

Abstract
In recent years, highly sensitive mass spectrometry–based phosphoproteomic analysis is beginning to be applied to identification of protein kinase substrates altered downstream of increased cAMP. Such studies identify a very large number of phosphorylation sites regulated in response to increased cAMP. Therefore, we now are tasked with the challenge of determining how many of these altered phosphorylation sites are relevant to regulation of function in the cell. This minireview describes the use of phosphoproteomic analysis to monitor the effects of cyclic nucleotide phosphodiesterase (PDE) inhibitors on cAMP-dependent phosphorylation events. More specifically, it describes two examples of this approach carried out in the authors’ laboratories using the selective PDE inhibitor approach. After a short discussion of several likely conclusions suggested by these analyses of cAMP function in steroid hormone–producing cells and also in T-cells, it expands into a discussion about some newer and more speculative interpretations of the data. These include the idea that multiple phosphorylation sites and not a single rate-limiting step likely regulate these and, by analogy, many other cAMP-dependent pathways. In addition, the idea that meaningful regulation requires a high stoichiometry of phosphorylation to be important is discussed and suggested to be untrue in many instances. These new interpretations have important implications for drug design, especially for targeting pathway agonists.
SIGNIFICANCE STATEMENT
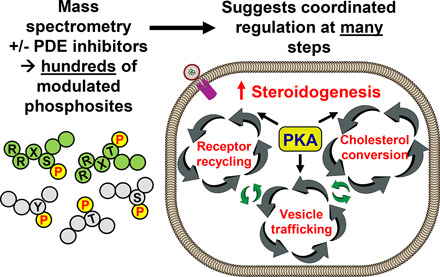
Phosphoproteomic analyses identify thousands of altered phosphorylation sites upon drug treatment, providing many possible regulatory targets but also highlighting questions about which phosphosites are functionally important. These data imply that multistep processes are regulated by phosphorylation at not one but rather many sites. Most previous studies assumed a single step or very few rate-limiting steps were changed by phosphorylation. This concept should be changed. Previous interpretations also assumed substoichiometric phosphorylation was not of regulatory importance. This assumption also should be changed.
Introduction
Why Use Nonbiased Phosphoproteomics to Interrogate Drug Mechanisms?
This minireview stems from a symposium held in 2019 on “Phosphoproteomic Analysis of G protein-coupled Pathways.”1 Since the first descriptions of the cAMP second messenger system by Sutherland and Rall (1958) over 60 years ago, scientists have been trying to elucidate pathways by which this small second messenger works in the cell. Over the years, consensus has arisen that most effects of cAMP are secondary to interactions with cAMP-dependent protein kinases (PKAs) (Beavo and Brunton, 2002), exchange factors activated by cAMP (Wehbe et al., 2020), or cyclic nucleotide–gated channels (Manoury et al., 2020). More recently, cAMP binding sites have been noted on Popeye domain–containing proteins that may also be cAMP effectors (Brand and Schindler, 2017). Of these mechanisms, those downstream of PKA activation are thought to be quantitatively most important at least in the cell types explored in these studies. However, all of these pathways likely have the ability to alter protein phosphorylation directly or indirectly, and therefore, cAMP-dependent phosphorylation events downstream of these effectors would be noted in phosphoproteomic studies. Until recently, nearly all studies on cAMP-dependent phosphorylation have picked a candidate kinase substrate and then investigated its functional roles in more detail. However, recent advances in high-resolution mass spectrometry have allowed identification of thousands of phosphoproteins in most cells and, equally importantly, allowed relative quantitation before and after cellular manipulation. Therefore, this method is now being used to interrogate how different hormones and drugs can alter these phosphorylation events downstream of specific signal-transduction pathways. This minireview focuses on the use of isozyme-selective, cyclic nucleotide phosphodiesterase (PDE) inhibitors as specific tools to elicit changes in cAMP that in turn alter phosphorylation of specific proteins. However, the approach is not, of course, limited to this cellular manipulation. Although the approach does not by itself necessarily identify the pathway(s) between PDE inhibitor–induced increases in cAMP and the phosphorylation response, it does provide new knowledge about a very large number of possible candidate regulatory sites. More generally, the identification and quantification (in a relative way) of these phosphoproteins in response to specific drug manipulation provide an unbiased approach to identifying possible new mechanisms by which the drug and cAMP function in the cell. All studies described were carried out without a phosphatase inhibitor present to avoid masking the response to the PDE inhibitors. Analogous approaches are beginning to be used by a number of other investigators for other pathways coupled to G-proteins or cAMP (Williams et al., 2016; Smith et al., 2016; Liu et al., 2018; Makhoul et al., 2018; Deshpande et al., 2019; Schleicher and Zaccolo, 2020). There are several other minireviews in this series that also address this general topic (von Zastrow, 2020; Salhadar et al., 2020; Schleicher and Zaccolo, 2020).
For this review, two specific examples of the PDE inhibitor–modulated phosphoproteomic approach are presented along with a brief introduction to each topic. First, the regulation of steroid hormone secretion by cAMP as modulated by PDE inhibitors selective for PDE4 and PDE8 is discussed. The model used for these experiments is the MA-10 cell line derived from a mouse Leydig cell tumor. Second, a shorter discussion is provided for similar studies on the regulation by cAMP of a Jurkat cell line used as a model of T-cell function. These studies used a similar approach, except that different combinations of PDE inhibitors appropriate for the T-cells were used. The reader is directed to the minireview in this series by Tasken and colleagues (Tasken et al., 2021) for more background on cAMP regulation of T-cell function. The phosphoproteomic analyses illustrated by these examples were carried out in the authors’ laboratories and have been published (Golkowski et al., 2016; Beltejar et al., 2017). It is hoped that they also will be instructive for those studying other similar processes. For additional background, we also have provided short historical sections on cAMP/PDE/PKA and on cAMP-regulated steroidogenesis to put the studies in context.
General Background for cAMP and cAMP-Dependent Phosphorylation as Regulators of Cellular Metabolism.
Since the discovery of cAMP by Sutherland and colleagues (1958) over 60 years ago and of cGMP a few years later (Ashman et al., 1963), many scientists have worked to determine how these cyclic nucleotides are regulated and what they do mechanistically and functionally in the cell. It soon became clear that cyclic nucleotide synthesis was controlled by multiple adenylyl and guanylyl cyclases, and their degradation was governed by multiple cyclic nucleotide PDEs. The early literature on this topic and many more recent advances on cyclic nucleotide mechanisms and functions have been reviewed, and readers are directed to these and other reviews (Beavo and Brunton, 2002; Maurice et al., 2014; Chen and Yan, 2021; Hofmann, 2020). We now know that many of the effects of cyclic nucleotides are mediated directly or indirectly by changes in the phosphorylation of key proteins in each pathway. The proximal kinase that is modulated by cAMP is PKA. However, modulation of kinase activity downstream of the cAMP-binding protein exchange factor activated by cAMP is also to be expected in most cells (Robichaux and Cheng, 2018). Moreover, a series of different protein phosphatases can reverse these phosphorylations, some of which can be modulated directly by cAMP-dependent phosphorylation (Leslie and Nairn, 2019; Osawa et al., 2020). Finally, there are many different substrates for PKA, some of which are other kinases. These substrates include many different types of regulatory proteins, including enzymes, protein modulators of enzymes, structural or scaffolding proteins, transporters or ion pumps, and likely many others yet to be defined. As mentioned, perhaps the greatest advantage of phosphoproteomic analysis is its sensitive unbiased nature. Such studies are only just beginning for cAMP-mediated events and show great promise for elucidating many new and unexpected mechanisms by which cAMP affects cellular function.
Of the many different processes regulated by cAMP, some are present in all cells, but many will be specific to a given cell type. For this reason, phosphoproteomic studies will need to be carried out in many cell types with many different modulators. Nearly all of these cAMP-regulated processes are mechanistically complex and contain multiple steps. Often these steps are sequestered in functional compartments of the cell delineated by specific organelles or scaffolding proteins (Esseltine and Scott, 2013). Commonly, the molecules being modified by the cellular process move from one compartment to the next, often in a reversible or cyclical manner. In other cases, they move progressively along a scaffolding system. These attributes allow the cell an enormous diversity for sites of control. Indeed, they require it. Given the many control sites available for each process, wide-scale phosphoproteomic analysis is one of the few currently available approaches able to address this issue.
Use of Selective Cyclic Nucleotide Phosphodiesterase Inhibitors as a Method for Raising cAMP and Probing the Mechanism(s) of cAMP Action in Cells.
One of the few ways to selectively stimulate cAMP or cGMP-dependent processes in the cell but not to stimulate other signaling pathways is to inhibit one or more cAMP- or cGMP-selective phosphodiesterases. Most cells express several different PDE genes, and often different PDEs are enriched in separate compartments. Eleven different gene families of PDEs exist, and most families are encoded by several closely related genes. These genes are expressed in cell type–selective manners to provide different isozymes of PDE, which exhibit unique localization, different cyclic nucleotide specificities, and different regulatory features. Mechanistically, this diversity is accomplished by utilizing alternate transcriptional start sites, alternate splicing, and/or alternate post-translational processing. Sometimes, individual compartments will contain the same PDE isozymes, and in other cases, a compartment will contain different PDEs (Maurice et al., 2014; Chen and Yan, 2021). In general, our understanding of which compartments contain what PDEs is only just beginning to be elucidated. Again, phosphoproteomic analysis allows a wide array of experimental design that can address these questions.
Over the last few years, scientists at both academic institutions and pharmaceutical companies have been identifying and characterizing compounds that are effective selective inhibitors of the various PDE isozyme families (Zuo et al., 2019; Lugnier et al., 2020; Chen and Yan, 2020). These compounds exhibit a wide variety of PDE isozyme selectivity. Many of the early agents would inhibit most PDE isozymes, albeit with different affinities for each of them. Until quite recently, most companies have directed their discovery efforts toward identifying compounds that are highly selective (often more than 100-fold) for each PDE gene family. Although these efforts have been largely successful for identifying gene family–selective agents, much less success has been reported for identifying compounds showing selectivity between members of any single PDE family. Nevertheless, these chemical agents have been very useful aids for identifying what processes in the cell are regulated by which PDE and for defining functional pools of cAMP within the cell. Most recently, with the understanding that many cAMP-dependent processes are regulated at multiple steps by different combinations of PDEs, there has been renewed interest in examining the efficacy of various combinations of PDE-selective inhibitors as modulators of specific processes and pathways. The studies described in this minireview make use of selective PDE inhibitors, which are sometimes used in combination, as probes to alter cAMP-dependent phosphorylation in different functional compartments of the cell that regulate complex, multistep metabolic pathways.
