dissociation causes a “structural rearrangement” in the residues of the activation loop that brings the PIP2 in proximity for catalysis. Here, we provide evidence for a change in the conformational state of the activation-loop residues and propose that their effect is allosteric, is population-based, and influences the chemistry of catalysis rather than the movement of the PIP2 lipid. In the simulations containing cognate pY peptide, only K776 in the active-site P loop was found to interact with the O3′ oxygen of the substrate, di-C4 PIP2, at low frequency, while none of the other activation or catalytic loop residues were observed in close proximity to the lipid substrate.
In this study, we have demonstrated nSH2 domain removal or cognate pY peptide binding allosterically alter the conformational ensemble of the activation loop, shifting the relative populations toward an increased proportion of catalytically competent states. Our findings show that contributions from multiple domains, and dynamic fluctuations of residues in spatially distant regions of the protein, effect this population shift. The identification of such contributions provides new insight into the regulation of large, multi-subunit protein complexes by motions that are not manifested as a change in the overall conformation of the ground state of the protein. In addition, the identification of new, albeit transient, structures provides novel targets for the design and discovery of new inhibitors.
STAR★METHODS
LEAD CONTACT AND MATERIALS AVAILABILITY
Further information and requests for resources and reagents should be directed to and will be fulfilled by the Lead Contact, L Mario Amzel (mamzel@jhmi.edu).
Materials Availability Statement
This study did not generate new unique reagents.
METHOD DETAILS
Structures
Careful consideration was given to the choice of starting structures for our simulations. For the simulations of PI3Kα p110-niSH2 (p110α residues 4 – 1062 and p85α residues 326 – 579, hereafter referred to as WT), PDB ID 3HHM (Mandelker et al., 2009) was used for the starting coordinates. The H1047R mutation in that structure was reverted to wild-type, and missing loop residues were rebuilt, using the loop modeling tool of Modeller (Šali and Blundell, 1993). To place the phosphatidylinositol 4,5-bis-phosphate diC4 (di-C4 PIP2), the resulting structure was aligned to PDB ID 4OVV (Miller et al., 2014). The ATP molecule and the Mg2+ ions in the active site were positioned by subsequent alignment to PDB ID 1e8x (Walker et al., 2000) (a structure of PI3Kγ). The coordinating Mg2+ ions were placed at the positions of the Lu3+ ions in PDB ID 1E8X. Similarly, for the simulations of PI3Kα p110-iSH2 (p110α residues 4 – 1062 and p85α residues 448 – 598, hereafter referred to as ΔnSH2), PDB ID 4A55 (Hon et al., 2012) was used for the starting coordinates, the di-C4 PIP2 was positioned by alignment to PDB ID 4OVV, and the ATP and Mg2+ ions were placed by alignment of the resulting structure to PDB ID 1E8X. The sequence of the p110 subunit of PDB ID 4A55, which derives from a murine model, was reverted to the human sequence during the model building process. Binding of a cognate pY peptide to PI3Kα was modelled using an available crystal structure of an activating peptide bound to the nSH2 domain of the p85α subunit (PDB ID: 2IUH) (Nolte et al., 1996). The peptide (TNE-pY-MDMK) is derived from CD117, a type of RTK (Nolte et al., 1996). The structure (PDB ID: 2IUH) was aligned to the p85α nSH2 domain of wild-type PI3Kα (PDB ID: 4OVV) and initial clashes were resolved through slight perturbations of the nSH2 position with respect to the helical domain; subsequent energy minimization and equilibration were performed for production runs.
