

ABSTRACT
Osteopetrosis (OP) is a group of rare genetic bone disorders. Osteoclast-poor form of osteopetrosis is much rarer in humans and represents a small percentage of the total cases of autosomal recessive osteopetrosis presenting with impaired bone remodeling due to defective osteoclastic activity and is characterized by distinctive increase in bone density and high bone fragility. Reduction in marrow spaces with decreased vasculature to the bone owing to increased bone mass makes the bones vulnerable for varied infections resulting in osteomyelitis. This case report discusses challenges in management of recalcitrant osteomyelitis of mandible developed as a complication in an 8-year-old girl child identified with rare, dual heterozygous mutations in RANKL (TNFSF11) gene and COL5A1 gene with uncertain significance responsible for osteoclast-poor osteopetrosis and Classic Ehlers-Danlos, respectively.
How to cite this article
Sharma A, Ingole SN, Deshpande MD, et al. A Rare Case of Osteoclast-poor Osteopetrosis (RANKL Mutation) with Recurrent Osteomyelitis of Mandible: A Case Report. Int J Clin Pediatr Dent 2020;13(6):717–721.
Keywords: Osteoclast, Osteoclast-poor, Osteomyelitis, Osteopetrosis.
INTRODUCTION
Osteopetrosis (OP) encompasses a group of rare genetic disease that affects bone remodeling, resulting in increased bone density. There are three genetically distinct forms of osteopetrosis, autosomal dominant, autosomal recessive, and X-linked, having variable phenotypes and prognosis owing to the heterogeneity of genetic defects.1 All forms are characterized by defective bone resorption due to mutation in the genes rendering osteoclasts either deficient or nonfunctional and results in disruption of normal bone remodeling. Based on osteoclast phenotype, ARO has two types: Osteoclast-rich has normal or more but nonfunctional osteoclast; this includes majority of patients. Osteoclast-poor exhibits no osteoclasts. Osteoclast-poor is a very rare form of autosomal recessive osteopetrosis that occurs due to loss-of-function mutations in the RANKL gene (TNFSFS11) and RANK gene (TNFRSF11A) and results in an osteoclast-poor form.2,3 This report provides a comprehensive review of osteoclast-poor osteopetrosis and management of jaw osteomyelitis, a well-known and often troublesome complication of osteopetrosis.
CASE DESCRIPTION
An 8-year-old girl had reported to the Department of Oral and Maxillofacial Surgery with the complaint of swelling and pus discharge on the left side of mandible for 4 months.
Medical History
The patient was born at 38 weeks’ of gestation by vacuum-assisted vaginal delivery and stayed at neonatal intensive care unit for the initial few days. At first months of birth, she suffered from lower respiratory tract infection. She reportedly had rapidly increasing head circumference at 9 months of age with hydrocephalus. Ventriculo-peritoneal shunt was established to relive the intracranial pressure at 11 months of age. She developed umbilical hernia at 2 years of age for which she was operated. From birth to 7 years of age, the patient had fracture of both the fore arm, ankle, humerus, and hip bone at various intervals with delayed healing. She suffered from visual disturbances and lost vision from left eye. She had chronic nasal stuffiness and recurrent lower respiratory tract infection. The patient had delayed motor milestones and reportedly started walking at the age of 3 years. The family history is positive for parental consanguinity. Both the parents are healthy, and she has a healthy sister 2 years elder to her. History of person with developmental disorder was present in both maternal and paternal families.
Physical Findings
The patient had a short stature, high forehead, bossing of frontal and parietal bones, blue sclera, and eyes prominent bilaterally with bilateral proptosis. Chest shape was abnormal, and pectus carinatum deformity was present (Fig. 1A). Facial examination revealed facial asymmetry with facial dysmorphism. Diffuse swelling of the mandible from chin region extending to the left mandibular body region(Fig. 2A) with multiple draining fistula and intermittent pus discharge (Fig. 2B). Anodontia with multiple draining sinus on mandibular alveolar ridge was present (Fig. 2C).