Cyclic AMP Regulation of Steroidogenesis: Background and Early Studies.
It has been known for many years that agents that increase cAMP in most steroid-producing cells can increase the output of steroid hormone both acutely and chronically (Sharma et al., 1974; Wong et al., 1986). “Acutely” in this case means in the minute-to-hour time frame, and “chronically” means in the hour-to-day time frame. Most studies have emphasized the conversion of cholesteryl esters stored in lipid droplets into the final steroid hormone. Much of this process occurs in the mitochondria and endoplasmic reticulum, and important regulatory steps involving the transport of cholesterol into the mitochondria via the steroidogenic acute regulatory (StAR) protein and possibly other proteins have been described. Longer-term cAMP-dependent induction of some of the P450-converting enzymes has also been established. A general outline showing some of the regulated steps in this process is shown in Fig. 1.
Fig. 1.

Model depicting parts of the classic cAMP-dependent, hormone-stimulated steroid-secretion pathway. In this case, the hormone is luteinizing hormone (LH) binding to the LH receptor. Receptor activation causes a G-protein–dependent activation of cAMP synthesis. The cAMP diffuses to many different compartments in the cell, where it interacts with PKA. The cAMP concentration is also modulated by several different PDEs. Once activated, PKA will phosphorylate and activate several specific proteins in the steroidogenic pathway. One of the best studied is hormone-sensitive lipase (HSL/Lipe), also originally known as CEH, which, acting with PAT proteins, such as perilipin (Plin), stimulates the production of free cholesterol from cholesteryl esters stored in lipid droplets. Another important PKA target is StAR, which assists cholesterol entry into the mitochondria, where much of the conversion of cholesterol to hormone occurs. On a longer time scale, PKA also stimulates the synthesis of several other steroidogenic enzymes, including P450 steroid 17 α-monooxygenase (c17) and 17β-hydroxysteroid dehydrogenase (17b-HSD). Many other enzymes and steps are required for synthesis and may be regulated but are not depicted in this classic model. AC, adenylyl cyclase; SCC, Side Chain Cleavage (Also referred to as P450scc); PAT, acronym for perilipin.
Most textbooks state that free cholesterol entry into the mitochondria is dependent on the StAR protein and that this protein is a major one regulated by cAMP-dependent phosphorylation. This step, therefore, has been assumed to be the most important “acute” cAMP-dependent regulatory point of the overall process. However, some researchers have suggested that additional and multiple mechanisms and sites are likely to be involved in cAMP control of steroid production (Stocco et al., 2005). For example, there are also many other longer-term cAMP-dependent changes in the steroidogenic machinery, including translocator protein (18 kDa), various P450 enzymes involved in the transformation of the cholesterol molecule into the final steroid, and voltage-dependent anion channels that are thought to be intimately involved in the regulation of steroidogenesis and therefore might be targets of cAMP-dependent changes (Manna and Stocco, 2005; Midzak and Papadopoulos, 2016). Similarly, changes in the endoplasmic reticulum (ER) cholesterol sensory machinery and especially in sterol regulatory element–binding protein (SREBP) 2, other StAR isozymes, and small G proteins are thought to be capable of playing important cAMP-dependent roles since much of the cholesterol that eventually ends up as hormone likely passes through the ER on its way to synthesis in the mitochondria (Shimizu-Albergine et al., 2016). In all of these studies it was assumed that much if not all of the regulation was controlled by cAMP activation of PKA either directly or indirectly. Nevertheless, there have been no generally accepted single mechanism or series of mechanisms that as yet fully explain all effects of increased cAMP in the steroidogenic cells. Part of the problem has been that results have depended in large part on exactly how steroid output was measured, whether the measurements were done in vitro or in vivo, and whether the measurements were acute or chronic in nature. Often only part of the steroidogenic pathway was investigated. Especially lacking have been measurements of steroid output that include all of the transport and processing steps from extracellular LDL or high-density lipoprotein lipids to final synthesis in the ER and mitochondria.
It is now generally agreed that the complete steroidogenic pathway involves the movement and transformation of cholesterol (in the form of lipoproteins: LDL or high-density lipoprotein, depending on species) from the blood stream ultimately to an active steroid hormone secreted back into the blood. This complete process involves many additional steps and probably several alternative pathways within the cell depending on the cell type and the amount of endogenous cholesterol stored within the cell. The analogy of the steroid substrate cholesterol moving through the cell like river water through a delta may be apt. In theory, any or all steps in these transport processes, including LDL entry, packaging and transport into various vesicles, conversion of LDL cholesteryl ester into free cholesterol, storage in lipid droplets, mobilization from droplets, movement into and out of the endoplasmic reticulum, and retransport of the newly synthesized cholesteryl ester in the ER back to either the Golgi apparatus or lipid storage droplets, could be important sites of regulation. Regulation of each of these processes is not mutually exclusive. Ultimately cholesterol must be moved into and out of the mitochondria, where many of the important steroid hormone synthetic steps occur. In some cases, different steroid hormone intermediates must be transported out and back into the mitochondrial matrix to complete final hormone synthesis and secretion.
Rationale for a Phosphoproteomic Approach to cAMP Effects on Steroid Hormone Synthesis.
With this history as background, we decided to directly test in an unbiased manner which proteins in a hormone-responsive, highly steroidogenic cell have sites phosphorylated on them in a cAMP-dependent manner. We were also interested in determining whether there might be different pools of cAMP that contributed to this regulation and, if so, which PDEs were regulating each of these pools. Given that there were so many different possible steps in this complex process known to exist in different physical compartments, this approach seemed worthwhile. However, it was also known that most cell types contain several different isoenzymes of PDE that are thought to coordinate the regulation of cAMP levels in different functional compartments in the cell. We assumed that a selective inhibitor of any particular family of PDEs would acutely increase cAMP in each functional compartment in which the PDE resided, and therefore, the studies might also provide initial information of which PDEs subserved what function(s) in the cell. In the case of steroidogenesis, it previously had been shown that to fully stimulate steroid hormone production by PDE inhibitors, a combination of PDE4 and PDE8 inhibition was required (Tsai et al., 2011; Shimizu-Albergine et al., 2012). Either inhibitor alone was much less effective than the combination, presumably because more than one PDE regulates many of the functional compartments.
Data Availability and Protocols for Phosphoproteomic Analysis of cAMP-Dependent Pathways.
Time to quenching is a major issue for studying all cAMP-regulated reactions since this cyclic nucleotide can turn over so quickly in the cell (Walseth et al., 1983). Similarly, phosphorylation/dephosphorylation of many sites may occur within milliseconds of stimulus (Catterall, 2015). All of the studies reported here were carried out at multiple time points after addition of the stimulus. The reactions were quenched with either ice-cold PBS and freshly prepared Tris-buffered 8-M urea containing a protease and phosphatase inhibitor cocktail (MA-10 cells) or boiling 6-M guanidinium hydrochloride (Jurkat cells). Nevertheless, given that kinases, phosphatases, and phosphodiesterases can be scaffolded together often in stoichiometric amounts with the cAMP and that kinase, phosphatase, cyclase, and phosphodiesterase activity must be quenched equally quickly, one is never quite sure whether stimulus and quench conditions are sufficient. Correspondingly, some of the interpretations of most phosphoproteomic results, including these, likely need to be understood with these caveats in mind. This is particularly true for modulation of pathways that cycle rapidly. Most of the data discussed in this review are recorded in public data bases. The MA-10 data mass spectrometry raw files and MaxQuant/Andromeda search results were deposited in the publicly available mass spectrometry data repositories Mass Spectrometry Interactive Virtual Environment (MSV000079412; http://www.massive.ucsd.edu.offcampus.lib.washington.edu/) and ProteomeXchange (PXD003280; http://www.proteomexchange.org/).
Leydig MA-10 Cell Results.
Using a stable isotope labeling by amino acids in cell culture approach (Ong et al., 2002), we identified over 28,000 unique phospho-peptide sites (Golkowski et al., 2016). Most importantly for the present discussion, of these sites approximately 750 were consistently altered by the combination of PDE4/8 inhibitors used. Individual inhibitors alone had a much smaller effect, as seen for the effects of PDE inhibitors on steroid production (Shimizu et al., 2012). Many of the 750 altered protein phosphosites contained a so-called “PKA consensus phosphorylation sequence” (Arg/Lys, Arg/Lys, X, SerP) and were therefore likely to be direct substrates of PKA. However, many did not contain this sequence, and since activation of cAMP/PKA can directly (and indirectly) regulate other kinases and also protein phosphatases of mixed specificity, it was expected that many nonconsensus PKA sites would also be altered by the PDE inhibitors and that these also might be important to the cAMP response. This is a strength of the phosphoproteomic approach. As expected, some sites were increased in phosphorylation, and others decreased depending on the time point measured. Note that PKA can activate some phosphatases, thereby providing a mechanism for PDE inhibitors to decrease phosphorylation on some sites. All data from all sites identified are available to the public on the ProteomeXchange web site listed above.
We were initially rather surprised that so many total phosphorylated peptides were identified and particularly that so many (∼750) were directly (or indirectly) altered by the combination of PDE inhibitors, agents that are thought to act only by direct effects on local cAMP levels. For illustration purposes, a list of some of the proteins showing the largest reproducible increases in consensus PKA sites are shown in Tables 1 and 2. The sequence of each site is given as well as the fold increase in response to the PDE inhibitors. Gene symbols identified in bold underlined type are particularly relevant to the steroidogenic pathway. Others may be involved in other processes in the cell or modulate unknown pathways related to steroidogenesis. The full list of phosphosites is too long for this short minireview but is available online and in the original publication (Golkowski et al., 2016). Even this abbreviated list of 60 altered consensus proteins is very large, and one immediately wonders which, if any, might be important sites for regulation of cAMP-stimulated steroid hormone production. This was particularly true since only two of these sites, hormone-sensitive lipase and perilipin, had been widely reported to be regulators of steroid production. Some of the other proteins phosphorylated have been associated in previous studies with aspects of steroid handling or synthesis. Many, however, had not previously been associated with this process. Nevertheless, since these phosphosites reproducibly responded to cAMP, the most straightforward interpretation of the data were that several and perhaps many of these altered protein phosphosites were likely to be involved in either the rapid or long-term increases in steroid hormone biosynthesis by these cells in response to cAMP.
TABLE 1.