MD Simulations
The CHARMM36 force field (July 2017 revision) was used for all systems (Huang et al., 2017), and simulations were performed with Gromacs 2016.4 (Abraham et al., 2015). Parameters for non-protein components were obtained as follows. The ATP ligand parameters are directly available in CHARMM36. For di-C4 PIP2, the coordinates were first converted into ‘mol2’ format using OpenBabel (O’Boyle et al., 2011) and hydrogen atoms were added using the PRODRG server (Schüttelkopf and van Aalten, 2004), followed by parameterization using the CGenFF forcefield (Vanommeslaeghe et al., 2009). The TIP3P water model was used for the solvent. Simulations of wild-type PI3Kα including the nSH2 domain had ~ 232,400 atoms and were performed in a rhombic dodecahedron box with a distance of 12 Å between the solute and the box. Simulations of wild-type PI3Kα with the nSH2 domain removed had ~ 242,800 atoms and the same aforementioned box specifications. The systems containing the cognate pY peptide had ~233,230 atoms and the same box specifications. All simulations were performed with a salt concentration of 0.15 M NaCl. In simulations performed with distance restraints, restraints were applied using a harmonic potential between NE2 of H936 and O3’ of diC4-PIP2, NE2 of H917 and O3G of ATP, NE2 of H917 and O41 of diC4-PIP2, and PG of ATP and O3’ of diC4-PIP2. All restraints were set to a distance of 0.35 nm and a force constant of 836.8 kJ/(mol $ nm2). In all simulations, the temperature was maintained at 310 K using V-rescale, a temperature coupling algorithm using velocity rescaling with a stochastic term, with a time constant of 0.1 ps. The pressure was isotropically maintained at 1 atm using the Parrinello-Rahman barostat with a time constant of 2 ps and a compressibility of 4.5 × 10−5 bar−1. Long-range electrostatic interactions were modeled using Smooth Particle Mesh Ewald (SPME) electrostatics, and non-bonded interactions were modeled with the Verlet cutoff scheme. The cutoff distance for short-range electrostatic and van der Waals interactions was set to 1.4 nm. A timestep of 2 fs was used, with atomic coordinates saved every 10 ps, and bonds containing hydrogen atoms were constrained using the LINCS algorithm (Hess et al., 1997). Before all simulations, structures were energy minimized with the steepest descent algorithm, with an initial step size of 0.01 nm and a tolerance of 10 kJ/mol $ nm. Positional restraints were enforced during NVT and NPT equilibration for 2 ns each, followed by unbiased MD simulations. A summary of the simulations described here is available in Supplemental Information, Table 1.
Histogram Analysis
A time-series of distances between ATP PG and the O3’ of di-C4 PIP2 was generated using Gromacs 2016.4 tools (distance module). Distances were divided into forty-eight bins, with the frequency in each bin normalized such that the sum of all bin frequencies added up to one. Plotting was performed using Matplotlib (Hunter, 2007), using the matplotlib.pyplot.hist function (where matplotlib.pyplot was imported as plt) in an interactive Python notebook. According to the information provided in this function, the frequency is normalized by dividing the bin count by the number of observations times the bin width, instead of dividing by the total number of observations. The following command was used to generate the histograms:
n, bins, patches = plt.hist(distances, 48, density=True, facecolor=‘blue’, edgecolor=‘black’, alpha=0.75)
where distances is an array of aggregate timeseries data.
Dynamic Cross Correlation Maps
Gromacs trajectories were first converted into the binary ‘dcd’ file format using the mdconvert script in MDTraj (McGibbon et al., 2015). The Cα atoms of the protein were selected as input to the EDA module of ProDy (Bakan et al., 2011) and used to generate a matrix of cross-correlation values. The syntax used for generating the cross-correlation matrix was as follows:
prody eda md_noPBC.dcd –pdb origin.pdb -s “protein and chain A and name CA” -r
where md_noPBC.dcd is the name of the file converted into ‘dcd’ format and origin.pdb is the reference PDB file for the trajectory. The resulting cross-correlation file was plotted with the R Statistical Computing package (R Core Team, 2018) using the lattice library (Sarkar, 2008). The following commands were performed in the R terminal to obtain the Dynamic Cross Correlation Maps:
> x <- scan(“md_noPBC_p110_pca_cross-correlations.txt”)
> x <- matrix(x,sqrt(length(x)))
> library(lattice)
> library(bio3d)
> levelplot(x[1:ncol(x),1:ncol(x)], region = TRUE, labels=FALSE, col.regions = bwr.colors(2000), at=c(−1, −0.9, −0.8, −0.7, −0.6, −0.5, −0.4, −0.3, −0.2, −0.1, 0.1, 0.2, 0.3, 0.4, 0.5, 0.6, 0.7, 0.8, 0.9, 1),
xlab=“Residue No.”, ylab=“Residue No.”,
main=“Dynamic Cross Correlation Matrix”)
Network Analysis
A ‘psf’ file in Visual Molecular Dynamics (VMD, (Humphrey et al., 1996)) X-PLOR format was generated from a PDB file of the PI3Kα system. First, the PDB file was processed so that HIS residues were assigned to one of the HSD (neutral histidine, proton on ND1), HSE (neutral histidine, proton on ND2), or HSP (protonated histidine) forms according to their protonation state. This file was split into the two chains, and all hydrogen atoms were removed. Next, in VMD, a series of Tcl commands (Data S1) were issued inside the Tcl console. Using the generated psf file and the DCD file generated from mdconvert (see Dynamic Cross Correlation Maps section above), a file specifying the configuration, network.config, was created, as described in the VMD Dynamical Network Analysis tutorial (Eargle et al., 2012). (For the parameters used see Supplemental Information, Data S2) In the following step, pairwise cross-correlations were generated in VMD by running networkSetup network.config in the VMD console. The resulting contact.corr file was processed using R and the Bio3D module (Grant et al., 2006) after removing the header line (Data S3).