Figs 1A and B.

(A) Pectus carinatum chest deformity with; (B) Sclerotic clavicles and ribs, arrow showing fracture at proximal end left humerus
Figs 2A to C.

(A) Frontal view: frontal and parietal bossing with bilateral proptosis, diffuse swelling of the left mandibleb; (B) Extraoral draining sinus; (C) Intraoral draining sinus
Neurological Findings
Patient had normal intellectual level with no symptoms of mental retardation. She suffered from visual disturbances and lost vision from the left eye. Audiometry had not been performed, but she seemed to hear well and had clear speech. Weakness of facial nerve on left side of the face was present.
Radiological Findings
Computed tomography (CT) of the face showed diffuse thickening and sclerosis of all skull and facial bones sclerotic skull base and maxilla and mandible as well as cervical vertebrae with obliteration of the medullary spaces and loss of corticomedullary differentiation. (Fig. 3A). Poorly pneumatized paranasal sinuses were seen. CT brain revealed a bur hole defect in the right parietal bone with VP shunt in situ (Fig. 3B). Chest X-ray revealed the sclerotic clavicles with widened medial ends and broad and diffusely sclerotic ribs (Fig. 1B). Panoramic radiograph revealed dense sclerotic maxilla and mandible, loss of corticomedullary differentiation, and poorly pneumatized maxillary sinus. Generalized alveolar bone loss and multiple deformed tooth bud with reduced follicle space. Regions of ill-defined mixed radio-opaque radiolucency in mandibular symphysis parasymphysis region appearing as moth-eaten appearance (Fig. 4).
Figs 3A and B.

(A) Frontal view CT face showing diffuse thickening and sclerosis of all skull and facial bones; (B) Axial view CT brain showing a VP shunt in situ
Fig. 4.
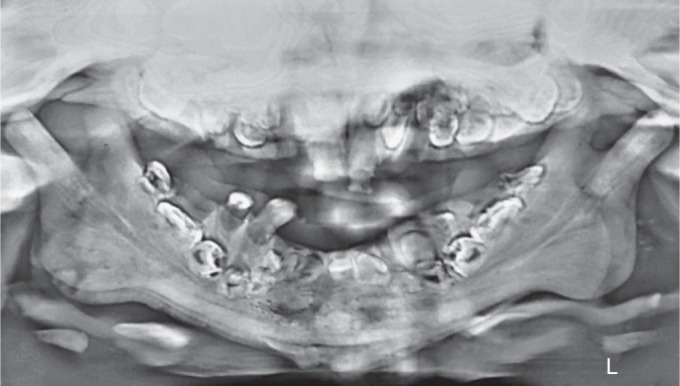
Panoramic radiograph showing multiple deformed tooth bud with reduced follicle space. Osteomyelitis appearing as ill-defined mixed radio-opaque radiolucency in mandibular symphysis parasymphysis
Biochemical Studies
Blood studies showed hemoglobin ranging between 8 and 9 g%, TLC 6780 cu/mm (neutrophils 58 %, lymphocytes 34%), and alkaline phosphatase 8.9 K.A.U/100 mL.
Mutation Analyses
Based on the clinical feature and medical history, she was evaluated for pathogenic variations in multiple genes. Targeted gene sequencing method was used for analyzing specific mutations and to detect variants present in multiple genes that have known or suspected associations with the disease or phenotype under study. A heterozygous missense variation in exon 5 of the TNFSF11 (RANKL) gene was detected. This variation is classified as a variant of osteoclast-poor form of osteopetrosis. A heterozygous missense variation in exon 23 of the COL5A1 gene was detected. This COL5A1 variation is classified as a variant of Ehlers-Danlos syndrome of uncertain significance. Specific phenotype due to mutation in COL5A1 gene was not prominent in this patient.