List of protein phosphosites in MA-10 cells that are increased by a combination of the PDE4 inhibitor rolipram and PDE8 inhibitor PF-04957325
| Fold Incr. | Protein Name/Function | Gene Symbol | P04 sequence | |
|---|---|---|---|---|
| 1 | 18.7 | CAP-Gly domain–containing linker protein 1/2; | Clip1/2/Restin | _KIS(ph)GTTALQEALK_ |
| 2 | 12.8 | Regulator of microtubule dynamics protein 2 | Fam82a1 | _KFGS(ph)LTLPEESHSAQ |
| 3 | 12.3 | Rho guanine nucleotide exchange factor 11 | Arhgef11 | _KVS(ph)LLPGGGVGAAK_ |
| 4 | 12.2 | Transcoblamin uptake receptor (CD320 antigen) | Cd320 | _ESLLLSERKTS(ph)LI_ |
| 5 | 12.2 | DNA-binding protein A | Csda | _RRRS(ph)RPLNAVSQDGK |
| 6 | 12.1 | Acyl-CoA–binding domain–containing protein 5 | Acbd5 | _GERWGS(ph)RGSLNEQIA |
| 7 | 11.2 | Phosphatidylinositol 4-kinase β | Pi4kb | _RRLS(ph)EQLAHTPTAFK_ |
| 8 | 11.0 | Interferon regulatory factor 2–binding protein–like | Irf2bp2/Irf2bpl | _RQS(ph)CYLCDLPR_ |
| 9 | 9.8 | Nesprin-2 (Nuc envel spectrin-repeat proteins) | Syne2 | _RRES(ph)EEPTSPQSLCHL |
| 10 | 9.8 | Bcl-2–related ovarian killer protein | Bok | _RS(ph)SVFAAEIMDAFDR |
| 11 | 9.8 | RAF proto-oncogene Ser/Thr-protein kinase | Raf1 | -IVQQFGYQRRAS(ph)DDG |
| 12 | 9.0 | Carbamyl phosphate synthetase 2 | Cad | _RLS(ph)SFVTK_ |
| 13 | 9.0 | Bcl-2–related ovarian killer protein | Bok | _RSS(ph)VFAAEIMDAFDR |
| 14 | 8.9 | Serine/threonine-protein kinase WNK1 | Wnk1 | _KFS(ph)APGQLCVPMTSN |
| 15 | 8.2 | Cytoplasmic protein NCK1 | Nck1 | _RKPS(ph)VPDTASPADDS |
| 16 | 7.9 | 182-kDa tankyrase-1–binding protein | Tnks1bp1 | _RFS(ph)EGVLQPPSQDQE |
| 17 | 7.9 | Inositol 1,4,5-trisphosphate receptor type 3 | Itpr3 | _KQS(ph)VFGASSLPAGVG |
| 18 | 7.7 | Ras-spec guanine nuc-release factor RalGPS2 | Ralgps2 | _KS(ph)SAAAAAAAAAEGA |
| 19 | 7.4 | Heat shock protein HSP 90-β | Hsp90ab1 | _RLS(ph)ELLR_ |
| 20 | 7.3 | Zinc finger FYVE domain–containing protein 16 | Endofin | _RCS(ph)KPVCDLISDMGN |
| 21 | 7.3 | Protein G-protein–coupled receptor 107 | Gpr107 | _KVS(ph)NGAVEPQGSWE |
| 22 | 7.1 | Horm-sensitive lipase/Cholest ester hydrolase | Lipe | _RSS(ph)QGVLHMPLYTSPI |
| 23 | 6.8 | Phos b kinase reg subunit α, liver isoform | Phka2 | _GHRKS(ph)LNLVDSPQPL |
| 24 | 6.8 | Endoribonuclease Dicer | Dicer1 | _KIS(ph)LSPFSASDSAYEW |
| 25 | 6.7 | Ras-specific guan nuc-release factor RalGPS2 | Ralgps2 | _KSS(ph)AAAAAAAAAEGA |
| 26 | 6.5 | SH3 and PX domain-containing protein 2A | Sh3pxd2a | _RGS(ph)ADIIPLTATTPPCV |
| 27 | 6.2 | Carbohydrate-responsive element–binding protein | ChREBP | _RLS(ph)GDLNSIQPSGALS |
| 28 | 5.8 | Oxysterol-binding protein–related protein 11 | Osbpl11 | _RPS(ph)QNAMSFFNVGH |
| 29 | 5.8 | Serine/threonine-protein kinase WNK1 | Wnk1/Osr1 | _LQKSIS(ph)NPPGSNLR_ |
| 30 | 5.7 | Liprin-β-1 | Ppfibp1 | _RRPS(ph)DENSITPSEVQ |
Sites are ranked 1–30 based on fold response to the PDE inhibitor treatment. The underlined gene symbols in bold type face represent some of the phosphoproteins shown in Figs. 2–4. Each PDE inhibitor has an isozyme selectivity of at least 30-fold if used at an appropriate concentration. Data are from the 1-h time point (Golkowski et al., 2016).
TABLE 2.
Continuation of list from Table 1 of protein phosphosites in MA-10 cells that are increased by a combination of PDE4 and PDE8 inhibitors
| Fold Incr. | Protein Name/Function | Gene Symbol | P04 sequence | |
|---|---|---|---|---|
| 31 | 5.7 | γ-Taxilin–ATF4/Creb2-binding protein | Txlng | _KHS(ph)LEGDEGSDFITK_ |
| 32 | 5.6 | Serine/threonine-protein kinase SIK3 | Sik3/QSK | _RFS(ph)DGAASIQAFK_ |
| 33 | 5.4 | Neuron navigator 1 | Nav1 | _KTS(ph)LDVSNSVEPGFLA |
| 34 | 5.4 | cAMP-regulated phosphoprotein 19 | Arpp19 | DHIPTPQDLPQRKPS(ph)L |
| 35 | 5.1 | Autophagy-related protein 16-1 | Atg16l1 | _RLS(ph)QPAGGLLDSITNI |
| 36 | 5.1 | Protein ETHE1, mitochondrial | Ethe1 | _RLS(ph)QQSASGAPVLLR |
| 37 | 4.9 | Perilipin-1 | Plin1 | _RLS(ph)TQFTAANELACR |
| 38 | 4.7 | ADP-ribosylation factor GTPase-activating prot 1 | Arfgap1 | _RSS(ph)DSWDVWGSGSA |
| 39 | 4.7 | SLAIN motif–containing protein 2 | Slain2 | _LS(ph)LQGHPTDLQTSNV |
| 40 | 4.7 | TBC1 domain family member 10B | Tbc1d10b | _RAS(ph)AGPVPGAVVIAE |
| 41 | 4.6 | A-kinase anchor prot 1, mitochondrial, Dakap1 | Akap1 | _RLS(ph)EEACPGVLSVAPT |
| 42 | 4.5 | RNA polymerase II nuclear localization protein | Slc7a6os | _KTS(ph)DPDVILCNSVELIR |
| 43 | 4.4 | TBC1 domain family member 25 | Tbc1d25; | _RSS(ph)LTTAALPFTQSILS |
| 44 | 4.4 | Vesicle-trafficking protein SEC22b | Sec22b | _NLGS(ph)INTELQDVQR_ |
| 45 | 4.2 | Low-affinity cationic amino acid transporter 2 | Slc7a2 | _NLS(ph)LPFILHEK_ |
| 46 | 4.1 | Golgin subfamily A member 5 | Golga5 | _KS(ph)EPDDELLFDFLNSS |
| 47 | 4.0 | CAP-Gly domain–containing linker protein 2 | Clip2 | _RYS(ph)LIDPASPPELLK_ |
| 48 | 4.0 | Nuclear factor related to κ-B–binding protein | Nfrkb/INO80 | _KGS(ph)LAALYDLAVLKK_ |
| 49 | 3.9 | Pleckstrin homol domain family F m 2 (Phafin2) | Plekhf2 | _RIS(ph)IVESCFGAAGQPL |
| 50 | 3.9 | Cytoskeleton-associated protein 5 | Ckap5 | _KYS(ph)DTDIEPFLK_ |
| 51 | 3.8 | Cytospin-A | Specc1l | _KGS(ph)SGNASEVSVACL |
| 52 | 3.8 | Kinesin-associated protein 3 | Kifap3/Kap3 | _LSEVEQLLYYLQNRRDS(p |
| 53 | 3.8 | GRB14 adapter protein | Grb14 | _RVT(ph)LPAITPIVLQK_ |
| 54 | 3.7 | Rho guanine nucleotide exchange factor 2 | Arhgef2 | _S(ph)LPAGDALYLSFNP |
| 55 | 3.7 | Phospholipase DDHD2 | Ddhd2 | _KNS(ph)VSINRPAM(ox)S |
| 56 | 3.5 | Protein FAM54B | Fam54b | _AS(ph)FETLPNISDLCLK_ |
| 57 | 3.5 | Rho family GTPase activator (Alsin) | Als2 | _RLS(ph)LPGLLSQVSPR_ |
| 58 | 3.5 | Reg of microtubule dynamics prot 3 (PTPIP51) | Fam82a2 | _SHS(ph)LPNSLDYAQASE |
| 59 | 3.5 | Serine/arginine repetitive matrix protein 2 | Srrm2 | _RSS(ph)SELSPEVVEK_ |
| 60 | 3.4 | Low-density lipoprot receptor adapter protein 1 | Ldlrap1/ARH | _NQEGGDVPGTRRDS(ph) |
At a minimum, the data showed that many phosphorylation events can and do occur at many places along the steroidogenic handling pathway and could therefore also be important to regulation of hormone production. This conclusion also is consistent with more recent data in the same cells using a different approach (Golkowski et al., 2020). One possible model illustrating how several of the multiple sites identified in the phosphoproteomic screen might act to alter steroid hormone production is shown in Fig. 2. The phosphoproteins shown are taken largely from the list of proteins having PKA consensus sites (Tables 1 and 2) and having the greatest increased phosphorylation in response to the PDE inhibitor treatment. Unexpectedly, a great many of them were known or highly implicated as being important to either vesicle formation or transport or are regulators of microtubule function (Fig. 2). Since many vesicles are thought to move via microtubules the processes are likely connected.
Fig. 2.

Phosphoproteomic identification of sites increased by cAMP in MA-10 cells. Model showing some of the proximal cAMP-dependent phosphorylation events occurring in cells treated with a combination PDE4 and PDE8 inhibitors. Arrows indicate movement of cholesterol and cholesteryl esters to provide substrate for the eventual mitochondrial synthesis of steroid hormone. Each protein listed in red type is consistently increased in phosphorylation at a PKA consensus site by treatment with the combination of PDE inhibitors. Notable is the fact that many of these proteins modulate small G-protein–dependent events and therefore are likely to be important regulators of cholesterol handling. Many of these processes involve formation or movement of microvesicles along microtubules. CCV, clathrin-coated vesicle; HDL, high-density lipoprotein; SR-B1, scavenger receptor B1; Gol, Golgi apparatus; mito, mitochondria. Other acronyms are gene symbols or names.