Node Betweenness
“Node betweenness” (Tse and Verkhivker, 2015) is a specific network metric that quantifies the involvement of residues in mediating correlated motions within the protein. This metric is defined as , a normalized sum over the fraction of shortest paths between all pairs of residues that pass through a node. Here, gjk is the number of shortest paths between nodes j and k, gjk(i) is the number of shortest paths between nodes j and k that pass through node ni, and N is the total number of nodes. The path length between any two nodes j and k is given by , the negative log of the cross-correlation between the nodes.
Clustering Analysis
Clustering was performed with the Gromacs 2016.4 tools (cluster module), which creates clusters by finding neighboring structures within an RMSD cutoff between trajectory frames. Specifically, we used the Gromos algorithm with a cutoff of 2.5 Å. This algorithm counts the number of neighboring structures for a given trajectory frame based on the RMSD cutoff, takes the structure with the largest number of neighbors with all of its neighbors as a cluster, and eliminates it from the cluster pool, repeating the procedure for all remaining clusters. Frames were selected every 80 ps. Structures were visualized in PyMOL (Delano, 2002).
RMSF and SASA Calculations
RMSF and SASA profiles were computed using Gromacs 2016.4 tools (rmsf and sasa modules, respectively). The specific command line arguments used were as follows:
RMSF: gmx rmsf -f md_noskip.xtc -s pi3k_WT_equil_ref.gro -n index.ndx -res -fit -dir
SASA: gmx sasa -f md_noskip.xtc -s pi3k_WT_equil_ref.gro -n index.ndx -o area.xvg -or resarea.xvg -surface -output < sele.txt
where md_noskip.xtc refers to the concatenated MD trajectory with all frames present, and pi3k_WT_equil_ref.gro is a Gromacs coordinate file of the reference protein structure.
For the SASA calculations, the default probe size of 1.4 Å was used, as well as the default number of dots per sphere (24), and as such these parameters were not specified in the command line. The file sele.txt contained selections for the -surface and -output flags, which were the protein itself and the SASA calculations for several user-specific index entries. The results from the output file area.xvg were then used to plot the average solvent-accessible surface area on a per-residue basis.
Porcupine Plots
Files for the eigenvalues and eigenvectors (eigenval.xvg and eigenvec.trr) based on the computed covariance matrix were generated using the covar module of Gromacs 2016.4. These were provided as inputs to the anaeig module of Gromacs, and used to generate *.pdb files for the first and second eigenvectors. The structures were visualized in an interactive Python notebook using the mdtraj and nglview (Nguyen et al., 2018) modules.