Management
Initially, she was prescribed oral systemic antibiotic regimen (amoxiclav, Metronidazol). Discharge from the draining sinus was sent for culture and sensitivity which was inconclusive. Limited surgical intervention was planned, and curettage and excision of the intra-roral and extraoral draining sinus were done. Patient was kept on follow-up, and after 4 months she again reported with draining sinus from the same region (Fig. 5A). She was then submitted to four sessions of hyperbaric oxygen (HBO) therapy each lasting for about 30–45 minutes at 1.5-atmosphere pressure and surgical debridement of the lesion and daily irrigation of the localized region with antibiotic (Fig. 5B). Subsequently, the patient received the same antibiotic by oral administration for 7 days followed by four sessions of HBO therapy. She also received hematinic, vitamin D, and calcitriol on pediatric medicine and endocrinology consultation. Remission achieved for a brief period (Fig. 5C). However, the patient experienced frequent exacerbation and remission, and the condition maintained a chronic course. After 7 months acute exacerbation occurred following which she was administered intravenous linezolid on, and again corticotomy and sequestrectomy with debridement of the necrotic bone were performed (Fig. 6). Currently, the patient is under constant follow-up.
Figs 5A to C.

(A) Recurrence of draining sinus after excision; (B) Excision of cutaneous fistula with drain in situ; (C) Remission after excision of cutaneous fistula and hyperbaric oxygen therapy
Figs 6A and B.

(A) Debridement with corticotomy and sequestrectomy; (B) Necrotic bone
DISCUSSION
Osteoclast-poor form of individuals present with severe osteopetrosis, but the disease progression is slower in comparison to classical ARO. In the absence of functional osteoclasts, unhindered osteoblastic activity results into exceedingly dense bone architecture that eventually results in bone marrow failure; however, surprisingly those affected with osteoclast-poor form still show evidence of active hematopoiesis, which explains their longer survival despite severe osteopetrosis.4 Hematopoietic stem cell transplantation (HSCT) early in life is reported to be beneficial in classical ARO,5 but in RANKL deficiency, the hematopoietic stem cells cannot replace the stromal/osteoblast cells that produce the mutant RANKL; hence, the role of HSCT in osteoclast-poor osteopetrosis (RANKL deficiency ) is still under investigation in humans and presently has no therapeutic value.6 Alternative therapies are used as HSCT is not possible. However, because they are not curative interventions, they cannot replace HSCT. Corticosteroids, high-dose calcitriol with low calcium diet, erythropoietin, 1.25-dihydroxy vitamin D, and interferon-γ are used alone or in combination to improve hematological function, reduce osteosclerotic disease, and improve immune function.7 Jaw osteomyelitis especially in mandible is a significant and often troublesome complication, especially in osteoclast-poor osteopetrosis, as it attains chronic course and is stubborn against all the therapies. The primary contributing factors to the development of osteomyelitis are polymicrobial odontogenic infections. The odontogenic pathogens usually consist chiefly of Staphylococcus aureus, and other organisms like bacteroides, Peptostreptococcus, and microaerophilic Streptococcus species have been isolated.8 Selection and administration of appropriate antibiotic therapy are crucial, as these patients require prolonged systemic antibiotic therapy for maintenance of therapeutic levels in overly dense bone. Commonly used antibiotic regimens include amoxicillin and clavulanic acid, metronidazole, levofloxacin, and clindamycin. Resistant infection refractory to common antibiotics requires addition of higher antibiotics such as ciprofloxacin, linezolid, vancomycin, etc.9 Adjuvant use of antimicrobial therapy along with hyperbaric oxygen reportedly has very beneficial role in recalcitrant osteomyelitis. An increased tension of oxygen decreases the required effective concentration of antibiotics. Increased blood oxygen tension enhances osteomyelitis healing through a direct bactericidal effect and increases fibroblastic and leukocytic activity.10 Osteopetrotic patient should be carefully and judicially subjected to surgical interventions. The wound healing properties of bone in these patients are limited, since the primary blood supply to the mandible comes from the periosteum, and it should be minimally disturbed to prevent further complication. In the present case, in order to limit further damage, conservative dissection and surgical debridement with sequestrectomy of necrotic bone was done. Despite administrations of appropriate antimicrobials, and hyperbaric oxygen therapy, and surgical debridement, the patient experienced frequent exacerbation and remission of osteomyelitis of the mandible for the last 12 months.