However, most of the sites identified usually have not been thought of as regulators in PKA-dependent steroid hormone synthesis. For example, several small G-proteins of the Rab family are well established as regulators of vesicle formation and fusion (Lamber et al., 2019; Homma et al., 2020). Of note are the large number of small G-protein guanine nucleotide protein exchange factors (Gefs) and guanine nucleotide protein activation factors (Gaps) among the identified phosphoproteins that appear to be PKA substrates. Therefore, it apparently is not the G-proteins themselves that are PKA targets but rather their regulatory partners. A few studies of regulation of Gefs or Gaps by phosphorylation have been published in other systems (Guidetti et al., 2013; Lutz et al., 2013; Yang and Terman, 2013; Nagy et al., 2015; Kulasekaran et al., 2015; Novick, 2016; Adame-García et al., 2019). However, neither Gefs nor Gaps previously have been considered to be important regulators of steroidogenesis. Nevertheless, they fit very well with the ideas that the transport and availability of cholesterol substrate to the mitochondria may constitute an important general control process for steroid hormone production by the cell and that this process is regulated at multiple steps along the pathway from cell surface to the final synthetic step.
A large number of the other proteins phosphorylated are involved in microtubule transport (Fig. 2; Tables 1 and 2). More generally, the data illustrate the likely importance of vesicle formation, processing, and transport via microtubules as a cAMP-regulated process that helps to control steroid hormone production. Many of the identified phosphoproteins are known to modulate these processes. By analogy, one might expect that many of the other identified proteins could also be modulators.
This hypothesis is strengthened when one considers all of the sites altered by the PDE inhibitors and not just PKA consensus sites. An independent way to analyze protein phosphorylation function uses the method of gene ontology (GO) analysis on the 750 modulated sites (Pomaznoy et al., 2018); some of these sites increase in phosphorylation, and some decrease. Figures 3 and 4 illustrate some of these associations. Two GO terms stand out: “Endocytosis” and “Vesicle Transport.” GO analysis only suggests that an association somehow exists between the proteins in question and the GO process (based on literature citations). But coupled with the changes in phosphorylation in response to a specific stimulus, it further connects the observed changes in phosphorylation with regulation of the process. Gene ontology analysis, of course, does not make any prediction about how the GO term (e.g., endocytosis) relates to the larger process of cAMP-regulated steroid production. However, it does make use of literature correlations from many different laboratory groups investigating a large number of different cell systems.
Fig. 3.

Endocytosis in MA-10 cells. Gene ontology analysis of phosphoproteomic data showing protein phosphorylation events modulated in cAMP-dependent control of endocytosis. Note that the PDE inhibitors caused a decrease in phosphorylation on several of these phosphosites possibly by activating a phosphatase. Also note the large number of potential regulatory proteins involved. Few have been previously implicated as being regulatory sites for cAMP control of steroid hormone secretion [original data from Golkowski et al. (2016)]. CDK, Cyclin-Dependent Kinase; LSR, Lipolysis-stimulated lipoprotein receptor; EGF, Epidermal Growth Factor; ERK, Extracellular-Signal-Regulated Kinase.
Fig. 4.

Vesicle transport in MA-10 cells. Gene ontology analysis of phosphoproteomic data showing protein phosphorylation events possibly involved in cAMP-dependent control of vesicle transport in MA-10 cells. Note that the PDE inhibitors caused a decrease in phosphorylation on several of these phosphosites possibly by activating a phosphatase. Also note the large number of potential regulatory proteins involved. Few have been previously implicated as being regulatory sites of cAMP control of steroid hormone secretion [original data from Golkowski et al. (2016)].
Arguments for Importance of Cholesterol Entry and Transport “Mini-Cycles” as Regulators of Steroid Hormone Production.
It has been known for some time that external cholesterol availability can limit steroid hormone biosynthesis (Capponi, 2002). Therefore, the argument that a relatively small shift in phosphorylation of a regulatory protein involved in the rate at which a cycling transport system operates could lead to profound changes in the speed or efficacy of at least part of the cycle seems reasonable. For example, the data in Tables 1 and 2 show that G-protein–coupled receptor 107 and low-density lipoprotein receptor adapter protein 1 each increase in phosphorylation in response to a combination of PDE4/8 inhibitors. Both of these proteins are known to be important regulators of initial clathrin-assisted internalization of LDL-containing vesicles (Soutar and Naoumova, 2007; Zhou et al., 2014), as shown in Figs. 2 and 3. The human homolog for scavenger receptor B1 has also been shown to be induced by PKA (Imachi et al., 1999). Once formed, the endocytic vesicles reorganize and move via microtubules toward more internal regions of the cell. Here they are transformed eventually into cholesteryl ester–loaded endosomes. This occurs with the assistance of several regulatory small G-proteins, including Rab5 and Rab7. Although phosphopeptides for Rab5 and Rab7 are not seen in the data, both Gefs that regulate these Rabs and Gaps that also regulate them are changed in their phosphorylation status by the PDE inhibitors. Similarly, cytoskeleton-associated protein 5 and Clip1/2 are microtubule-associated proteins known to be important for their polymerization function. In fact, Clip1/2 consistently showed the greatest increase in phosphorylation of all phosphopeptides in these data sets. Finally, we know that once formed, part of the endocytic vesicles containing, for example, the LDL receptor are returned to the membrane so that they can be reused. This return step is known to be a Rab35-dependent process (Chaineau et al., 2013). Again, Rab35 phosphopeptides are not found in the data set, but Dennd1a (a Rab35 Gef) and Tbc1d (an Rab35 Gap) both show increased phosphorylation in response to the PDE inhibitor treatment. Thus, all aspects of the cycling (or recycling) of this part of cholesterol entry and movement machinery have multiple cAMP-dependent phosphorylation events. It would appear that to speed up overall cholesterol delivery into the cell, the rate at which the cycle operates must be increased. A diagram illustrating how of such a regulatory “mini-cycle” involving these proteins might operate is illustrated in Fig. 5.
Fig. 5.

Cholesterol transport mini-cycle. Model for one of the first “mini-cycles” involved in the provision of cholesterol substrate for steroid hormone biosynthesis. The model illustrates some of the initial steps of cholesterol uptake—in this case from LDL—and its initial vesicular transport and processing. Once at the endosome, the cholesterol is further transported to other organelles in the cell, including lipid storage droplets, endoplasmic reticulum, and mitochondria. Upper panel shows cycle going slower in absence of PDE inhibitors; lower panel shows cycle going faster in presence of PDE inhibitors because of increased phosphorylation at many of the proteins indicated in red type. Analogous cycles operate at several steps further downstream in the steroid hormone biosynthetic process. LDLR, LDL receptor; PM, Plasma Membrane.
General Implications of Multisite Phosphorylation.
If we assume that at least several of the many identified phosphorylation sites are important to the control of cAMP-regulated steroid hormone production, then we need to consider the likelihood that there are multiple regulatory sites throughout this pathway/process. In fact, the concept that cAMP/PKA regulates only a single or small number of “rate-limiting” steps does not seem viable. Upon reflection, if indeed much of the machinery for overall steroid biosynthesis and release resides in compartments or on scaffolds and especially if it involves speeding up of transport cycles, it seems more likely that regulation must occur in a coordinated manner at multiple locations. The purpose of regulated reversible phosphorylation at multiple sites would be to “coordinate” these steps. Although the concept of regulation at a single step or a limited number of “rate-limiting” steps may work satisfactorily for solution-based biochemical enzymatic reactions, it has much less appeal for explaining the regulation of such directional, compartmentalized, and scaffolded pathways.
Issues of Interpretation of These and Other Phosphoproteomic Studies—Does a Low Level of Phosphorylation Stoichiometry Alter This Conclusion?
One general problem with the mass-spectrometry approach described is that as usually implemented, it only gives correlative data relative to a “biological” control condition. This makes it difficult to assign a causal mechanistic relationship to any given phosphorylation event (see Fig. 6). Usually, the method gives little information on the quantitative aspects of the identified phosphorylation event (unless a control peptide for each site can be included as a standard). This is because each value obtained from the mass spectrometer is determined relative to the biologic control for the experiment since the “yield” of phosphopeptide varies greatly among individual peptides. Therefore, any results are necessarily relative to the same peptide in the biologic control and not to an absolute control sample. In this case, it is a comparison of the results with and without cAMP PDE inhibitors. For example, a 10-fold increase in phosphopeptide might be from 0.001 to 0.01 mol per mol or it might be from 0.1 to 1.0 mol per mol (i.e., complete phosphorylation). At least one study has begun to address this issue experimentally by applying a nonspecific phosphatase treatment arm to the phosphoproteomic protocols (Wu et al., 2011). However, in general, this type of experiment has not yet been widely adopted.
Fig. 6.

Criteria for determining phosphorylation function. A series of criteria developed over 40 years ago helped guide our thinking about the physiologic relevance of a phosphorylation event (Krebs and Beavo, 1979). These criteria were based largely on what was known about regulation of metabolic cascades at that time. The subject was revisited in 2009 with emphasis on the use of mass spectrometry as a tool to address these criteria (Rider et al., 2009). More recently, our increased understanding that many metabolic and signaling pathways are compartmentalized and often cyclic in nature has raised questions about the generality and usefulness of these criteria. In particular, the improved sensitivity and resolving power of the mass spectrometer has improved vastly since 2009, greatly increasing the number of known phosphosites. The identification of thousands of phosphorylation sites on many different proteins suggests that an expanded, more nuanced set of criteria for the importance of any phosphorylation site needs to be established. Although most of the original criteria are still quite relevant, our newer understanding that many cellular processes are compartmentalized and cycling in nature requires a new set of guidelines. These new, revised criteria, therefore, do not include requirements for stoichiometric phosphorylation nor do they refer only to enzymatic reactions. It is realized that in many cases, measurement of the most immediately affected step/reaction will be challenging, particularly at scale. As with the original criteria, the expanded/revised criteria need to be shown for each phosphosite on the protein in question.