Projection of Individual Frames onto Principal Components (PCs)
The PI3Kα ΔnSH2 trajectory (153,000 frames; one frame every 10 ps) and the trajectory of the PI3Kα with cognate pY peptide bound (153,000 frames; one frame every 10 ps) were concatenated into a single trajectory file that included only the x, y, and z coordinates of the common backbone atoms (N, Cα, C) of the residues of the p110 subunit. Backbone trajectories of the p110 subunit for the different systems were obtained using the trjconv module of Gromacs 2016.4. The two trajectories were concatenated and superimposed onto a common reference frame using the Python mdtraj module. Using the equilibrated structure of the first frame from the PI3Kα ΔnSH2 trajectory as a reference, the displacements of the 306,000 sets of coordinates ( where “s” is the frame number, “i” is the residue number, and “j” is the backbone atom numberidentifier for N, Cα and C) were used to generate a covariance matrix of size 9 N × 9 N, where N is the number of residues in p110α. The matrix was subjected to PCA and the coordinates of the individual frames were projected onto each of the first two principal components (PC1 and PC2) using the equation , i.e., the scalar product of the displacement vector of frame “s” times the eigenvector of the principal component “n”. The projection of frames onto the first and second principal components was performed using the sklearn.decomposition module of the scikit-learn library in Python, using the PCA fit_transform method. The results were visualized in an interactive Python notebook with the kdeplot function of the seaborn library in Python.
QUANTIFICATION AND STATISTICAL ANALYSIS
Histograms were plotted using timeseries data from each simulation frame, yielding a total of n = 459,003 frames that were binned using Matplotlib (Hunter, 2007). RMSF calculations were performed after least-squares fit to a reference structure and are reported as the average for each residue. Similarly, SASA calculations are reported as an average area per residue. Cross-correlation matrices were computed using only Cα atoms of the protein. The covariance matrix used to obtain eigenvectors for principal component analysis was similarly computed using only the Cα atoms of the protein.
DATA AND CODE AVAILABILITY
Timeseries data for the distance between ATP PG and PIP2 O3 atoms in all simulations conducted in this work are provided in Tables S3, S4, S5, S6, S7, S8, S9, S10, and S11 as comma-separated value (*.csv) files. Scripts used to generate files used for network analysis (Figure 6) are provided in Data S1–S3. The MD trajectories and structures used in this study are available from the corresponding author upon request.
Supplementary Material
KEY RESOURCES TABLE
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Software and Algorithms | ||
| Modeller 9v22 | Šali and Blundell, 1993 | https://salilab.org/modeller/ |
| CHARMM36 forcefield (July 2017 revision) | Huang et al., 2017 | http://mackerell.umaryland.edu/charmm_ff.shtml |
| Gromacs 2016.4 | Abraham et al., 2015 | http://manual.gromacs.org/documentation/ |
| OpenBabel 2.4.1 | O’Boyle et al., 2011 | http://openbabel.org/wiki/Main_Page |
| PRODRG server | Schüttelkopf and van Aalten, 2004 | http://davapc1.bioch.dundee.ac.uk/cgi-bin/prodrg |
| CGenFF forcefield | Vanommeslaeghe et al., 2009 | https://cgenff.umaryland.edu/ |
| Matplotlib | Hunter, 2007 | https://matplotlib.org/ |
| MDTraj 1.9.3 | McGibbon et al., 2015 | http://mdtraj.org/1.9.3/ |
| ProDy 1.10.10 | Bakan et al., 2011 | http://prody.csb.pitt.edu/ |
| R 3.6.0 | R Core Team (2018) | https://www.r-project.org/ |
| Lattice library (R 3.6.0 package) | Sarkar, 2008 | https://cran.r-project.org/web/packages/lattice/index.html |
| Visual Molecular Dynamics 1.9.3 (VMD) | Humphrey et al., 1996 | https://www.ks.uiuc.edu/Research/vmd/ |