CONCLUSION
Since osteopetrosis is not just limited to the bones and rather affect other structures structurally and functionally that may result into complications that are difficult to manage, it is necessary to individualize effective therapies to prevent them. In order to maintain their oral health status and prevent devastating complications such as osteomyelitis, these patients should receive increased attention with frequent oral hygiene procedures and prophylactic dental treatment. Management of jaw osteomyelitis in these patients is challenging owing to the altered bone morphology. Appropriate antibiotic therapy combined with hyperbaric oxygen therapy to potentiate the effect of antibiotic therapy is the main stay treatment. Recurrent infection often makes surgical intervention necessary.
Footnotes
Source of support: Nil
Conflict of interest: None
REFERENCES
- 1.Del Fattore A, Cappariello A, Teti A. Genetics, pathogene sis and complications of osteopetrosis. Bone. 2008;;42((1):):19––29.. doi: 10.1016/j.bone.2007.08.029. DOI: [DOI] [PubMed] [Google Scholar]
- 2.Villa A, Guerrini MM, Cassani B, et al. Infantile malignant, autosomal recessive osteopetrosis: the rich and the poor. Calcif Tissue Int. 2009;;84((1):):1.. doi: 10.1007/s00223-008-9196-4. DOI: [DOI] [PubMed] [Google Scholar]
- 3.Teti A, Econs MJ. Osteopetroses, emphasizing potential approaches to treatment. Bone. 2017;;102::50––59.. doi: 10.1016/j.bone.2017.02.002. DOI: [DOI] [PubMed] [Google Scholar]
- 4.Sobacchi C, Frattini A, Guerrini MM, et al. Osteoclast-poor human osteopetrosis due to mutations in the gene encoding RANKL. Nat Genet. 2007;;39((8):):960.. doi: 10.1038/ng2076. DOI: [DOI] [PubMed] [Google Scholar]
- 5.Driessen GJ, Gerritsen EJ, Fischer A, et al. Long-term outcome of haematopoietic stem cell transplantation in autosomal recessive osteopetrosis: an EBMT report. Bone Marrow Transplant. 2003;;32((7):):657.. doi: 10.1038/sj.bmt.1704194. DOI: [DOI] [PubMed] [Google Scholar]
- 6.Pangrazio A, Cassani B, Guerrini MM, et al. RANK-dependent autosomal recessive osteopetrosis: characterization of five new cases with novel mutations. J Bone Miner Res. 2012;;27((2):):342––351.. doi: 10.1002/jbmr.559. DOI: [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Wu CC, Econs MJ, DiMeglio LA, et al. Diagnosis and management of osteopetrosis: consensus guidelines from the osteopetrosis working group. J Clin Endocrinol Metab. 2017;;102((9):):3111––3123.. doi: 10.1210/jc.2017-01127. DOI: [DOI] [PubMed] [Google Scholar]
- 8.Tabrizi R, Arabi AM, Arabion HR, et al. Jaw osteomyelitis as a complication in osteopetrosis. J Craniofac Surg. 2010;;21((1):):136––141.. doi: 10.1097/SCS.0b013e3181c46df2. DOI: [DOI] [PubMed] [Google Scholar]
- 9.Spellberg B, Lipsky BA. Systemic antibiotic therapy for chronic osteomyelitis in adults. Clin Infect Dis. 2011;;54((3):):393––407.. doi: 10.1093/cid/cir842. DOI: [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Chen CE, Shih ST, Fu TH, et al. Hyperbaric oxygen therapy in the treatment of chronic refractory osteomyelitis: a preliminary report. Chang Gung Med J. 2003;;26((2):):114––121.. [PubMed] [Google Scholar]