Nevertheless, for many years it has been a common assumption that a “trace” phosphorylation is not likely to be of regulatory significance. This assumption was initially put forward in a review of PKA-dependent reactions published over 40 years ago (Krebs and Beavo, 1979) (see Fig. 6). As alluded to in the previous paragraphs, however, given our current understanding that most regulatory steps are highly compartmentalized, that many proteins participate in multiple pathways, and that most cellular systems are not fully synchronized at the time of measurement, this assumption is likely to be inappropriate for many biologic processes and should be re-evaluated. Therefore, the common objection of using relative values that might reflect very low phosphorylation stoichiometry for interpretation of phosphoproteomic results also may not be applicable.
It is true that with most phosphoproteomic experiments we do not know the phosphorylation stoichiometry for any of the sites identified nor do we know the possible effect(s) of phosphorylation (at the identified sites) on activity of these proteins. Nevertheless, given the cellular functions of the proteins identified and the fact that the major process regulated by cAMP in these cells is an increase in steroid hormone output, it is highly suggestive that some and perhaps each of these sites could be important for hormone regulation. This conclusion is strengthened by the consistent large fold increases in phosphorylation seen for many of the key sites. Importantly, since there are many molecules of each of these proteins in each cell and they reside in multiple places in the cell, only a few might be expected to be at the same stage of phosphorylation/dephoshorylation at any one time. What is important to the cell is to speed up the process or make it more efficient in response to the surge in cAMP. The only way to do this is to either initiate more cholesterol transport “events” or to make each cycle go faster some way (for example, to recruit more LDL receptors to start more transport cycles). Again, this argues for multiple sites of regulation and for phosphorylation/dephosphorylation at many different parts of each cycle.
If phosphorylation increases the speed or efficacy for each of the functions of these phosphoproteins, we might expect the cAMP response to be balanced by an increase in phosphatase activity in order for the cycle to operate more rapidly. Again, this argues for less than full stoichiometry of phosphorylation at any given time even at full agonist stimulus if only for the reason that any individual molecule will likely be at a different part of the cycle. In fact, if phosphatase activity is also increased by cAMP, one might expect to see cAMP-dependent decreases in phosphorylation of some sites at some time points (see Fig. 3), and this is what is seen for many sites [data not shown here but presented in Golkowski et al. (2016)].
In summary, full stoichiometric phosphorylation of any particular site is not required for the idea that many regulatory signaling processes can be regulated at multiple sites by cAMP. It is also notable that any given protein in the cell can be involved in several completely disparate processes. For example, all tubulin molecules undoubtedly are not dedicated to moving Rab5 vesicles around the cell, nor are all Rab5 molecules likely to be dedicated only to this same process. So, if compartmentalization of cAMP occurs, and it undoubtedly does (Bock et al., 2020), then when measured at the whole cell level, one might expect only a partial stoichiometric phosphorylation response for most substrates. A 10% or even 1% phosphorylation response at the whole cell level may reflect a much greater response in a given functional compartment.
Other Processes in MA-10 Cells Influenced by cAMP.
The large number of phosphosites detected suggests that there are many other processes in MA-10 (and other cells) that also are likely to be regulated by cAMP/PKA. Several of the proteins also have a transport component or even a cyclical nature. For example, lipolysis likely occurs in several compartments as cholesteryl esters make their way through these cells. Hormone-sensitive lipase, a substrate known to be regulated by PKA in other cells, turns out to be the same protein initially known as one of the cholesteryl ester hydrolases (CEHs). Indeed, CEH was one of the first identified substrates of PKA (Krebs and Beavo, 1979).
Similarly, the ER carries out several functions relating to the handling of the cholesteryl esters used as substrate for steroid hormone biosynthesis as well as lipid handling and protein synthesis and transfer (Sewer and Li, 2008; Ortiz Sandoval and Simmen, 2012; Fryer et al., 2014; Pfisterer et al., 2016; Wilhelm et al., 2017; Gao et al., 2019; Progida, 2019; Volkmar et al., 2019). These include regulation of de novo cholesterol synthesis, resynthesis of cholesteryl esters from free cholesterol that arrives at the ER, sensing of free cholesterol levels in the cell, transport of newly synthesized cholesterol ultimately to the mitochondria or lipid storage droplets, transfer of cholesterol synthetic proteins via the Golgi apparatus, and many others. The ER also performs some of the final enzymatic steps of steroid hormone biosynthesis. There are close contacts between the ER, Golgi, plasma membrane, and mitochondria. A number of the regulatory proteins known to be involved in these processes are changed in their phosphorylation status upon treatment with PDE inhibitors or other agents that increase cAMP. They include some of the Gefs and Gaps that likely regulate the process. For example, the CopI arm of the vesicular transport system between the ER and the Golgi is thought to be regulated by the small G-protein Arf, and the Cop2 arm is thought to be regulated by Sar1. The GTPase activity of Arf is regulated by both the Gefs and Gaps (ArfGef1/2 and ArfGap1/17) for these G-proteins. Each of these proteins are phosphorylated on PKA sites in response to the PDE inhibitors (see Fig. 2).
One of the proteins transported by the ER is SREBP2. This protein is transported to the Golgi after interaction with SREBP cleavage–activating protein (SCAP) 1. SCAP1 acts as a sensor for free cholesterol levels in the ER. Upon binding to SREBP2, the complex is translocated to the Golgi where SREBP2 goes through a series of cleavages to form an active helix-loop-helix transcription factor that in turn is transported back to the nucleus where it activates transcription of several key proteins in the cholesterol biosynthetic pathway. SCAP1 is phosphorylated in the WD domain in response to increased cAMP. Much of this data plus descriptions of several other protein phosphorylation reactions likely to be important to steroid hormone biosynthesis have been described (Shimizu-Albergine et al., 2016).
Finally, in analysis of the results, one needs to consider whether the apparent complete absence of a protein phosphorylation site has any significance. Absence of a particular protein phosphosite signal might mean that the protein (and therefore the process that it catalyzes or regulates) is not present in the cell type. However, it also might just mean that this particular peptide was degraded by the proteases used in generating the mass-spectrometry sample, that it was not recovered during the initial fractionation steps, or that it was “hiding” under some other larger phosphopeptide peak. So, lack of a signal does not necessarily mean that there is not an important phosphorylation regulatory step at a particular site. A good example in the present case may be S195 of the StAR protein, which has been shown in many other studies to be an important regulatory phosphosite but was not detected in these studies.
How Does One Determine the Effects of Phosphorylation on Function?
Classically, one of the problems in determining the importance of phosphorylation on any given site to regulation of a process has been the difficulty in correlating the effect of the phosphorylation on function (i.e., in this case on steroid hormone production) (see Fig. 6). In this case, if we assume that regulation of steroid hormone production consists of a series of mini-cycles or steps, in which each mini-cycle hands off substrate (cholesterol) to the next, then this suggests a different approach for determining function of a phosphorylation event. That is, one needs to quantify each of the “partial reactions” of the process as close to the site and as close in time to the phosphorylation as possible. For example, if one can test the effects of phosphorylation on Gef or Gap function directly (ideally at the exact location of the event) and measure a response to this part of the process, then this would go a long way toward critically testing the hypothesis that Rab5 Gef or Gap activity is an important regulator. The trick is to be able to measure a small part of the process—say, production of endocytic vesicles—rather than the overall process of steroid output. One might not expect to be able to show an effect of this phosphorylation on overall steroid hormone production regardless of stoichiometry because later steps in the cycle likely would still limit overall flux in the system. But, if one can figure out how to measure the various “partial reactions,” it should be possible to make a correlation of phosphorylation with function. The beauty of a small soluble molecule like cAMP that responds to specific hormone stimuli in a cell type–specific manner is that it is perfectly designed to coordinate the regulation of a large number of different proteins that in turn act together to regulate many different steps of a cellular process.
Genetic Approaches—Gene Disruption and Gene Editing as Another Possible Approach for Determining Phosphosite Function.
Practically speaking, it is difficult for any single laboratory to have the expertise and facilities available to test partial reactions for each of the steps in a multistep regulatory pathway or even several of them. In the last several years, by using various forms of CRISPR gene editing it has been possible to selectively modify expression of or edit nearly any gene in a cell type–selective manner (Banan, 2020). For the last several decades, one of the most successful methods for determining the role(s) for any protein in a cellular process has been to selectively alter the activity of that protein either with drugs or genetic manipulation. Although selective activators can be used to measure rates of partial reactions, interpretation of data from use of selective inhibitors (or gene knockout) is more problematic since a decrease in any critical step of a process would necessarily limit the overall rate of the process. However, using CRISPR technology, it is increasingly possible to genetically manipulate DNA in almost any cell type. These methods include disruption of the gene either in the germ line or in a cell type–specific manner. More interesting for the present discussion has been the possibility in most cell types to alter single amino acids in any protein, including substitution of serine for an alanine (inability to phosphorylate a site) or for a glutamic acid (often acting as a phospho-mimetic). An advantage of an alanine mutation knock-in is that one can potentially study only the effect of being unable to phosphorylate a specific site, whereas a whole gene knockout tells one only that the protein is obligatorily required for the pathway. One study from the authors’ laboratories has evaluated steroidogenesis in this manner (Shimizu-Albergine et al., 2016). In this study, SCAP1 was knocked out in the MA-10 cell line and as predicted decreased production of key steroidogenic proteins. The appropriate phosphorylated serine was also changed to alanine or glutamic acid. However, this approach illustrated one of the major problems with this approach, i.e., no major changes in overall steroid hormone production could be demonstrated (unpublished data). Unfortunately, what is most commonly seen in this type of study is a very small effect or no effect on the overall process being measured since measuring partial responses is usually not attempted. It is likely that in the case of SCAP1, the fact that partial reactions could not be evaluated influenced the outcome. As a result, it is difficult to decide whether the lack of effect is due to the process not being regulated at this step or whether the step is just one of many regulatory steps, none of which individually have a large effect on the overall process unless paired with other regulatory steps upstream and/or downstream of the step being investigated. This is a real concern especially given the very large number of phosphorylation sites usually measured because of changes in cAMP. Even for the experiment of looking for a putative increase in function by knocking in a glutamic acid (phospho-mimetic substitution), it is not likely that a single phospho-mimetic substitution would have a large effect on overall steroid production. Again, if one can measure the effect of such a knock-in on formation of product immediately downstream of the step being manipulated, then one might expect a much larger percentage effect. Although multiple knock-ins of glutamic acids on multiple proteins in a pathway are technically possible, difficulties in interpretation of such studies accrue with each genetic manipulation, particularly with regard to what control to use. Clearly more work needs to be done in this regard, which will likely require collaborations among multiple laboratories.