| Bio3D v. 2.3–4 (R 3.6.0 package) | Grant et al., 2006 | http://thegrantlab.org/bio3d/index.php |
| PyMOL | Delano, 2002 | https://pymol.org/2/ |
| nglview module (IPython) | Nguyen et al., 2018 | https://github.com/arose/nglview |
| Deposited Data | ||
| Crystal structure of p110alpha H1047R mutant in complex with niSH2 of p85alpha and the drug wortmannin | Mandelker et al., 2009 | PDB: 3HHM |
| Crystal structure of p110alpha in complex with iSH2 of p85alpha and the inhibitor PIK-108 | Hon et al., 2012 | PDB: 4A55 |
| Crystal Structure of PI3Kalpha in complex with diC4-PIP2 | Miller et al., 2014 | PDB: 4OVV |
| Crystal structure of PI3Kgamma bound to ATP | Walker et al., 2000 | PDB: 1E8X |
| Crystal structure of the PI3-kinase p85 N-terminal SH2 domain in complex with c-Kit phosphotyrosyl peptide | Nolte et al., 1996 | PDB: 2IUH |
| Timeseries data for the distance between the ATP PG and di-C4 PIP2 O3’ for PI3Kα WT simulation, first replicate | This paper | Table S3 |
| Timeseries data for the distance between the ATP PG and di-C4 PIP2 O3’ for PI3Kα WT simulation, second replicate | This paper | Table S4 |
| Timeseries data for the distance between the ATP PG and di-C4 PIP2 O3’ for PI3Kα WT simulation, third replicate | This paper | Table S5 |
| Timeseries data for the distance between the ATP PG and di-C4 PIP2 O3’ for PI3Kα simulation without nSH2, first replicate | This paper | Table S6 |
| Timeseries data for the distance between the ATP PG and di-C4 PIP2 O3’ for PI3Kα simulation without nSH2, second replicate | This paper | Table S7 |
| Timeseries data for the distance between the ATP PG and di-C4 PIP2 O3’ for PI3Kα simulation without nSH2, third replicate | This paper | Table S8 |
| Timeseries data for the distance between the ATP PG and di-C4 PIP2 O3’ for PI3Kαsimulation with pY peptide and no active site restraints, first replicate | This paper | Table S9 |
| Timeseries data for the distance between the ATP PG and di-C4 PIP2 O3’ for PI3Kαsimulation with pY peptide and no active site restraints, second replicate | This paper | Table S10 |
| Timeseries data for the distance between the ATP PG and di-C4 PIP2 O3’ for PI3Kαsimulation with pY peptide and active site restraints | This paper | Table S11 |
Highlights.
Our MD simulations suggest catalytically competent conformations of PI3Kα
The nSH2 domain and phosphotyrosine ligands allosterically modulate PI3Kα activity
Dynamic allostery can increase the population of catalytically competent states
Our findings have implications for the development of new PI3K therapeutics
ACKNOWLEDGMENTS
We thank Dr. Yunlong Liu for helpful discussions. We acknowledge the use of the computational resources and scientific computing services at the Maryland Advanced Research Computing Center. This research was funded in part by the US Department of Defense, DOD CDMRP BC151831 (S.B.G.).
Footnotes
SUPPLEMENTAL INFORMATION
Supplemental Information can be found online at https://doi.org/10.1016/j.str.2020.01.010.
DECLARATION OF INTERESTS
The authors declare no competing interests.
REFERENCES
- Abraham MJ, Murtola T, Schulz R, Páll S, Smith JC, Hess B, and Lindahl E. (2015). GROMACS: high performance molecular simulations through multi-level parallelism from laptops to supercomputers. SoftwareX 1–2, 19–25. [Google Scholar]