Several studies have evaluated the effects of disruption of kinase genes using phospho-proteomic approaches for G protein–modulated pathways. One recent study examined the effect of deletion of PKA on vasopressin V2 receptor–mediated phosphorylation responses (Datta et al., 2020). Another approach used kinase inhibitors bound to Sepharose beads to enrich all kinases and their binding partners in MA-10 cells (Golkowski et al., 2020). Only these latter studies were immediately relevant to cAMP-dependent steroidogenesis, and they implicated many of the same pathways discussed in this review. Similar studies using different kinases and different kinase inhibitors bound to Sepharose beads are quite possible in principle, and the field looks forward to the results of such experiments. See Fig. 7 for a further general discussion of how to evaluate the functional significance of any particular phosphorylation.
Fig. 7.

Approaches for determining phosphorylation function. A number of experimental approaches for determination of the function of a phosphorylation event at any specific site on a protein are listed in this figure. Included is a brief discussion of each approach and some of their limits.
Various combinations of CRISPR-mediated knockout and knock-in models can in theory also be used to evaluate function of individual PDEs, particularly if conducted in a conditional manner. However, all of the caveats relating to knockout and knock-in of protein kinase substrates also apply to this approach.
Results in Jurkat Cells
Most of the data described in this short review are presented for only one cell type. Other minireviews in this volume describe similar phosphoproteomic analyses for G-protein–coupled pathways in different tissues and cell types. In each of those studies, many different proteins are phosphorylated in response to a stimulus. As would be expected, for each cell type and each stimulus a different pattern of phosphorylation is elicited. One additional cell type studied in the authors’ laboratories is the Jurkat T-cell line (Beltejar et al., 2017). In this cell type, a different combination of PDE inhibitors showed cAMP-regulated phosphorylation occurring on components of many different pathways, including RNA processing and transport, actin and microtubule cytoskeletal organization, DNA repair, histone methylation, and T-cell selection. PKA is well known as a major regulator of T-cell function [see chapter by Tasken and colleagues in the series, (Tasken et al., 2021], but exactly how many regulatory mechanisms are modulated by cAMP remains to be determined. Phosphoproteomic analysis is therefore a good approach to answering this question, and several other groups have begun to study the system using a phosphoproteomic approach (Giansanti et al., 2013; Wehbi and Taskén, 2016; Ross and Cantrell, 2018).
As with the MA-10 Leydig cells, in Jurkat cells many different proteins were phosphorylated in response to the increase in cAMP elicited by the PDE inhibitors. Using a nonlabeled phosphoproteomic protocol, over 13,000 phosphopeptides in ∼3400 proteins were identified. These ∼600 phosphopeptides distributed among 340 proteins were substantially regulated by different combinations of PDE inhibitor treatment (Beltejar et al., 2017). Shown in Table 3 is a sample of some of the consensus PKA phosphosites increased by different combinations of PDE inhibitors. As with the MA-10 cells, most of these sites had not previously been reported as potential downstream targets of cAMP action in these cells, although some (e.g., diacylglycerol kinase ζ) are known control sites for regulation of T-cell function (Yang et al., 2021). As might be expected in a cell as complex as a T-cell, the phosphoproteins were distributed among a large number of different pathways that likely benefit from coordination by cAMP/PKA. Note that different combinations of PDE inhibitors gave different quantitative changes in phosphorylation.
TABLE 3.
List of protein phosphosites in Jurkat cells that are increased by different combinations of PDE inhibitors
| Jurkat Cell Data - PGE2 + PDE inhibitors | Fold increase over control | |||||||||
|---|---|---|---|---|---|---|---|---|---|---|
| Gene | Description | Site# | Sequence | PGE2 1 nM | CIL 5 µM | Rol 10 µM | IC BRL PF8 | CIL & ROL | IBMXPF8 50 µM | IBMX PF8 200 µM |
| RANBP2 | E3 Sumo-prot lig RanBP2 | 1509 | PRKQSLPA | 1.7 | 1.2 | 2.4 | 2.2 | 4.5 | 5.0 | 5.2 |
| HIST1H1C | HistoneH1.2 | 36 | PRKASGPP | 1.3 | 1.2 | 2.4 | 1.2 | 4.1 | 3.2 | 3.9 |
| MAGED2 | Melanoma-assoc antig D2 | 200 | ARRASRG | 1.4 | 1.2 | 1.9 | 0.7 | 3.8 | 1.0 | 1.5 |
| SEC22B | Vesicle-traffic prot SEC22b | 137 | RNLGSINT | 1.3 | 1.2 | 1.5 | 1.4 | 3.4 | 3.5 | 3.5 |
| PWP1 | Periodic trypt prot 1 homolog | 485 | ARNSSISGP | 1.5 | 1.2 | 1.8 | 1.3 | 3.3 | 4.4 | 3.2 |
| STMN1 | Stathmin | 63 | ERRKSHEA | 1.2 | 1.2 | 2.2 | 0.4 | 3.3 | 1.6 | 1.9 |
| HIST1H1E | Histone H1.4 | 37 | KRKASGP | 1.6 | 1.2 | 1.6 | 1.9 | 3.2 | 5.6 | 5.4 |
| NUMA1 | Nucl mitotic apparatus pro1 | 1955 | LRRASMQ | 1.3 | 1.2 | 1.4 | 1.5 | 2.9 | 3.6 | 3.6 |
| NFRKB | Nuc factor rel to kappa-B BP | 310 | GRKGSLA | 1.4 | 1.2 | 1.7 | 1.3 | 2.9 | 2.2 | 2.3 |
| CAD | CAD protein - Dihydroorotase | 1343 | GRRLSSFV | 1.3 | 1.2 | 1.5 | 1.4 | 2.9 | 5.2 | 4.1 |
| MKI6 | Antigen KI-67 | 538 | TKRKSLV | 1.0 | 1.0 | 1.3 | 1.1 | 2.5 | 3.3 | 3.0 |
| DGK2 | Diacylglycerol kinase zeta | 163 | LRRASSHL | 1.1 | 1.6 | 1.6 | 0.9 | 2.6 | 1.9 | 1.8 |
The fold response of different inhibitors or combinations of inhibitors on the same site in each protein is shown. All but one protein has a good consensus PKA site. Each PDE inhibitor has an isozyme selectivity of at least 30–200-fold if used at appropriate concentrations. From (Beltejar et al., 2017).
BRL, BRL50481, a PDE7-selective inhibitor; Cil, cilostamide, a PDE3-selective inhibitor; IBMX, isobutyl-methyl-xanthine, a nonselective inhibitor that targets all PDEs except PDE8; IC, ITI-078, a PDE1-selective inhibitor; PF8, PF-04957325, a selective PDE8 inhibitor; PGE2, prostaglandin E2; Rol, rolipram, a PDE4 selective inhibitor.
Figure 8 shows a STRING/GO analysis of the phosphoproteins modulated by PDE inhibitor treatments (Szklarczyk et al., 2019). A major conclusion from these analyses is that many different sites potentially regulating a number of different processes are phosphorylated in response to the increase in cAMP levels. As with steroidogenesis, it appears that not one site but rather multiple sites within a pathway or process are regulated. However, all of the same concerns and caveats discussed in relation to cAMP control of steroidogenesis are relevant to this cell type, including proof of the effects of phosphorylation on function. Phosphoproteomic raw data from these studies have been deposited with the Mass Spectrometry Interactive Virtual Environment maintained by the Center for Computational Mass Spectrometry in the Computer Science and Engineering Department of the University of California, San Diego (MSV000081115).
Fig. 8.

Vesicle transport in Jurkat cells. Gene ontology analysis of phosphoproteomic data showing protein phosphorylation events likely involved in cAMP-dependent control of vesicle transport. Note that the combination of PDE inhibitors indicated caused a change in each of these phosphosites. Only a few have been previously implicated as being regulatory sites for cAMP control of the processes listed. The filled circles indicate the process likely being regulated. The boxes indicate the gene/protein being altered. Original data from Beltejar et al. (2017).
Implications of Data for Mechanisms of Drug Action and Drug Design.
If one accepts that many metabolic processes and signaling pathways really are regulated by the actions of cAMP/PKA at multiple (perhaps many) different sites, then this concept has important implications for understanding the actions of many drugs. Perhaps most importantly, it suggests that to stimulate a process with an agonist, one must target either a regulatory signaling pathway that coordinates multiple regulatory sites, or possibly one could stimulate the last step of the pathway if enough substrate is available. Even this latter approach likely will not work if the previous steps of the pathway already limit the final output of the process. This would be true for either drug or genetic manipulation. Of course, an antagonist can make any step in the process rate-limiting and regulatory. Perhaps it is no wonder that most current drugs are antagonists and not direct pathway agonists.
To What Metabolic and Regulatory Pathways Do the Phosphoproteomic Studies and Analyses Described in This Minireview Most Relate?
Almost all of the data selected for examples in this minireview have been taken from metabolic or regulatory pathways and processes that are carried out on intracellular organelles, such as, for example, the endosomes, endoplasmic reticulum, Golgi, etc. In part this was because many of the sites with the greatest changes in phosphorylation occurred in peptides associated with these pathways. However, such unbiased studies are equally useful for analyses of more “classical” pathways, many of which are located in other organelles, such as the nucleus or on the ribosome, or are thought to be “soluble” in nature. Of particular relevance are those likely to be organized into discrete functional compartments by anchoring proteins or other mechanisms. For example, in the MA-10 cell studies, over 700 different phosphosites were modulated by one or more PDE inhibitors. In the Jurkat cells, over 600 sites were modulated, albeit by different PDE inhibitors. Moreover, most of these sites were different than those seen in the MA-10 cells, as might be expected for a different cell type. Eventually, when more studies have been carried out using the same standard stimuli and the same mass-spectrometry protocols, it will be of interest to determine whether there are sets of phosphosites common to each PDE inhibitor (or combination of inhibitors). Similarly, it will be of great interest to see how many sites are common among different cell types stimulated by the same drugs. At present, there are just not enough published data to evaluate these questions properly.
Summary
It is hoped that this short review encourages the reader to consider use of stimulus-driven phosphoproteomic analysis of the signaling and metabolic pathways that they are most interested in understanding. It is also hoped that the discussions of the advantages and difficulties of this approach will inform future studies not just on cAMP and PDE regulation of cellular function but also on many other regulatory processes. From the initial phosphoproteomic analyses performed in our and other laboratories, the authors have proposed that many metabolic processes do not have single, rate-limiting steps. This is particularly true when many of the pathways identified are cyclical in nature or are compartmentalized or scaffolded. Rather, the cell appears to have chosen to coordinate regulation of many, many steps along the pathway. The use of wide-scale, unbiased phosphoproteomic analysis has allowed a first step in identifying some of these previously unidentified regulatory steps. It is gratifying that initial studies have already detected a large number of new candidate regulatory sites. It is now our responsibility to determine which of these sites are really most important for cellular regulation. It may be that many of the steps in many pathways are rate-limiting until changed in their phosphorylation state by kinase or phosphatase activity.