- Bakan A, and Bahar I. (2011). Computational generation inhibitor-bound conformers of p38 MAP kinase and comparison with experiments. Pac. Symp. Biocomput 181–192, 10.1142/9789814335058_0020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bakan A, Meireles LM, and Bahar I. (2011). ProDy: protein dynamics inferred from theory and experiments. Bioinformatics 27, 1575–1577. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Burke JE, Perisic O, Masson GR, Vadas O, and Williams RL (2012). Oncogenic mutations mimic and enhance dynamic events in the natural activation of phosphoinositide 3-kinase p110 (PIK3CA). Proc. Natl. Acad. Sci. U S A 109, 15259–15264. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Campitelli P, Guo J, Zhou H-X, and Ozkan SB (2018). Hinge-shift mechanism modulates allosteric regulations in human Pin1. J. Phys. Chem. B 122, 5623–5629. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Capdevila DA, Braymer JJ, Edmonds KA, Wu H, and Giedroc DP (2017). Entropy redistribution controls allostery in a metalloregulatory protein. Proc. Natl. Acad. Sci. U S A 114, 4424–4429. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carpenter CL, Auger KR, Chanudhuri M, Yoakim M, Schaffhausen B, Shoelson S, and Cantley LC (1993). Phosphoinositide 3-kinase is activated by phosphopeptides that bind to the SH2 domains of the 85-kDa subunit. J. Biol. Chem 268, 9478–9483. [PubMed] [Google Scholar]
- Carson JD, Van Aller G, Lehr R, Sinnamon RH, Kirkpatrick RB, Auger KR, Dhanak D, Copeland RA, Gontarek RR, Tummino PJ, et al. (2008). Effects of oncogenic p110α subunit mutations on the lipid kinase activity of phosphoinositide 3-kinase. Biochem. J 409, 519–524. [DOI] [PubMed] [Google Scholar]
- Cooper A, and Dryden DTF (1984). Allostery without conformational change: a plausible model. Eur. Biophys. J 11, 103–109. [DOI] [PubMed] [Google Scholar]
- Delano W. (2002). The PyMOL Molecular Graphics System. Version 2.4.0 (Delano Scientific, LLC; ). [Google Scholar]
- Dhand R, Hara K, Hiles I, Bax B, Gout I, Panayotou G, Fry MJ, Yonezawa K, Kasuga M, and Waterfield MD (1994). PI 3-kinase: structural and functional analysis of intersubunit interactions. EMBO J. 13, 511–521. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eargle J, Li L, and Luthey-Shulten Z. (2012). Dynamical network analysis. http://faculty.scs.illinois.edu/schulten/tutorials/network/network_tutorial.pdf.
- Echeverria I, Liu Y, Gabelli SB, and Amzel LM (2015). Oncogenic mutations weaken the interactions that stabilize the p110α-p85α heterodimer in phosphatidylinositol 3-kinase a. FEBS J. 282, 3528–3542. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Engelman JA, Luo J, and Cantley LC (2006). The evolution of phosphatidylinositol 3-kinases as regulators of growth and metabolism. Nat. Rev. Genet 7, 606–619. [DOI] [PubMed] [Google Scholar]
- Floyd RW (1962). Algorithm 97: shortest path. Commun. ACM 5, 345. [Google Scholar]
- Fuentes EJ, Der CJ, and Lee AL (2004). Ligand-dependent dynamics and intramolecular signaling in a PDZ domain. J. Mol. Biol 335, 1105–1115. [DOI] [PubMed] [Google Scholar]
- Grant BJ, Rodrigues APC, ElSawy KM, McCammon JA, and Caves LSD (2006). Bio3d: an R package for the comparative analysis of protein structures. Bioinformatics 22, 2695–2696. [DOI] [PubMed] [Google Scholar]
- Hess B, Bekker H, Berendsen HJC, and Fraaije JGEM (1997). LINCS: a linear constraint solver for molecular simulations. J. Comput. Chem 18, 1463–1472. [Google Scholar]
- Hon W-C, Berndt A, and Williams RL (2012). Regulation of lipid binding underlies the activation mechanism of class IA PI3-kinases. Oncogene 31, 3655–3666. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang C-H, Mandelker D, Schmidt-Kittler O, Samuels Y, Velculescu VE, Kinzler KW, Vogelstein B, Gabelli SB, and Amzel LM (2007). The structure of a human p110alpha/p85alpha complex elucidates the effects of oncogenic PI3Kalpha mutations. Science 318, 1744–1748. [DOI] [PubMed] [Google Scholar]
- Huang C-H, Mandelker D, Gabelli SB, and Amzel LM (2008). Insights into the oncogenic effects of PIK3CA mutations from the structure of p110α/p85α. Cell Cycle 7, 1151–1156. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang J, Rauscher S, Nawrocki G, Ran T, Feig M, de Groot BL, Grubmüller H, and MacKerell AD (2017). CHARMM36m: an improved force field for folded and intrinsically disordered proteins. Nat. Methods 14, 71–73. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Humphrey W, Dalke A, and Schulten K. (1996). VMD: Visual molecular dynamics. J. Mol. Graph 14, 33–38. [DOI] [PubMed] [Google Scholar]