Acknowledgments
The authors would like to thank Le-Chun Lisa Tsai, Brian van Yserloo, Ho-Tak Lau, and Danny Suh for their assistance with some of the described studies.
Abbreviations
- CEH
cholesteryl ester hydrolase
- ER
endoplasmic reticulum
- GAP
guanine nucleotide protein activation factor
- GEF
guanine nucleotide protein exchange factor
- GO
gene ontology
- LDL
low-density lipoprotein
- P450
cytochrome P450
- PDE
cyclic nucleotide phosphodiesterase
- phospho-
phosphorylated
- PKA
cAMP-dependent protein kinase
- SCAP
SREBP cleavage–activating protein
- SREBP
sterol regulatory element–binding protein
- StAR
steroidogenic acute regulatory protein
Authorship Contributions
Participated in research design: Beavo, Golkowski, Shimizu-Albergine, Beltejar, Bornfeldt, Ong.
Conducted experiments: Golkowski, Shimizu-Albergine, Beltejar.
Contributed new reagents or analytic tools: Golkowski, Shimizu-Albergine, Beltejar, Bornfeldt, Ong.
Performed data analysis: Beavo, Golkowski, Shimizu-Albergine, Beltejar, Bornfeldt, Ong.
Wrote or contributed to the writing of the manuscript: Beavo, Golkowski, Beltejar, Bornfeldt.
Footnotes
This work was supported by National Institutes of Health National Institute of General Medical Sciences [Grant R01-GM083926] (to J.B.); National Heart, Lung, and Blood Institute [Grant R35-HL150754] and National Institute of Diabetes and Digestive and Kidney Diseases [Grant P30-DK017047] (to K.B. and M.S.); and National Institute of General Medical Sciences [Grants R01-GM086858 and R01-GM129090], National Institute of Arthritis and Musculoskeletal and Skin Diseases [Grant R01-AR065459], National Institute of Biomedical Imaging and Bioengineering [Grant R21-EB018384], and National Cancer Institute [Grants R21-CA177402 and K22-CA201229-01] (to S.O. and M.G.).
1ASPET Axelrod Symposium 2019, Phosphoproteomic analysis of G-protein coupled pathways. Experimental Biology Meeting, Orlando, FL.
Much of this work was presented at the following meeting: 2019 Axelrod Symposium of the American Society of Pharmacology and Experimental Therapeutics, Experimental Biology Meeting; 2019; Orlando, FL. “Phosphoproteomic analysis of G-protein coupled pathways.”
References
- Adame-García SR, Cervantes-Villagrana RD, Orduña-Castillo LB, Del Rio JC, Gutkind JS, Reyes-Cruz G, Taylor SS, Vázquez-Prado J (2019) cAMP-dependent activation of the Rac guanine exchange factor P-REX1 by type I protein kinase A (PKA) regulatory subunits. J Biol Chem 294:2232–2246. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ashman DF, Lipton R, Melicow MM, Price TD (1963) Isolation of adenosine 3′, 5′-monophosphate and guanosine 3′, 5′-monophosphate from rat urine. Biochem Biophys Res Commun 11:330–334. [DOI] [PubMed] [Google Scholar]
- Banan M (2020) Recent advances in CRISPR/Cas9-mediated knock-ins in mammalian cells. J Biotechnol 308:1–9. [DOI] [PubMed] [Google Scholar]
- Beavo JA, Brunton LL (2002) Cyclic nucleotide research -- still expanding after half a century. Nat Rev Mol Cell Biol 3:710–718. [DOI] [PubMed] [Google Scholar]
- Beltejar MG, Lau HT, Golkowski MG, Ong SE, Beavo JA (2017) Analyses of PDE-regulated phosphoproteomes reveal unique and specific cAMP-signaling modules in T cells. Proc Natl Acad Sci USA 114:E6240–E6249. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bock A, Annibale P, Konrad C, Hannawacker A, Anton SE, Maiellaro I, Zabel U, Sivaramakrishnan S, Falcke M, Lohse MJ (2020) Optical mapping of cAMP signaling at the nanometer scale. Cell 182:1519–1530.e17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brand T, Schindler R (2017) New kids on the block: the Popeye domain containing (POPDC) protein family acting as a novel class of cAMP effector proteins in striated muscle. Cell Signal 40:156–165. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Capponi AM (2002) Regulation of cholesterol supply for mineralocorticoid biosynthesis. Trends Endocrinol Metab 13:118–121. [DOI] [PubMed] [Google Scholar]
- Catterall WA (2015) Regulation of cardiac calcium channels in the fight-or-flight response. Curr Mol Pharmacol 8:12–21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chaineau M, Ioannou MS, McPherson PS (2013) Rab35: GEFs, GAPs and effectors. Traffic 14:1109–1117. [DOI] [PubMed] [Google Scholar]
- Chen S, Yan C (2021) An update of cyclic nucleotide phosphodiesterase as a target for cardiac diseases. Expert Opin Drug Discov 16(2):183–196. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Datta A, Yang CR, Limbutara K, Chou CL, Rinschen MM, Raghuram V, Knepper MA (2020) PKA-independent vasopressin signaling in renal collecting duct. FASEB J 34:6129–6146. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Deshpande V, Kao A, Raghuram V, Datta A, Chou CL, Knepper MA (2019) Phosphoproteomic identification of vasopressin V2 receptor-dependent signaling in the renal collecting duct. Am J Physiol Renal Physiol 317:F789–F804. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Esseltine JL, Scott JD (2013) AKAP signaling complexes: pointing towards the next generation of therapeutic targets? Trends Pharmacol Sci 34:648–655. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fryer LG, Jones B, Duncan EJ, Hutchison CE, Ozkan T, Williams PA, Alder O, Nieuwdorp M, Townley AK, Mensenkamp AR, et al. (2014) The endoplasmic reticulum coat protein II transport machinery coordinates cellular lipid secretion and cholesterol biosynthesis. J Biol Chem 289:4244–4261. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gao M, Huang X, Song BL, Yang H (2019) The biogenesis of lipid droplets: lipids take center stage. Prog Lipid Res 75:100989. [DOI] [PubMed] [Google Scholar]
- Giansanti P, Stokes MP, Silva JC, Scholten A, Heck AJR (2013) Interrogating cAMP-dependent kinase signaling in Jurkat T cells via a protein kinase A targeted immune-precipitation phosphoproteomics approach. Mol Cell Proteomics 12:3350–3359. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Golkowski M, Shimizu-Albergine M, Suh HW, Beavo JA, Ong SE (2016) Studying mechanisms of cAMP and cyclic nucleotide phosphodiesterase signaling in Leydig cell function with phosphoproteomics. Cell Signal 28:764–778. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Golkowski M, Vidadala VN, Lau HT, Shoemaker A, Shimizu-Albergine M, Beavo J, Maly DJ, Ong SE (2020) Kinobead/LC-MS phosphokinome profiling enables rapid analyses of kinase-dependent cell signaling networks. J Proteome Res 19:1235–1247. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guidetti GF, Manganaro D, Consonni A, Canobbio I, Balduini C, Torti M (2013) Phosphorylation of the guanine-nucleotide-exchange factor CalDAG-GEFI by protein kinase A regulates Ca(2+)-dependent activation of platelet Rap1b GTPase. Biochem J 453:115–123. [DOI] [PubMed] [Google Scholar]
- Hofmann F (2020) The cGMP system: components and function. Biol Chem 401:447–469. [DOI] [PubMed] [Google Scholar]
- Homma Y, Hiragi S, Fukuda M (2020) Rab family of small GTPases: an updated view on their regulation and functions. FEBS J 288:36–55. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Imachi H, Murao K, Sayo Y, Hosokawa H, Sato M, Niimi M, Kobayashi S, Miyauchi A, Ishida T, Takahara J (1999) Evidence for a potential role for HDL as an important source of cholesterol in human adrenocortical tumors via the CLA-1 pathway. Endocr J 46:27–34. [DOI] [PubMed] [Google Scholar]
- Krebs EG, Beavo JA (1979) Phosphorylation-dephosphorylation of enzymes. Annu Rev Biochem 48:923–959. [DOI] [PubMed] [Google Scholar]
- Kulasekaran G, Nossova N, Marat AL, Lund I, Cremer C, Ioannou MS, McPherson PS (2015) Phosphorylation-dependent regulation of connecdenn/DENND1 guanine nucleotide exchange factors. J Biol Chem 290:17999–18008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lamber EP, Siedenburg AC, Barr FA (2019) Rab regulation by GEFs and GAPs during membrane traffic. Curr Opin Cell Biol 59:34–39. [DOI] [PubMed] [Google Scholar]
- Leslie SN, Nairn AC (2019) cAMP regulation of protein phosphatases PP1 and PP2A in brain. Biochim Biophys Acta Mol Cell Res 1866:64–73. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu JJ, Sharma K, Zangrandi L, Chen C, Humphrey SJ, Chiu YT, Spetea M, Liu-Chen LY, Schwarzer C, Mann M (2018) In vivo brain GPCR signaling elucidated by phosphoproteomics. Science 360:eaao4927. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lugnier C, Meyer A, Talha S, Geny B (2020) Cyclic nucleotide phosphodiesterases: new targets in the metabolic syndrome? Pharmacol Ther 208:107475. [DOI] [PubMed] [Google Scholar]
- Lutz S, Mohl M, Rauch J, Weber P, Wieland T (2013) RhoGEF17, a Rho-specific guanine nucleotide exchange factor activated by phosphorylation via cyclic GMP-dependent kinase Iα. Cell Signal 25:630–638. [DOI] [PubMed] [Google Scholar]
- Makhoul S, Walter E, Pagel O, Walter U, Sickmann A, Gambaryan S, Smolenski A, Zahedi RP, Jurk K (2018) Effects of the NO/soluble guanylate cyclase/cGMP system on the functions of human platelets. Nitric Oxide 76:71–80. [DOI] [PubMed] [Google Scholar]
- Manna PR, Stocco DM (2005) Regulation of the steroidogenic acute regulatory protein expression: functional and physiological consequences. Curr Drug Targets Immune Endocr Metabol Disord 5:93–108. [DOI] [PubMed] [Google Scholar]
- Manoury B, Idres S, Leblais V, Fischmeister R (2020) Ion channels as effectors of cyclic nucleotide pathways: functional relevance for arterial tone regulation. Pharmacol Ther 209:107499. [DOI] [PubMed] [Google Scholar]