- Hunter JD (2007). Matplotlib: a 2D graphics environment. Comput. Sci. Eng 9, 90–95. [Google Scholar]
- Leontiadou H, Galdadas I, Athanasiou C, and Cournia Z. (2018). Insights into the mechanism of the PIK3CA E545K activating mutation using MD simulations. Sci. Rep 8, 15544. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maheshwari S, Miller MS, O’Meally R, Cole RN, Amzel LM, and Gabelli SB (2017). Kinetic and structural analyses reveal residues in phosphoinositide 3-kinase α that are critical for catalysis and substrate recognition. J. Biol. Chem 292, 13541–13550. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Malay AD, Watanabe M, Heddle JG, and Tame JRH (2011). Crystal structure of unliganded TRAP: implications for dynamic allostery. Biochem. J 434, 427–434. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mandelker D, Gabelli SB, Schmidt-Kittler O, Zhu J, Cheong I, Huang C-H, Kinzler KW, Vogelstein B, and Amzel LM (2009). A frequent kinase domain mutation that changes the interaction between PI3K and the membrane. Proc. Natl. Acad. Sci. U S A 106, 16996–17001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McGibbon RT, Beauchamp KA, Harrigan MP, Klein C, Swails JM, Hernández CX, Schwantes CR, Wang L-P, Lane TJ, and Pande VS (2015). MDTraj: a modern open library for the analysis of molecular dynamics trajectories. Biophys. J 109, 1528–1532. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miled N, Yan Y, Hon W-C, Perisic O, Zvelebil M, Inbar Y, Schneidman-Duhovny D, Wolfson HJ, Backer JM, and Williams RL (2007). Mechanism of two classes of cancer mutations in the phosphoinositide 3-kinase catalytic subunit. Science 317, 239–242. [DOI] [PubMed] [Google Scholar]
- Miller MS, Schmidt-Kittler O, Bolduc DM, Brower ET, Chaves-Moreira D, Allaire M, Kinzler KW, Jennings IG, Thompson PE, Cole PA, et al. (2014). Structural basis of nSH2 regulation and lipid binding in PI3Kα. Oncotarget 5, 5198–5208. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nguyen H, Case DA, and Rose AS (2018). NGLview—interactive molecular graphics for Jupyter notebooks. Bioinformatics 34, 1241–1242. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nolte RT, Eck MJ, Schlessinger J, Shoelson SE, and Harrison SC (1996). Crystal structure of the PI 3-kinase p85 amino-terminal SH2 domain and its phosphopeptide complexes. Nat. Struct. Biol 3, 364. [DOI] [PubMed] [Google Scholar]
- O’Boyle NM, Banck M, James CA, Morley C, Vandermeersch T, and Hutchison GR (2011). Open Babel: an open chemical toolbox. J. Cheminform 3, 33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Philp AJ, Campbell IG, Leet C, Vincan E, Rockman SP, Whitehead RH, Thomas RJS, and Phillips WA (2001). The phosphatidylinositol 3′-kinase p85α gene is an oncogene in human ovarian and colon tumors. Cancer Res. 61, 7426. [PubMed] [Google Scholar]
- Pirola L, Zvelebil MJ, Bulgarelli-Leva G, Van Obberghen E, Waterfield MD, and Wymann MP (2001). Activation loop sequences confer substrate specificity to phosphoinositide 3-kinase α (PI3Kα): functions of lipid kinase-deficient PI3Kα in signaling. J. Biol. Chem 276, 21544–21554. [DOI] [PubMed] [Google Scholar]
- Popovych N, Sun S, Ebright RH, and Kalodimos CG (2006). Dynamically driven protein allostery. Nat. Struct. Mol. Biol 13, 831–838. [DOI] [PMC free article] [PubMed] [Google Scholar]
- R Core Team (2018). R: A Language and Environment for Statistical Computing (R Foundation for Statistical Computing; ). [Google Scholar]
- Šali A, and Blundell TL (1993). Comparative protein modelling by satisfaction of spatial restraints. J. Mol. Biol 234, 779–815. [DOI] [PubMed] [Google Scholar]
- Sarkar D. (2008). Lattice: Multivariate Data Visualization with R (Springer; ). [Google Scholar]
- Schüttelkopf AW, and van Aalten DMF (2004). PRODRG: a tool for high-throughput crystallography of protein-ligand complexes. Acta Crystallogr. D Biol. Crystallogr 60, 1355–1363. [DOI] [PubMed] [Google Scholar]
- Tsai C-J, del Sol A, and Nussinov R. (2008). Allostery: absence of a change in shape does not imply that allostery is not at play. J. Mol. Biol 378, 1–11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tse A, and Verkhivker GM (2015). Molecular dynamics simulations and structural network analysis of c-Abl and c-Src kinase core proteins: capturing allosteric mechanisms and communication pathways from residue centrality. J. Chem. Inf. Model 55, 1645–1662. [DOI] [PubMed] [Google Scholar]
- Vadas O, Burke JE, Zhang X, Berndt A, and Williams RL (2011). Structural basis for activation and inhibition of class I phosphoinositide 3-ki-nases. Sci. Signal 4, re2, 10.1126/scisignal.2002165. [DOI] [PubMed] [Google Scholar]
- Vanommeslaeghe K, Hatcher E, Acharya C, Kundu S, Zhong S, Shim J, Darian E, Guvench O, Lopes P, Vorobyov I, et al. (2009). CHARMM general force field: a force field for drug-like molecules compatible with the CHARMM all-atom additive biological force fields. J. Comput. Chem 31, 671–690. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vogt PK, Kang S, Elsliger M-A, and Gymnopoulos M. (2007). Cancer-specific mutations in phosphatidylinositol 3-kinase. Trends Biochem. Sci 32, 342–349. [DOI] [PubMed] [Google Scholar]
- Walker EH, Perisic O, Ried C, Stephens L, and Williams RL (1999). Structural insights into phosphoinositide 3-kinase catalysis and signalling. Nature 402, 313–320. [DOI] [PubMed] [Google Scholar]
- Walker EH, Pacold ME, Perisic O, Stephens L, Hawkins PT, Wymann MP, and Williams RL (2000). Structural determinants of phosphoinositide 3-kinase inhibition by wortmannin, LY294002, quercetin, myricetin, and staurosporine. Mol. Cell 6, 909–919. [DOI] [PubMed] [Google Scholar]
- Weinkam P, Pons J, and Sali A. (2012). Structure-based model of allostery predicts coupling between distant sites. Proc. Natl. Acad. Sci. U S A 109, 4875–4880. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu H, Shekar SC, Flinn RJ, El-Sibai M, Jaiswal BS, Sen KI, Janakiraman V, Seshagiri S, Gerfen GJ, Girvin ME, et al. (2009). Regulation of Class IA PI 3-kinases: C2 domain-iSH2 domain contacts inhibit p85/p110 and are disrupted in oncogenic p85 mutants. Proc. Natl. Acad. Sci. U S A 106, 20258–20263. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yu J, Zhang Y, McIlroy J, Rordorf-Nikolic T, Orr GA, and Backer JM (1998a). Regulation of the p85/p110 phosphatidylinositol 3′-kinase: stabilization and inhibition of the p110α catalytic subunit by the p85 regulatory subunit. Mol. Cell. Biol 18, 1379. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yu J, Wjasow C, and Backer JM (1998b). Regulation of the p85/p110α phosphatidylinositol 3′-kinase: distinct roles for the N-terminal and C-terminal SH2 domains. J. Biol. Chem 273, 30199–30203. [DOI] [PubMed] [Google Scholar]
- Zhang M, Jang H, and Nussinov R. (2019). The mechanism of PI3Kα activation at the atomic level. Chem. Sci 10, 3671–3680. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
Timeseries data for the distance between ATP PG and PIP2 O3 atoms in all simulations conducted in this work are provided in Tables S3, S4, S5, S6, S7, S8, S9, S10, and S11 as comma-separated value (*.csv) files. Scripts used to generate files used for network analysis (Figure 6) are provided in Data S1–S3. The MD trajectories and structures used in this study are available from the corresponding author upon request.