- Maurice DH, Ke H, Ahmad F, Wang Y, Chung J, Manganiello VC (2014) Advances in targeting cyclic nucleotide phosphodiesterases. Nat Rev Drug Discov 13:290–314. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Midzak A, Papadopoulos V (2016) Adrenal mitochondria and steroidogenesis: from individual proteins to functional protein assemblies. Front Endocrinol (Lausanne) 7:106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nagy Z, Wynne K, von Kriegsheim A, Gambaryan S, Smolenski A (2015) Cyclic nucleotide-dependent protein kinases target ARHGAP17 and ARHGEF6 complexes in platelets. J Biol Chem 290:29974–29983. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Novick P (2016) Regulation of membrane traffic by Rab GEF and GAP cascades. Small GTPases 7:252–256. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ong SE, Blagoev B, Kratchmarova I, Kristensen DB, Steen H, Pandey A, Mann M (2002) Stable isotope labeling by amino acids in cell culture, SILAC, as a simple and accurate approach to expression proteomics. Mol Cell Proteomics 1:376–386. [DOI] [PubMed] [Google Scholar]
- Ortiz Sandoval C, Simmen T (2012) Rab proteins of the endoplasmic reticulum: functions and interactors. Biochem Soc Trans 40:1426–1432. [DOI] [PubMed] [Google Scholar]
- Osawa J, Akizuki K, Kashimura A, Ueta S, Nakatani M, Inui Y, Shigeri Y, Ishida A, Kameshita I, Sueyoshi N (2020) Dual phosphorylation of protein phosphatase PPM1H promotes dephosphorylation of Smad1 in cellulo. Biochem Biophys Res Commun 530:513–519. [DOI] [PubMed] [Google Scholar]
- Pfisterer SG, Peränen J, Ikonen E (2016) LDL-cholesterol transport to the endoplasmic reticulum: current concepts. Curr Opin Lipidol 27:282–287. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pomaznoy M, Ha B, Peters B (2018) GOnet: a tool for interactive Gene Ontology analysis. BMC Bioinformatics 19:470. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Progida C (2019) Multiple roles of Rab GTPases at the Golgi. Results Probl Cell Differ 67:95–123. [DOI] [PubMed] [Google Scholar]
- Rider MH, Waelkens E, Derua R, Vertommen D (2009) Fulfilling the Krebs and Beavo criteria for studying protein phosphorylation in the era of mass spectrometry-driven kinome research. Arch Physiol Biochem 115:298–310. [DOI] [PubMed] [Google Scholar]
- Robichaux WG 3rd, Cheng X (2018) Intracellular cAMP sensor EPAC: physiology, pathophysiology, and Therapeutics development. Physiol Rev 98:919–1053. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ross SH, Cantrell DA (2018) Signaling and function of interleukin-2 in T lymphocytes. Annu Rev Immunol 36:411–433. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Salhadar K, Matthews A, Raghuram V, Limbutara K, Yang CR, Datta A, Chou CL, Knepper MA (2021) Phosphoproteomic identification of vasopressin/cAMP/PKA-dependent signaling in kidney. Mol Pharmacol 99(5):358–369. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schleicher K, Zaccolo M (2021) Defining a cellular map of cAMP nanodomains. Mol Pharmacol 99(5):383–391. [DOI] [PubMed] [Google Scholar]
- Sewer MB, Li D (2008) Regulation of steroid hormone biosynthesis by the cytoskeleton. Lipids 43:1109–1115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sharma RK, Ahmed NK, Sutliff LS, Brush JS (1974) Metabolic regulation of steroidogenesis in isolated adrenal cells of the rat. ACTH regulation of cGMP and cAMP levels and steroidogenesis. FEBS Lett 45:107–110. [DOI] [PubMed] [Google Scholar]
- Shimizu-Albergine M, Tsai LC, Patrucco E, Beavo JA (2012) cAMP-specific phosphodiesterases 8A and 8B, essential regulators of Leydig cell steroidogenesis. Mol Pharmacol 81:556–566. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shimizu-Albergine M, Van Yserloo B, Golkowski MG, Ong SE, Beavo JA, Bornfeldt KE (2016) SCAP/SREBP pathway is required for the full steroidogenic response to cyclic AMP. Proc Natl Acad Sci USA 113:E5685–E5693. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smith LC, Ralston-Hooper KJ, Ferguson PL, Sabo-Attwood T (2016) The G protein-coupled estrogen receptor agonist G-1 inhibits nuclear estrogen receptor activity and stimulates novel phosphoproteomic signatures. Toxicol Sci 151:434–446. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Soutar AK, Naoumova RP (2007) Mechanisms of disease: genetic causes of familial hypercholesterolemia. Nat Clin Pract Cardiovasc Med 4:214–225. [DOI] [PubMed] [Google Scholar]
- Stocco DM, Wang X, Jo Y, Manna PR (2005) Multiple signaling pathways regulating steroidogenesis and steroidogenic acute regulatory protein expression: more complicated than we thought. Mol Endocrinol 19:2647–2659. [DOI] [PubMed] [Google Scholar]
- Sutherland EW, Rall TW (1958) Fractionation and characterization of a cyclic adenine ribonucleotide formed by tissue particles. J Biol Chem 232:1077–1091. [PubMed] [Google Scholar]
- Szklarczyk D, Gable AL, Lyon D, Junge A, Wyder S, Huerta-Cepas J, Simonovic M, Doncheva NT, Morris JH, Bork P, et al. (2019) STRING v11: protein-protein association networks with increased coverage, supporting functional discovery in genome-wide experimental datasets. Nucleic Acids Res 47:D607–D613. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tsai LC, Shimizu-Albergine M, Beavo JA (2011) The high-affinity cAMP-specific phosphodiesterase 8B controls steroidogenesis in the mouse adrenal gland. Mol Pharmacol 79:639–648. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Volkmar N, Thezenas ML, Louie SM, Juszkiewicz S, Nomura DK, Hegde RS, Kessler BM, Christianson JC (2019) The ER membrane protein complex promotes biogenesis of sterol-related enzymes maintaining cholesterol homeostasis. J Cell Sci 132:jcs223453. [DOI] [PMC free article] [PubMed] [Google Scholar]
- von Zastrow M (2021) Proteomic approaches to investigate regulated trafficking and signaling of GPCRs. Mol Pharmacol 99(5):392–398. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Walseth TF, Gander JE, Eide SJ, Krick TP, Goldberg ND (1983) 18O labeling of adenine nucleotide alpha-phosphoryls in platelets. Contribution by phosphodiesterase-catalyzed hydrolysis of cAMP. J Biol Chem 258:1544–1558. [PubMed] [Google Scholar]
- Wehbe N, Nasser SA, Al-Dhaheri Y, Iratni R, Bitto A, El-Yazbi AF, Badran A, Kobeissy F, Baydoun E, Eid AH (2020) EPAC in vascular smooth muscle cells. Int J Mol Sci 21:5160. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wehbi VL, Taskén K (2016) Molecular mechanisms for cAMP-mediated immunoregulation in T cells - role of anchored protein kinase A signaling units. Front Immunol 7:222. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wilhelm LP, Wendling C, Védie B, Kobayashi T, Chenard MP, Tomasetto C, Drin G, Alpy F (2017) STARD3 mediates endoplasmic reticulum-to-endosome cholesterol transport at membrane contact sites. EMBO J 36:1412–1433. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Williams GR, Bethard JR, Berkaw MN, Nagel AK, Luttrell LM, Ball LE (2016) Exploring G protein-coupled receptor signaling networks using SILAC-based phosphoproteomics. Methods 92:36–50. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wong M, O’Brien D, Schimmer BP (1986) The roles of cAMP and cAMP-dependent protein kinase in the regulation of adrenocortical functions: analysis using DNA-mediated gene transfer. Biochem Cell Biol 64:1066–1071. [DOI] [PubMed] [Google Scholar]
- Wu R, Haas W, Dephoure N, Huttlin EL, Zhai B, Sowa ME, Gygi SP (2011) A large-scale method to measure absolute protein phosphorylation stoichiometries. Nat Methods 8:677–683. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang J, Wang HX, Xie J, Li L, Wang J, Wan ECK, Zhong XP (2020) DGK α and ζ activities control TH1 and TH17 cell differentiation. Front Immunol 10:3048. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang T, Terman JR (2013) Regulating small G protein signaling to coordinate axon adhesion and repulsion. Small GTPases 4:34–41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhou GL, Na SY, Niedra R, Seed B (2014) Deficits in receptor-mediated endocytosis and recycling in cells from mice with Gpr107 locus disruption. J Cell Sci 127:3916–3927. [DOI] [PubMed] [Google Scholar]
- Zuo H, Cattani-Cavalieri I, Musheshe N, Nikolaev VO, Schmidt M (2019) Phosphodiesterases as therapeutic targets for respiratory diseases. Pharmacol Ther 197:225–242. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Data Availability Statement
Time to quenching is a major issue for studying all cAMP-regulated reactions since this cyclic nucleotide can turn over so quickly in the cell (Walseth et al., 1983). Similarly, phosphorylation/dephosphorylation of many sites may occur within milliseconds of stimulus (Catterall, 2015). All of the studies reported here were carried out at multiple time points after addition of the stimulus. The reactions were quenched with either ice-cold PBS and freshly prepared Tris-buffered 8-M urea containing a protease and phosphatase inhibitor cocktail (MA-10 cells) or boiling 6-M guanidinium hydrochloride (Jurkat cells). Nevertheless, given that kinases, phosphatases, and phosphodiesterases can be scaffolded together often in stoichiometric amounts with the cAMP and that kinase, phosphatase, cyclase, and phosphodiesterase activity must be quenched equally quickly, one is never quite sure whether stimulus and quench conditions are sufficient. Correspondingly, some of the interpretations of most phosphoproteomic results, including these, likely need to be understood with these caveats in mind. This is particularly true for modulation of pathways that cycle rapidly. Most of the data discussed in this review are recorded in public data bases. The MA-10 data mass spectrometry raw files and MaxQuant/Andromeda search results were deposited in the publicly available mass spectrometry data repositories Mass Spectrometry Interactive Virtual Environment (MSV000079412; http://www.massive.ucsd.edu.offcampus.lib.washington.edu/) and ProteomeXchange (PXD003280; http://www.proteomexchange.org/).